DOI:
10.1039/D0RA06682F
(Paper)
RSC Adv., 2020,
10, 38583-38587
Preparation of Ag nanoparticles by spark ablation in gas as catalysts for electrocatalytic hydrogen production†
Received
4th August 2020
, Accepted 1st October 2020
First published on 19th October 2020
Abstract
Spark ablation in gas (SAG) technology has the characteristics of being green, fast quenching, fast dynamics and specializes in producing metallic nanoparticles with a clean surface, small size, and abundant defects. In this study, Ag nanoparticles were prepared via SAG and in situ loaded on a carbon fiber through nitrogen flow. The effect of the carrier gas flow rate and deposition time on the particle size and the dispersibility of the as-prepared Ag nanoparticles on the carbon fiber by SAG were investigated, and the hydrogen evolution reaction (HER) performances of the samples in acidic media were further studied. When the carrier gas flow rate and deposition time are controlled at 5 L min−1 and 120 min, respectively, the sample displays an optimal activity with an overpotential of 362 mV at 10 mA cm−2, which is superior to commercial Ag nanoparticles on carbon fibers. Accordingly, this synthetic technology provides a new way to obtain efficient metallic nano-catalysts and is expected to achieve large-scale application.
1 Introduction
Metallic nanoparticles abundant in low-coordinated atomic sites1–3 have attracted extensive attention in numerous applications, such as catalysis,4–6 optoelectronics7,8 and drug delivery.9–11 Furthermore, decreasing the size of metallic nanoparticles can effectively increase the ratio of surface-to-bulk atoms, resulting in better interaction between nanoparticles and other reactive substances, and thus enhancing their performance.12 Therefore, it is of great significance to find a cost-effective, green and rapid method to prepare metallic nanoparticles with enhanced properties for catalytic applications. Among the reported synthetic methods, physical methods, such as liquid/gas phase ablation technology, which utilizes high energy beams to bombard metallic targets to form plasma or aerosol and is then quenched to form metallic nanoparticles, are usually adopted to prepare highly efficient metallic catalysts.13–16 Spark ablation in gas (SAG) is characterized as a low-cost, green, facile and efficient gas-phase ablation method to prepare metallic nanoparticles, where an inert gas is used as the reaction medium to avoid the restriction of the liquid medium on vapor group.17–19 Moreover, the carrier gas not only provides a clean environment for the formation of nanoparticles but also has a rapid quenching effect, which is beneficial to the formation of metallic nanoparticles with smaller sizes.20–22 Moreover, compared with the chemical method, the SAG method also avoids the influence of other chemical reagents on the surface of the product, such as the surfactants, making the product with a clean surface, which is favorable to catalytic reactions.23–25
HER is a half-reaction of water electrolysis,26 usually seen as an effective way for hydrogen generation, but with a fatal flaw: the most efficient catalysts are Pt-based materials, its high price and scarcity limit their commercial applications.5,27 On the contrary, Ag possesses abundant reserves, low cost and the highest conductivity in all metallic elements, but its d10 electronic structure results in weak intermediate H* adsorption and poor HER performance in acidic media.13,28,29 Therefore, many efforts have been devoted to regulating pure Ag, including increasing its surface area,29 creating more unsaturated coordination atoms6 and introducing tensile stress13 to cause upshift of the d-band center, so that the HER performance of Ag can approach or exceed that of commercial Pt and make Ag a potential alternative for Pt.30 Therefore, combined with the characteristics of the SAG method, it is feasible to prepare Ag nanoparticles with various features, such as small size and stacking fault to adjust the d-band electronic structure of Ag and thus improve the acidic HER performance.6,29
Herein, Ag nanoparticles with high purity and clean surface are prepared by the SAG method. Carbon fibers serve as a platform for the in situ deposition of Ag nanoparticles to suppress the aggregation of Ag nanoparticles and improve charge transfer. The effect of the carrier gas flow rate and deposition time on the particle size, dispersibility and electrocatalytic HER performance of the Ag catalysts were studied, respectively. The Ag catalyst obtained at a carrier gas flow rate of 5 L min−1 and a deposition time of 120 min shows an optimized HER performance with an overpotential of 362 mV at a current density of 10 mV cm−2 in 0.5 M H2SO4, superior to that of commercial Ag power on the carbon fiber (645 mV at 10 mA cm−2). Interestingly, when the carrier gas flow rate was increased, the HER properties of the three samples prepared showed a trend of gradual improvement (618 mV > 580 mV > 462 mV at 10 mA cm−2 vs. RHE). In addition, as the deposition time was extended, the HER overpotential at 10 mA cm−2 exhibited the decreasing trend at first and then increasing (618 mV > 422 mV > 362 mV < 455 mV < 638 mV vs. RHE). This work provides a guideline to predict the catalytic properties of metallic nano-catalysts synthesized by the SAG method.
2 Experimental
2.1 Synthesis and deposition of Ag nanoparticles
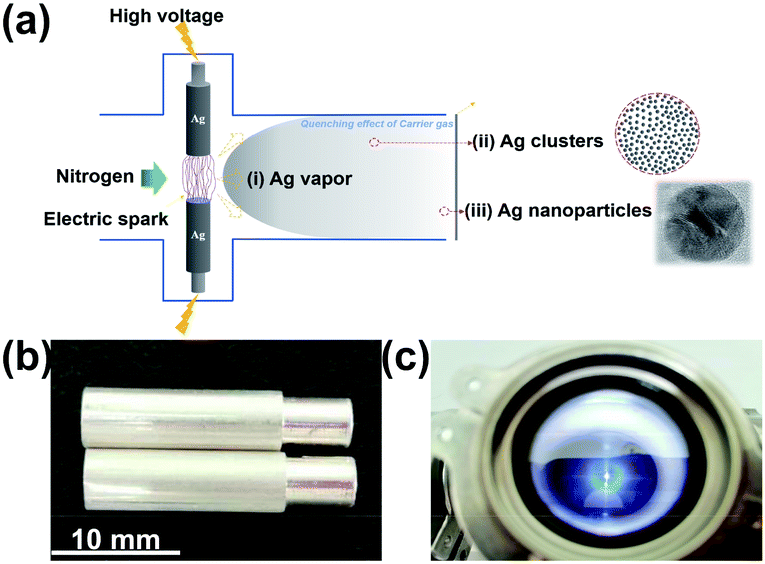
The synthetic process of Ag nanoparticles via the SAG is shown in Fig. 1a. The spark ablation generator (VSP-G1, VSPARTICLE B.V., Delft, The Netherlands) was used and the operating voltage and current were 1.35 kV and 7 mA, respectively. In this process, nitrogen (99.999%) used as the carrier gas and bulk Ag electrodes (99.99%) with a fixed size (length = 20 mm and diameter = 6 mm) were purchased from Tianjin Incole Union Technology Co. Ltd (Fig. 1b). Before starting the deposition process, opening the power switch, the equipment was preheated by nitrogen flow for 30 min. As shown in Fig. 1c, the electric spark was generated when initial electrons collide and ionize with background gas atoms in the process of the electrons moving to the anode under the action of high voltage.20,31 In the spark discharge process, Ag electrodes were first partially vaporized by the electric spark with a high temperature, and then accompanying the quenching effect of flowing carrier gas, Ag nanoparticles were formed32 and deposited on a carbon fiber. Ag nanoparticles with different particle sizes were produced by varying the carrier gas flow at 5 L min−1, 15 L min−1 and 25 L min−1, and the deposition time was fixed at 30 min, and were named as V-Ag-5 L min−1, V-Ag-15 L min−1, V-Ag-25 L min−1, respectively. A carbon fiber with 50 mm in diameter was used to collect and disperse Ag nanoparticles for subsequent electrochemical tests. Then, the carrier gas flow rate was fixed at 5 L min−1, different deposition time (0, 30, 60, 120, 240 and 360 min) were selected to produce Ag nanoparticles, termed as V-Ag-x min (x = 0, 30, 60, 120, 240, 360), respectively.
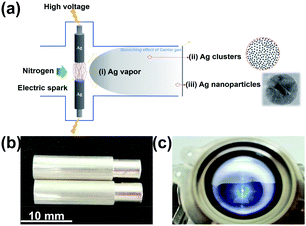 |
| | Fig. 1 (a) Schematic illustrating a simple mechanism for the formation of Ag nanoparticles on a carbon fiber. Photographs of (b) a pair of pure Ag electrodes and (c) electric spark machining process. | |
2.2 Samples characterizations
The morphology and size of Ag nanoparticles were determined via transmission electron microscopy (TEM, JEM-2100F, 200 kV) and the structure of the deposited Ag nanoparticles on the carbon fiber was observed by scanning electron microscopy (SEM, Hitachi S-4800). X-ray diffraction (XRD) patterns of the samples were acquired on Bruker D8 Advance with Cu Kα radiation (λ = 1.54056 Å) and Ni filter. The XRD specimens prepared by the in situ deposition of Ag nanoparticles on the carbon fiber by the SAG method were directly used for the XRD test and the characterization of crystallinity was performed in the range of 35° ≤ 2θ ≤ 85°.
2.3 Electrochemical measurements
The carbon fiber deposited with Ag nanoparticles was used as a catalyst, which was cut to a fixed size (10 mm × 20 mm) as the working electrode. Commercial Ag powder was loaded on carbon fiber as comparison sample was named as the C-Ag/carbon fiber, and its load was 0.245 mg cm−2, which was consistent with the deposition amount by the SAG method at the same deposition time. Loads of commercial Ag powers on carbon fiber was calculated by the theoretical production (2.4 mg h−1) of the SAG technology. All electrochemical measurements were carried out in an Ar-saturated 0.5 M H2SO4 aqueous solution in a three-electrode system at room temperature on a CHI 760E electrochemical workstation (CH Instruments, Shanghai). A graphite rod was used as the counter electrode and an Hg/Hg2Cl2 (in saturated KCl solution) electrode as the reference electrode. The potentials can be converted into a reversible hydrogen electrode (RHE): ERHE (V) = ESCE (V) + 0.242 V + 0.0592 V × pHelectrolyte.33 Before each linear sweep voltammetry (LSV) test at a scan rate of 5 mV s−1, 30 cycles of cyclic voltammetry (CV) curves with 50 mV s−1 were swept to activate the catalyst. Finally, the Tafel slope can be derived from the Tafel equation: η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(j), where a is the intercept, η is the overpotential, j is the current density and b is the Tafel slope.34
log(j), where a is the intercept, η is the overpotential, j is the current density and b is the Tafel slope.34
3 Results and discussion
3.1 Effect of the carrier gas flow rate on the Ag nanoparticle size
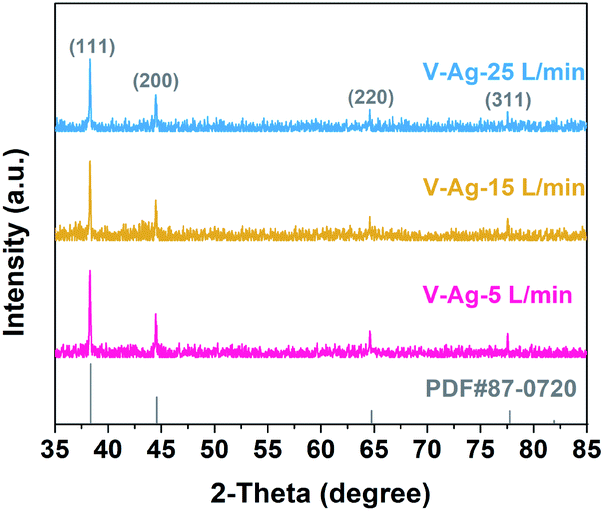
Fig. 2a–c show the typical TEM images of Ag nanoparticles at different gas flow rates. All the Ag nanoparticles are spherical with a clean surface, uniformly dispersed and homogenous in size. Moreover, with the increase in the carrier gas velocity, the size of Ag nanoparticles decreases gradually. The size distribution of Ag nanoparticles prepared at three different carrier gas flow rates were executed on low-magnification TEM images and shown in the insets, the size distribution of the three samples are relatively narrow and with the average sizes of V-Ag-5 L min−1, V-Ag-15 L min−1 and V-Ag-25 L min−1 are 4.73, 3.79 and 2.41 nm, respectively. In addition, the particle size of the three samples shows the typical Gaussian distribution. This phenomenon is mainly attributed to the fact that a higher carrier gas flow rate will facilitate the cooling rate and transport velocity of primary particles, which weakens the agglomeration of Ag nanoparticles and causes smaller particle sizes.22 The high-resolution TEM images also further confirmed the microstructure and crystallinity of as-prepared Ag nanoparticles that the lattice spacing of 0.235 nm corresponds to the (111) planes of pure Ag (Fig. 2d–f). The XRD analysis was performed on the three kinds of Ag nanoparticles deposited onto the carbon fiber, and the results shown in Fig. 3, indicating that all the diffraction peaks of the three samples correspond to the typical characteristic of pure Ag (JCPDS card #04-0783) with diffraction peaks of the Ag samples at 2θ angles of 38.12°, 44.31°, 64.45°, 77.41°, which correspond to the (111), (200), (220), and (311) crystal planes. The intensity peaks at the 2θ values confirm the face-centered cubic nature of Ag nanoparticles.35 Meanwhile, no peaks of other impurity crystalline phases can be detected. Furthermore, at 2θ = 28.12°, the Ag (111) peak of the three samples show the highest intensity, illustrating the successful materialization and small size of the crystals.36
 |
| | Fig. 2 Characterization of V-Ag nanoparticles at different carrier gas flow rates. Low-magnification TEM images and high-resolution TEM images for (a and d) 5 L min−1, (b and e) 15 L min−1 and (c and f) 25 L min−1, the insets are corresponding the size distribution. | |
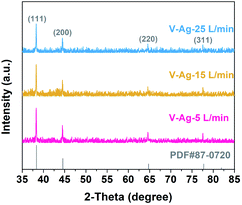 |
| | Fig. 3 XRD patterns of V-Ag-5 L min−1, V-Ag-15 L min−1, and V-Ag-25 L min−1 samples. | |
3.2 Effect of the deposition time on dispersibility of Ag nanoparticles on the carbon fiber
The schematic in Fig. 4a shows the change in the nanostructure of V-Ag nanoparticles deposited on the carbon fiber with prolonging deposition time. To further investigate the effect of the deposition time on the dispersibility of V-Ag on the carbon fiber nanoparticles, scanning electron microscopy (SEM) was conducted. As shown in Fig. 4b–d, when the deposition time was below 120 min, the Ag nanoparticles are deposited evenly on the carbon fiber and no agglomeration was found. However, the agglomeration of Ag nanoparticles was found when the deposition time increased to 120 min (Fig. 4e). When the deposition time is longer than 120 min, the V-Ag nanoparticles grow gradually to form larger Ag nanoparticles and new Ag nanoparticles also have gradually covered above the Ag nanoparticles on the carbon fiber to agglomerate. Thus, the deposition time is critical for obtaining uniformly ultrafine Ag nanoparticles on the substrate. To corroborate that the elemental analysis of the samples were performed using EDS mapping, which demonstrates that the presence of elemental Ag on the carbon fiber and when the deposition time is between 0 and 120 min, Ag elements distribute evenly across the carbon fiber, indicating that the Ag nanoparticles are small and embed evenly on the carbon fiber (Fig. S1†). However, with the extension of the deposition time (greater than 120 min), the Ag nanoparticles agglomerate and grow gradually, which are attributed to the quantity of Ag nanoparticles increasing over time.
 |
| | Fig. 4 (a) A schematic of the deposition process of Ag nanoparticles on the carbon fiber with the extension of deposition time. SEM images of V-Ag nanoparticles (5 L min−1) deposited on the carbon fiber at different deposited time with a blank carbon fiber as a reference. (b) Blank carbon fiber. (c) 30 min. (d) 60 min. (e) 120 min. (f) 240 min. (g) 360 min. | |
3.3 Electrocatalytic HER activity of different samples obtained under different experimental parameters (carrier gas flow rate and deposition time)
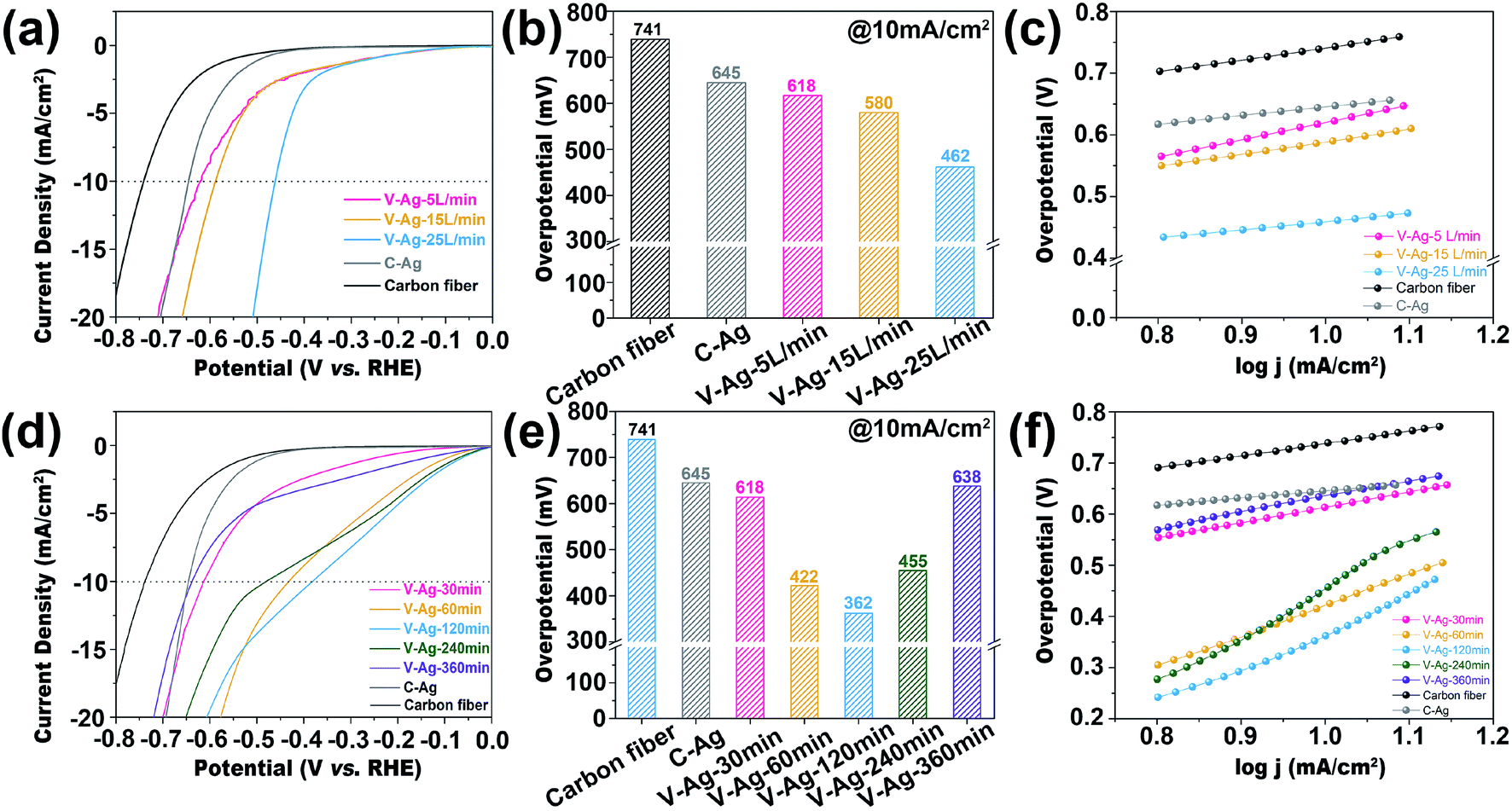
In order to explore the advantage of the SAG technology, electrocatalytic acidic HER performances of samples obtained at different carrier gas flow rates and deposition time were analyzed. As shown in Fig. 5a, the overpotential at 10 mA cm−2 of V-Ag-25 L min−1 (462 mV vs. RHE) is superior to that of V-Ag-5 L min−1 (618 mV vs. RHE), V-Ag-15 L min−1 (580 mV vs. RHE) and C-Ag (645 mV vs. RHE) (Fig. 5b). Moreover, V-Ag-25 L min−1 shows a lower Tafel slope (136 mV dec−1) than V-Ag-5 L min−1 (279 mV dec−1) and V-Ag-15 L min−1 (201 mV dec−1) (Fig. 5c). The electrocatalytic HER improvement is mainly attributed to the reduction in the size of Ag nanoparticles along with the increase in the carrier gas flow rate, which causes Ag nanoparticles to have a larger surface area, exposing more surface unsaturated coordination atoms, including edge and corner atoms, and facilitating the adsorption of intermediates.6,13,29
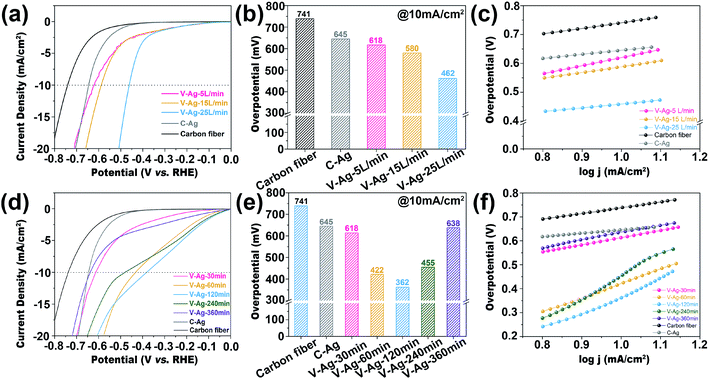 |
| | Fig. 5 (a) HER polarization curves of C-Ag and V-Ag nanoparticles on the carbon fiber at a fixed deposition time (30 min) and different carrier gas flow rates (5 L min−1, 15 L min−1, 25 L min−1) in a 0.5 M H2SO4 solution. (b) The overpotential comparison at 10 mA cm−2. (c) Tafel slopes. (d) HER polarization curves of C-Ag and V-Ag nanoparticles on the carbon fiber at a fixed carrier gas flow rate (5 L min−1) and different deposited time (30 min, 60 min, 120 min, 240 min, 360 min) in 0.5 M H2SO4 solution. (e) The overpotential comparison at 10 mA cm−2. (f) Tafel slopes. | |
By comparing the LSV curves obtained at different deposition times with a fixed carrier gas flow rate (5 L min−1), the HER overpotential showed a decreasing trend at first and then increasing, as shown in Fig. 5d. The sample of V-Ag-120 min shows the highest activity (362 mV vs. RHE at the current of 10 mA cm−2). Furthermore, when changing the deposition time, the whole trend of the electrocatalyst activity is V-Ag-120 min (362 mV vs. RHE) < V-Ag-60 min (422 mV vs. RHE) < V-Ag-240 min (455 mV vs. RHE) < V-Ag-30 min (614 mV vs. RHE) < V-Ag-360 min (638 mV vs. RHE) < C-Ag (645 mV vs. RHE) (Fig. 5e). The change in electrocatalytic performance is mainly due to the agglomeration and growth of Ag nanoparticles on the carbon fiber with the increasing deposition time, which reduces the exposed active sites and decreases the surface area of Ag nanoparticles.37 Meanwhile, it can be observed from the Tafel slope of all samples that the HER reaction kinetics of the samples are relatively weaker (Fig. 5f), which is attributed to the nature of the Ag metal itself, which has a weak capacity to absorb H*.6 Thus, the catalytic activities of the samples are indeed significantly improved by reasonably tuning the deposition time.
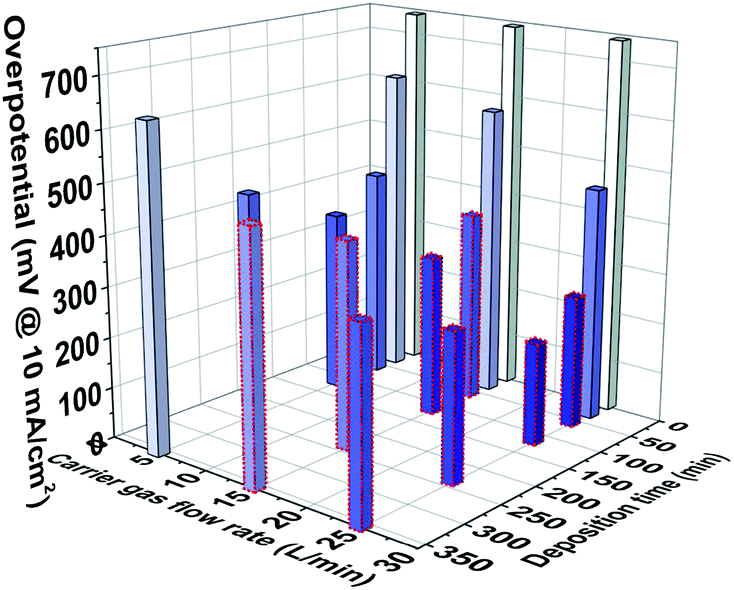
Combined with the above comparison of the electrocatalytic HER performance of the samples under acidic conditions, the performance variation trend is predicted under a higher flow rate, as shown in Fig. 6. The HER overpotential of the samples was obtained when the carrier flow rate was 15 and 25 L min−1, and the deposition time was higher than 30 min, which was based on the equal scaling of HER overpotential obtained by adjusting the different carrier gas flow rate at 5 L min−1 and tuning the deposition time at 30–360 min. The purpose is to obtain the overall performance change trend caused by controlling the two process parameters, which reveals that the metallic nano-catalysts with optimal performance can be realized by precisely adjusting the carrier gas flow rate and deposition time. When the carrier gas flow rate reaches 25 L min−1, the size of Ag nanoparticles can reach about 2.41 nm, which is more conducive to the catalytic reaction due to the size effect.6,29 Moreover, controlling the deposition time at 120 min, the optimal nanoparticle size of the payload without agglomeration can be achieved on the carbon fiber that further optimize the catalytic performance.
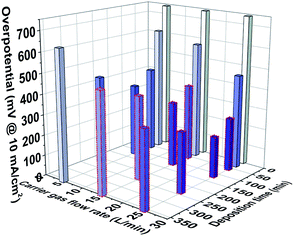 |
| | Fig. 6 Histogram of the change trend of the HER performance when the carrier gas flow rate is controlled in the range of 0 to 25 L min−1 and the deposition time is controlled in the range of 0–360 min (the column enclosed by red dashed represents the predicted change in higher carrier gas flow rate over deposition time). | |
4 Conclusions
In summary, uniformly dispersed spherical Ag nanoparticles with small size and clean surface supported on a carbon fiber are successfully prepared by the SAG technology. In this study, the carrier gas flow rate and deposition time as the important parameters are regulated to control the size and nanostructure Ag nanoparticles on the carbon fiber. Furthermore, the influence of the carrier gas flow rate and deposition time was verified by electrochemical tests, and Ag nanoparticles with optimal HER performance can be obtained by precisely controlling the carrier gas flow and deposition time. Thus, this synthesis method has the potential to be further applied in electrocatalytic fields and the production of ultrafine metallic nano-catalysts. At the same time, only electrical energy and metal electrodes are consumed in the process and the experimental period is short, which makes this process easier to commercialize and mass produce.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51771067, 51771068, 51671079).
Notes and references
- X. H. Liu, J. Luo and J. Zhu, Nano Lett., 2006, 6, 408–412 CrossRef CAS.
- K. N. Thakkar, S. S. Mhatre and R. Y. Parikh, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 257–262 CrossRef CAS.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS.
- T. Y. Kou, D. W. Li, C. Zhang, Z. H. Zhang and H. Yang, J. Mol. Catal. A: Chem., 2014, 382, 55–63 CrossRef CAS.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- W. J. Kang, C. Q. Cheng, Z. Li, Y. Feng, G. R. Shen and X. W. Du, ChemCatChem, 2019, 11, 5976–5981 CrossRef CAS.
- T. H. Lee, J. I. Gonzalez, J. Zheng and R. M. Dickson, Acc. Chem. Res., 2005, 38, 534–541 CrossRef CAS.
- J. Ma and S. Gao, ACS Nano, 2019, 13, 13658–13667 CrossRef CAS.
- D. Lee, R. E. Cohen and M. F. Rubner, Langmuir, 2005, 21, 9651–9659 CrossRef CAS.
- E. J. Kang, Y. M. Baek, E. Hahm, S. H. Lee, X.-H. Pham, M. S. Noh, D.-E. Kim and B.-H. Jun, Int. J. Mol. Sci., 2019, 20(2), 315 CrossRef.
- M. P. Patil and G.-D. Kim, Colloids Surf. B Biointerfaces, 2018, 172, 487–495 CrossRef CAS.
- S. Gharpure, A. Akash and B. Ankamwar, J. Nanosci. Nanotechnol., 2020, 20, 3303–3339 CrossRef.
- Z. Li, J.-Y. Fu, Y. Feng, C.-K. Dong, H. Liu and X.-W. Du, Nat. Catal., 2019, 2, 1107–1114 CrossRef CAS.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- J. Y. Lin, C. Xi, Z. Li, Y. Feng, D. Y. Wu, C. K. Dong, P. Yao, H. Liu and X. W. Du, Chem. Commun., 2019, 55, 3121–3123 RSC.
- H. B. Wang, J. Q. Wang, N. Mintcheva, M. Wang, S. Li, J. Mao, H. Liu, C. K. Dong, S. A. Kulinich and X. W. Du, Materials, 2019, 12, 10 CrossRef.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2013, 15, 3027–3046 RSC.
- M.-R. Kalus, V. Reimer, S. Barcikowski and B. Goekce, Appl. Surf. Sci., 2019, 465, 1096–1102 CrossRef CAS.
- H. Zeng, X.-W. Du, S. C. Singh, S. A. Kulinich, S. Yang, J. He and W. Cai, Adv. Funct. Mater., 2012, 22, 1333–1353 CrossRef CAS.
- N. S. Tabrizi, M. Ullmann, V. A. Vons, U. Lafont and A. Schmidt-Ott, J. Nanoparticle Res., 2009, 11, 315–332 CrossRef CAS.
- M. Domaschke, M. Schmidt and W. Peukert, J. Aerosol Sci., 2018, 126, 133–142 CrossRef CAS.
- S. Ahmad, P. Laiho, Q. Zhang, H. Jiang, A. Hussain, Y. Liao, E.-X. Ding, N. Wei and E. I. Kauppinen, J. Aerosol Sci., 2018, 123, 208–218 CrossRef CAS.
- S. Peng, J. M. McMahon, G. C. Schatz, S. K. Gray and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14530–14534 CrossRef CAS.
- H. Li, H. Xia, D. Wang and X. Tao, Langmuir, 2013, 29, 5074–5079 CrossRef CAS.
- M. S. Bootharaju, V. M. Burlakov, T. M. D. Besong, C. P. Joshi, L. G. AbdulHalim, D. M. Black, R. L. Whetten, A. Goriely and O. M. Bakr, Chem. Mater., 2015, 27, 4289–4297 CrossRef CAS.
- Y. M. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- T. Uchida, H. Mogami, A. Yamakata, Y. Sasaki and M. Osawa, J. Am. Chem. Soc., 2008, 130, 10862 CrossRef CAS.
- F. W. Campbell, S. R. Belding, R. Baron, L. Xiao and R. G. Compton, J. Phys. Chem. C, 2009, 113, 14852–14857 CrossRef CAS.
- Y. N. Xia, Chem.–Eur. J., 2019, 25, 4244 CrossRef CAS.
- X. Wang, Z. Liu, M. Qiu, Z. Hui, Z. Tian and Y. Huang, Mater. Manuf. Processes, 2014, 29, 1367–1373 CrossRef CAS.
- T. E. Itina and A. Voloshko, Appl. Phys. B: Lasers Opt., 2013, 113, 473–478 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29(14), 1605838 CrossRef.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef.
- M. A. M. Khan, S. Kumar, M. Ahamed, S. A. Alrokayan and M. S. AlSalhi, Nanoscale Res. Lett., 2011, 6, 434 CrossRef.
- S. Some, B. Sarkar, K. Biswas, T. K. Jana, D. Bhattacharjya, P. Dam, R. Mondal, A. Kumar, A. K. Deb, A. Sadat, S. Saha, A. Kati, I. Ocsoy, O. L. Franco, A. Mandal, S. Mandal, A. K. Mandal and I. A. Ince, RSC Adv., 2020, 10, 22742–22757 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06682f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Zunming Lu
*a and
Zunming Lu