
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective and metal-free reduction of α,β-unsaturated ketones by in situ produced benzeneselenol from O-(tert-butyl) Se-phenyl selenocarbonate†
Andrea
Temperini
 *a,
Marco
Ballarotto
a and
Carlo
Siciliano
b
*a,
Marco
Ballarotto
a and
Carlo
Siciliano
b
aDipartimento di Scienze Farmaceutiche, Università di Perugia, Consorzio C.I.N.M.P.I.S., Via del Liceo 1, 06123 Perugia, Italy. E-mail: andrea.temperini@unipg.it
bDipartimento di Farmacia e Scienze della Salute e della Nutrizione, Università della Calabria, Edificio Polifunzionale, 87030 Arcavacata di Rende, Cosenza, Italy
First published on 11th September 2020
Abstract
The carbon–carbon double bond of arylidene acetones and chalcones can be selectively reduced with benzeneselenol generated in situ by reacting O-(tert-butyl) Se-phenyl selenocarbonate with hydrochloric acid in ethanol. This mild, metal-free and experimentally simple reduction procedure displays considerable functional-group compatibility, products are obtained in good to excellent yields, and the use of toxic Se/CO mixture and NaSeH, or the smelly and air-sensitive benzeneselenol, is avoided.
Introduction
Organoselenium compounds have attracted considerable interest as intermediates and reagents in organic synthesis during the past years.1 Among these reagents, benzeneselenol was utilized for the reductive cleavage of nitrogen-containing functional groups such as nitro, azo and hydrazino groups,2 giving the corresponding amino group, or for the reductive amination of aldehydes,3 and the reduction of aromatic aldehydes to the corresponding alcohols.4Moreover, examples of reduction of the C–C double bond of α,β-unsaturated carbonyl compounds (Scheme 1(i)) by benzeneselenol under photochemical5 or oxygen-induced activation6 have seldom been reported. Besides, poisonous hydrogen selenide generated from Se/CO/H2O7 and Se/NaBH4 mixtures8 have been used for the selective reduction of the olefinic linkage of α,β-unsaturated ketones, esters, and acids. More recently reduction of chalcones with 3 equiv. of H2Se produced from the reaction of elemental selenium, DMF and a base at 150 °C in the presence of water has been also proposed9 (Scheme 1(ii)). These reduction systems possess limitation due to the use of poisonous elemental selenium and CO, the formation of highly toxic hydrogen selenide, handling of smelly and sensitive reagents, no selenium recovery, harsh reaction conditions as well as low chemoselectivity.
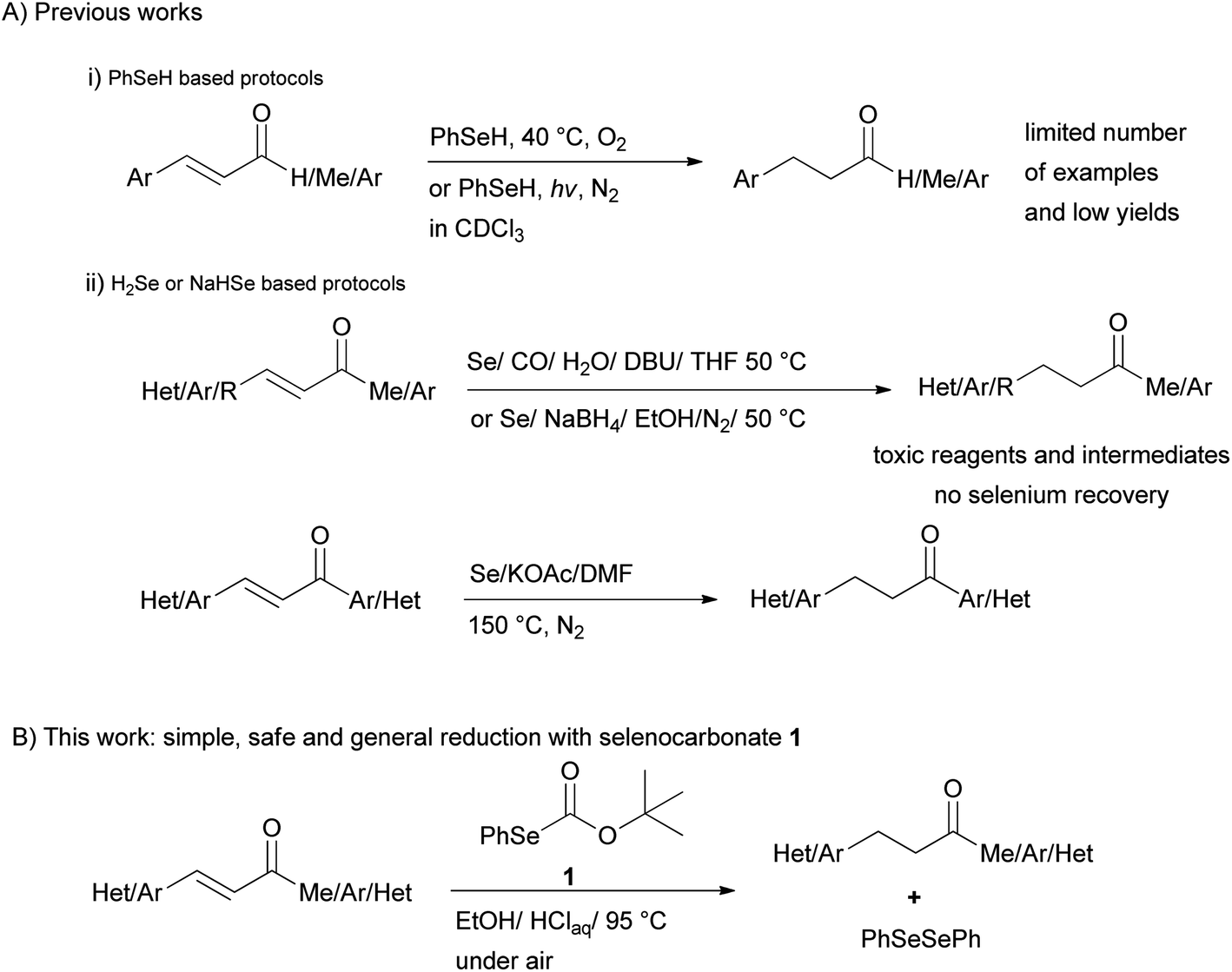 | ||
| Scheme 1 Methods for the selenium mediated selective reduction of enones. | ||
Other ways to reduce the carbon–carbon double bond of α,β-unsaturated ketones are known and most of them have their own merits and demerits. Transition-metal catalyzed transfer hydrogenation,10 metal-catalyzed hydrogenation,11 dissolving metal reduction,12 use of hydrides13 and metal-free reductive processes14 are methods for selective reduction of the C–C double bond. Notwithstanding these methods suffer from their limitation as handling of sensitive reagents or catalysts, relatively expensive metal complexes, long reaction time, poor yields, and metal contamination of the products. Thus a simple, mild, efficient, and metal-free approach to the 1,4-chemoselective reduction of α,β-unsaturated ketones is still challenging.
For years, we have been engaged in the selenofunctionalization of organic molecules15 and the development of new synthetic methodologies involving organoselenium species.16 In continuation of our research on the preparation of potentially useful selenium derivatives, we recently described17 the use of O-(tert-butyl) Se-phenyl selenocarbonate 1 as a safer, practical, and efficient reagent enabling the “in situ” generation of benzeneselenol under advantageous transition-metal free conditions. Furthermore, the nucleophilic benzeneselenol thus obtained can be trapped with different electrophiles, affording alkyl phenyl selenides, β-hydroxyselenides, β-phenylseleno carbonyl derivatives, and vinyl selenides (Scheme 2).
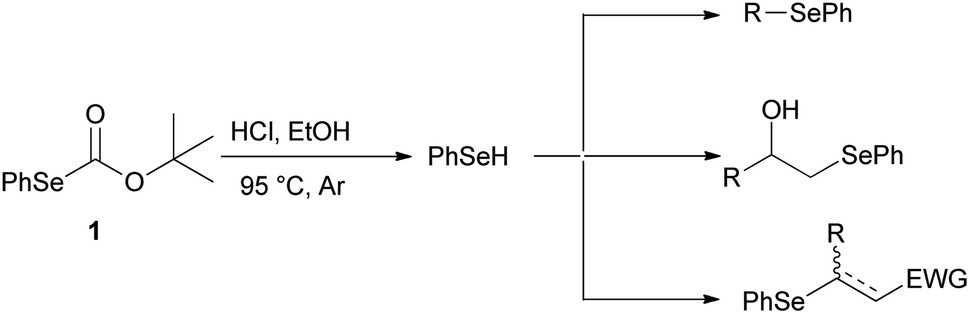 | ||
| Scheme 2 Alkyl phenyl selenides, β-hydroxyselenides, β-phenylseleno carbonyl derivatives and vinyl selenides from O-(tert-butyl) Se-phenyl selenocarbonate (1). | ||
Results and discussion
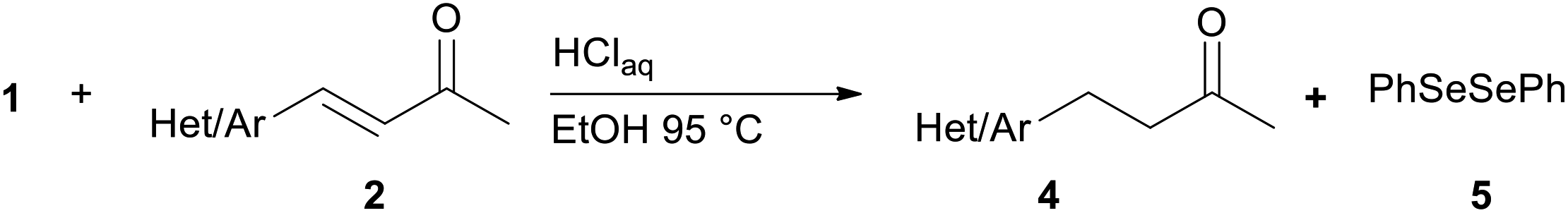
We report a facile and selective reduction of the C–C double bond of arylidene acetones and chalcones by using benzeneselenol prepared via the acidic decomposition of O-(tert-butyl) Se-phenyl selenocarbonate 1 (Scheme 1B). Previous results from our laboratory17 reported on the facile 1,4-addition of benzeneselenol to 1-phenyl-but-3-en-2-one, methyl vinyl ketone, and some alkylidene acetones with 1 in ethanol, at 95 °C, and under an inert atmosphere. Thus we initially exploited the reaction of benzylidene acetone 2a under these standard conditions. The predominant formation of the 1,4-addition product 3a (33% yield) together with minor amounts of the reduced product 4a (15% yield), was observed (Scheme 3). Unreacted 2a was also isolated from the reaction mixture. Modification of the stoichiometric amounts of selenocarbonate 1 (e.g. from 1.2 to 2 equiv.) resulted in only a mild enhancement in the yield of product 4a. Similar results were previously observed for the 1,4-addition of pure benzeneselenol to 2a, chalcone, and a β,β-dimethyl enone at room temperature and under air atmosphere.18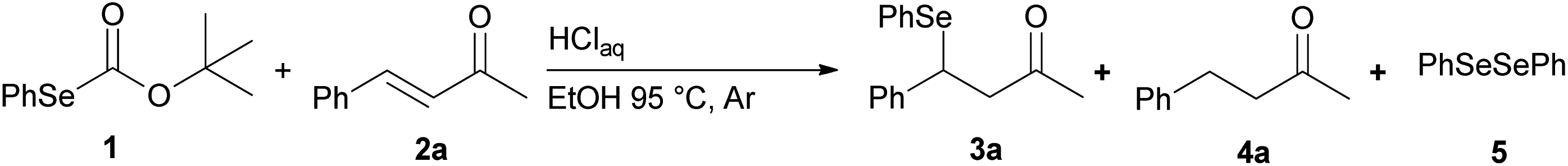 | ||
| Scheme 3 Preliminary experiments. | ||
When the reaction was performed under air (closed glassware apparatus) the reduction was not complete, and 4a was obtained as the major product in a 28% yield. This result was supposed due to the fact that the benzeneselenol intermediate, produced during the decomposition of 1, might further be oxidized to diphenyl diselenide 5. To our delight, when the reaction was carried out in the presence of 3 equiv. of selenocarbonate 1, in no degassed ethanol and under air, compound 4a was obtained in a maximal 48% yield.
To investigate the substrate scope of this carbon–carbon double bond selective reduction of β-aryl substituted enones, we first tested various (hetero)arylidene acetones under optimized conditions (Table 1). Good to excellent yields in the respective products were observed when electron-donating (Table 1, entries 1–3), electron-withdrawing (Table 1, entries 5 and 6), or both kinds of substituents (Table 1, entry 4) were placed on the aromatic ring of the arylidene acetone.
|
|
||||
|---|---|---|---|---|
| Entry | Enones 2 | Time (h) | Products 4 | Yielda (%) |
| a Reaction scale: enone 2 (0.3 mmol), selenocarbonate 1 (0.9 mmol), 35% HCl (6 equiv.) in technical grade ethanol, 95 °C under air atmosphere. | ||||
| 1 | 2b | 9 |
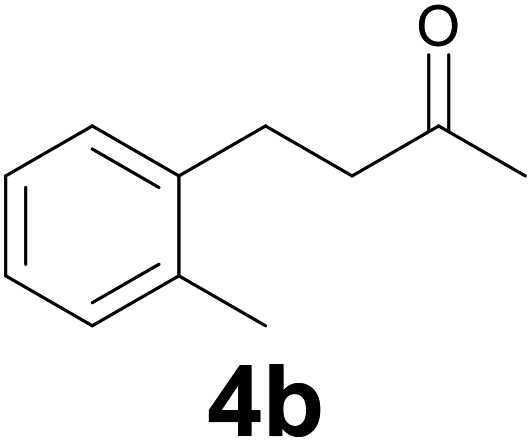
|
77 |
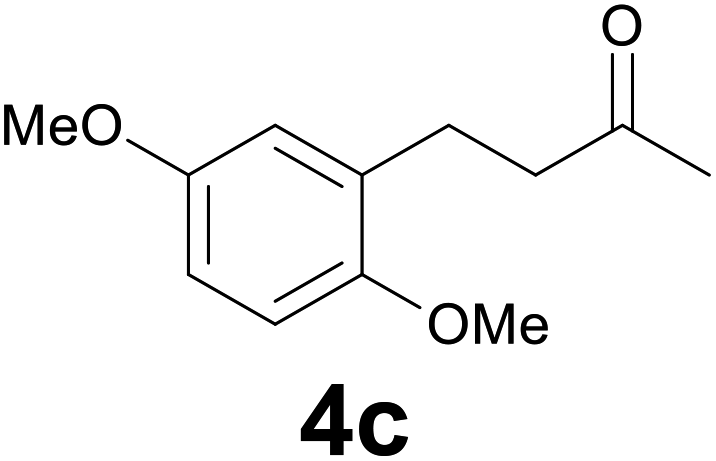
| 2 | 2c | 9 |
|
89 |
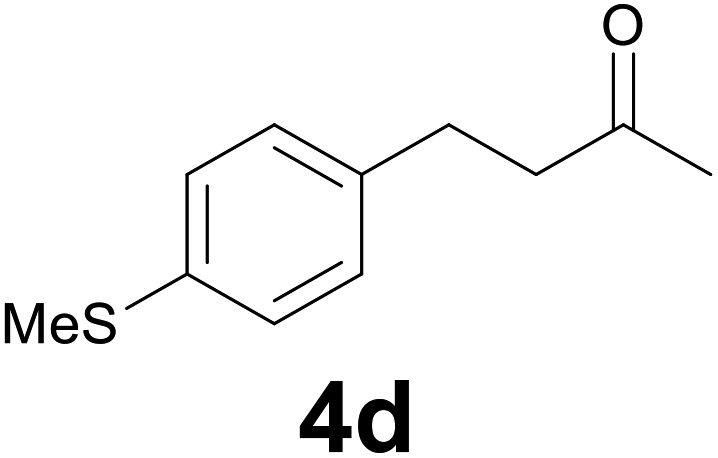
| 3 | 2d | 7 |
|
93 |
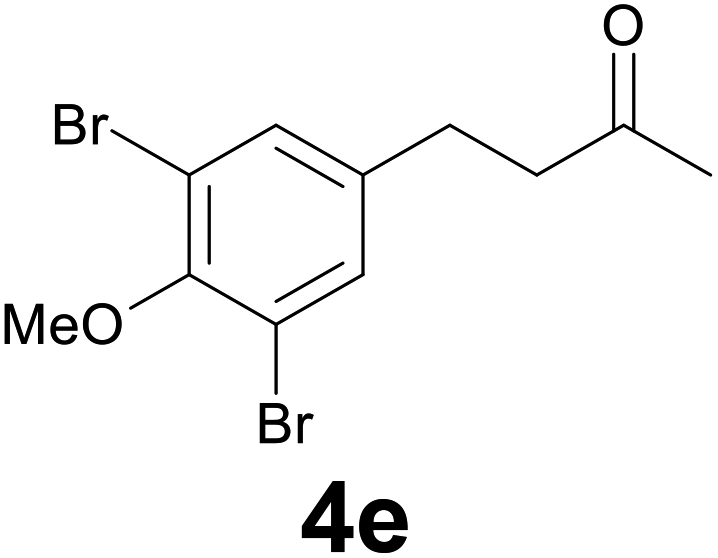
| 4 | 2e | 8 |
|
79 |
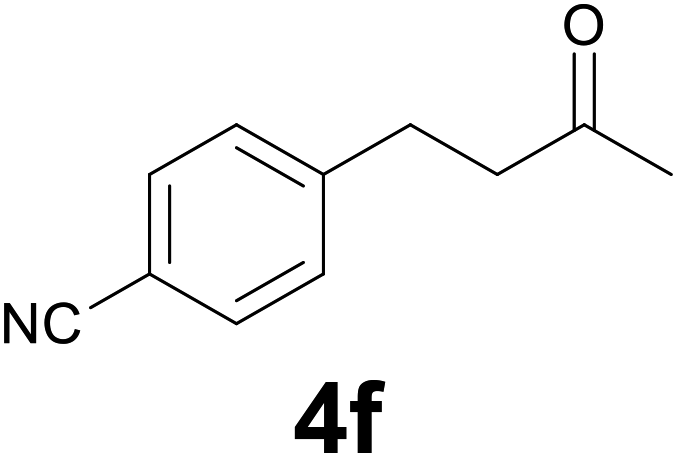
| 5 | 2f | 8 |
|
90 |
| 6 | 2g | 10 |
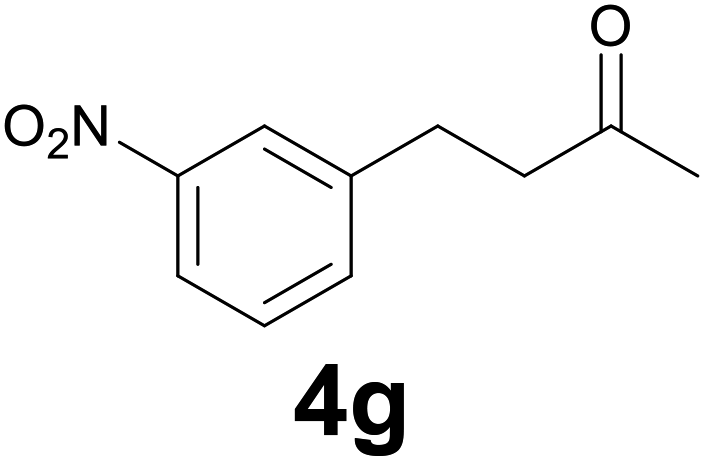
|
78 |
| 7 | 2h | 8 |
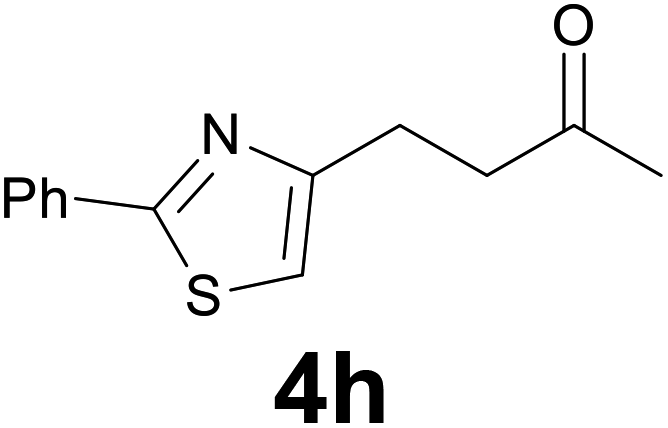
|
86 |
| 8 | 2i | 8 |
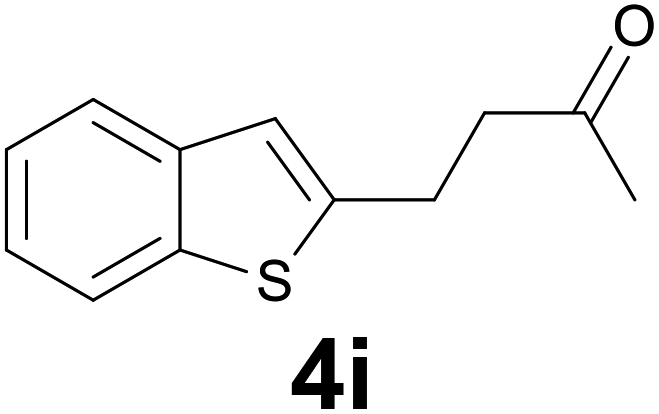
|
85 |
| 9 | 2j | 7 |
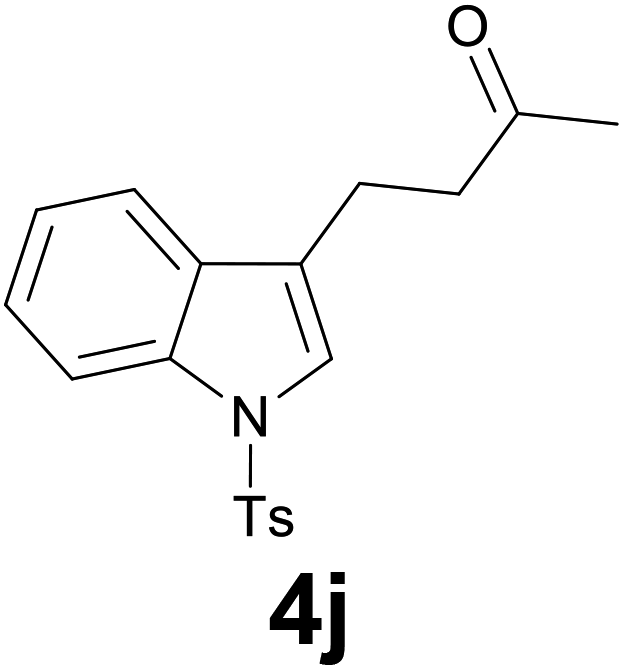
|
91 |
| 10 | 2k | 6 |
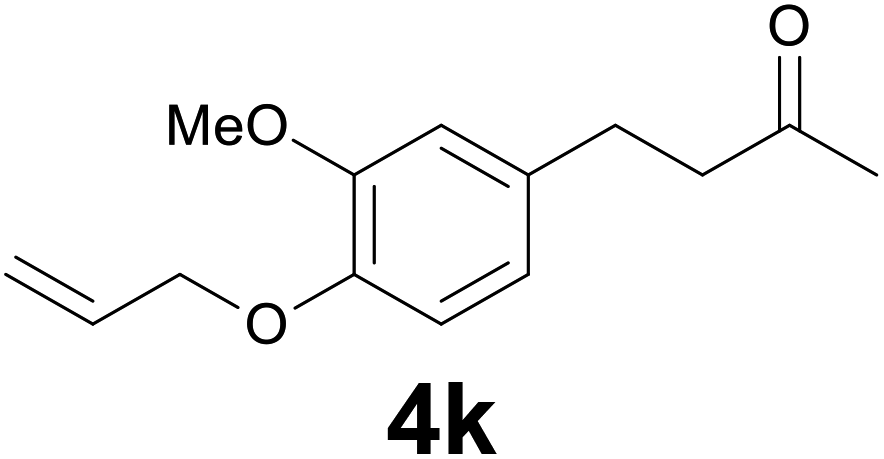
|
78 |
| 11 | 2l | 6 |
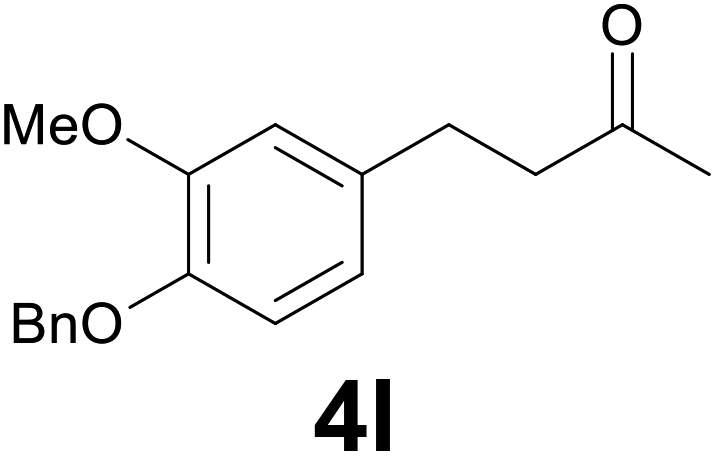
|
76 |
| 12 | 2m | 8 |
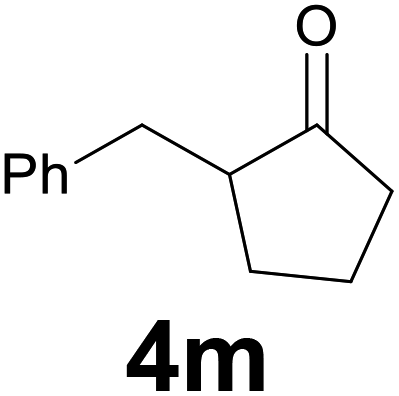
|
66 |
Satisfying yields and selectivity were observed starting from heteroarylidene acetones (Table 1, entries 7–9). Thiazole, benzothiophene, and indole containing enones gave the expected reduced products with total selectivity and in excellent yields. It should be underlined that our protocol resulted to be compatible with the presence of a methoxy group in the precursors, unlikely from the previously reported method,14c in which the demethylation products are unwantedly formed during the reduction process. Moreover, methylthio, cyano, nitro, and tosyl groups on the aromatic ring displayed high tolerance to the reaction conditions (Table 1, entries 3, 5, 6 and 9). Moreover, it was remarkable to observe that no cleavage of the allyl- and benzyl ether protecting groups occurred (Table 1, entries 10 and 11). The presence of an alkyl group at the C–C double bond α-position, as in the cyclic unsaturated ketone 2f, resulted in a slight decrease in yield, proving that steric effects on the α-carbon might negatively influence this transformation.
We further desired to check the performance and robustness of our method in the simple two-step synthesis of 4-(4-hydroxy-3-methoxyphenyl)-2-butanone 7 (zingerone),19 the spicy principle of ginger (Scheme 4). To fulfill our aim, this objective vanillin was condensed with acetone to afford the vanillylidenacetone 6 intermediate in 89% yield.20 The chemoselective reduction of 6 with 1 under the optimized conditions, furnished zingerone 7 in 80% excellent yield.
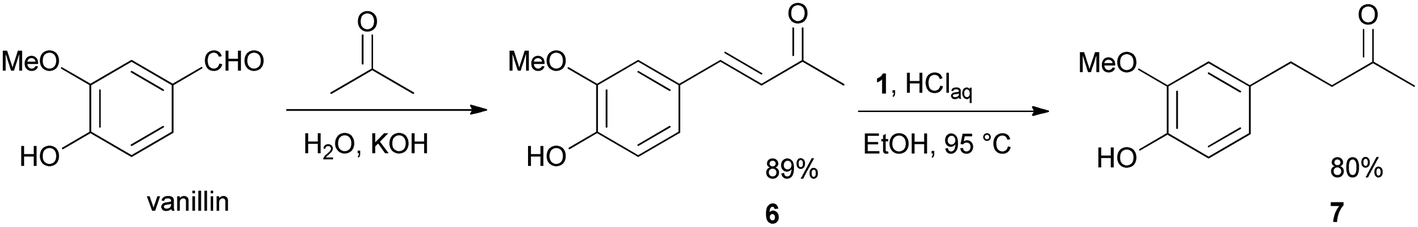 | ||
| Scheme 4 The two steps synthesis of zingerone (7) from vanillin. | ||
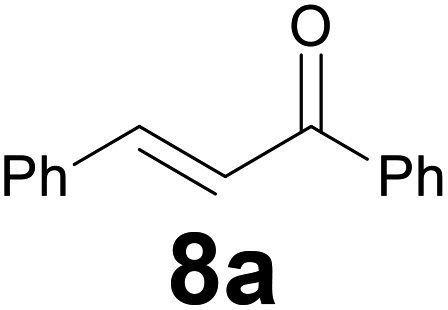
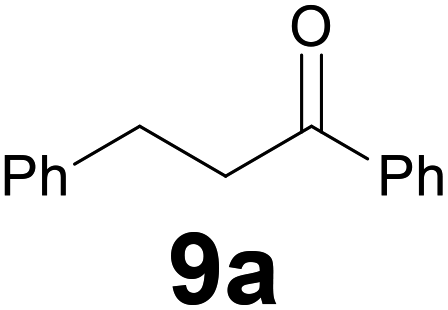
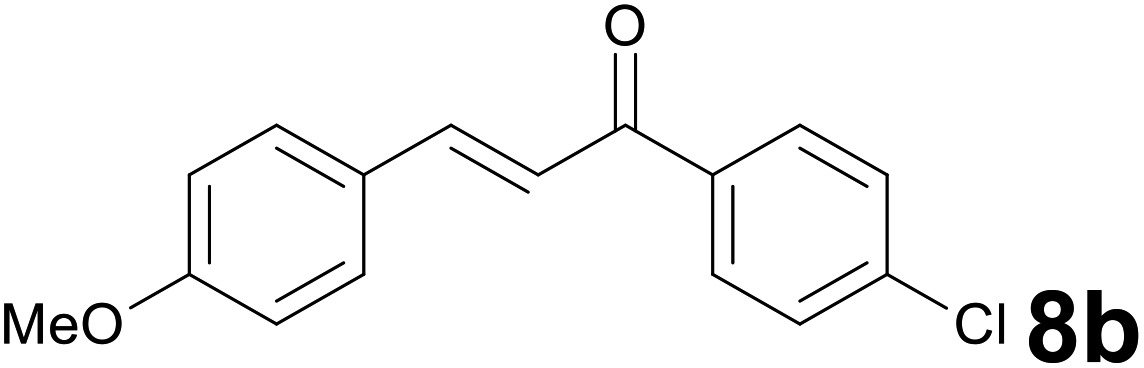
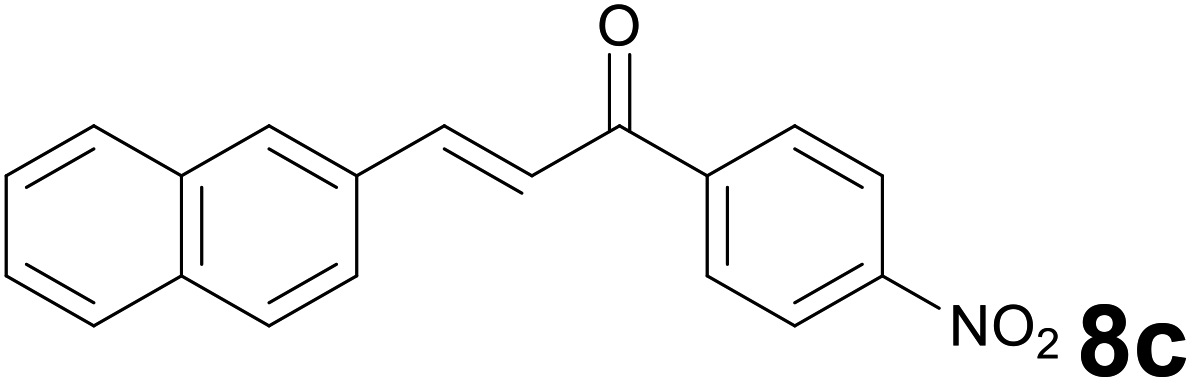
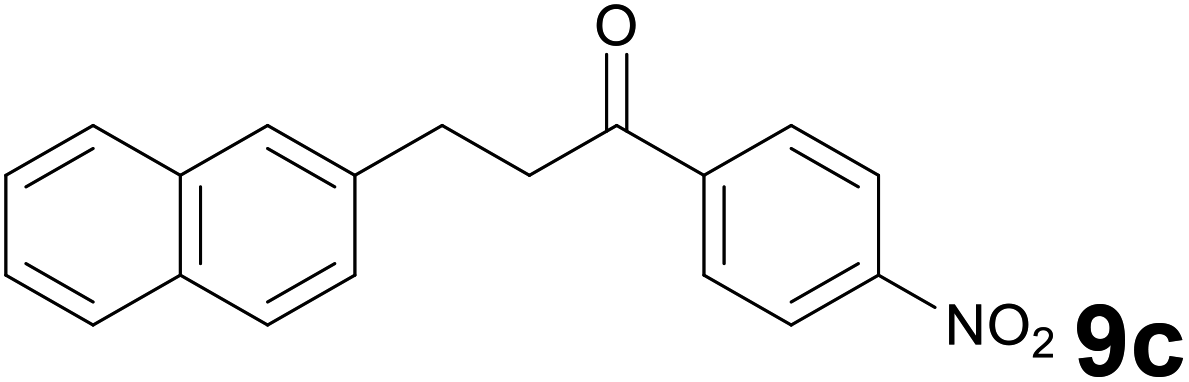
Successively, we verified the versatility of the presented method by subjecting to the reduction of the C–C double bond various chalcones (Table 2). Reduction of chalcones successfully afforded the corresponding 1,3-diarylpropan-1-ones, which were obtained in good to excellent yields. Both electron-donating or -withdrawing substituents on the phenyl, or heteroaryl ring, had no significant influence on the yield. Substrates with thiophene, pyrazole, and benzothiazole rings were also suitable under the employed reaction conditions. Our protocol showed high chemoselectivity, as established when the carbon–carbon double bond in substrate 8c was reduced selectivity in the presence of nitro group (Table 2, entry 3), unlikely the already reported reduction of p-nitrotoluene to p-toluidine with benzeneselenol.2
|
|
||||
|---|---|---|---|---|
| Entry | Chalcones 8 | Time (h) | Products 9 | Yielda (%) |
| a Reaction scale: chalcone 8 (0.2 mmol), selenocarbonate 1 (0.6 mmol), 35% HCl (6 equiv.) in technical grade ethanol, 95 °C under air atmosphere. b The corresponding deacetylated product was isolated. c A gram scale reaction with 2.5 mmol of 8g was also carried out. d Selenocarbonate 1 (1.2 mmol) and 35% HCl (12 equiv.) were employed. | ||||
| 1 |
|
3.5 |
|
79 |
| 2 |
|
8.5 |
|
96 |
| 3 |
|
3 |
|
77 |
| 4 |
|
3.5 |
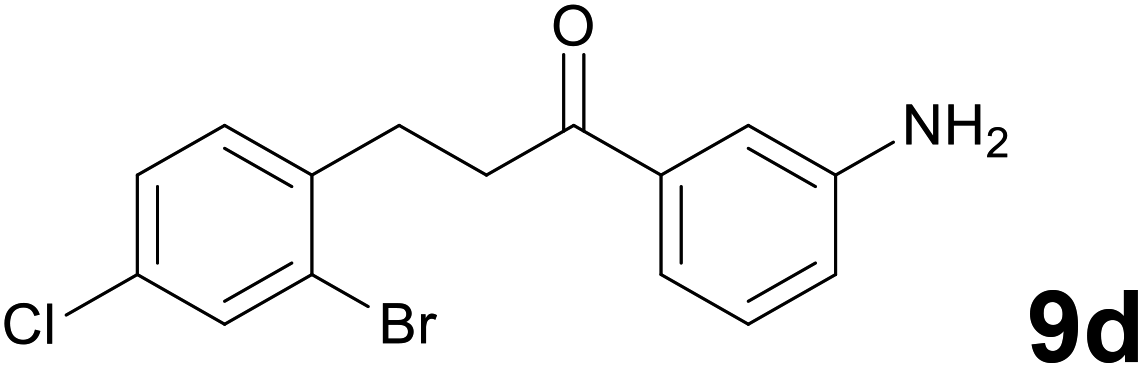
|
63b |
| 5 |
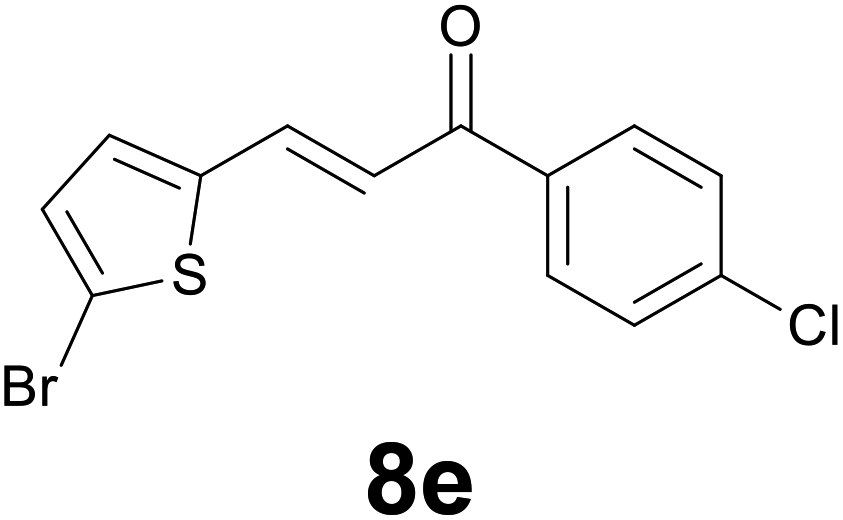
|
9 |
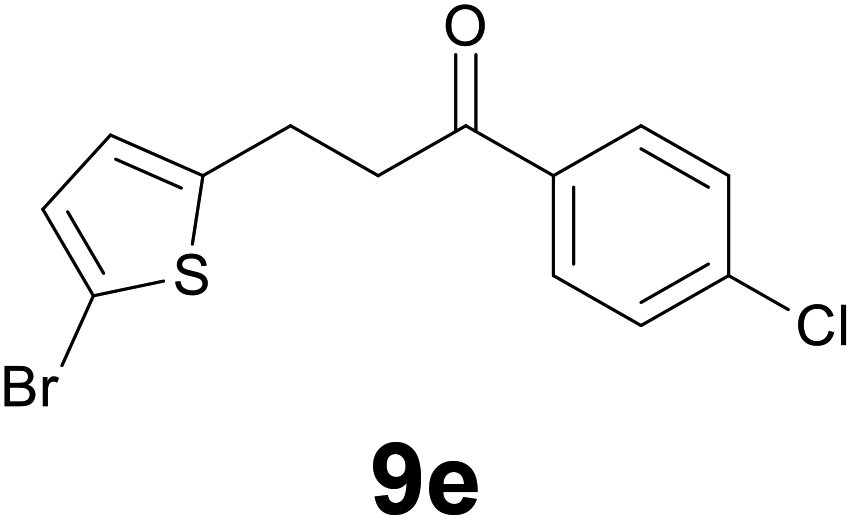
|
72 |
| 6 |
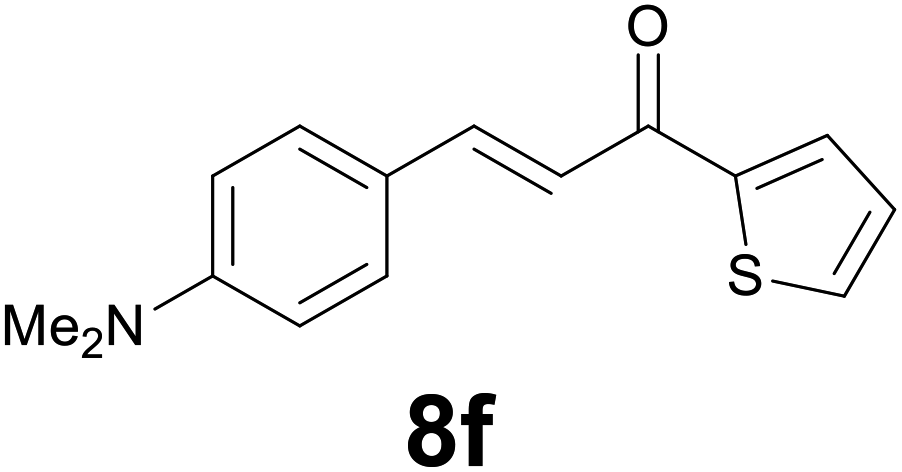
|
3 |
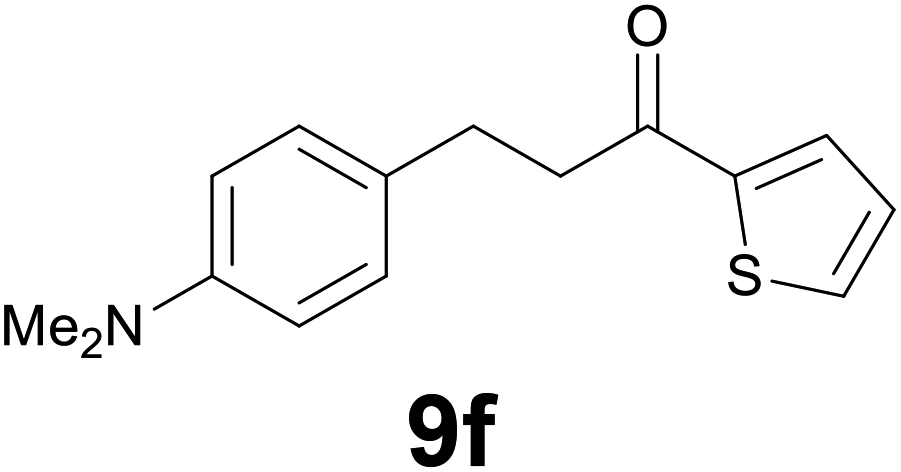
|
86 |
| 7 |
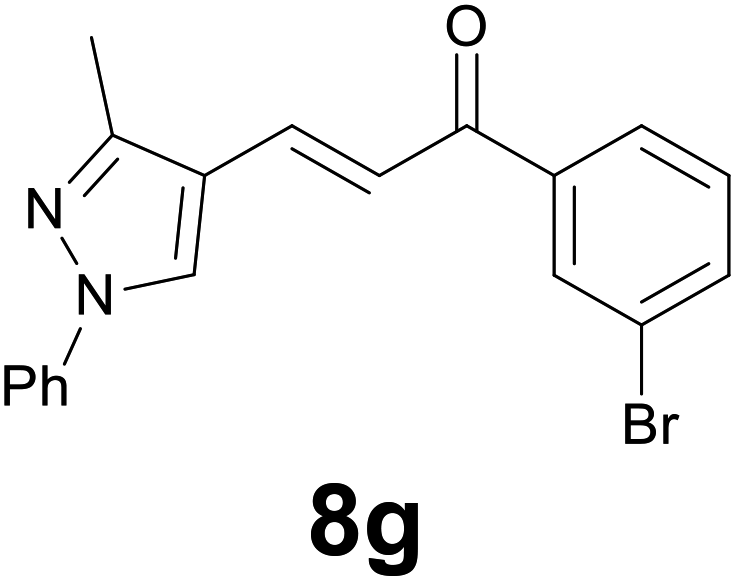
|
3.5 |
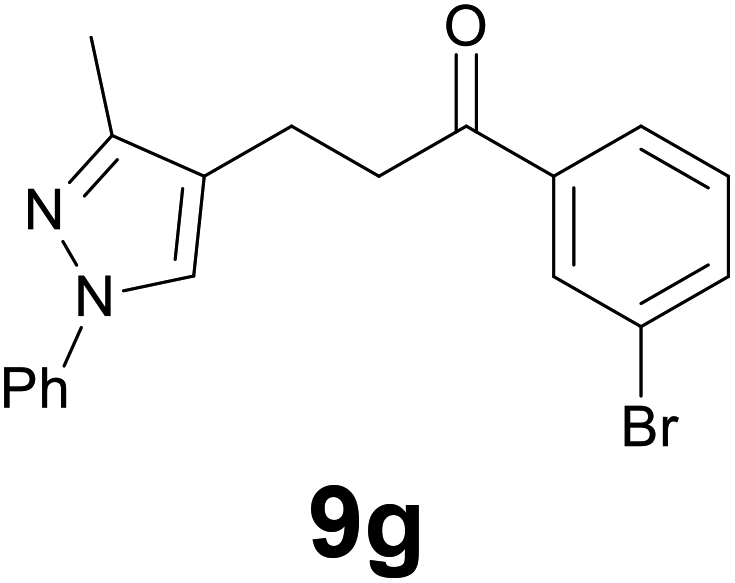
|
89c |
| 8 |
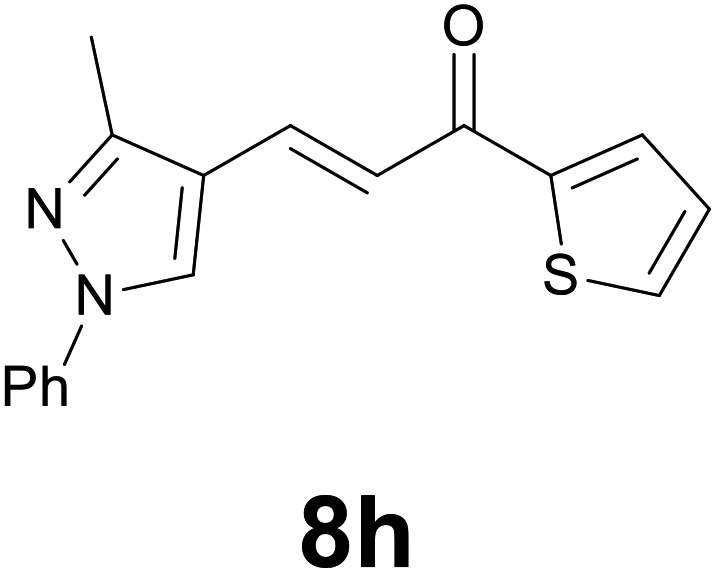
|
3 |
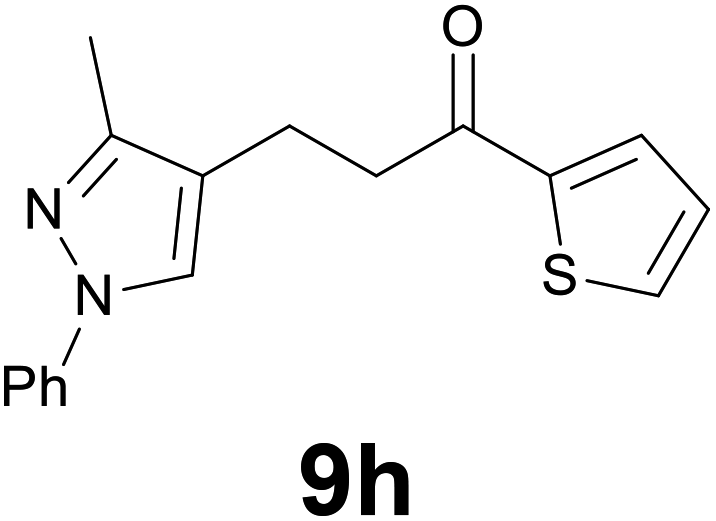
|
88 |
| 9 |
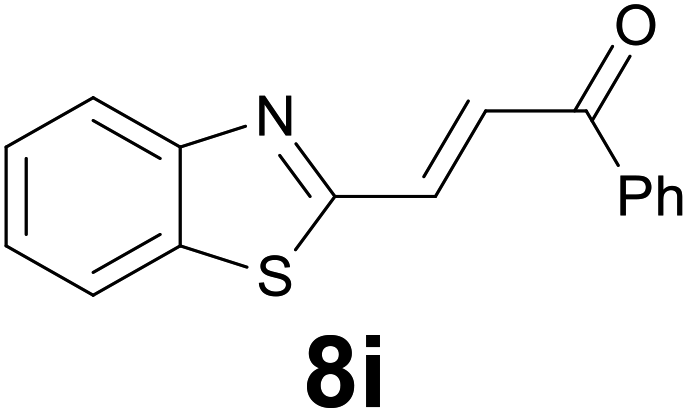
|
3 |
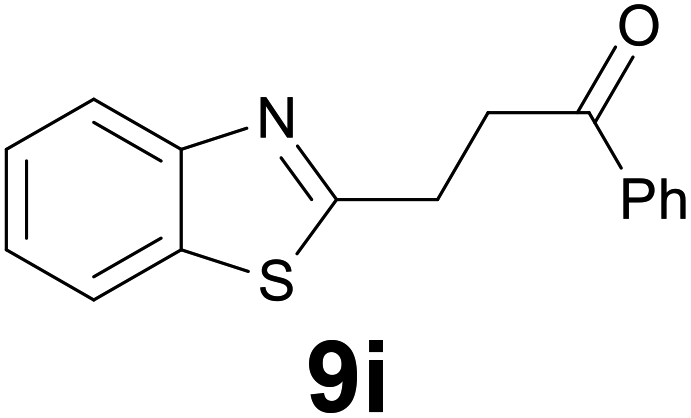
|
92 |
| 10 |
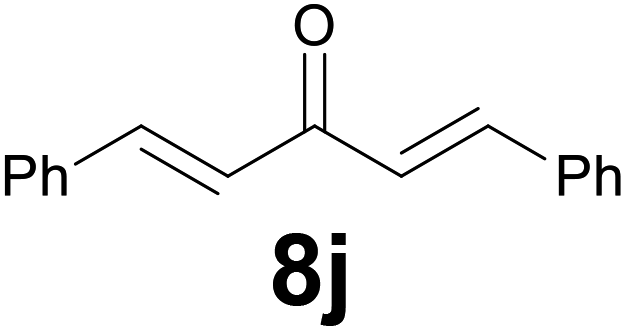
|
9 |
|
74d |
| 11 |
|
9 |
|
53 |
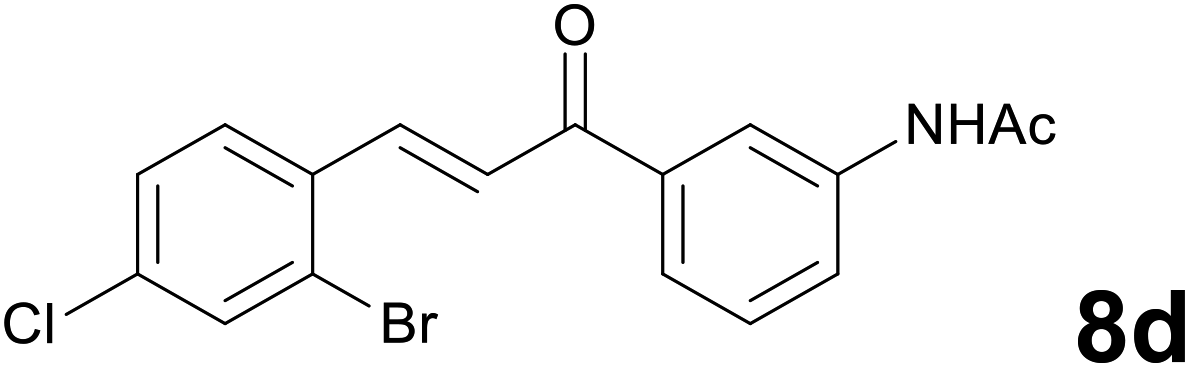
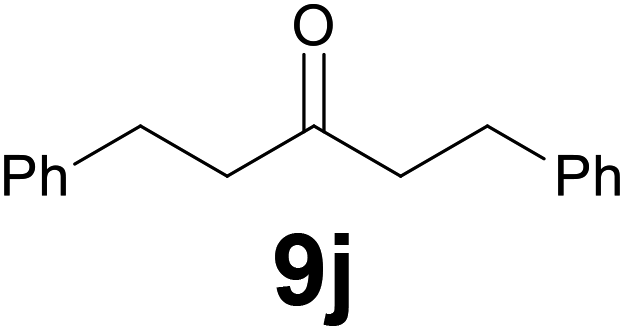
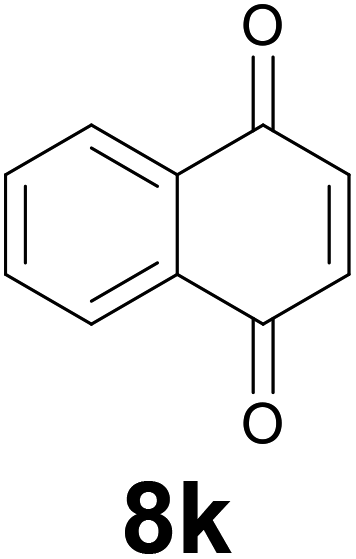
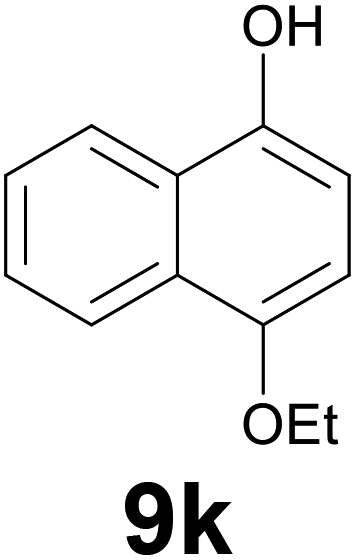
Noteworthy, in the case of chalcone 8d bearing the N-acetyl group, the reduction was accompanied by hydrolysis of the amide function to afford the corresponding aniline derivative 9d (Table 2, entry 4). The N,N-dimethylamino group was also well tolerated (Table 2, entry 6), although the reaction proceeded slower, and the addition of selenocarbonate 1, in four discrete portions, was necessary to obtain product 9f in excellent yield. Moreover, the reduction of dibenzylidene acetone 8j required the use of 6 equiv. of 1 to yield the corresponding saturated ketone 9j in good amounts. Another noteworthy result was obtained by treating the ene-dione 1,4-naphthoquinone 8k with selenocarbonate 1. The expected reduced product 1,4-naphthalenediol was not isolated. In its place, the 1-ethoxy-4-hydroxynaphthalene derivative 9k was obtained in an appreciable yield. Most probably the initially formed 1,4-naphthalenediol reacts with ethanol under acidic conditions to give 9k as previously reported by Laatsh.21
Finally, we checked the reduction procedure on a gram scale. In that case, the reaction was conducted, as usual, using 7.5 mmol of the reagent 1 and 2.5 mmol of the chalcone 8g obtaining the expected ketone 9g in an 82% yield. Moreover, although it was necessary to use 3 equiv. of 1, almost all of the phenylseleno unit was recovered as diphenyl diselenide at the end of the process (1.05 g, 89% yield), and recycled to prepare compound 1, demonstrating the atom economy of the procedure. We also tested the possibility to run the reduction of an arylidenacetone in the presence of an α,β-unsaturated ester moiety, given the known difficulty preparation of an enone featuring the two above mentioned functional groups. Thus, an equimolar amount (0.2 mmol) of enone 2c and ethyl cinnamate was reacted with 3 equiv of 1 under the standard conditions (Scheme 5).
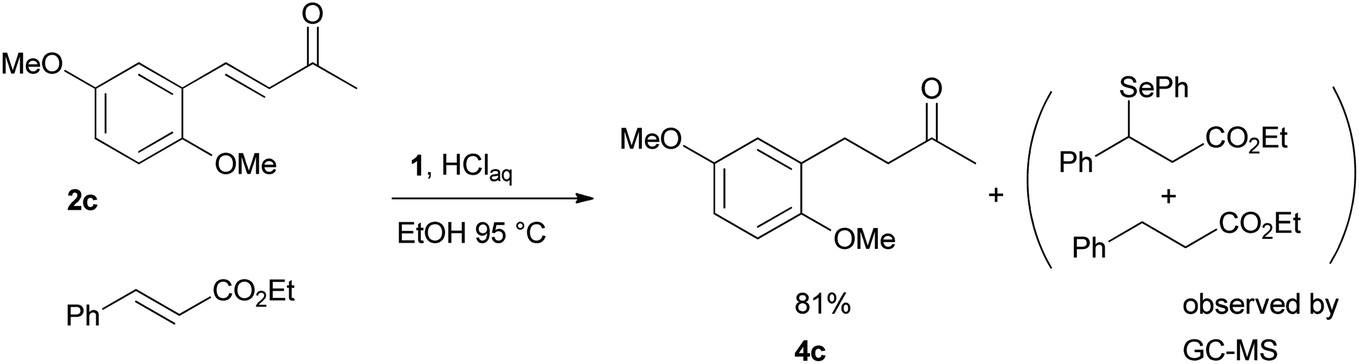 | ||
| Scheme 5 Competitive reduction of the C–C double bond of α,β-unsaturated carbonyl compounds. | ||
After 7 hours the reaction outcome was analyzed using GC-MS. The result indicated that the enone 2c was completely consumed to give the reduced product 4c whereas the ethyl cinnamate remained unchanged; only negligible amounts of 1,4-phenylseleno adduct to ethyl cinnamate and reduced ethyl cinnamate were observed by GC-MS analysis. Ketone 4c was then isolated in 81% yield confirming the mildness and selectivity of the proposed method.
A plausible mechanism of this oxygen-induced reduction of arylidenacetones and chalcones with benzeneselenol generated from 1 suggests the reaction to proceed via a free radical pathway in which the benzeneselenenyl radical might be formed in the reaction system by the reaction of benzeneselenol with oxygen accordingly to the mechanism previously described by Sonoda6 for similar substrates working at 40 °C in 1,2,3,4-tetrahydronaphthalene.
The role played by a benzeneselenenyl radical in the reduction process was supported by the reaction of methyl propiolate 13 with 1, in methanol under the proposed reaction conditions (Scheme 6). A 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (E/Z) mixture of bis-selenides 15 and 16 contaminated by methyl β-phenylselenoacrylate 17 was isolated in a good amount. Bis-selenides 15 and 16 were firstly obtained by Back22 through the photolysis of diphenyl diselenide, carried out in the presence of methyl propiolate, via a free-radical chain addition mechanism of benzeneselenenyl radical.
:9 (E/Z) mixture of bis-selenides 15 and 16 contaminated by methyl β-phenylselenoacrylate 17 was isolated in a good amount. Bis-selenides 15 and 16 were firstly obtained by Back22 through the photolysis of diphenyl diselenide, carried out in the presence of methyl propiolate, via a free-radical chain addition mechanism of benzeneselenenyl radical.
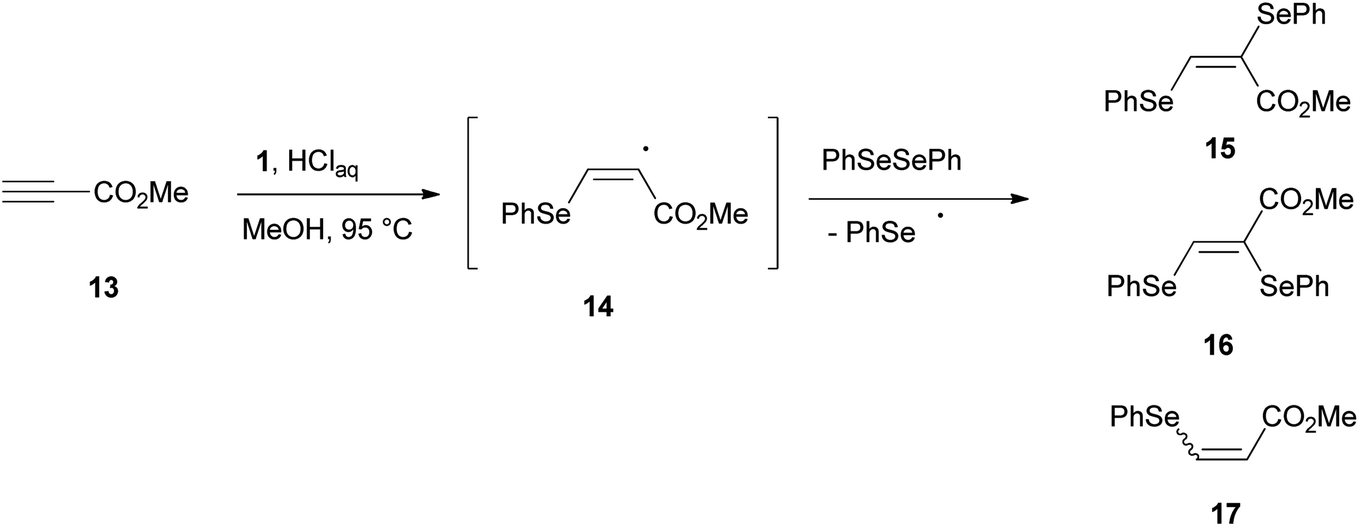 | ||
| Scheme 6 Supporting benzeneselenenyl radical mediated mechanism experiment. | ||
Most likely, the benzeneselenenyl radical, formed under our reaction conditions, gives addition to the triple bond of 13, resulting in the formation of the alkenyl radical intermediate 14 (Scheme 6). This intermediate undergoes chain transfer to a molecule of diphenyl diselenide, formed in the reaction mixture by oxidation of benzeneselenol with oxygen, to afford the corresponding (E)-bis-selenide 15. The formation of 16 suggests that the intermediate vinyl radical 14 is able to equilibrate prior to the chain-transfer step. Moreover, if the radical intermediate 14 abstracts an hydrogen atom from benzeneselenol, the β-phenylseleno derivative 17 can be formed.
Conclusions
A mild, efficient, and high chemoselective C–C double bond reduction of (hetero)arylidene acetones and chalcones with O-(tert-butyl) Se-phenyl selenocarbonate 1 as the precursor of benzeneselenol was established. At the same time, this safer procedure uses ethanol as an environmentally friendly solvent, and successfully works at moderate temperature (95 °C), in the presence of air. This method showed high functional group tolerance, and a wide substrate scope representing a valuable choice in alternative to the known traditional methods.Experimental section
General information
Reactions were performed under atmosphere. Compounds purification by column chromatography was executed on silica gel (Merck 60, 70–230 mesh), and analytical TLC was carried out on pre-coated silica gel plates (Merck 60 F254, 0.25 mm) using UV light and 0.5% w/v KMnO4 aqueous solution (followed by gentle heating) for visualisation. Melting points were measured on a hot plate apparatus and are uncorrected. Proton magnetic resonance (1H-NMR) spectra were recorded at 200 and 400 MHz. Proton-decoupled carbon magnetic resonance (13C-NMR) spectra were recorded at 50.3 and 100.6 MHz. Chemical shifts (δ) are reported in parts per million (ppm). The NMR spectra were calibrated using the proton or carbon signals of residual non-deuterated solvent peaks: δH 7.27 and δC77.0 for CDCl3. Infrared (IR) spectra were recorded with a FT-IR instrument, using a diamond ATR sample interface. Absorption maxima (νmax) are reported in wavenumbers (cm−1). GC-MS analysis were obtained with a gas chromatograph (HP-5MS capillary column 29.0 m, ID 0.25, film 0.25 μm) equipped with a mass selective detector at an ionising voltage of 70 eV. Combustion analyses were carried out on an elemental analyser. IUPAC names of compounds were generated with ACD/Name. When the E and Z isomers were not separated (compounds 15, 16 and 17), the relative spectra were assigned by comparison with previously reported data. The reported yields were calculated based on the amount of isolated products. Unless otherwise noted, all reagents and solvents were obtained commercially and used without further purification. Compound 1 was synthesized in gram-scale by the method described in the literature.17,23 Enones 2b–2e were synthesized by aldol condensation of the corresponding aldehyde with acetone, in accord with previously reported procedures.24 The enones 2f–j were prepared by Wittig reaction of the corresponding aldehydes with 1-(triphenylphosphoranylidene)propan-2-one.25 The remaining α,β-unsaturated ketones 2k,262l,272m,288b,298c,308e,318f,32 and 8h33 were prepared according to literature procedures.Preparation of ketones 8d and 8g (general procedure)
To an ethanolic solution (30 mL) of substituted aryl methyl ketone (2.2 mmol) and substituted aromatic aldehyde (2 mmol), 8.0 M aqueous potassium hydroxide (1 mL) was added dropwise with stirring at room temperature. After completion of the reaction (48 h), 2.0 M aqueous hydrochloric acid (5 mL) was added and the crude product was isolated by filtration and washed with water, dried, and purified by chromatography using a petroleum ether–dichloromethane mixture (1:9) as eluant.
Preparation of ketone 8i
A mixture of benzothiazole-2-carboxaldehyde (0.49 g, 3 mmol) and acetophenone (0.36 g, 3.3 mmol) was dissolved in 50% aqueous sulfuric acid (25 mL) and then heated at 50 °C (oil bath). After 48 h the mixture was cooling down and water (50 mL) was added. The mixture was extracted three times with 20 mL of CH2Cl2. The combined extracts were washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. The crude mixture was purified by column chromatography on silica gel with a mixture of petroleum ether–dichloromethane mixture (2:8) as eluant.
Preliminary experiment for the reduction of 2a
Reaction conditions described in previous work17 have been used. Thus aqueous 37% hydrochloric acid (0.1 mL, 1.2 mmol) was added to a solution of 1 (0.16 g, 0.6 mmol) and the α,β-unsaturated ketone 2a (0.5 mmol) in 8 mL of degassed ethanol. The mixture was heated at 95 °C (oil bath) under argon atmosphere until the selenocarbonate 1 disappeared on TLC monitoring (5 h). After the addition of water (10 mL) the mixture was extracted three times with 10 mL of EtOAc. The combined extracts were washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. The crude mixture was purified by column chromatography on silica gel with a 95:5 mixture of petroleum ether and ethyl ether as eluant to give the corresponding β-phenylseleno adduct 3a and the reduced ketone 4a.
General procedure for chemoselective reduction of (hetero)arylidene acetones 2a–o
A 25 mL round-bottom flask equipped with a stir bar and a condenser was charged with α,β-unsaturated ketone 2 (0.3 mmol), selenocarbonate 1 (0.24 g, 0.9 mmol) and ethanol (6 mL). The open end of the condenser was fitted with a rubber septum and then aqueous 37% hydrochloric acid (0.15 mL, 1.8 mmol) was added to the flask. The reaction mixture was stirred at 95 °C (oil bath) and the progress of the reaction was monitored by TLC (3–10 h). After cooling down, a saturated aqueous sodium hydrogencarbonate solution was carefully added dropwise (for ketone 4d, saturated aqueous sodium carbonate solution was added). The mixture was extracted with three 10 mL portions of EtOAc. The combined extracts were washed with brine (10 mL), dried (MgSO4), filtrated and concentrated in vacuo. The residue was then purified by flash chromatography on silica gel to provide the corresponding reduced product 4.:9) as eluent. Yield: 50 mg (77%); 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.18–7.08 (m, 4H), 2.96–2.82 (m, 2H), 2.78–2.65 (m, 2H), 2.32 (s, 3H), 2.18 (s, 3H).
:6) as eluent to give 4c. Yield: 56 mg (89%); colourless oil. 1H NMR (200 MHz, CDCl3) δ (ppm) 6.82–6.63 (m, 3H), 3.77 (s, 3H), 3.75 (s, 3H), 2.94–2.80 (m, 2H), 2.79–2.62 (m, 2H), 2.15 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 208.6, 153.3, 151.6, 130.4, 116.3, 111.2, 111.0, 55.7, 55.6, 43.6, 29.9, 25.1; FTIR νmax/cm−1 2938, 1712, 1498, 1220, 1039, 800, 712; m/z (E/I): 208 (M+, 100%), 165 (31), 151 (51), 121 (24), 91 (13), 77 (15). Found: C, 68.97; H, 7.31. Calc. for C12H16O3: C, 69.21; H, 7.04.
:9) as eluent to give 4d. Yield: 52 mg (93%); 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.18 (d, J = 8.3, Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 2.85 (t, J = 7.5 Hz, 2H), 2.73 (t, J = 7.5 Hz, 2H), 2.46 (s, 3H), 2.13 (s, 3H). m/z (E/I): 194 (M+, 69%), 137 (100), 122 (15), 91 (9).
:9) as eluent to give 4e. Yield: 53 mg (79%); colourless oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.32 (s, 2H), 3.85 (s, 3H), 2.90–2.65 (m, 4H), 2.16 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 206.9, 152.3, 139.8, 132.5 (2C), 117.9 (2C), 60.5, 44.5, 30.0, 28.1; FTIR νmax/cm−1 2928, 1773, 1422, 1259, 993, 737; m/z (E/I): 336 (M+, 100%), 278 (87), 266 (18), 214 (42), 185 (11), 118 (11). Found: C, 39.37; H, 3.84. Calc. for C11H12Br2O2: C, 39.32; H, 3.60.
:6) as eluent to give 4f. Yield: 47 mg (90%); light yellow solid, mp 44–46 °C. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.55 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 3.04–2.86 (m, 2H), 2.85–2.68 (m, 2H), 2.15 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 206.8, 146.7, 132.1 (2C), 129.1 (2C), 118.9, 109.8, 44.0, 29.9, 29.4; FTIR νmax/cm−1 2962, 2228, 1708, 1376, 1167, 827, 560; m/z (E/I): 173 (M+, 100%), 130 (96), 116 (51), 103 (32), 89 (21), 77 (19). Found: C, 76.01; H, 6.60; N, 7.97. Calc. for C11H11NO: C, 76.28; H, 6.40; N, 8.09.
:6) as eluent to give 4g. Yield: 60 mg (78%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 8.13–7.95 (m, 2H), 7.62–7.3 (m, 2H), 3.06–2.92 (m, 2H), 2.90–2.78 (m, 2H), 2.17 (s, 3H).
:6) as eluent to give 4h. Yield: 40 mg (86%); colourless oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 8.01–7.82 (m, 2H), 7.50–7.37 (m, 3H), 6.92 (s, 1H), 3.28–3.01 (m, 2H), 3.00–2.85 (m, 2H), 2.19 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 207.9, 167.8, 156.6, 133.6, 129.9, 128.9 (2C), 126.4 (2C), 113.7, 42.8, 30.0, 25.5; FTIR νmax/cm−1 2918, 1713, 1356, 1159, 1003, 764, 690; m/z (E/I): 231 (M+, 6%), 188 (100), 158 (12), 78 (20). Found: C, 67.39; H, 5.91; N, 5.90. Calc. for C13H13NOS: C, 67.50; H, 5.66; N, 6.06.
:8) as eluent to give 4i. Yield: 36 mg (85%); colourless oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.77 (dd, J = 6.9, 1.6 Hz, 1H), 7.68 (dd, J = 6.6, 2.1 Hz, 2H), 7.38–7.21 (m, 2H), 7.03 (s, 1H), 3.20 (t, J = 7.2 Hz, 2H), 2.89 (t, J = 7.2 Hz, 2H), 2.19 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 207.0, 144.6, 140.0, 139.2, 124.2, 123.6, 122.8, 122.1, 121.1, 44.5, 30.1, 24.6; FTIR νmax/cm−1 2914, 1712, 1354, 1097, 813, 749, 484; m/z (E/I): 204 (M+, 51%), 161 (100), 147 (59), 128 (20), 115 (12). Found: C, 70.45; H, 5.90. Calc. for C12H12OS: C, 70.55; H, 5.92.
:6) as eluent to give 4j. Yield: 64 mg (91%); colourless oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.98 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.3 Hz, 1H), 7.37–7.13 (m, 5H), 3.01–2.89 (m, 2H), 2.88–2.67 (m, 2H), 2.39 (s, 3H), 2.16 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 207.5, 144.8, 135.2, 135.1, 130.6, 129.8 (2C), 126.7 (2C), 124.7, 123.0, 122.7, 121.9, 119.3, 113.7, 42.6, 38.0, 21.5, 18.7; FTIR νmax/cm−1 3106, 2921, 1706, 1370, 1169, 1129, 752, 702, 607, 534; m/z (E/I): 341 (M+, 100%), 298 (45), 284 (48), 186 (25), 155 (47), 144 (77), 115 (16), 91 (49), 65 (139). Found: C, 66.68; H, 5.87; N, 4.15. Calc. for C19H19NO3S: C, 66.84; H, 5.61; N, 4.10.
:99) as eluent to give 4k. Yield: 54 mg (78%); colourless oil. 1H NMR (200 MHz, CDCl3) δ (ppm) 6.79 (d, J = 8.1 Hz, 1H), 6.71 (d, J = 2.0 Hz, 1H), 6.68 (dd, J = 8.1, 2.0 Hz, 1H), 6.13–6.01 (m, 1H), 5.38 (dq, J = 17.3, 1.5 Hz, 1H), 5.26 (dq, J = 10.5, 1.5 Hz, 1H), 4.58 (dt, J = 5.4, 1.5 Hz, 2H), 3.85 (s, 3H), 2.88–2.79 (m, 2H), 2.78–2.70 (m, 2H), 2.13 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 208.1, 149.3, 146.2, 134.0, 133.4, 119.9, 117.8, 113.5, 111.9, 69.9, 55.8, 45.3, 30.1, 29.3; FTIR νmax/cm−1 2925, 1713, 1512, 1139, 1020, 803; m/z (E/I): 234 (M+, 88%), 193 (100), 161 (25), 151 (15), 133 (61), 119 (79), 102 (21), 91 (36), 77 (21). Found: C, 71.22; H, 7.84. Calc. for C14H18O3: C, 71.37; H, 7.74.
:99) as eluent to give the known36 ketone 4l. Yield: 40 mg (71%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.46–7.40 (m, 2H), 7.39–7.33 (m, 2H), 7.32–7.26 (m, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H), 6.65 (dd, J = 8.1, 2.0 Hz, 1H), 5.13 (s, 2H), 3.88 (s, 3H), 2.87–2.79 (m, 2H), 2.77–2.71 (m, 2H), 2.13 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 208.2, 149.5, 146.4, 137.3, 134.2, 128.5, 127.7, 127.2, 120.0, 114.1, 112.1, 71.1, 55.9, 45.3, 30.1, 29.3.
:97) as eluent to give the commercially available ketone 4m. Yield: 38 mg (66%).
Two steps synthesis of zingerone 7.19
To a solution of vanillin (0.5 g, 3.3 mmol) in 2 mL of acetone was added gradually 2.5 M aqueous sodium hydroxide (2 mL) at room temperature. The mixture was stirred for 48 h and then diluted with 2.0 M aqueous hydrochloric acid (5 mL) and EtOAc (10 mL). The organic and aqueous layers were separated, and the aqueous layer was extracted with EtOAc (3× 20 mL). The combined extracts were washed with brine (10 mL), dried (MgSO4), filtrated and concentrated in vacuo. Purification by silica gel column chromatography using an ethyl ether–hexane mixture (6:4) as eluent afforded the known20 (3E)-4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one (6) intermediate (0.56 g, 89% yield). m/z (E/I): 192 (M+, 92%), 177 (100), 145 (81), 117 (37), 89 (26), 77 (18).
Then compound 6 (0.3 mmol) was reduced as reported above for other arylidene acetones. After 7 h the mixture was cooling down and water (10 mL) was added. The mixture was extracted with three 10 mL portions of EtOAc. The combined extracts were washed with brine (10 mL), dried (MgSO4), filtrated and concentrated in vacuo. The crude ketone was purified by chromatography using a ethyl ether–hexane mixture (1:9) as eluent to furnish the commercially available ketone 4-(4-hydroxy-3-methoxyphenyl)butan-2-one (7). Yield: 47 mg (80%).
General procedure for chemoselective reduction of chalcones 8a–l and naphthoquinone 8m
A 25 mL round-bottom flask equipped with a stir bar and a condenser was charged with chalcone 8 (0.2 mmol), selenocarbonate 1 (0.16 g, 0.6 mmol) and ethanol (4 mL). The condenser was fitted with a rubber septum and then aqueous 37% hydrochloric acid (0.1 mL, 1.2 mmol) was added to the flask. The reaction mixture was stirred at 95 °C (oil bath) and the progress of the reaction was monitored by TLC (3–9 h). After cooling down to room temperature, a saturated aqueous sodium hydrogencarbonate solution was carefully added dropwise (for chalcone 8d, saturated aqueous sodium carbonate solution was added). To obtain ketone 9f in high yield it was necessary to dissolve the selenocarbonate 1 in ethanol (1 mL) and then added this solution in four portions during the first six hours and finally quenched the reaction with saturated aqueous sodium carbonate solution. The mixture was extracted with three 10 mL portions of EtOAc. The combined extracts were washed with brine (10 mL), dried (MgSO4), filtrated and concentrated in vacuo. The residue was then purified by flash chromatography on silica gel to provide the corresponding ketone 9.:94) as eluent to give the commercially available ketone 9a. Yield: 33 mg (79%).
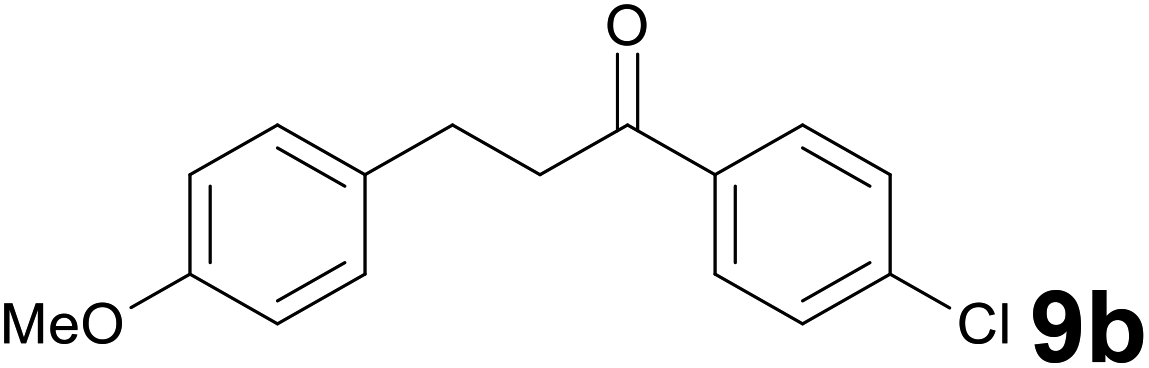
:85) as eluent to give ketone 9b. Yield: 53 mg (95%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.92–7.86 (m, 2H), 7.46–7.40 (m, 2H), 7.20–7.13 (m, 2H), 6.88–6.82 (m, 2H), 3.80 (s, 3H), 3.24 (t, J = 7.5 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H).
:1) as eluent to give 9c. Yield: 47 mg (77%); light yellow solid, mp 132–134 °C. 1H NMR (CDCl3, 200 MHz) δ (ppm) 8.30 (d, J = 8.8 Hz, 2H), 8.10 (d, J = 8.8 Hz, 2H), 7.89–7.73 (m, 3H), 7.72 (s, 1H), 7.55–7.34 (m, 3H), 3.52–3.37 (m, 2H), 3.35–3.20 (m, 2H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 197.5, 150.2, 114.1, 138.0, 133.5, 132.1, 129.0 (2C), 128.3, 127.6, 127.4, 126.9, 126.6, 125.5, 123.8 (2C), 40.9, 29.9; FTIR νmax/cm−1 3113, 2897, 1784, 1517, 1319, 1195, 807, 476; m/z (E/I): 305 (M+, 81%), 155 (100), 141 (62), 115 (19), 104 (139), 76 (10). Found: C, 74.78; H, 4.89; N, 4.35. Calc. for C19H15NO3: C, 74.74; H, 4.95; N, 4.59.
:96) as eluent to give 9d. Yield: 42 mg (63%); light yellow oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.56 (d, J = 1.6 Hz, 1H), 7.40–7.16 (m, 5H), 6.91 (d, J = 7.8 Hz, 1H), 7.30 (br s, 2H), 3.31–3.20 (m, 2H), 3.19–3.08 (m, 2H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 198.7, 146.2, 139.1, 137.7, 132.6, 132.3, 131.5, 129.5, 127.7, 124.5, 120.0, 118.7, 114.1, 38.3, 30.1; FTIR νmax/cm−1 3468, 3364, 2932, 1683, 1585, 1469, 1313, 1037, 732, 684; m/z (E/I): 339 (M+, 29%), 258 (89), 120 (100), 92 (53), 65 (29). Found: C, 52.93; H, 3.99; N, 4.25. Calc. for C15H13BrClNO: C, 53.20; H, 3.87; N, 4.14.
:86) as eluent to give 9e. Yield: 48 mg (72%); yellow solid, mp < 40 °C. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.89 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 3.7 Hz, 1H), 6.62 (d, J = 3.7 Hz, 1H), 3.32–3.26 (m, 2H), 3.24–3.18 (m, 2H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 196.9, 145.3, 139.7, 134.8, 129.5, 129.4 (2C), 129.0 (2C), 125.2, 109.4, 40.0, 24.4; FTIR νmax/cm−1 2896, 1699, 1587, 1206, 1090, 962, 796, 473; m/z (E/I): 330 (M+, 54%), 330 (54), 177 (56), 139 (100), 111 (40). Found: C, 47.17; H, 3.28. Calc. for C13H10BrClOS: C, 47.37; H, 3.06.
:2) as eluent to give 9f. Yield: 45 mg (86%); light yellow oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.70 (dd, J = 3.8, 1.1 Hz, 1H), 7.63 (d, J = 4.9, 1.1 Hz, 1H), 7.22–7.08 (m, 3H), 6.78 (d, J = 8.6 Hz, 2H), 3.30–3.13 (m, 2H), 3.08–2.90 (m, 2H), 2.94 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 192.5, 148.8, 144.2, 133.4, 131.7, 129.7, 129.1 (2C), 128.0, 113.4 (2C), 41.6, 41.1 (2C), 29.5; FTIR νmax/cm−1 3089, 2907, 1652, 1519, 1415, 1347, 1203, 811, 723; m/z (E/I): 259 (M+, 49%), 134 (100), 118 (14), 111 (10). Found: C, 69.38; H, 6.89; N, 5.55. Calc. for C15H17NOS: C, 69.46; H, 6.61; N, 5.40.
:99) as eluent to give 9h. Yield: 53 mg (88%); colourless oil. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.78–7.68 (m, 2H), 7.67–7.52 (m, 3H), 7.47–7.33 (m, 2H), 7.27–7.16 (m, 1H), 7.13 (dd, J = 5.0, 3.8 Hz, 1H), 3.20 (t, J = 7.2 Hz, 2H), 2.93 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H); 13C NMR (CDCl3, 50 MHz) δ (ppm) 192.0, 149.0, 144.1, 140.0, 132.6, 131.8, 129.3 (2C), 128.1, 125.7, 125.6, 120.2, 118.4 (2C), 39.8, 18.4, 11.9; FTIR νmax/cm−1 3100, 2922, 1652, 1503, 1415, 1221, 1058, 724; m/z (E/I): 296 (M+, 86%), 185 (30), 171 (100), 158 (35), 111 (24), 77 (30). Found: C, 68.65; H, 5.70; N, 9.33. Calc. for C17H16N2OS: C, 68.89; H, 5.44; N, 9.45.
:9) as eluent to give the commercially available ketone 9j. Yield: 35 mg (74%).
Competitive Michael-addition experiment
According to general procedure an aqueous 37% hydrochloric acid solution (0.1 mL, 1.2 mmol) was added to a solution of 2c (41 mg, 0.2 mmol), ethyl cinnamate (34 mL, 0.2 mmol) and the selenocarbonate 1 (0.16 g, 0.6 mmol) in 4 mL of ethanol. The mixture was heated at 95 °C (oil bath) under atmosphere (condenser fitted with a rubber septum) and the progress of the reaction was monitored by TLC (7 h). After cooling to room temperature a saturated aqueous sodium hydrogencarbonate solution was carefully added dropwise. The mixture was extracted with three 10 mL portions of EtOAc. The combined extracts were washed with brine (5 mL), dried (MgSO4), filtrated and concentrated in vacuo. Purification of the crude product by chromatography (SiO2, diethyl ether/hexane 4:6) afforded 34 mg (81% yield) of ketone 4c.
Benzeneselenenyl radical-mediated addition of diphenyldiselenide to alkyne 13
A 50 mL round-bottom flask equipped with a stir bar and a condenser was charged with methyl propiolate 13 (0.2 mmol), selenocarbonate 1 (0.16 g, 0.6 mmol) and methanol (4 mL). In this experiment methanol was employed to eliminate the potential transesterification reaction of methyl propiolate with ethanol. The condenser was fitted with a rubber septum and then aqueous 37% hydrochloric acid (0.1 mL, 1.2 mmol) was added to the flask. The reaction mixture was stirred at 95 °C (oil bath) and the progress of the reaction was monitored by TLC. Reaction quenching was achieved after 4 h by simply adding a saturated aqueous sodium hydrogencarbonate solution. The mixture was extracted with three 10 mL portions of EtOAc. The combined extracts were washed with brine (10 mL), dried (MgSO4), filtrated and concentrated in vacuo. The residue was then purified by flash chromatography on silica gel using a diethyl ether/hexane 5:95 mixture as eluant. Unfortunately an inseparable 91:9 (E/Z) mixture of bis-selenides 15 and 16 respectively (60 mg, 75% yield) contaminated by β-phenylselenoacrylate 17 was isolated.
Another fraction containing β-phenylselenoacrylate 17 (95:5 Z/E stereoisomeric mixture) was also obtained (10 mg, 20% yield).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the Università degli Studi di Perugia within financing program “Progetto Sviluppo di metodologie innovative per la sintesi efficiente di composti eterociclici, molecole drug-like e intermedi sintetici ad alto valore aggiunto, finanziato con il Fondo Ricerca di Base, 2018, dell’Università degli Studi di Perugia”.References
- (a) C. Paulmier, Selenium Reagents and Intermediates in Organic Synthesis, Pergamon, Oxford, 1986 Search PubMed; (b) Organoselenium Chemistry, ed. D. C. Liotta, John Wiley & Sons, New York, 1987 Search PubMed; (c) Organoselenium Chemistry - A Pratical Approach, ed. T. G. Back, Oxford, UK, New York, 2000 Search PubMed; (d) Organoselenium Chemistry – Synthesis and Reactions, ed. T. Wirth, Wiley WCH, Weinheim, 2011 Search PubMed.
- K. Fujimori, Y. Yoshimoto and S. Oae, Tetrahedron Lett., 1979, 20, 4397 CrossRef.
- K. Fujimori, Y. Yoshimoto and S. Oae, Tetrahedron Lett., 1980, 21, 3385 CrossRef CAS.
- T. Masawaki, A. Ogawa, N. Kambe, S. Murai and N. Sonoda, J. Phys. Org. Chem., 1988, 1, 119 CrossRef CAS.
- J. Perkins, B. V. Smith and E. S. Turner, J. Chem. Soc., Chem. Commun., 1980, 977 RSC.
- T. Masawaki, Y. Uchida, A. Ogawa, N. Kambe, N. Miyoshi and N. Sonoda, J. Phys. Org. Chem., 1988, 1, 115 CrossRef CAS.
- Y. Nishiyama, Y. Makino, S. Hamanaka, A. Ogawa and N. Sonoda, Bull. Chem. Soc. Jpn., 1989, 62, 1682 CrossRef CAS.
- Y. Nishiyama, M. Yoshida, S. Ohkawa and S. Hamanaka, J. Org. Chem., 1991, 56, 6720 CrossRef CAS.
- H.-C. Li, C. An, G. Wu, G.-X. Li, X.-B. Huang, W.-X. Gao, J.-C. Ding, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Lett., 2018, 20, 5573 CrossRef CAS.
- (a) S. Sakaguchi, T. Yamaga and Y. Ishii, J. Org. Chem., 2001, 66, 4710 CrossRef CAS; (b) A. Pelšs, E. T. Kumpulainen and A. M. P. Koshinen, J. Org. Chem., 2009, 74, 7598 CrossRef PubMed; (c) A. Boffi, S. Cacchi, P. Ceci, R. Cirilli, G. Fabrizi, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, ChemCatChem, 2011, 3, 347 CrossRef CAS.
- H. Hagiwara, Nat. Prod. Commun., 2018, 13, 489 CrossRef.
- (a) T. Ling, C. Chowdhury, B. A. Kramer, B. G. Vong, M. A. Palladino and E. A. Theodorakis, J. Org. Chem., 2001, 66, 8843 CrossRef CAS PubMed; (b) H. Y. Lee and M. An, Tetrahedron Lett., 2003, 44, 2775 CrossRef CAS; (c) J.-P. Li, Y.-X. Zhang and Y. Ji, J. Chin. Chem. Soc., 2008, 55, 390 CrossRef CAS.
- (a) J. M. Khurana and P. Sharma, Bull. Chem. Soc. Jpn., 2004, 77, 549 CrossRef CAS; (b) S. Kotani, K. Osakama, M. Sugiura and M. Nakajima, Org. Lett., 2011, 13, 3968 CrossRef CAS PubMed; (c) J. Shang, F. Li, X. Bai, J. Jiang, K. Yang, G. Lai and L. Xu, Eur. J. Org. Chem., 2012, 14, 2809 CrossRef.
- (a) K. G. Akamanchi, H. C. Patel and R. Meenakshi, Synth. Commun., 1992, 22, 1655 CrossRef CAS; (b) S. A. Van Arman, A. J. Zimmet and I. E. Murray, J. Org. Chem., 2016, 81, 3528 CrossRef CAS; (c) R. Prasanna, S. Guha and G. Sekar, Org. Lett., 2019, 21, 2650 CrossRef CAS.
- (a) M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini and C. Santi, Synlett, 2001, 1767 CrossRef CAS; (b) M. Tiecco, L. Testaferri, L. Bagnoli, C. Scarponi, A. Temperini, F. Marini and C. Santi, Tetrahedron: Asymmetry, 2006, 17, 2768 CrossRef CAS; (c) A. Temperini, R. Terlizzi, L. Testaferri and M. Tiecco, Chem.–Eur. J., 2009, 15, 7883 CrossRef CAS; (d) A. Temperini, A. Barattucci, P. M. Bonaccorsi, O. Rosati and L. Minuti, J. Org. Chem., 2015, 80, 8102 CrossRef CAS PubMed.
- (a) M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini and C. Santi, Synth. Commun., 1998, 28, 2167 CrossRef CAS; (b) M. Tiecco, L. Testaferri, F. Marini, S. Sternativo, C. Santi, L. Bagnoli and A. Temperini, Tetrahedron: Asymmetry, 2003, 14, 1095 CrossRef CAS; (c) M. Tiecco, L. Testaferri, F. Marini, S. Sternativo, C. Santi, L. Bagnoli and A. Temperini, Tetrahedron: Asymmetry, 2003, 14, 2651 CrossRef CAS; (d) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, S. Santoro, F. Marini, L. Bagnoli and A. Temperini, Tetrahedron, 2007, 63, 12373 CrossRef CAS; (e) A. Temperini, A. Capperucci, A. Degl'Innocenti, R. Terlizzi and M. Tiecco, Tetrahedron Lett., 2010, 51, 4121 CrossRef CAS.
- A. Temperini and C. Siciliano, Tetrahedron, 2020, 76, 131311, DOI:10.1016/j.tet.2020.131311.
- C.-M. Chu, S. Gao, M. N. V. Sastry, C.-W. Kuo, C. Lu, J.-T. Liu and C.-F. Yao, Tetrahedron, 2007, 63, 1863 CrossRef CAS.
- W. J. Sanders and J. L. Seidel, J. Agric. Food Chem., 1992, 40, 263 CrossRef CAS.
- E. M. Guantai, K. Ncokazi, T. J. Egan, J. Gut, P. J. Rosenthal, R. Bhampidipati, A. Kopinathan, P. J. Smith and K. Chibale, J. Med. Chem., 2011, 54, 3637 CrossRef CAS PubMed.
- H. Laatsch, Liebigs Ann. Chem., 1980, 140 CrossRef CAS.
- T. G. Back and M. V. Krishna, J. Org. Chem., 1988, 53, 2533 CrossRef CAS.
- A. Temperini, F. Piazzolla, L. Minuti, M. Curini and C. Siciliano, J. Org. Chem., 2017, 82, 4588 CrossRef CAS PubMed.
- (a) L. Minuti, A. Temperini and E. Ballerini, J. Org. Chem., 2012, 77, 7923 CrossRef CAS PubMed; (b) L. Minuti, E. Ballerini, A. Barattucci, P. M. Bonaccorsi, M. L. Di Gioia, A. Leggio, C. Siciliano and A. Temperini, Tetrahedron, 2015, 71, 3253 CrossRef CAS.
- L. Minuti, F. Piazzolla and A. Temperini, Eur. J. Org. Chem., 2017, 36, 5370 CrossRef.
- J. Tatsuzaki, K. F. Bastow, K. Nakagawa-Goto, S. Nakamura, H. Itokawa and K.-H. Lee, J. Nat. Prod., 2006, 69, 1445 CrossRef CAS.
- Z. Wang, G. Yin, J. Qin, M. Gao, L. Cao and A. Wu, Synthesis, 2008, 3675 CAS.
- S.-M. Lu and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 8920 CrossRef CAS.
- J. Montes-Avila, S. P. Díaz-Camacho, J. Sicairos-Félix, F. Delgado-Vargas and I. A. Rivero, Bioorg. Med. Chem., 2009, 17, 6780 CrossRef CAS.
- L. D. Chiaradia, A. Mascarello, M. Purificação, J. Vernal, M. N. Cordeiro, M. E. Zenteno, A. Villarino, R. J. Nunes, R. A. Yunes and H. Terenzi, Bioorg. Med. Chem. Lett., 2008, 18, 6227 CrossRef CAS PubMed.
- B. Mathew, A. Haridas, G. Uçar, I. Baysal, M. Joy, G. E. Mathew, B. Lakshmanan and V. Jayaprakash, ChemMedChem, 2016, 11, 1161 CrossRef CAS PubMed.
- A. Thangamani, Eur. J. Med. Chem., 2010, 45, 6120 CrossRef CAS PubMed.
- M. Kinger, J. H. Park, M. G. Hur, S. W. Kim and S. D. Yang, Med. Chem., 2013, 9, 1035 CrossRef CAS PubMed.
- D. Zhang, T. Iwai and M. Sawamura, Org. Lett., 2019, 21, 5867 CrossRef CAS.
- J.-J. Cao, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4976 CrossRef CAS PubMed.
- D. Enders, H. Eichenauer and R. Pieter, Chem. Ber., 1979, 112, 3703 CrossRef CAS.
- B. Scheiper, M. Bonnekessel, H. Krause and A. Fürstner, J. Org. Chem., 2004, 69, 3943 CrossRef CAS.
- S.-C. Lu, H.-S. Li, S. Xu and G.-Y. Duan, Org. Biomol. Chem., 2017, 15, 324 RSC.
- V. F. Ferreira and F. J. Schimitz, J. Organomet. Chem., 1998, 571, 1 CrossRef CAS.
- V. P. Ananikov, D. A. Malyshev and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 1475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07128e |
| This journal is © The Royal Society of Chemistry 2020 |