
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective methods for the synthesis of hydrazones, quinazolines, and Schiff bases: reaction monitoring using a chemometric approach†
Jana Pisk a,
Ivica Đilovića,
Tomica Hrenara,
Danijela Cvijanovićb,
Gordana Pavlovićc and
Višnja Vrdoljak*a
a,
Ivica Đilovića,
Tomica Hrenara,
Danijela Cvijanovićb,
Gordana Pavlovićc and
Višnja Vrdoljak*a
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: visnja.vrdoljak@chem.pmf.hr
bUniversity of Zagreb, School of Medicine, Department of Chemistry and Biochemistry, Šalata 3, 10000 Zagreb, Croatia
cUniversity of Zagreb, Faculty of Textile Technology, Division of Applied Chemistry, Prilaz baruna Filipovića 28a, 10000 Zagreb, Croatia
First published on 20th October 2020
Abstract
Synthesis of hydrazones (1a–4a and 1b–4b), quinazolines (3c·MeOH and 3d·MeOH), and hydrazone-Schiff bases (4c and 4d) is achieved by combining suitable aldehydes (2,3- or 2,4-dihydroxybenzaldehyde) with four hydrazides (isonicotinic, nicotinic, and 2- or 4-aminobenzoic acid hydrazide). A suite of approaches for their preparation is described: solution-based synthesis, mechanosynthesis, and solid-state melt reactions. The mechanochemical approach is generally a better choice for the quinazolines, while the solid-state melt reaction is more efficient for derivatives of (iso)nicotinic based hydrazones. Crystalline amine-functionalised hydrazones 4a and 4b undergo post-synthetic modifications in reactions with 3- or 4-pyridinecarbaldehyde vapours to form hydrazone-Schiff bases 4a-3py, 4b-3py, 4a-4py, and 4b-4py. Mechanochemical and vapour-mediated reactions are followed by ex situ powder X-ray diffraction and IR-ATR methods, respectively. The chemometric analysis of these data using principal component analysis provided an insight into the reaction profiles and reaction times. Azines (5a and 5b), achieved from aldehydes and hydrazine, reversibly change colour in response to temperature changes. The structures of all products are ascertained by a combined use of spectroscopic and X-ray diffraction methods. The cytotoxic and antimicrobial activities of all compounds against selected human cancer cell lines and bacterial strains are evaluated.
Introduction
Hydrazide–hydrazones and quinazolin-4(3H)-ones are very attractive compounds in medicinal chemistry due to their broad spectrum of biological activities.1–14 In particular, quinazolin-4(3H)-ones were reported to possess high anti-inflammatory and analgesic activity.15 These properties have stimulated interest in developing methodologies for their synthesis. However, there is still an issue to find a simple and effective approach for their selective preparation.The most common pathway for the preparation of these hydrazide-based compounds includes the reaction of appropriate hydrazides with different aldehydes or ketones in various organic solvents. Different solution-based synthetic approaches have been tested and combined.16–19 Gudasi et al. described the ineffectiveness in synthesizing hydrazones by condensing pyridine-2-carbaldehyde with 2-aminobenzoylhydrazide.17–19 It was found that reactions of the corresponding hydrazide and 3-methoxybenzaldehyde, in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, led to the formation of quinazolin-4(3H)-one. Recently, the mechanochemical synthetic route provided an efficient solution for obtaining hydrazones20 and 3-arylideneaminoquinazolin-4(1H)-one compounds, from various aldehydes.21
:1, led to the formation of quinazolin-4(3H)-one. Recently, the mechanochemical synthetic route provided an efficient solution for obtaining hydrazones20 and 3-arylideneaminoquinazolin-4(1H)-one compounds, from various aldehydes.21
On account of it, 2,3- and 2,4-dihydroxybenzaldehyde were used as carbonyl forerunners, while isonicotinic and nicotinic acid hydrazides, were used as hydrazides for the hydrazones synthesis. Additionally, 2- and 4-aminobenzoic acid hydrazides were also investigated as compounds that provided classical hydrazones, quinazolinone or hydrazone-Schiff bases via controlled condensation. Conventional and a variety of green synthesis methodologies were used and critically compared (Fig. 1). We aimed to investigate their effect on reaction time, yield, as well as the crystallinity and purity of the prepared compounds. This study was also focused on applicability of the synthetic procedures and their selectivity towards the desired product. The synthetic path was refreshed by employing the “melt synthesis” technique.
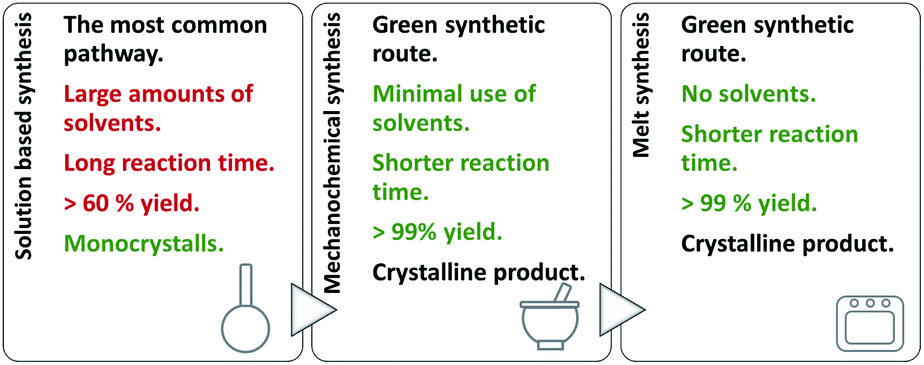 | ||
| Fig. 1 Synthetic procedures applied for the preparation of hydrazones, quinazolines, and hydrazine-Schiff bases. | ||
Also, post-synthetic condensation with aldehyde vapours was applied to yield hydrazone-Schiff bases. The chemometric study was used for monitoring the starting material conversion by mechanochemical and vapour-mediated synthesis by processing ex situ powder X-ray diffraction and ATR spectra, respectively. Principal component analysis of these data was applied as a data reduction technique where collection of data (diffractograms or spectra) were decomposed in order to provide linearly independent variables. These variables can be plotted in one or two dimensions giving a detailed insight into reaction profiles and allowing identification of the reaction end points. Adequacy of the used method was validated by visual inspection of the final PXRD diffractograms and ATR spectra with the final product and in each case, it was confirmed that reaction finished in the exact time obtained by PCA.
Crystal and molecular structures of hydrazones 1a·MeOH, 1b·H2O, 3a, 3b, 4a, and 4b·MeOH, quinazolinone 3c·MeOH, hydrazone-Schiff base 4c, and azine 5b·H2O were determined by the single crystal X-ray diffraction method, providing proposed investigation more comprehensive. It should be noted that in Cambridge Structural Database (CSD)22 there are few hydrazone structures reported with 2,3- and 2,4-dihydroxybenzaldehyde fragment.23–40 All of the investigated compounds were further characterized by elemental analysis, thermogravimetric and differential scanning calorimetry measurements, IR-ATR and NMR spectroscopy. The solid-state thermochromic changes of azines and influence of the central spacer and functional groups on the responsive properties were also explored.
To complement our study, the cytotoxicity of the compounds was evaluated against HepG2 and THP-1 cells. The compounds were also screened for their antimicrobial activity against S. aureus, E. faecalis, E. coli and M. catarrhalis to evaluate influence of the compound type and substituents on their activities.
Experimental section
General methods
Commercially available solvents were used without further purification. 2,3- and 2,4-dihydroxybenzaldehyde, isonicotinic and nicotinic acid hydrazide, and 2- and 4-aminobenzoic acid hydrazide, 3- and 4-pyridinecarboxaldehyde were used as received.Elemental analyses were provided by the Analytical Services Laboratory of the Rudjer Bošković Institute, Zagreb, Croatia. The powder X-ray diffraction data (PXRD) for qualitative phase analysis were collected on a Phillips X'Change powder diffractometer in the Bragg–Brentano geometry using CuKα radiation. The data were collected and visualized using the X'Pert programs Suite.41 IR-ATR spectra were recorded on a Perkin Elmer Spectrum One spectrometer. Thermogravimetric analyses (TGA) were conducted on a Mettler Toledo TG/DSC 3+ Stare System coupled with Thermo Fischer Nicolet iS50 FT-IR with Al2O3 crucibles under nitrogen stream in a temperature range between 25 °C and 400 °C, while the heating rate was adjusted to 10 °C min−1. Differential scanning calorimetry (DSC) measurements were performed under the nitrogen stream on the Mettler-Toledo DSC823e calorimeter with aluminium crucibles, in the temperature range 25–400 °C, with the heating rate 10 °C min−1. The results of both TGA and DSC experiments were evaluated using the Mettler STARe 9.01 software. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III HD 400 spectrometer operating at 400 MHz. Compounds were dissolved in dmso-d6 and measured in 5 mm NMR tubes at 298 K with TMS as an internal standard.
Single-crystal X-ray measurements and structure determinations
The crystals of compounds 1a·MeOH and 4b·MeOH were air-sensitive so the single crystal diffraction data were collected at 150 K. Diffraction intensity data were collected by ω-scans on an Oxford Diffraction Xcalibur 3 (all compounds except 4c and 5b·H2O) using graphite-monochromated MoKα radiation (λ = 0.71073 Å) and on an Oxford Diffraction Nova (4c and 5b·H2O) using micro-focus sealed X-ray tube with CuKα radiation (λ = 1.54184 Å). The data were reduced using the CrysAlis42 program package. A summary of general and crystallographic data parameters is presented in Table S1, see ESI†.The structures were solved by direct methods using SHELXT.43 The refinement procedure by full-matrix least-squares methods based on F2 values against all reflections included anisotropic displacement parameters for all non-H atoms. The positions of H atoms each riding on carbon atoms were determined on stereochemical grounds. The positions of other H atoms were determined from the difference Fourier map but were refined using the riding model. In the asymmetric unit of 3c·MeOH, the methanol molecule exhibits a two-site (57:43) disorder. The restraints on geometrical and displacement parameters (DFIX, DELU, SIMU and ISOR) were applied for that molecule. Refinements were performed using SHELXL-97. The SHELX programs operated within the Olex2 v1.2 (ref. 44) suite. Geometrical calculations and molecular graphics were done with PLATON, MERCURY,45 ORTEP,46 PyMOL47 and POV-Ray.48
Synthesis of 1a, 1b, 2a, and 2b. A mixture of 2,3- or 2,4-dihydroxybenzaldehyde (0.35 g, 2.5 mmol) and isonicotinic or nicotinic hydrazide (0.34 g, 2.5 mmol) in 50 mL of methanol was refluxed with continuous stirring for 3 h. The obtained solution was cooled and after several days colourless crystalline product 1a, 1b, 2a, or 2b was filtered and dried in a desiccator up to a constant weight. Yield: 0.53 g, 83% (1a); 0.59 g, 92% (1b); 0.52 g, 81% (2a); and 0.55 g, 86% (2b). Upon recrystallization and slow evaporation isoniazid-based hydrazones were obtained as solvates 1a·MeOH (from methanol) and 1b·H2O (from acetone). Analytical, DSC, IR. and NMR spectral data are given in the ESI, Tables S2 and S3.†
Synthesis of 3a, 3b, 4a, and 4b. 2,3- or 2,4-Dihydroxybenzaldehyde (0.35 g, 2.5 mmol) dissolved in 20 mL of methanol was added dropwise to a solution of 2- or 4-aminobenzhydrazide (0.80 g, 2.5 mmol in 40 mL of ethanol) under stirring at room temperature. The mixture was continuously stirred for 30 min and then gently heated at 40 °C for 2 h. The resulting product 4a precipitated during heating the reaction mixture. In the case of hydrazones 3a, 3b, and 4b, the obtained solution was concentrated under vacuum to one-quarter of its volume and left at room temperature for several days. The obtained colourless crystalline product was filtered and dried in a desiccator up to a constant weight. Yield: 0.31 g, 46% (3a); 0.36 g, 53% (3b); 0.55 g, 82% (4a); and 0.53 g, 78% (4b). Crystals of 4b·MeOH were obtained if the methanolic solution was evaporated slowly. Analytical, DSC, IR. and NMR spectral data are given in the ESI, Tables S4 and S5.†
Synthesis of 3c·MeOH, 4c, and 4d. 2- or 4-Aminobenzhydrazide (0.80 g, 2.5 mmol) was added to a methanolic solution of 2,3-dihydroxybenzaldehyde (0.69 g, 5.0 mmol in 30 mL). The solution was refluxed for 3 h and then, in the case of 3c, concentrated under vacuum to one-quarter of its volume and left at room temperature for several days. The obtained precipitate was filtered and dried. Yield: 0.74 g, 76% (3c). Colourless crystals of 3c·MeOH were obtained upon recrystallization from methanol. The resulting products 4c and 4d precipitated during heating the reaction mixture. They were isolated from hot solutions, rinsed and dried. Yield: 0.89 g, 91% (4c, a dark red powder); 0.79 g, 81% (4c, a yellowish-orange powder). Analytical, DSC, IR. and NMR spectral data are given in the ESI, Tables S6 and S7.†
Synthesis of 5a and 5b. A solution of hydrazine monohydrate (0.062 mL, 1.25 mmol) was added to an ethanolic solution of 2,3- or 2,4-dihydroxybenzaldehyde (0.35 g, 2.5 mmol in 15 mL) and the resulting solution was heated at 55 °C for 2 h. It afforded a crystalline product which was filtered, washed with ethanol and dried. Yield: 0.29 g, 84% (5a, a beige powder); 0.29 g, 84% (5b, a yellow powder). Crystals of 5b·H2O were obtained upon recrystallization of 5b from acetonitrile. Analytical, DSC, IR. and NMR spectral data are given in the ESI, Table S8.†
Synthesis of 1a, 2b, 3a, 3b, 4a, and 4b. Liquid assisted grinding (LAG) experiments were performed using a Retsch MM200 ball mill. 0.1500 g (1 mmol) of an appropriate dihydroxybenzaldehyde, 1 mmol of an appropriate hydrazide and 50 μL of methanol were placed with one 10 mm milling ball in a 10 mL Teflon jar. A sample of the powder obtained after 60 min, milled at 25 Hz, was immediately analysed by PXRD. No purification of the sample was performed.
Synthesis of 3c and 3d. 0.1500 g (1 mmol) of an appropriate dihydroxybenzaldehyde, 0.05 mmol of an appropriate hydrazide and 50 μL of methanol were used. A sample of the powder obtained after 60 min, milled at 25 Hz, was immediately analysed by PXRD. No purification of the sample was performed.
Synthesis of 1a, 2a, 1b, and 2b. A mixture of 2,3- or 2,4-dihydroxybenzaldehyde (0.035 g, 0.25 mmol) and isonicotinic or nicotinic hydrazide (0.034, 0.25 mmol) were placed in a glass tube with glass stopper. The tube was placed in Kugelror oven and heated for one hour at 110 °C for 1a and 2a and 140 °C for 1b and 2b. Afterwards, samples were analysed by PXRD. No purification of the sample was performed.
Mechanochemical synthesis of 1a, 2b, 3a, 3b, 4a, and 4b. The syntheses were performed several times by using the same experimental conditions (10 mL Teflon jar with one 10 mm milling ball) each time starting from the reagents and grinding liquid. Reactions were conducted at 25 Hz frequency and the time was set to 2.5 min, 5 min, 10 min, 20 min, 30 min, 40 min, or 1 h. Afterwards, each sample was immediately analysed ex situ by PXRD. The sample was contained on a Si sample holder, patterns were collected in the range of 2θ = 5–50°, and chemometric data analysis was performed.
Vapour-mediated synthesis of 4a-3py, 4b-3py, 4a-4py, and 4b-4py. The reactions were performed in several independent sets at 25 °C. 10 mL of 3- or 4-pyridinecarbaldehyde was left overnight in a beaker inside a desiccator. A sample of 4a or 4b (10 mg) was placed on a watch glass, which was then put into the desiccator saturated with 3- or 4-pyridinecarbaldehyde vapour. The sample of each set was taken out after a certain period: 2,5; 5; 10; 20 min, and then every 30 min, and was subsequently analysed ex situ by IR-ATR spectroscopy, and then principal component analysis (PCA) was performed. The exposure time necessary to afford hydrazone-Schiff bases was determined: 4a-3py (100 min), 4b-3py (90 min), 4a-4py (70 min), or 4b-4py (120 min).
Chemometric data analysis
Statistical numerical analyses were performed using the second-order tensor decomposition tool principal component analysis, PCA. In PCA, the data matrix with mean-centered columns X of rank r is decomposed as a sum of r matrices tipτi each one with rank 1.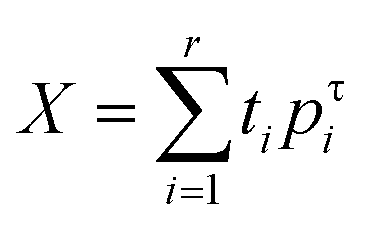
In one case the data matrix X was composed of PXRD diffractograms whereas in the other the data matrix was composed of ATR spectra. PCA enables one to find the best linear projections for a high dimensional set of data in the least-squares sense. Scores ti represent projections of the original points on the principal component (PC) direction and can be used for classification or building of probability distributions,49 whereas loadings56 represent the eigenvectors of data covariance (or correlation) matrix and can be used for the identification of variability among the data. The initial development of PCA goes back to Beltrami50 and Pearson,51 whereas the name was introduced by Hotelling.52 More details on PCA can be found in the literature.53 Principal component analysis was used as a dimensionality reduction tool for a set of PXRD diffractograms and for a set of ATR spectra. In each cae PCA was performed using a NIPALS algorithm54 implemented in our own program moonee.55,56
In vitro cytotoxic and antibacterial activity
The synthesized compounds were tested in vitro against human acute monocytic leukemia (THP-1, ATCC TIB-202) and hepatocellular carcinoma (HepG2, ATCC HB-8065) cells. The cells were grown as reported.57 The cytotoxicity studies were performed according to the protocol described previously.57 In brief, the cytotoxicity of the tested compounds was evaluated at concentrations ranging from 0.05 to 100 μmol L−1 (THP-1) or 0.80 to 100 μmol L−1 (HepG2). The cells were seeded in 96-well microplates and incubated overnight before they were treated with varying concentrations of the tested compounds. The viability of cells after the compound treatment was determined using the MTS assay.58 IC50 (the concentration of the compound required to decrease cell viability for 50%) values were calculated for all tested compounds from dose–response curves. All experiments were performed in duplicate.In vitro antibacterial activity of the investigated compounds was evaluated against Gram-positive, namely Staphylococcus aureus (ATCC 13709) and Enterococcus faecalis (ATCC 29212), and Gram-negative, namely Escherichia coli (ECM 1556) and Moraxella catarrhalis (ATCC 23246), bacterial strains by broth microdilution according to the CLSI guidelines.59 Details of the bacterial growth and the method used have been described previously.57 Briefly, the bacteria were seeded in 96-well microplates and incubated overnight before they were treated with varying concentrations of the tested compounds. After the treatment, the lowest concentrations at which no bacterial growth was observed were determined by visual inspection and reported as minimum inhibitory concentration (MIC) values. All experiments were performed in duplicate.
Results and discussion
Reactions of aldehydes with hydrazides in the molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 – synthesis and structural studies of hydrazones
:1 – synthesis and structural studies of hydrazones
:1, in methanol or ethanol. In the case of 2- or 4-aminobenzhydrazide-based hydrazones the crucial step was maintaining the room temperature and dropwise addition of aldehyde. Products obtained in such a way were taken as reference compounds to those obtained under green reaction conditions. Solvates were isolated upon recrystallization from methanol (1a·MeOH, 3c·MeOH, 3d·MeOH, and 4b·MeOH) or acetone (1b·H2O).
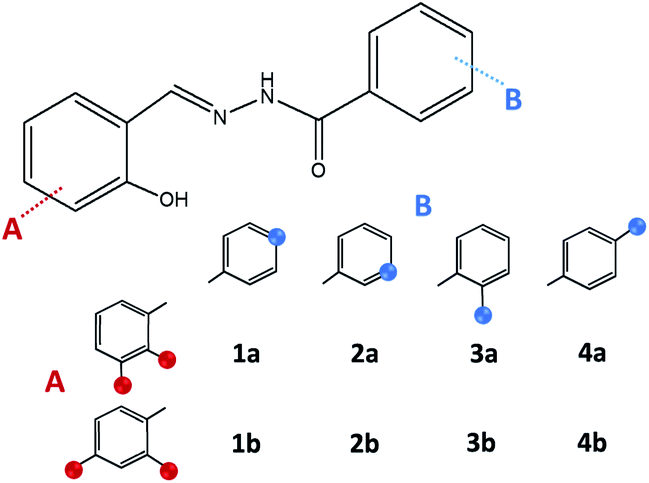 | ||
| Fig. 2 Hydrazones 1a–4a and 1b–4b. Blue sphere presents N atom in the case of isonicotinic or nicotinic acid hydrazide, or NH2 group in the case of 2- or 4-aminobenzoic acid hydrazide. Red sphere presents OH group. | ||
Like in many similar compounds, the keto form dominates in the solid state (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is around 1.2 Å). An extensive survey of the Cambridge Structural Database shows that hydrazones derived from salicylaldehyde derivatives predominantly crystallize in the amide tautomeric form with E configuration. This appears to be the case in all the investigated hydrazones (1a·MeOH, 1b·H2O, 3a, 3b, 4a, and 4b·MeOH): all of them have an intramolecular six-membered heteronuclear resonance-assisted hydrogen bond (Fig. 3). The cis configuration of the O1 atom with respect to N2 is present in all the examined molecules.
O bond is around 1.2 Å). An extensive survey of the Cambridge Structural Database shows that hydrazones derived from salicylaldehyde derivatives predominantly crystallize in the amide tautomeric form with E configuration. This appears to be the case in all the investigated hydrazones (1a·MeOH, 1b·H2O, 3a, 3b, 4a, and 4b·MeOH): all of them have an intramolecular six-membered heteronuclear resonance-assisted hydrogen bond (Fig. 3). The cis configuration of the O1 atom with respect to N2 is present in all the examined molecules.
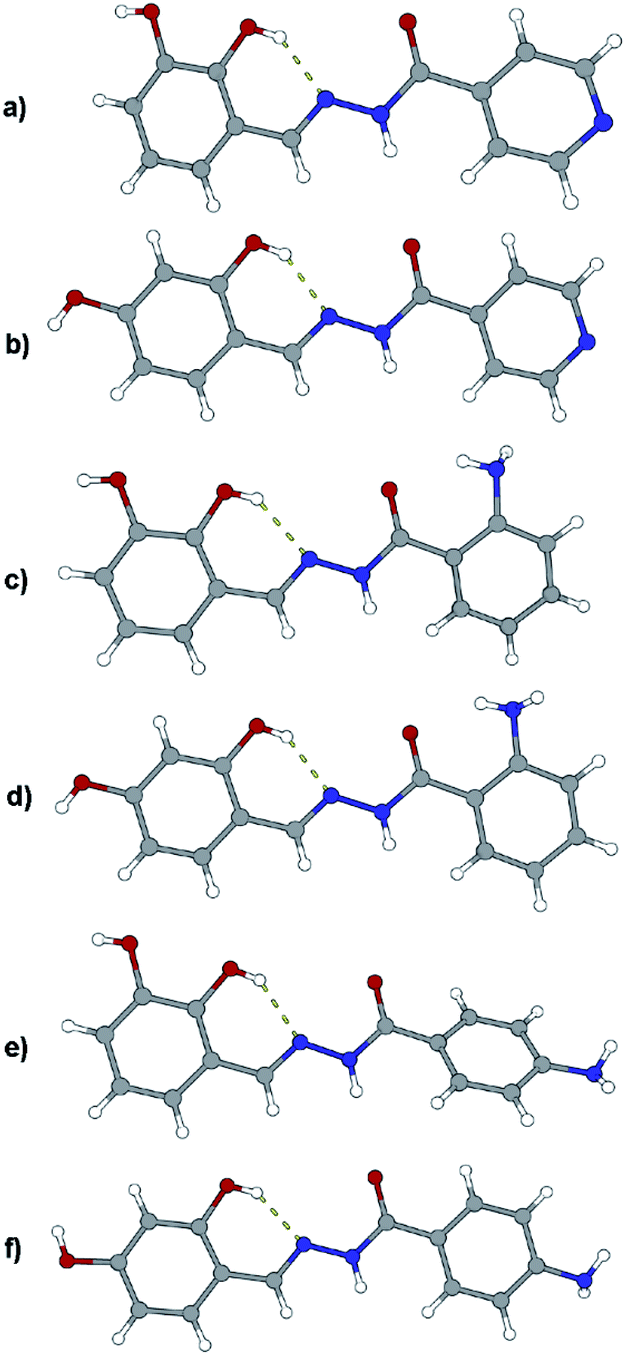 | ||
| Fig. 3 Molecular structures of compounds: (a) 1a·MeOH, (b) 1b·H2O, (c) 3a, (d) 3b, (e) 4a, and (f) 4b·MeOH. Solvent molecules were excluded because of clarity. Atoms are shown as spheres of arbitrary small radii. The intramolecular hydrogen bonds are indicated as an array of yellow cylinders. In all the structures carbonyl oxygen atom has label O1, neighboring nitrogen atoms N1 and N2 labels (the latter participates in intramolecular hydrogen bond). | ||
In all the crystal structures, molecules are almost planar or slightly inclined in a way to maximize their intermolecular hydrogen bonding potential (the planarity is disrupted in the case of solvated compounds). Planarity enhances the delocalization of π electrons through the spacer unit between two aromatic fragments, regardless of the position of substituents. This can be easily seen from the bond length distribution in the central parts of molecules (which fits well by already mentioned tautomeric form). The other, endocyclic, amino or hydroxyl, bond lengths and angles are comparable to those found in the CSD database for similar structures.
The crystal structures of the prepared hydrazones unveil that in all cases molecules are mutually connected with an extensive hydrogen bonding (Fig. 3, 4, 5, and S1–S6, see ESI†). The pyridine N atoms are connected with hydroxyl H atoms, carbonyl O atoms and amido H atoms are usually employed in hydrogen bonding with solvent molecules.
 | ||
| Fig. 4 Crystal packing of compound 1b·H2O. Molecules of 1b are arranged in arrays which are mutually connected with hydrogen bonds (water molecules serve as a bridge) forming sheets. Sheets of molecules are held by stacking interactions and hydrogen bonds. | ||
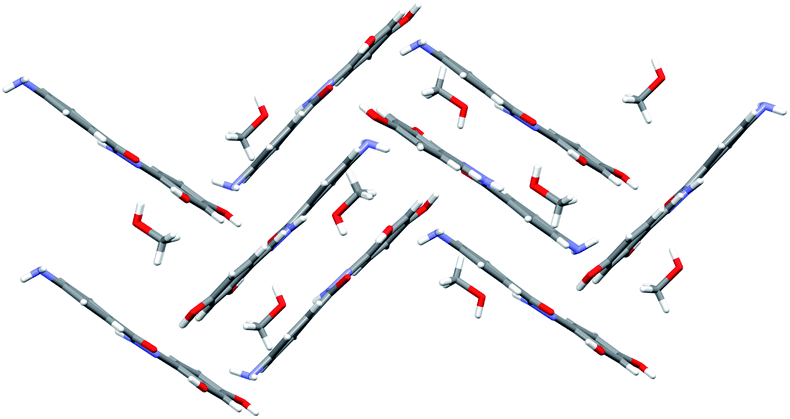 | ||
| Fig. 5 Crystal packing of compound 4b·MeOH. Molecules are connected with an extensive network of hydrogen bonds. Methanol molecules serve as a bridge between sheets of hydrazone molecules. The view is projected down the crystallographic c axis. | ||
All solvates lose their solvent upon prolonged standing at room temperature. A comparison of PXRD patterns was used to identify obtained forms, Fig. S7, see ESI†. Structures of 1a and 2a were reported previously (CSD code WAFVEG and WOFYUN, respectively) but their synthesis was performed differently.23,24
Even-though a variety of products can be obtained by a solution-based method, large amounts of alcohol was employed. To minimize the use of solvent, and turn the synthetic protocols towards more sustainable ones: mechanochemical and solid-state melt-synthesis were engaged. The idea was to explore pros and cons of the methods as well as their selectivity towards the desired product.
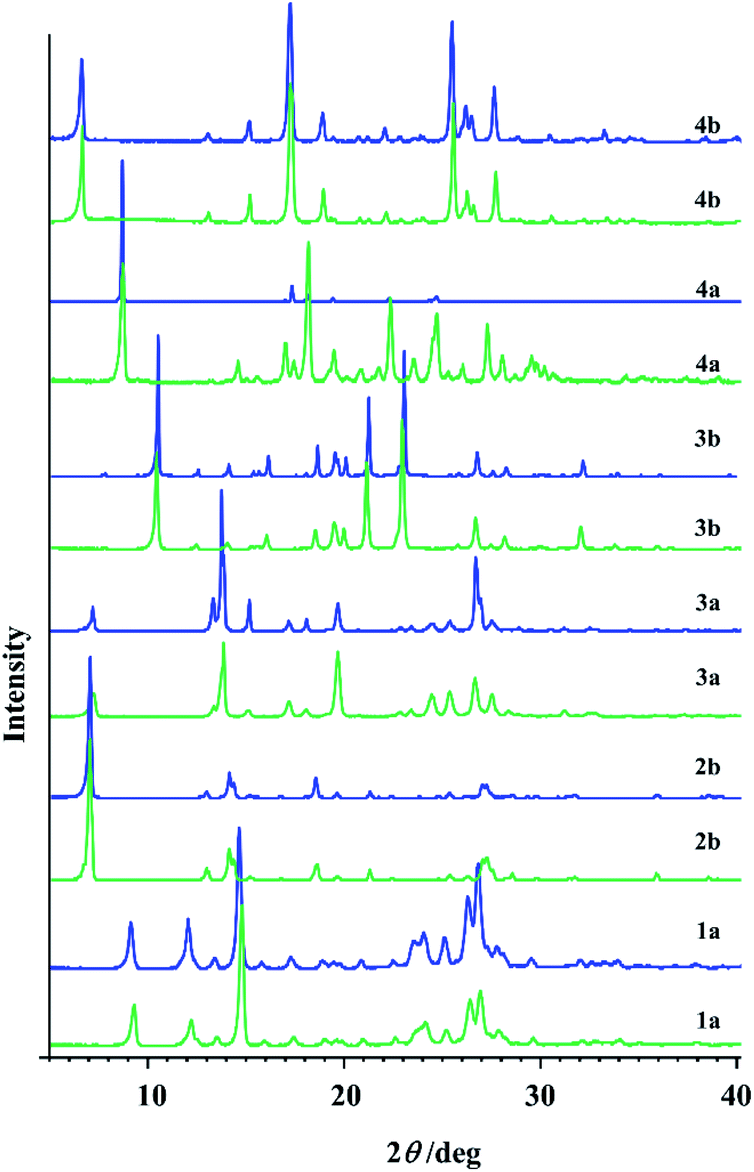 | ||
| Fig. 6 PXRD patterns of hydrazones obtained by the solution-based method (blue line) and by the mechanosynthesis (green line). | ||
In the case of 1b and 2a longer milling was necessary which doesn't follow the sustainable conditions. Besides that, under the prolonged grinding the original PXRD patterns were found to be altered and the amorphization degree increased with milling time. Similarly, PXRD diffractograms of 1a, 2b, 3a, 3b, 4a, and 4b additionally milled for 30 min showed great amount of amorphous phase.
The first idea of the proposed research was to follow the formation of all hydrazones by ex situ PXRD and application of principal component analysis for the mechanochemical synthesis monitoring. PXRD were measured and collected in a data matrix X that was decomposed using the PCA. The duration of the milling was systematically increased up to 60 min to observe the influence of the milling duration on the reaction conversion. Even though it seemed that the PXRD patterns of the samples obtained after a particular reaction time fit well (Fig. S8, see ESI†), PCA revealed the problems in this kind of analysis for monitoring the reaction profiles and detection of reaction end.
Satisfactory results were not obtained in majority of cases due to the several causes: (a) mechanochemical reactions were too fast, (b) amorphization of the reaction components with milling time and reduction in overall quality of the data obtained, and (c) continuation of the reaction after the milling was stopped. In this way, after the reduction of data matrix composed of PXRD diffractograms and plotting the scores values in the dependence on time, the time scale on the ordinate was shifted by an unpredictable amount. This amount of time was different for each point in the reaction because the reaction was continued for different time.
Nevertheless, the PCA was able to provide some additional insight into the reaction mechanisms that we encountered and explained in our previous work.57 For 4a ligand the first principal component described only 41.56% of the total variance whereas the second described 27.57%. This low value of variance for the PC1 and relatively high value of variance for PC2 are indicators of many simultaneous reactions (at least two).57 To confirm that fact, principal component loadings were plotted in 2- and 3-dimensional space and inspected visually. 2D and 3D loadings display a particular symmetrical “butterfly” pattern (Fig. 7a and b).57
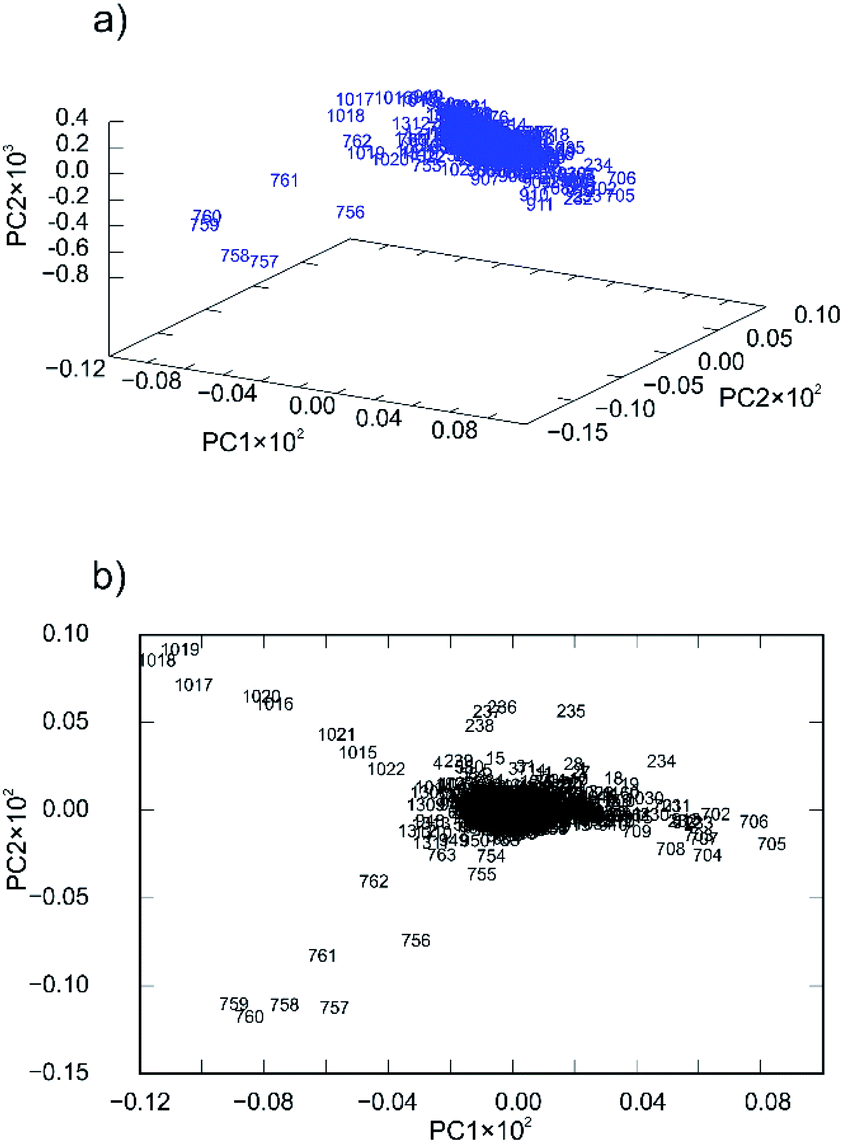 | ||
| Fig. 7 Principal component loadings spanned by (a) three and (b) two principal components calculated for a set of PXRD data collected through mechanochemical synthesis of 4a. | ||
This symmetrical pattern spanned along the first principal component (PC1) suggests that at least two orthogonal eigenvectors extracted from the original PXRD data are necessary for good description of the reaction profile. Since this arrangement is almost symmetrical along PC1 (with symmetry plane cutting the PC2-ordinate at 0.0) it is reasonable to expect that two or even more reactions are occurring simultaneously. To further confirm this fact, we investigated loadings in three dimensions (Fig. 7a). The pattern indeed looks two-dimensional where each branch of the loadings falls symmetrically on the side thus confirming two reaction steps.
However, 2- or 4-aminobenzhydrazide-based hydrazones were not accessible in this manner, and each time reactions gave a mixture of products. This suggests higher reactivity of the amino groups at the 2- and 4- position under these conditions.
Reactions of aldehydes with hydrazides in the molar ratio 2:1 – synthesis and structural studies of quinazolines and hydrazone-Schiff bases
To explore a more profoundly variety of reaction routes, the synthesis was performed by changing the ratio of starting reagents (dihydroxybenzaldehydes and aminobenzhydrazides) to 2:1. The combination of 2-aminobenzhydrazide and 2,3-dihydroxybenzaldehyde in methanol gave crystals of 3c·MeOH, Fig. 8. Formation of 3d was not observed either under neutral conditions or in the presence of an acid, but instead, the 1:1 cyclic intermediate 3-amino-2-(2,4-dihydroxyphenyl)-4(3H)-quinazolinone (Scheme S1, see ESI†) was isolated from methanolic solution and characterized by NMR analysis. This result indicated that the reaction path was remarkably influenced by the position of the OH-group.
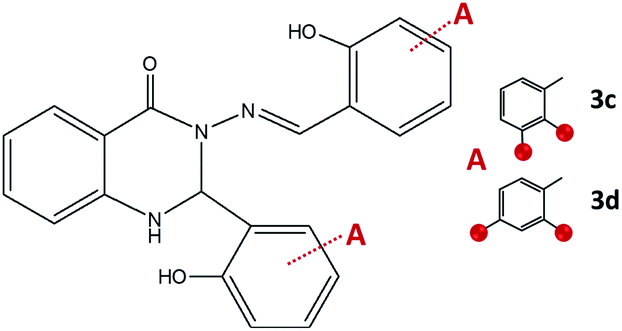 | ||
| Fig. 8 2,3-Dihydroquinazolinones obtained by the reaction of 2-aminobenzhydrazide and 2,3-dihydroxybenzaldehyde, 3c, and 2,4-dihydroxybenzaldehyde, 3d. Red spheres present OH groups. | ||
When employing 2-aminobenzhydrazide, the best results were obtained by using LAG mechanochemical method, and disubstituted quinazolin-4(3H)-one products 3c·MeOH and 3d·MeOH were prepared, Fig. 8. The heterocyclic product dominates in both cases, and purity of products was confirmed also by NMR analysis.
It seems, when 1:1 condensation product under solution-based or mechanochemical conditions was formed in the first step, the quinazoline product was not achieved in the next step even if 2,3- or 2,4-dihydroxysalicylaldehyde was added in excess (Scheme S1, see ESI†).
On the other hand, the condensation of 4-aminobenzhydrazide and 2,3- or 2,4-dihydroxybenzaldehyde provided hydrazone-Schiff bases 4c and 4d only by the solution-based method (Fig. 9). The LAG route did not afford pure compounds and according to NMR, a significant quantity of reagents remained post milling. Most probably hydrazone-Schiff base molecules assemble and afterwards disassemble under mechanical force.
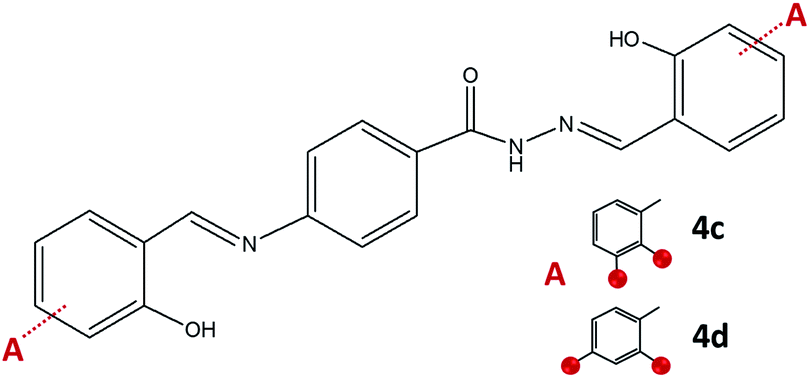 | ||
| Fig. 9 Hydrazone-Schiff bases obtained by the reaction of 4-aminobenzhydrazide and 2,3-dihydroxybenzaldehyde, 4c, and 2,4-dihydroxybenzaldehyde, 4d. Red sphere presents OH group. | ||
Solutions of quinazolines and hydrazone-Schiff bases at room temperature were stable and no traces of the starting aldehydes, hydrazide or azine (vide infra) were detected as a result of the hydrolysis.60 Their absence is proven by the XRD method.
Compound 3c crystallizes as methanol solvate and its molecular structure contrasts the most among the other molecular structures. The reason emerges from the fact that during synthesis both terminal NH2 groups were condensed with 2,3-dihydroxysalicylaldehyde. The resulting molecule consists of four cyclic fragments, out of four only one is heterocyclic and non-planar, Fig. 10. Since molecules crystallize in the centrosymmetric space group and sp3 hybridized carbon atom is chiral, both stereoisomers can be found in the crystal structure. A relatively similar thing happened in the case with 4c where two salicylaldehyde molecules condensed to both terminal NH2 groups, Fig. 10. Molecules are mutually connected with an extensive hydrogen bonding (Fig. S9 and S10, see ESI†).
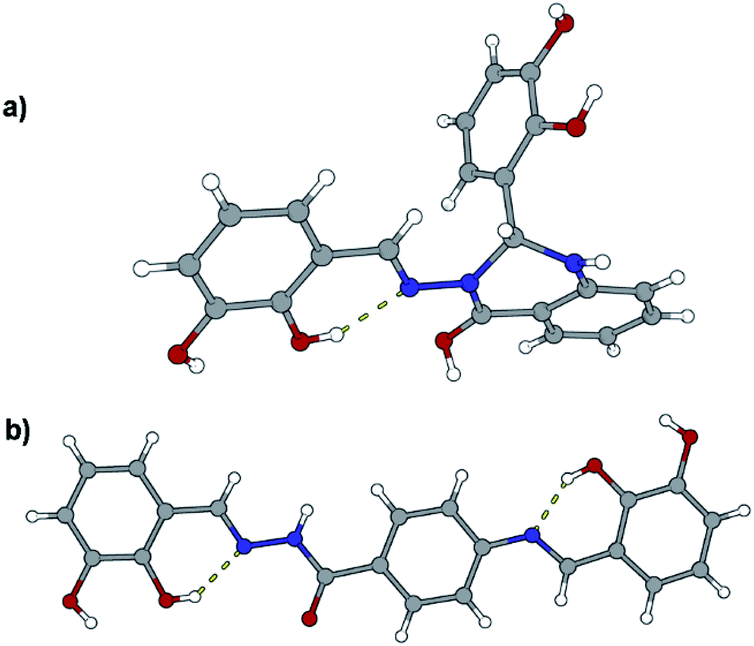 | ||
| Fig. 10 Molecular structures of compounds: (a) 3c·MeOH and (b) 4c. Solvent molecules were excluded because of clarity. Atoms are shown as spheres of arbitrary small radii. The intramolecular hydrogen bonds are indicated as an array of yellow cylinders. | ||
A comparison of reaction yields obtained by different approaches is given in Table 1. Besides high yields, the advantages of solid-state procedures also include operational simplicity and use of environmentally benign conditions.
| Ligand | Synthetic procedure | ||
|---|---|---|---|
| Solution-based | Mechanochemistry | Melt synthesis | |
| 1a | 83 | >99 | >99 |
| 1b | 92 | — | >99 |
| 2a | 81 | — | >99 |
| 2b | 86 | >99 | >99 |
| 3a | 46 | >99 | — |
| 3b | 53 | >99 | — |
| 3c | 76 | >99 | — |
| 3d | — | >99 | — |
| 4a | 82 | >99 | — |
| 4b | 78 | >99 | — |
| 4c | 91 | — | — |
| 4d | 81 | — | — |
Spectroscopic characterization
The IR-ATR spectra of all the compounds (except quinazolines 3c·MeOH and 3d·MeOH) confirmed the presence of the carbonyl group of the amide tautomer, indicated by sharp and intense absorption band in the range of 1680–1620 cm−1. The corresponding band in 3c·MeOH and 3d·MeOH is overlapped with CN band and due to it cannot be easily distinguished. All the compounds exhibited a vibration band at ca. 1610 cm−1 belonging to CNimine group along with an absorption band at ca. 2850–2830 cm−1 attributed to C–H stretching of the –(CO)–NH–NCH– moiety. The band at ca. 1150 cm−1 is assigned to N–N stretching vibrations. Additionally, the spectra of solvates 3c·MeOH and 3d·MeOH showed the absorption band at around 1022 cm−1 implying the presence of the crystalline methanol.
The proton and carbon NMR chemical shifts of all compounds in dmso-d6 solution were deduced by combined use of one (1H, 13C APT) and two-dimensional NMR techniques (COSY, HMQC and HMBC), Tables S2–S7 and Fig. S11–S22, in ESI.† The 1H NMR spectra exhibited downfield singlet signals in the range from 9.11 to 13.32 ppm attributed to NNH and phenolic OH protons, respectively. The broad OH and NH signals and chemical shift values indicated the presence of hydrogen-bonding interactions. The azomethine CHN proton and carbon signals were observed in the range from 8.29 to 9.00 ppm and 149.39 to 165.64 ppm. In the 13C NMR spectra, the low-field amide CO carbon signals appeared in the range from 160.24 to 165.40 ppm.
Additionally, singlets observed around 6.4 ppm and 5.8 ppm were assigned to –NH2 protons in the spectra of 3a–3b and 4a–4b, respectively. Quinazolines 3c and 3d showed the characteristic doublet peaks at 6.61 and 7.28 ppm, and 6.87 and 7.44 ppm, respectively in the 1H NMR spectra. They were assigned to CH and NH protons of the quinazoline ring. The CH carbon atom of the quinazoline ring was observed around 66 ppm.
Vapour-mediated synthesis – yield optimization by a chemometric approach
Based on previously described results where 2:1 solution-based direct reaction provided the route to hydrazone-Schiff bases, we were interested to explore the possibility of altering hydrazones in the presence of aldehyde vapours. We exposed the crystals of synthesized aminobenzhydrazone-based derivatives 4a, and 4b to 3 or 4-pyridinecarboxaldehyde (3py or 4py) vapours. After a certain period, the sample was taken out and was immediately analysed ex situ by IR-ATR spectroscopy. Long exposure time was necessary to complete the condensation reaction at 25 °C and to afford hydrazone-Schiff bases 4a-3py, 4b-3py, 4a-4py, and 4b-4py. It should be noted that these compounds could not be obtained pure in such manner from solution most probably due to the capacity of hydrazones to readily undergo transimination reaction.
Application of principal component analysis to IR-ATR spectral data obtained by reaction monitoring provided a detailed insight into the reaction profiles and the detection of reaction end. These profiles can be well represented using only the 1st principal component (Fig. 11–14).
In each case the PC1 describes more than 70% of the total variance (Table 2). These high values of explained variances in the principal component ensure the proper description of the reaction with only the first principal component. The first component picks up the most prominent changes during reactions. The reaction profiles nicely show the progress and the end of the reaction. For the vapour-mediated synthesis of 4a-3py and 4a-4py the end of the reaction can be estimated to be 100 and 90 min (Fig. 11 and 12), respectively. In the case of 4b-3py and 4b-4py these values were estimated to 70 and 120 minutes (Fig. 13 and 14). These values were confirmed by comparison of the spectra measured in these times with the spectrum of final product. Loadings vectors for monitored reactions are presented on Fig. S23–S26.† Investigation of loadings vectors confirms the creation of products during reactions.
| Component | 4a-3py | 4a-4py | 4b-3py | 4b-4py |
|---|---|---|---|---|
| PC1 | 76.62 | 86.42 | 92.65 | 74.58 |
| PC2 | 15.08 | 8.08 | 4.47 | 16.03 |
| PC3 | 3.33 | 2.82 | 1.26 | 4.62 |
| PC4 | 2.04 | 1.40 | 0.70 | 3.36 |
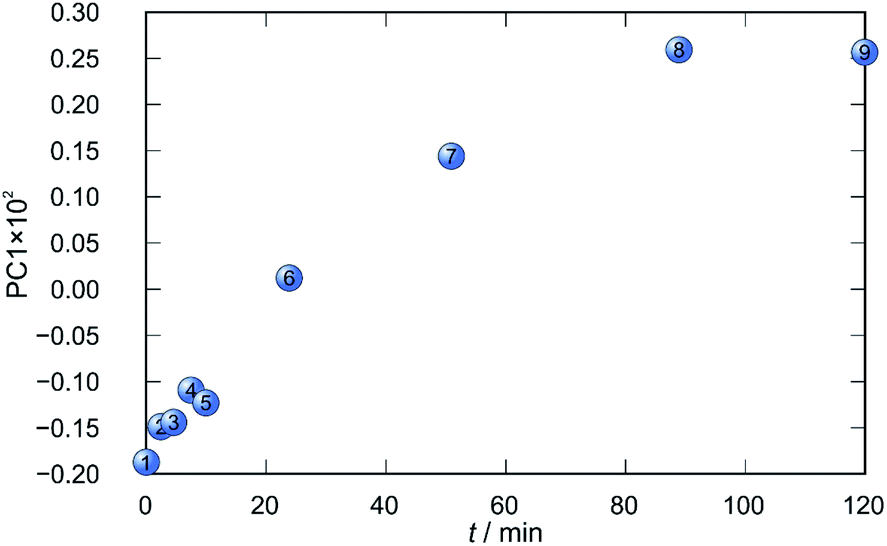 | ||
| Fig. 11 Time dependence of PC1 scores calculated for a set of IR-ATR data collected through a synthesis of 4a-3py. | ||
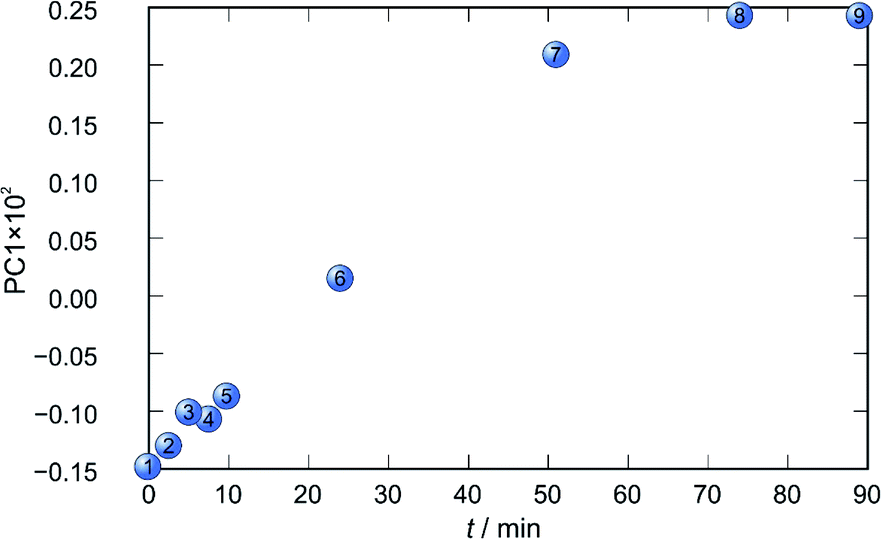 | ||
| Fig. 12 Time dependence of PC1 scores calculated for a set of IR-ATR data collected through a synthesis of 4a-4py. | ||
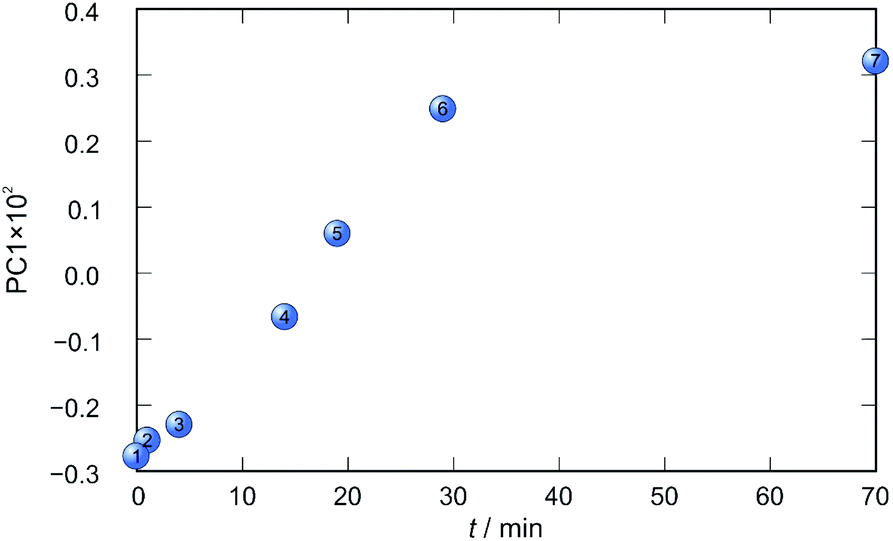 | ||
| Fig. 13 Time dependence of PC1 scores calculated for a set of IR-ATR data collected through a synthesis of 4b-3py. | ||
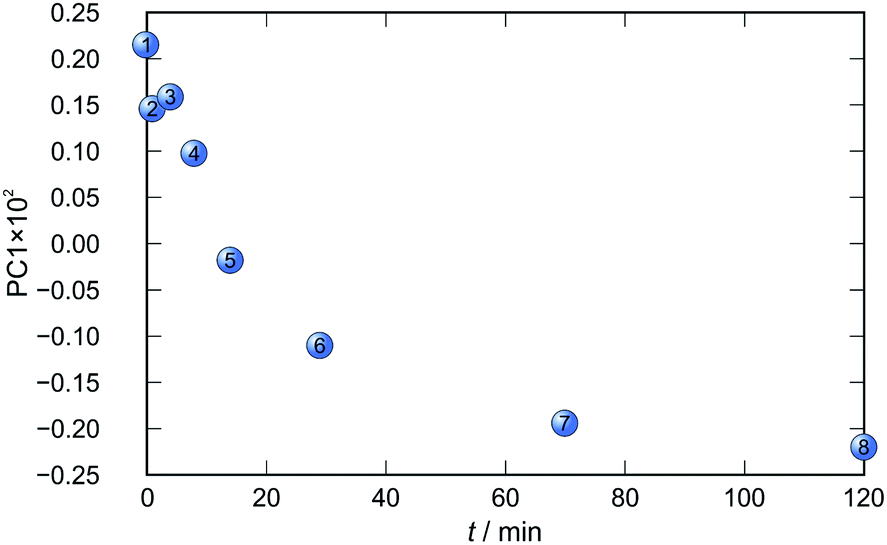 | ||
| Fig. 14 Time dependence of PC1 scores calculated for a set of IR-ATR data collected through a synthesis of 4b-4py. | ||
Investigation of spectral patterns confirmed that reactions were completed in the time predicted by the PC1 reaction profiles. A comparison of the NMR spectra is given in Fig. S27, see ESI†. Singlets observed around 5.8 ppm, assigned to –NH2 protons in the spectra of 4a and 4b, are missing in the spectra of the hydrazone-Schiff bases.
Reactions of aldehydes with hydrazine in the molar ratio 2:1 – synthesis of azines
To gain more insight into a variety of synthetic outcomes for the reactions carried out in the molar ratio of 2:1, hydrazine was used instead of hydrazides. The azine derivatives 5a and 5b (Fig. 15) were obtained by condensing 2,3- and 2,4-dihydroxybenzaldehyde with hydrazine, respectively (yield: 84%). Crystals of 5b·H2O were obtained upon crystallization from acetonitrile (Fig. 16). In 5b·H2O parallel sheets are mutually interconnected via water molecules (Fig. 17). NMR spectra with the NMR numbering scheme are given in Fig. S28–S30 and Table S8, see ESI†
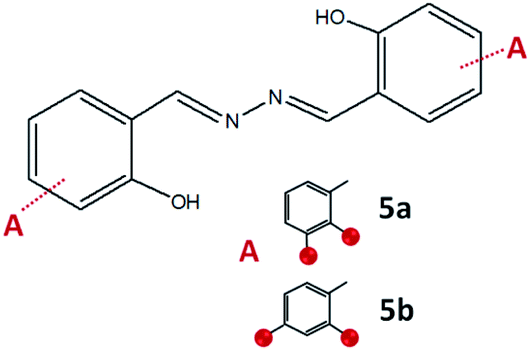 | ||
| Fig. 15 Compounds obtained by the reaction of azine and 2,3-dihydroxybenzaldehyde, 5a, and 2,4-dihydroxybenzaldehyde, 5b. Red sphere presents OH group. | ||
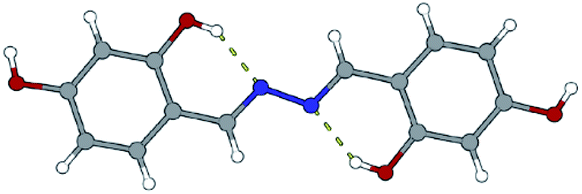 | ||
| Fig. 16 Molecular structures of compound 5b·H2O. The solvent molecule was excluded because of clarity. Atoms are shown as spheres of arbitrary small radii. The intramolecular hydrogen bonds are indicated as an array of yellow dotted lines. | ||
 | ||
| Fig. 17 Crystal packing of compound 5b·H2O. Molecules are connected with an extensive network of hydrogen bonds. Parallel sheets are mutually interconnected via water molecules. The view is projected down the crystallographic c axis. | ||
Thermal behaviour
All compounds, except solvates, remained stable in the solid state for a longer time at RT. The azine 5b demonstrated the largest thermal stability and the lowest one was detected for the quinazolines 3c and 3d (see ESI†). All compounds were analysed with TG-FTIR through heating the samples from 25 to 400 °C. It is seen that methanol is present in the structure of quinazolines 3c·MeOH and 3d·MeOH, confirming the previous assumption seen through IR-ATR.TG-FTIR analysis of compounds 4a–4d, as well as 5a and 5b, indicates absorption bands characteristic for ammonia in the IR spectra during decomposition of the mentioned compounds caused by heating. On the other hand, the TG-FTIR spectra of the compounds 1a–3a and 1b–3b did not provide results that could be easily interpreted.
The solid-state thermochromic behaviour
Since hydrazone-Schiff bases are intensively coloured we aimed to investigate thermochromic behavior of all 2:1 compounds. Interestingly, pronounced solid-state thermochromic changes in the solid state were observed only for azine 5a. The beige crystals of 5a, were found to turn orange-red when heated slowly above room temperature up to 225 °C (Fig. 18). The initial colour reverted on cooling to room temperature. The gradual colour changes were observed repeatedly on heating and subsequent cooling.
 | ||
| Fig. 18 The solid-state thermochromic behaviour of 5a. The images of the temperature effects on the appearance of azine 5a were taken when sample was heated in Kugelror oven. The beige colour is recovered after it was cooled to room temperature. | ||
To elucidate the thermal behaviour of 5a, several series of cyclic DCS experiments were conducted. The thermograms, during several heating runs from 25 °C to 225 °C, did not exhibit any peaks and therefore phase transformations could be excluded. It is, therefore, reasonable to assume that the gradual colour changes could be caused by changes in the intermolecular interactions.61
In all crystal structures, molecules are almost planar, and a similar scenario could be assumed also for 5a. We assume that at higher temperatures, the increased unit cell volume and the distance between assembled molecules could influence the charge delocalization.
The colour change of 5b·H2O upon heating up to 300 °C was less pronounced (from yellow to light orange). This is most probably influenced by the distance between molecules in the crystal structure. Thermochromic changes were not observed for hydrazone-Schiff bases, implying the importance of the aldazine –CN–NC– unit towards the external stimuli.
In vitro cytotoxic and antibacterial activity
The results obtained by the MTS assay are given in Table S9, see ESI†. Due to the IC50 values higher than 100 μmol L−1, all the tested compounds can be considered noncytotoxic against HepG2 cells. Only compounds 3c and 4d with the IC50 values of 69.18 and 22.46 μmol L−1 can be considered somewhat cytotoxic against THP-1 cells. No cytotoxic effect on HepG2 cells had also been established for the related aroylhydrazones derived from salicylaldehyde, 3- or 4-methoxysalicylaldehyde.57,62Unlike the tested hydrazones (1a–4a and 1b–4b), these related hydrazones had been reported to exhibit weak to moderate cytotoxicity against THP-1 cells.57,62 Several observations can be highlighted about the factors affecting the cytotoxic activity of hydrazones on THP-1 cells. To specify, introducing the hydroxy group on position 3- and 4- of the salicylaldehyde moiety significantly lowered the cytotoxic activity. Replacing the 3- or 4-methoxy group by a hydroxy one also decreased the hydrazone cytotoxicity. In addition, the cytotoxicity of hydrazones diminished upon introduction of nitrogen into the benzhydrazide moiety regardless of whether the nitrogen is a part of the ring or the amino group.
In general, as seen from the high MIC values (Table S10, see ESI†), the investigated compounds did not inhibit the growth of Gram-positive bacteria. Regarding the Gram-negative bacteria, the majority of tested compounds (excluding the azines) exhibited a weak growth-inhibitory effect on E. coli while most of them showed moderate antibacterial activity against M. catarrhalis. However, the observed activity of the tested compounds on M. catarrhalis was substantially lower compared to the reference antibiotic azithromycin.
The only hydrazone that exhibited broad-spectrum activity was 2a. Interestingly, hydrazone 2b that differs from 2a only in the position of a hydroxy group was found to be inactive against all selected bacterial strains. The influence of the hydroxy group position on the anti-M. catarrhalis activity observed in hydrazones derived from isonicotinic, 2- or 4-aminobenzoic acid hydrazide is not as pronounced as in hydrazones 2a and 2b. Specifically, the anti-M. catarrhalis activity of 4a is four times greater than the activity of 4b while the MIC values for 1a and 1b as well as for 3a and 3b differ even less. Also, a slightly greater inhibitory effect of 4- over 3-hydroxy isomer was observed only for 2-aminobenzoylhydrazones (3a and 3b).
The higher antibacterial activity against Gram-negative bacteria of the 3-hydroxy isomer, when compared to the corresponding 4-hydroxy isomer, was established not only for hydrazones but also for all other investigated types of compounds. Moreover, the azine containing the 3-hydroxy groups (5a) was one of the most efficient tested compounds while the azine with 4-hydroxy groups (5b) did not inhibit the growth of M. catarrhalis (Table S9†).
Quinazolinones 3c and 3d and Schiff bases 4c and 4d, were generally more active against M. catarrhalis than the corresponding hydrazones (3a, 3b, 4a, and 4b, respectively).
Conclusions
The results described in this paper demonstrate that a specific condensation reaction can take place preferentially by using different green synthetic methodologies. The use of mechanochemical synthesis is beneficial in terms of ease of work-up and high yields, in comparison to solution-based synthesis. It also represents a preferable synthetic procedure for obtaining 4(3H)-quinazolinone core products. But in the case of the hydrazones, it has a limited application. Nevertheless, the solid-state melt reaction offers an easy pathway to obtain hydrazones that cannot undergo further condensation reactions. However, in order to analyse the molecular and crystal structures, solution-based methods are still beneficial.Mild reaction conditions are necessary for the post-synthetic modification of 4-aminobenzhydrazones. Furthermore, vapour-mediated synthesis is the superior approach to convert the primary amine group into an imine functionality and to avoid transimination processes.
Mechanochemical and vapour-mediated reactions can be successfully followed by ex situ PXRD and IR-ATR methods thus avoiding the use of a solution-phase spectroscopic analysis and possibility of further reaction occurrence. Implementation of principal component analysis provided a detailed insight into the reaction profiles and detection of reaction end.
The azines exhibited temperature-responsive colour change. The central unit and position of functional groups in molecules affects their response.
Quinazolinones and Schiff bases are generally more active against M. catarrhalis than the corresponding hydrazones. Introducing the hydroxy group on position 3- and 4- of the salicylaldehyde moiety significantly lowered the cytotoxic activity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has been fully supported by Croatian Science Foundation under the project IP-2016-06-4221. The authors are grateful to Dr K. Molčanov for his assistance in performing some XRD experiments. We extend our thanks to students M. Razum, J. Mihalinec, and A. Bafti for conducting some experiments.Notes and references
- D. Kumar, N. Maruthi Kumar, S. Ghosh and K. Shah, Bioorg. Med. Chem. Lett., 2012, 22, 212–215 CrossRef CAS.
- B. Yadagiri, U. D. Holagunda, R. Bantu, L. Nagarapu, V. Guguloth, S. Polepally and N. Jain, Bioorg. Med. Chem. Lett., 2014, 24, 5041–5044 CrossRef CAS.
- S. S. Machakanur, B. R. Patil, D. S. Badiger, R. P. Bakale, K. B. Gudasi and S. W. A. Bligh, J. Mol. Struct., 2012, 1011, 121–127 CrossRef CAS.
- T. Nasr, S. Bondock and M. Youns, Eur. J. Med. Chem., 2014, 76, 539–548 CrossRef CAS.
- K. Vasantha, G. Basavarajaswamy, M. Vaishali Rai, P. Boja, V. R. Pai, N. Shruthi and M. Bhat, Bioorg. Med. Chem. Lett., 2015, 25, 1420–1426 CrossRef CAS.
- B. Çakır, Ö. Dağ, E. Yıldırım, K. Erol and M. F. Şahin, J. Fac. Pharm. Gazi., 2001, 18, 99–106 Search PubMed.
- S. Şenkardeş, N. Kaushik-Basu, İ. Durmaz, D. Manvar, A. Basu, R. Atalay and Ş. G. Küçükgüzel, Eur. J. Med. Chem., 2016, 108, 301–308 CrossRef.
- A. Inam, S. M. Siddiqui, T. S. Macedo, D. R. M. Moreira, A. C. L. Leite, M. B. P. Soares and A. Azam, Eur. J. Med. Chem., 2014, 75, 67–76 CrossRef CAS.
- R. J. Butcher, J. P. Jasinski, B. Narayana, K. Sunil and H. S. Yathirajan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o4025–o4026 CrossRef CAS.
- A. Mandal and B. K. Patel, J. Mol. Struct., 2018, 1155, 78–89 CrossRef CAS.
- X.-S. Wang, J. Sheng, L. Lu, K. Yang and Y.-L. Li, ACS Comb. Sci., 2011, 13, 196–199 CrossRef CAS.
- M. Ikram, S. Rehman, F. Subhan, M. N. Akhtar and M. O. Sinnokrot, Open Chem., 2017, 15, 308–319 CAS.
- D. Tinguiano, A. Sy, I. E. Thiam, M. Gaye and P. Retailleau, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2374–o2375 CrossRef CAS.
- K. B. Gudasi, S. A. Patil, R. S. Vadavi, R. V. Shenoy and M. Nethaji, Transition Met. Chem., 2006, 31, 586–592 CrossRef CAS.
- A. C. Tinker, H. G. Beaton, N. Boughton-Smith, S. L. Cooper, L. Fraser-Rae, K. Hallam, P. Hamley, T. McInally, D. J. Nicholls, A. D. Pimm and A. V. Wallace, J. Med. Chem., 2003, 46, 913–916 CrossRef CAS.
- F. Fülöp, M. F. Simeonov and K. Pihlaja, Tetrahedron, 1992, 48, 531–538 Search PubMed.
- K. B. Gudasi, R. S. Vadavi, R. V. Shenoy, S. A. Patil and M. Nethaji, Transition Met. Chem., 2006, 31, 374–381 CAS.
- K. B. Gudasi, R. S. Vadavi, R. V. Shenoy, S. A. Patil and M. Nethaji, Transition Met. Chem., 2006, 31, 135–145 CrossRef CAS.
- R. S. Hunoor, B. R. Patil, D. S. Badiger, R. S. Vadavi, K. B. Gudasi, C. V. Magannavar and I. S. Muchand, Chem. Pharm. Bull., 2010, 58, 712–716 CrossRef CAS.
- P. F. M. Oliviera, M. Baron, A. Chamayou, C. André-Barrès, B. Giidetti and M. Baltas, RSC Adv., 2014, 4, 56736 RSC.
- S. Kuntikana, C. Bhat, M. Kongot, S. I. Bhat and A. Kumar, ChemistrySelect, 2016, 1, 1723–1728 CrossRef CAS.
- F. H. Allen, Acta Crystallogr. B, 2002, 58, 380 CrossRef , The Cambridge Structural Database, V5.30.
- E. Tecer, N. Dege, A. Zülfikaroğlu, N. Senyüz and H. Batı, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o3369–o3370 CrossRef CAS.
- N. Dege, N. Senyüz, H. Batı, N. Günay, D. Avcı, Ö. Tamer and Y. Atalay, Spectrochim. Acta, Part A, 2014, 120, 323–331 CrossRef CAS.
- D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 1958–1972 CrossRef CAS.
- M. Taha, M. S. Baharudin, N. H. Ismail, S. A. Shah and S. Yousuf, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o3256 CrossRef CAS.
- M. Sutradhar, E. C. B. A. Alegria, K. T. Mahmudov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, RSC Adv., 2016, 6, 8079–8088 RSC.
- H. M. Ali, S. Puvaneswary, W. J. Basirun and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o1013–o1014 CrossRef CAS.
- H. M. Ali, S. Puvaneswary, W. J. Basirun and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o1083–o1084 CrossRef CAS.
- M. Albrecht, I. Latorre, Y. Liu and R. Fröhlich, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 311–316 CAS.
- P. Promdet, J. Horkaew, S. Chantrapromma and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o3224 CrossRef CAS.
- Y.-M. Chen, Q.-R. Zeng, Y. Li, J.-N. Lin, J.-M. Wang, B. Qu and Q.-F. Xie, Chin. J. Inorg. Chem., 2016, 1398–1404 CAS.
- M. Mohanraj, G. Ayyannan, G. Raja and C. Jayabalakrishnan, Mater. Sci. Eng. C, 2016, 69, 1297–1306 CrossRef CAS.
- C. Arunagiri, A. G. Anitha, A. Subashini and S. Selvakumar, J. Mol. Struct., 2018, 1163, 368–378 CrossRef CAS.
- M. Taha, N. H. Ismail, H. M. Zaki, A. Wadood, E. H. Anouar, S. Imran, B. M. Yamin, F. Rahim, M. Ali and K. M. Khan, Bioorg. Chem., 2017, 75, 235–241 CrossRef CAS.
- N. M. Lair, H. Khaledi and H. M. Ali, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o459 CrossRef CAS.
- M. Sánchez-Lozano, E. M. Vázquez-López, J. M. Hermida-Ramón and C. M. Estévez, Polyhedron, 2011, 30, 953–962 CrossRef.
- J. D. Siqueira, A. C. O. Menegatti, H. Terenzi, M. B. Pereira, D. Roman, E. F. Rosso, P. C. Piquini, B. A. Iglesias and D. F. Back, Polyhedron, 2017, 130, 184–194 CrossRef CAS.
- H. S. Naveenkumar, A. Sadikun, P. Ibrahim, W.-S. Loh and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o1202–o1203 CrossRef CAS.
- Z. Wei, J. Wang, X. Jiang, Y. Li, G. Chen and Q. Xie, Chin. J. Appl. Chem., 2015, 32, 1014–1021 CAS.
- X'Pert Software Suite, Version 1.3e, Panalytical B. V., Almelo, The Netherlands, 2001 Search PubMed.
- CrysAlis Software system, Version 1.171.33.32, Xcalibur CCD system, Oxford Diffraction Ltd, Abingdon, UK, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. A, 2008, 64, 112–122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst., 2006, 39, 453–457 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC Search PubMed.
- http://www.povray.org/.
- T. Hrenar, I. Primožič, D. Fijan and M. Majerić Elenkov, Phys. Chem. Chem. Phys., 2017, 19, 31706–31713 RSC.
- E. Beltrami, Giornale di Matematiche ad Uso degli Studenti Delle Università, 1873, vol. 11, pp. 98–106 Search PubMed.
- K. Pearson, Philos. Mag., 1901, 2, 559–572 Search PubMed.
- H. Hotelling, J. Educ. Psychol., 1933, 24, 417–441 CrossRef.
- I. T. Jolliffe, Principal Component Analysis, Springer, Berlin, 1986 Search PubMed.
- P. Geladi and B. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
- T. Hrenar, moonee, Program for Manipulation and Analysis of Multi- and Univariate Data, rev. 0.6827, 2017 Search PubMed.
- O. Jović, T. Smolić, I. Primožič and T. Hrenar, Anal. Chem., 2016, 88, 4516–4524 CrossRef.
- J. Pisk, T. Hrenar, M. Rubčić, G. Pavlović, V. Damjanović, J. Lovrić, M. Cindrić and V. Vrdoljak, CrystEngComm, 2018, 20, 1804–1817 RSC.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Eight edition, CLSI document M07-A8, Clinical and Laboratory Standards Institute, Wayne, USA, 2009 Search PubMed.
- V. Vrdoljak, B. Prugovečki, I. Primožič, T. Hrenar, D. Cvijanović, J. Parlov Vuković, R. Odžak, M. Skočibušić, S. Prugovečki, J. Lovrić, D. Matković-Čalogović and M. Cindrić, New J. Chem., 2018, 42, 11697–11707 RSC.
- P. Naumov, S. C. Lee, N. Ishizawa, Y. G. Jeong, I. H. Chung and S. Fukuzumi, J. Phys. Chem. A, 2009, 113, 11354–11366 CrossRef CAS.
- M. Cindrić, A. Bjelopetrović, G. Pavlović, V. Damjanović, J. Lovrić, D. Matković-Čalogović and V. Vrdoljak, New J. Chem., 2017, 41, 2425–2435 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Analytical and spectral data, (2) view of structures and packing diagrams, (3) crystallographic data, and (4) powder diffraction patterns. Crystallographic data sets for the structures 1a·MeOH, 1b·H2O, 3b, 3c, 4a, 4b·MeOH, 4c, and 5b·H2O. CCDC 1985275–1985283. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06845d |
| This journal is © The Royal Society of Chemistry 2020 |