
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light photooxidation in water by 1O2-generating supramolecular hydrogels†
Sankarsan
Biswas
 abc,
Mohit
Kumar
ab,
Andrew M.
Levine
abc,
Ian
Jimenez
ae,
Rein V.
Ulijn
*abcd and
Adam B.
Braunschweig
*abcd
abc,
Mohit
Kumar
ab,
Andrew M.
Levine
abc,
Ian
Jimenez
ae,
Rein V.
Ulijn
*abcd and
Adam B.
Braunschweig
*abcd
aAdvanced Science Research Center, Graduate Center, City University of New York, 85 St. Nicholas Terrace, New York, NY 10031, USA. E-mail: abraunschweig@gc.cuny.edu
bDepartment of Chemistry, Hunter College, 695 Park Avenue, New York, NY 10065, USA
cPhD Program in Chemistry, Graduate Center, City University of New York, New York, NY 10016, USA
dPhD Program in Biochemistry, Graduate Center, City University of New York, New York, NY 10016, USA
eThe High School for Math, Science, and Engineering, The City College of New York, 240 Convent Avenue, New York, NY 10031, USA
First published on 3rd April 2020
Abstract
We report supramolecular photocatalytic hydrogels, produced by the enzymatically driven self-assembly of low molecular weight gelators (LMWGs). These LMWG precursors are composed of the organic chromophore diketopyrrolopyrrole (DPP), which is bi-functionalized with a series of amino acid (Phe, Tyr, Leu) methyl esters. In situ enzymatic hydrolysis of these photoactive precursors results in supramolecular hydrogels that provide a high density of photocatalytic sites. Under visible light irradiation these hydrophobic fibers recruit the reaction substrates and also produce 1O2, which is used here for the photooxidation of thioanisole (aromatic substrate) and cyclohexyl methyl sulfide (aliphatic substrate), with yields as high as 100% and without over-oxidation. Finally, we demonstrate that the nature of the amino acids in the LWMGs has a central role in dictating J-/H-/mixed state aggregates, gel properties, and, hence, the efficiency of chemoselective photooxidation of thioanisole and cyclohexyl methyl sulfide inside these hydrogels.
Introduction
There is a growing need for new catalytic methods that proceed photochemically under visible light illumination, are compatible with aqueous solvents, and avoid the need for expensive heavy metals, thereby reducing detrimental environmental impacts.1–9 In this context, organic photocatalysts that operate in aqueous solvents have been the focus of considerable recent interest as sustainable alternatives to conventional photocatalysts.10–14 To date, these photocatalysts/photosensitizers are primarily porphyrin-based, and their photocatalytic applications are mostly limited to water-soluble substrates.There remain several substantial challenges that have precluded the broader adoption of aqueous photocatalysts, including their inability to act upon hydrophobic molecules in aqueous media and the lack of alternatives to porphyrin-based photocatalytic systems that work efficiently in water. Catalytic supramolecular hydrogels provide a promising solution to this challenge.15–19 Hydrogel networks can address the solubility problem because hydrophobic aromatic compounds often associate into the nanofiber network in water and become reactively accessible.20 For example, the Meijer21 and Escuder22 groups have shown that a self-assembled, 1D fiber network enhances catalytic activity compared to its non-assembled counterpart.23–28 Most recent examples of hydrogel photocatalysts29 involve co-assembling of the photocatalysts with gelators, where catalysts and gelators are two separate components. These hydrogel photocatalysts were used for the oxidation of iodide,30 hydrogen production,31 polymer crosslinking,32 and artificial photosynthesis.33,34 There remain, however, several drawbacks with hydrogel photocatalysts, including the limited control of positioning and accessibility of photoactive sites and the possibility of dissociation of non-covalently bound photoactive sites from the gel. We propose that by making the catalyst an integral and inseparable component of the gelator itself, supramolecular photocatalysts could be produced with increased number of photoactive sites and enhanced stability.
Here, we report a new approach to aqueous photocatalysis composed of organic chromophores that assemble into chiral supramolecular hydrogels, where the catalytic site is itself both a structural and functional component of the photoactive gel. The assembly of the gels is driven by aromatic amino acid bola-amphiphiles35 that form in a controlled manner through biocatalytic self-assembly that occurs following hydrolysis of methyl esters in the presence of α-chymotrypsin.36,37 This versatile and reproducible biocatalytic assembly approach previously provided functional hydrogels by avoiding productive kinetic aggregates, and is especially relevant to self-assembly involving poorly soluble functional components.38,39 Here, the precursors for these low molecular weight gelators (LMWGs) are composed of the chromophore diketopyrrolopyrrole (DPP) with amino acid methyl esters connected through a linker appended to the heterocyclic N (Fig. 1). The self-assembly is activated in situ through enzymatic ester hydrolysis leading to rebalancing of amphiphilicity to favor unidirectional assembly. Aromatic components provide hydrophobic contacts and π⋯π interactions, and the amino acid residues provide chirality and H-bonding, leading to the formation of 1D superstructures in water. The ability to vary the amino acid side chain provides a versatile route to explore different superstructures.40 DPP was chosen as the chromophore as it has been used previously to generate 1O2 upon irradiation with visible light,41–45 which is a powerful oxidizing agent in catalysis.46–48
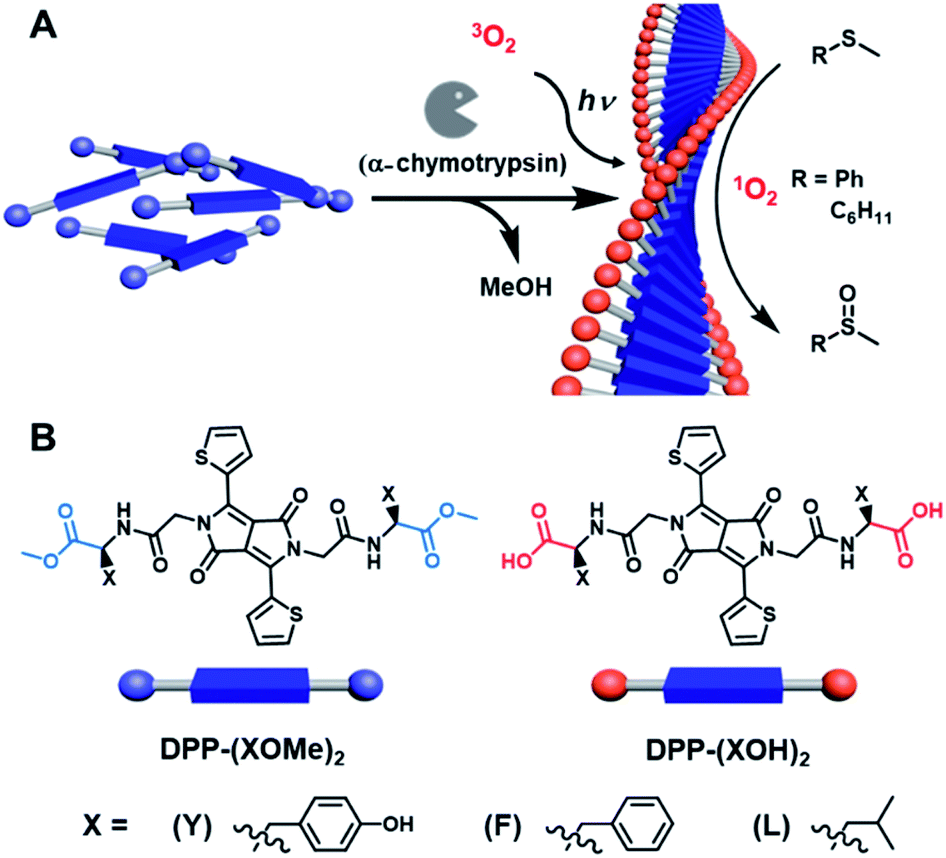 | ||
| Fig. 1 Visible-light photooxidation enabled by a supramolecular hydrogel. (A) Biocatalytic self-assembly of the LMWGs into superstructures via in situ enzymatic hydrolysis of DPP-(XOMe)2 with α-chymotrypsin. These superstructures were used for the photooxidation of thioanisole and cyclohexyl methyl sulfide via1O2 formation in water. (B) Photoactive precursors, DPP-(XOMe)2 and components, DPP-(XOH)2, of the supramolecular hydrogels. | ||
Here we demonstrate the utility of this catalytic platform by oxidizing thioanisole and cyclohexyl methyl sulfide to their corresponding sulfoxides in water, which is a relevant model system because sulfoxides are precursors in the synthesis of several pharmaceuticals and many common preparations suffer from overoxidation to the sulfone.46,49 We have assessed the catalytic activities of three such DPP-based supramolecular hydrogels that vary in their amino acid side chains, where they include either tyrosine (Y), phenylalanine (F) or leucine (L). The two aromatic amino acids were chosen as they are prone to supramolecular gelation,50 and aliphatic leucine (L) was selected as a control because it forms fibers but is not prone to gelation. We found that the hydrogel state plays a vital role in facilitating catalysis, presumably by sequestering the hydrophobic thioanisole and cyclohexyl methyl sulfide inside the fiber networks. This study demonstrates how the incorporation of photocatalysts in conjunction with hydrogel formation can play a vital role in photooxidation of hydrophobic substrates in water, towards sustainable and environmentally benign visible-light photocatalysis.
Results and discussion
The design of our LMWGs requires a chromophore that would produce long-lived triplets in response to light, which can generate 1O2, and to which amino acids could be readily appended to direct subsequent aqueous supramolecular assembly. Achieving controlled photooxidation by using dissolved oxygen in aqueous media poses a number of significant challenges because of the low solubility and short 1O2 lifetime.51,52 Photosensitizers with long excited state lifetimes facilitate efficient energy transfer, converting 3O2 into 1O2, thereby increasing catalytic efficiency. DPP was selected as the photosensitizer because it has been shown previously to generate 1O2 in the context of photodynamic therapy.45 It is also known that triplet lifetimes for DPP increase from ps53 to μs54 upon assembly because the excited state of DPP has the ability to undergo singlet fission. Although, DPP has been used in the context of organic semiconductors and solar cells,55,56 to our knowledge it has not yet been used for aqueous catalysis or photocatalysis.All three DPP-(XOMe)2 derivatives were synthesized in four steps following identical procedures. First, the DPP core was prepared by Dieckmann condensation of dimethyl succinate and thiophene-2-carbonitrile with 87% yield. The core was then functionalized at the heterocyclic N with tert-butyl acetate via an SN2 substitution with 47% yield. The diester was then hydrolyzed with trifluoroacetic acid in 97% yield, and the final diacid was then functionalized with different amino acids via HBTU coupling in 81% yield for DPP-(YOMe)2 (1Y), 84% yield for DPP-(FOMe)2 (1F), and 81% yield for DPP-(LOMe)2 (1L). L-Enantiomers of the amino acids were incorporated to facilitate subsequent α-chymotrypsin catalysed hydrolysis. All LMWG precousors were characterized by 1H NMR and 13C NMR spectroscopy, and high-resolution mass spectrometry, and all data were consistent with the proposed structures.
Changing amino acids provided access to distinct supramolecular structures for investigating how subtle changes in assembly affect the photophysical properties and catalytic performance of the hydrogels. The assembly was studied by high performance liquid chromatography (HPLC), UV-Vis, fluorescence, and circular dichroism (CD) spectroscopies, transmission electron microscopy (TEM), atomic force microscopy (AFM), and rheology. The DPP heterocycle is hydrophobic and insoluble in most organic and aqueous solvents, and, consequently, the DPP-(XOMe)2 derivatives have limited solubility in aqueous solvents. Thus to make the hydrogels, 200 mM stock solutions of LMWGs 1Y and 1L were prepared in DMSO and then diluted with 100 mM phosphate buffer (pH 8) containing 1 mg mL−1 α-chymotrypsin (Sigma, lyophilized power, ≥40 units per mg protein). 1F was sparingly soluble in DMSO at room temperature, so rather than creating a stock solution, it was directly weighed in a vial and DMSO was added followed by heating until solubilized to create a gel. To this hot DMSO solution, phosphate buffer was added and the solution was sonicated. After the solution reached room temperature, enzyme in phosphate buffer was added to a final concentration of 1 mg mL−1. 10 mM solutions of DPP-(YOH)2 (3Y) and DPP-(FOH)2 (3F) formed self-supporting hydrogels after 1 h for 3Y and 2 h for 3F, while the DPP-(LOH)2 (3L) did not gel. All enzymatic hydrolysis reactions were monitored by HPLC to assess the reaction kinetics and yields. The hydrolysis of 1Y took 7 h to complete (confirmed by LC-MS, Fig. 2C and S16†), whereas 3L and 1F took three days for complete hydrolysis, suggesting that gelation occurs well before hydrolysis is completed (Fig. S17 and S18†).
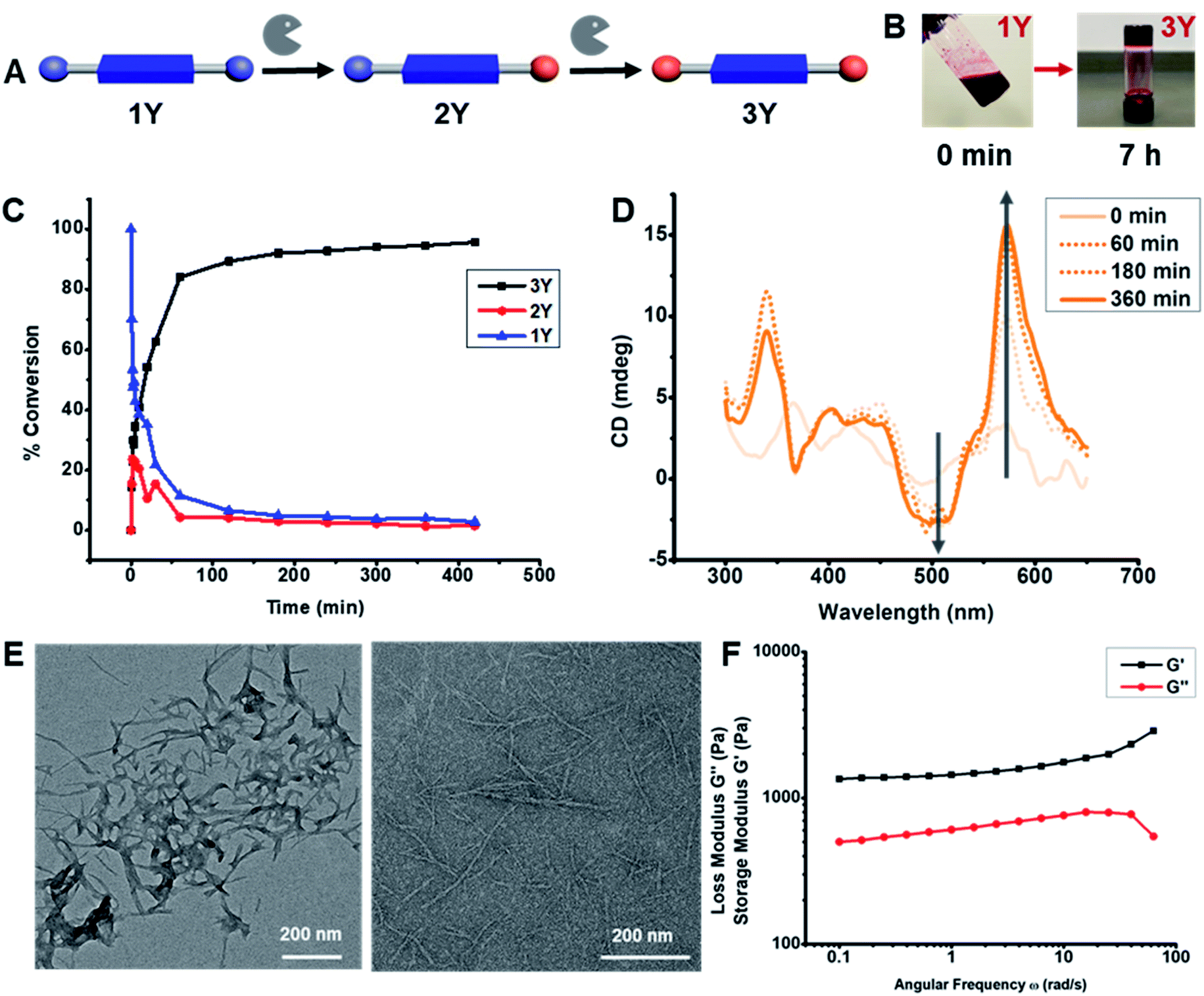 | ||
| Fig. 2 (A) Enzymatic hydrolysis of DPP-(YOMe)2 in phosphate buffer (pH 8, 100 mM, 5% DMSO) in the presence of α-chymotrypsin (1 mg mL−1). (B) Photograph of reaction vial at 0 min (left) and at 7 h (right). (C) HPLC analysis of enzymatic hydrolysis of DPP-(YOMe)2 at 10 mM. (D) CD spectra showing the formation of homochiral superstructure during the enzymatic hydrolysis of DPP-(YOMe)2 at 10 mM. (E) TEM images of DPP-(YOMe)2 (left) and DPP-(YOH)2 (right). (F) Oscillatory rheology of the DPP-(YOH)2 (10 mM) gel showing elastic modulus (G′) of 1.7 kPa (angular frequency = 10 rad s−1; frequency sweep). | ||
Because of its faster hydrolysis and gelation, 1Y was used to explore the kinetics and photophysical properties of gelation in greater detail. As 3Y formed a self-supporting gel at 10 mM, the characterization studies were performed at this concentration, unless otherwise noted. We first used HPLC to monitor the kinetics of enzymatic hydrolysis. After exposing 1Y to the enzyme, aliquots of the reaction mixtures were taken periodically and added to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN:H2O mixture for HPLC-MS analysis. All species present in the enzymatic reaction were characterized by high-resolution mass spectroscopy, which, together with HPLC chromatograms, confirmed the presence of 1Y, MeOY-DPP-YOH (2Y) and 3Y in the reaction mixtures. The relative peak integrations showed an increase of 3Y with a decay of 1Y over time (Fig. 2C). The intermediate 2Y was observed within the first 8 min of the reaction, after which 3Y became the dominating species in the reaction mixture. It was found that 92% conversion into 3Y was observed within 3 h, while complete hydrolysis (>96%) took 7 h. Superstructure formation during hydrolysis was monitored by CD spectroscopy. After exposing 1Y to the enzyme, aliquots of the reaction mixtures were taken periodically for CD measurements. A bisignated CD signal was observed with a positive signal from 650 nm to 550 nm followed by a negative signal from 550 nm to 450 nm (Fig. 2D), and signal intensity increased for all peaks over time. This bisignated cotton effect is a hallmark of dyes that are aggregated into homochiral helical superstructures.57,58 Signal intensity approached saturation after 3 h, when HPLC data suggests that 92% of the hydrolysis is completed. Similarly, aliquots of the reaction mixtures of 3F and 3L were analyzed by CD after 3 d of exposure to α-chymotrypsin. When comparing CD spectra for DPP-(XOH)2, it was found that 3L and 3Y have similar bisignated CD signals between 650–450 nm, whereas the CD signal of 3F had a negative signal from 650 nm to 560 nm followed by a positive signal from 560 nm to 525 nm (Fig. S20†). The different CD signal for 3F, confirms that superstructure geometry is dependent upon F, Y, L amino acid side chains.
:1 CH3CN:H2O mixture for HPLC-MS analysis. All species present in the enzymatic reaction were characterized by high-resolution mass spectroscopy, which, together with HPLC chromatograms, confirmed the presence of 1Y, MeOY-DPP-YOH (2Y) and 3Y in the reaction mixtures. The relative peak integrations showed an increase of 3Y with a decay of 1Y over time (Fig. 2C). The intermediate 2Y was observed within the first 8 min of the reaction, after which 3Y became the dominating species in the reaction mixture. It was found that 92% conversion into 3Y was observed within 3 h, while complete hydrolysis (>96%) took 7 h. Superstructure formation during hydrolysis was monitored by CD spectroscopy. After exposing 1Y to the enzyme, aliquots of the reaction mixtures were taken periodically for CD measurements. A bisignated CD signal was observed with a positive signal from 650 nm to 550 nm followed by a negative signal from 550 nm to 450 nm (Fig. 2D), and signal intensity increased for all peaks over time. This bisignated cotton effect is a hallmark of dyes that are aggregated into homochiral helical superstructures.57,58 Signal intensity approached saturation after 3 h, when HPLC data suggests that 92% of the hydrolysis is completed. Similarly, aliquots of the reaction mixtures of 3F and 3L were analyzed by CD after 3 d of exposure to α-chymotrypsin. When comparing CD spectra for DPP-(XOH)2, it was found that 3L and 3Y have similar bisignated CD signals between 650–450 nm, whereas the CD signal of 3F had a negative signal from 650 nm to 560 nm followed by a positive signal from 560 nm to 525 nm (Fig. S20†). The different CD signal for 3F, confirms that superstructure geometry is dependent upon F, Y, L amino acid side chains.
The biocatalytic self-assembly of 1Y was also monitored by UV-Vis (Fig. S21†) and fluorescence (Fig. S22†) spectroscopies to observe the photophysical changes that occurred upon their transformation into 1D-superstructures. To monitor the changes of the UV-Vis spectra over time, the reaction mixture was sonicated and vortexed for 10 s, and aliquots of 1Y (10 mM in 100 mM phosphate buffer pH 8 with 5% DMSO and 1 mg mL−1 enzyme) were periodically analyzed. Time dependent UV-Vis spectroscopy revealed a change in the intensity ratio of the characteristic DPP λmax at 512 nm and 540 nm, corresponding to the 0–1 and 0–0 transitions, respectively (Fig. S21C†). It has been previously reported that H- and J-aggregates can be differentiated for the DPP dyes based upon the ratio of 0–1/0–0 vibronic peaks, where a higher 0–1 peak indicates H-aggregates, and a higher 0–0 peak is suggestive of J-aggregates.59–61 By monitoring the change in this ratio during hydrolysis it was found that the ratio went from more than unity (t < 40 min) to less than unity (t > 40 min), suggesting that J-aggregates form upon biocatalytic hydrolysis and assembly. Comparative analysis of the UV-Vis spectra (Fig. S21D†) of all DPP-(XOH)2 showed that 3L has a sharper absorbance and less scattering compared to 3F and 3Y which have much broader features and more scattering in the UV-Vis spectrum which could be related to the relative degrees of gelation. The final 0–1/0–0 ratios of all DPP-(XOH)2 are also dependent upon the amino acids. These data suggest that 3L formed H-aggregates, whereas 3Y is a J-aggregate, and 3F is a mixed state. Time dependent fluorescence for the hydrolysis of 1Y (λex = 450 nm, 10 mM) showed that the intensity at 557 nm and 601 nm increased for the first 50 min, after which intensity gradually decreased until 100 min, when hydrolysis is nearly completed (Fig. S22†). Comparison of the fluorescence spectra of all three DPP-(XOH)2, taken after 1 d for 3Y and 3 d for 3F and 3L, showed that 3L has the highest emission intensity among all DPP-(XOH)2 (Fig. S23†), which can be explained because aggregation of DPP leads to quenching,62 and the most weakly assembled hydrogel has the least aggregation-induced quenching. 3F, in contrast showed the least emission intensity, suggesting the formation of a highly aggregated species (Fig. S23†).
Further evidence of superstructure formation was provided by TEM and AFM images of 1Y (Fig. 2E, S10 and S12†) and 3Y (Fig. 2E, S11 and S13†) that were taken at 0 min and at 7 h after exposure to the enzyme. AFM and TEM show fibers with 6.2 ± 1 nm diameters. This diameter is larger than that of the molecular dimension of 3Y (∼2 nm) suggesting bundle formation rather than single molecular stacks. Microscopic evidence of fiber formations for 3L and 3F were also obtained by TEM imaging. Although 1Y transformed into uniform fibers after enzymatic hydrolysis, 3L and 3F formed a distribution of both thin fibers and bundled fibers (Fig. S14 and S15†). These data confirm that all LMWGs formed 1D-superstructures upon enzymatic hydrolysis, although only 1Y and 1F formed hydrogels. Rheology measurements on 3Y and 3F were performed to define viscoelastic moduli and it was found that 3Y has an elastic moduli (G′) of 1.7 kPa (angular frequency = 10 rad s−1) in the frequency sweep (Fig. 2F). Rheology measurements on 3F showed the formation of a much stronger hydrogel (G′ of 21 kPa; angular frequency = 10 rad s−1) compared to 3Y (Fig. S26†).
Next, all these DPP-(XOH)2 were then tested for their ability to produce 1O2 upon photoirradiation. 1O2 yields for these DPP-(XOH)2 catalysts were determined by following a previously reported indirect method, where methylene blue (MB) was used as a standard photosensitizer and 1,3-diphenylisobenzofuran (DPBF) as an indicator.63 Please note that due to high extinction coefficient of DPP, we were unable to use this method in water at the gelation concentration. All catalysts (3Y, 3F, 3L) were first dissolved in DMSO, then DPBF and the catalysts were mixed in a 1:1 ratio and saturated with air. These mixtures were excited for 2 min (λex = 534 nm for 3L, λex = 535 nm for 3Y and λex = 535 nm for 3F) in a fluorimeter, and absorbance were taken immediately after excitation to monitor changes in DPBF intensity. This process was then repeated 5 more times. The decrease in absorbance at 418 nm from the photooxidation of DPBF by 1O2 was then compared with the decrease caused by MB under identical experimental conditions to provide 1O2 yields of 67% for 3Y and 71% for 3L, which is the highest 1O2 yield reported yet for DPP-based molecules,64 and 47% for 3F (Fig. S24†).
We then demonstrated the photooxidation of thioanisole and cyclohexyl methyl sulfide by these supramolecular hydrogels. The challenge associated with sulfoxidation in water is that the precursor sulfides are hydrophobic and generally insoluble in water. The nanofiber network of photocatalysts surrounding them could increase substrate accessibility towards the photocatalysts. The oxidation experiments were performed in 1 mL solutions of catalysts prepared in 5 mL vials via enzymatic hydrolysis of the corresponding DPP-(XOMe)2. Thioanisole and cyclohexyl methyl sulfide were then added into these systems after 7 h for 3Y and after 3 d for 3F and 3L and stirred under positive pressure of air for 3 days. A white light source (150 W Fiber Optic Dual Gooseneck Microscope Illuminator) was used for all photooxidation reactions. To explore the reaction scope and the effect of reaction conditions on yield, we varied solvent, and the amino acid substitutions chain and concentration of DPP-(XOH)2 (Table 1). For initial optimization of the reaction conditions, we used thioanisole as a model substrate. For 3Y it was found that yield increases with increasing concentration of supramolecular catalyst, with an increase in yield from 2% to 30% as [3Y] increases from 5 mM to 10 mM, and a similar trend was observed upon increasing [3F] from 5 mM to 10 mM giving rise to an increase from 14% to 39%. In both cases, gelation was only observed at the higher concentration, indicating that gelation and fiber formation have a positive role in catalysis. The observed yield decreased at [3Y] > 10 mM, which can be explained by the poor light penetration at higher [3Y] as a consequence of the high extinction coefficient of the LMWGs or because of slower diffusion of the reactants through this denser gel. For further confirmation of the positive effect of gelation on catalysis, 10 mM of 3L, which does not form a hydrogel, was examined as a catalyst for the photooxidation of thioanisole. Under the same reaction conditions, the yield at 10 mM (10%) was much lower compared to the gel-forming samples. In addition, a control experiment with soluble 10 mM DPP-acetate (7) was performed which gave rise to a much reduced oxidation yield of 3% compared to that observed in 3Y, 3F and 3L. These observations strongly suggest that both fiber formation and hydrogelation are crucial for the catalysis, presumably because the gel networks trap hydrophobic thioanisole molecules and improve the accessibility towards photoactive sites. All non-assembled/randomly assembled precursors (DPP-(XOMe)2) of these catalysts had lower yields than the assembled forms of these catalysts. Yields were further increased to 87% in 3Y (10 mM) and 67% in 3F (10 mM) by using D2O phosphate buffer as a solvent. It is known that 1O2 has a higher lifetime in D2O (68 μs) vs. H2O (4 μs).65 1:1 (v/v) mixture of D2O and H2O had half the yield (44%) compared to catalysis in 100% D2O. Photooxidation experiments in D2O were only performed for 3Y and 3F as these catalysts gave higher yields in H2O compared to 3L. In order to investigate if the higher yields for 3Y and 3F are related to the interaction of the substrate with aromatic cores of F/Y and thioanisole, we have carried out our photooxidation reaction with cyclohexyl methyl sulfide (aliphatic sulfide) with all three catalysts. These experiments resulted in photooxidation yields of 100%, 94%, and 88% for 3Y, 3F, and 3L respectively. This suggests that 3Y is a better catalyst than 3L for non-aromatic substrates as well, thus suggesting that hydrogelation, or more precisely, the favorable fiber/solvent interactions may indeed have a positive effect in catalysis, although the differences are more pronounced for aromatic substrates. The overall higher yield is not surprising as a similar trend has been reported previously for enantioselective sulfoxidation using a series of flavin–cyclodextrin conjugates of both aromatic and aliphatic suflides.66 To further confirm that this reaction is indeed photocatalyzed, we performed control experiments both with 10 mM of 3Y in dark and without any catalyst. Both of these failed to produce sulfoxide. Another control experiment for the photooxidation of thioanisole, using agarose hydrogel (5 mg mL−1), containing soluble DPP-acetate, 7 (see ESI†), was performed. This experiment resulted in 5% yield of the oxidized product, which is significantly lower than that of self-assembled DPP-hydrogelator photocatalysts. We believe that this experiment clearly suggests the advantages of hydrogelators, where the photocatalyst is itself an integral and inseparable component of the gelator. In addition, we found that the photooxidation proceeds optimally at room temperature with 2.5 mol% catalyst loading. Finally, the absence of any evidence of sulfone in HPLC data showed that this approach is also chemoselective and does not suffer from over oxidation.67–70
|
|
|||
|---|---|---|---|
| Catalyst | [Catalyst] (mM) | Substrate | Yield (%) |
| a Unless otherwise noted, all reactions were conducted with 1.0 mL of reaction solutions and were used for photooxidation after enzymatic hydrolysis of corresponding DPP-(XOMe)2 without any further purification. Thioanisole and cyclohexyl methyl sulfide were added to these solutions and stirred under positive pressure of air for 48 h. Final concentration of the sulfides were 0.4 mM in the hydrogel. The solution was irradiated with a white halogen light that was connected to the reaction chamber with an optical fiber and reactions were performed at rt. All yields were calculated from HPLC. | |||
| — | 0 | R = Ph | 0 |
| 7 | 10 | R = Ph | 5 |
| 3Y | 1 | R = Ph | 1 |
| 3Y | 5 | R = Ph | 2 |
| 3Y | 10 | R = Ph | 30 |
| 3Y | 10 (1:1 D2O/H2O) |
R = Ph | 44 |
| 3Y | 10 (D2O) | R = Ph | 86 |
| 3Y | 20 | R = Ph | 6 |
| 3Y | 30 | R = Ph | 5 |
| 3L | 10 | R = Ph | 10 |
| 3F | 5 | R = Ph | 14 |
| 3F | 10 | R = Ph | 39 |
| 3F | 10 (D2O) | R = Ph | 67 |
| 1Y | 10 | R = Ph | 9 |
| 1Y | 20 | R = Ph | 2 |
| 1L | 10 | R = Ph | 5 |
| 1F | 10 | R = Ph | 17 |
| 3Y | 10 | R = C6H11 | 100 |
| 3F | 10 | R = C6H11 | 94 |
| 3L | 10 | R = C6H11 | 88 |
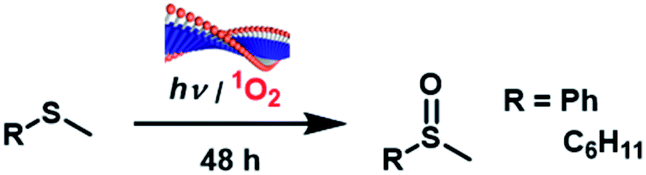
Conclusions
A new strategy for photooxidation that combines supramolecular gelation and catalysis in an effort to simultaneously achieve high yield, circumvent the need for toxic and expensive metals, and operate in water has been studied. A small library of supramolecular catalysts was obtained by varying amino acids appended to the heterocyclic N of the DPP core. Tuning the hydrophobicity of the amino acids enabled us to access different superstructures (H-/J-/mixed state) and photophysical properties. In studying the conditions for the catalytic photooxidation of thioanisole and cyclohexyl methyl sulfide in water, it was found that the favorable solvent/fiber interactions that cause hydrogelation, and supramolecular aggregation of the photocatalyst play a vital role in catalytic efficacy. The effect is more prominent in the case of aromatic substrates. Lower yields for the oxidation of thioanisole in H2O compared to D2O can be explained by the relative low 1O2 lifetimes in the former. This catalyst is designed following the principles of hierarchical structure and emergent photophysics that are common in nature, but not yet widely adopted by the catalytic community, and this manuscript shows how at least some of the major challenges in synthesis could be addressed by this approach.71 Finally, the high 1O2 yield in these DPP hydrogels and our design approach have potential further applications towards sustainable asymmetric visible-light photocatalysis, wastewater remediation, oxygen sensing, and photodynamic therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Air Force Office of Scientific Research (FA9550-19-1-0220), National Science Foundation (NSF) grant number CHE-1808143, CHE-1G10755, and CHE-2003847. National Science Foundation CREST Center for Interface Design and Engineered Assembly of Low Dimensional Systems (IDEALS), NSF grant number HRD-1547830. The NMR data (Bruker Avance 300 MHz) presented herein were collected in part at the CUNY ASRC Biomolecular NMR facility and the NMR facility of the City College of the CUNY. Mass spectra were collected at the CUNY ASRC Biomolecular Mass Spectrometry Facility and the Hunter College Mass Spectrometry Facility of the CUNY. We thank Dr Barney Yoo (Hunter College Mass Spectrometry Facility) for help in acquiring the spectral data and Dhwanit R. Dave for rheology measurements.Notes and references
- C. Ayed, L. Caire da Silva, D. Wang and K. A. I. Zhang, J. Mater. Chem. A, 2018, 6, 22145–22151 RSC.
- J. Byun, W. Huang, D. Wang, R. Li and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 2967–2971 CrossRef CAS PubMed.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. Zhang, Angew. Chem., Int. Ed. Engl., 2015, 54, 14549–14553 CrossRef CAS PubMed.
- W. Huang, Z. J. Wang, B. C. Ma, S. Ghasimi, D. Gehrig, F. Laquai, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2016, 4, 7555–7559 RSC.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- B. Long, Z. Ding and X. Wang, ChemSusChem, 2013, 6, 2074–2078 CrossRef CAS PubMed.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685 CrossRef CAS PubMed.
- K. T. Oppelt, J. Gasiorowski, D. A. M. Egbe, J. P. Kollender, M. Himmelsbach, A. W. Hassel, N. S. Sariciftci and G. Knör, J. Am. Chem. Soc., 2014, 136, 12721–12729 CrossRef CAS PubMed.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- H.-Y. Xie, L.-S. Han, S. Huang, X. Lei, Y. Cheng, W. Zhao, H. Sun, X. Wen and Q.-L. Xu, J. Org. Chem., 2017, 82, 5236–5241 CrossRef CAS PubMed.
- W. Chen, F. N. Rein and R. C. Rocha, Angew. Chem., Int. Ed., 2009, 48, 9672–9675 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kishi, H. Kotani, Y.-M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- T. Ishizuka, S. Ohkawa, H. Ochiai, M. Hashimoto, K. Ohkubo, H. Kotani, M. Sadakane, S. Fukuzumi and T. Kojima, Green Chem., 2018, 20, 1975–1980 RSC.
- Q. Fu, C. Duan, Z. Yan, Y. Li, Y. Si, L. Liu, J. Yu and B. Ding, Macromol. Rapid Commun., 2018, 39, 1800058 CrossRef PubMed.
- W. Jiang, Y. Zhu, G. Zhu, Z. Zhang, X. Chen and W. Yao, J. Mater. Chem. A, 2017, 5, 5661–5679 RSC.
- F. Zhao, J. Bae, X. Zhou, Y. Guo and G. Yu, Adv. Mater., 2018, 30, 1801796 CrossRef PubMed.
- M. C. Nolan, J. J. Walsh, L. L. E. Mears, E. R. Draper, M. Wallace, M. Barrow, B. Dietrich, S. M. King, A. J. Cowan and D. J. Adams, J. Mater. Chem. A, 2017, 5, 7555–7563 RSC.
- J. Byun, K. Landfester and K. A. I. Zhang, Chem. Mater., 2019, 31, 3381–3387 CrossRef CAS.
- N. Singh, M. P. Conte, R. V. Ulijn, J. F. Miravet and B. Escuder, Chem. Commun., 2015, 51, 13213–13216 RSC.
- L. N. Neumann, M. B. Baker, C. M. A. Leenders, I. K. Voets, R. P. M. Lafleur, A. R. A. Palmans and E. W. Meijer, Org. Biomol. Chem., 2015, 13, 7711–7719 RSC.
- M. Tena-Solsona, J. Nanda, S. Díaz-Oltra, A. Chotera, G. Ashkenasy and B. Escuder, Chem.–Eur. J., 2016, 22, 6687–6694 CrossRef CAS PubMed.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762–11774 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS PubMed.
- O. Zozulia, M. A. Dolan and I. V. Korendovych, Chem. Soc. Rev., 2018, 47, 3621–3639 RSC.
- C. Zhang, X. Xue, Q. Luo, Y. Li, K. Yang, X. Zhuang, Y. Jiang, J. Zhang, J. Liu, G. Zou and X.-J. Liang, ACS Nano, 2014, 8, 11715–11723 CrossRef CAS PubMed.
- K. Tao, B. Xue, S. Frere, I. Slutsky, Y. Cao, W. Wang and E. Gazit, Chem. Mater., 2017, 29, 4454–4460 CrossRef CAS PubMed.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS PubMed.
- B. Sun, K. Tao, Y. Jia, X. Yan, Q. Zou, E. Gazit and J. Li, Chem. Soc. Rev., 2019, 48, 4387–4400 RSC.
- S. Frühbeißer, G. Mariani and F. Gröhn, Polymers, 2016, 8, 180 CrossRef PubMed.
- J. Sun, B. V. K. J. Schmidt, X. Wang and M. Shalom, ACS Appl. Mater. Interfaces, 2017, 9, 2029–2034 CrossRef CAS PubMed.
- J. Liu, T. An, Z. Chen, Z. Wang, H. Zhou, T. Fan, D. Zhang and M. Antonietti, ACS Appl. Mater. Interfaces, 2017, 5, 8933–8938 CAS.
- K. Liu, X. Ren, J. Sun, Q. Zou and X. Yan, Adv. Sci., 2018, 5, 1701001 CrossRef PubMed.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., Int. Ed., 2012, 51, 517–520 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- Z. Feng, T. Zhang, H. Wang and B. Xu, Chem. Soc. Rev., 2017, 46, 6470–6479 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. J. M. Frederix and R. V. Ulijn, Angew. Chem., Int. Ed., 2014, 53, 5882–5887 CrossRef CAS PubMed.
- S. Bai, S. Debnath, N. Javid, P. W. J. M. Frederix, S. Fleming, C. Pappas and R. V. Ulijn, Langmuir, 2014, 30, 7576–7584 CrossRef CAS PubMed.
- X. Liang, Z. Guo, H. Wei, X. Liu, H. Lv and H. Xing, Chem. Commun., 2018, 54, 13002–13005 RSC.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13105 CrossRef CAS PubMed.
- K. R. Weishaupt, C. J. Gomer and T. J. Dougherty, Cancer Res., 1976, 36, 2326–2329 CAS.
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 5533–5539 CrossRef CAS PubMed.
- Y. Cai, P. Liang, Q. Tang, X. Yang, W. Si, W. Huang, Q. Zhang and X. Dong, ACS Nano, 2017, 11, 1054–1063 CrossRef CAS PubMed.
- R. Bentley, Chem. Soc. Rev., 2005, 34, 609–624 RSC.
- I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef PubMed.
- J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307–1350 RSC.
- I. Agranat and H. Caner, Drug Discovery Today, 1999, 4, 313–321 CrossRef CAS PubMed.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- X. Xie, C. Mao, X. Liu, Y. Zhang, Z. Cui, X. Yang, K. W. K. Yeung, H. Pan, P. K. Chu and S. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 26417–26428 CrossRef CAS PubMed.
- M. T. Jarvi, M. J. Niedre, M. S. Patterson and B. C. Wilson, Photochem. Photobiol., 2006, 82, 1198–1210 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1587–1595 CrossRef CAS.
- C. X. Guzman, R. M. K. Calderon, Z. Li, S. Yamazaki, S. R. Peurifoy, C. Guo, S. K. Davidowski, M. M. A. Mazza, X. Han, G. Holland, A. M. Scott and A. B. Braunschweig, J. Phys. Chem. C, 2015, 119, 19584–19589 CrossRef CAS.
- Y. Zhou, C. X. Guzman, L. C. Helguero-Kelley, C. Liu, S. R. Peurifoy, B. Captain and A. B. Braunschweig, J. Phys. Org. Chem., 2016, 29, 689–699 CrossRef CAS.
- S. Rieth, Z. Li, C. E. Hinkle, C. X. Guzman, J. J. Lee, S. I. Nehme and A. B. Braunschweig, J. Phys. Chem. C, 2013, 117, 11347–11356 CrossRef CAS.
- E. Schwartz, V. Palermo, C. E. Finlayson, Y.-S. Huang, M. B. J. Otten, A. Liscio, S. Trapani, I. González-Valls, P. Brocorens, J. J. L. M. Cornelissen, K. Peneva, K. Müllen, F. C. Spano, A. Yartsev, S. Westenhoff, R. H. Friend, D. Beljonne, R. J. M. Nolte, P. Samorì and A. E. Rowan, Chem.–Eur. J., 2009, 15, 2536–2547 CrossRef CAS PubMed.
- S. Militzer, T. M. P. Tran, P. J. Mésini and A. Ruiz-Carretero, ChemNanoMat, 2018, 4, 790–795 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef.
- D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, J. Am. Chem. Soc., 2014, 136, 7809–7812 CrossRef CAS PubMed.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC.
- M. Lan, S. Zhao, W. Liu, C.-S. Lee, W. Zhang and P. Wang, Adv. Healthcare Mater., 2019, 8, 1900132 CrossRef PubMed.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- T. Hartman, V. Herzig, M. Buděšínský, J. Jindřich, R. Cibulka and T. Kraus, Tetrahedron: Asymmetry, 2012, 23, 1571–1583 CrossRef CAS.
- A. Casado-Sánchez, R. Gómez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2016, 52, 9137–9140 RSC.
- J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620–623 CrossRef.
- D. Latassa, O. Enger, C. Thilgen, T. Habicher, H. Offermanns and F. Diederich, J. Mater. Chem., 2002, 12, 1993–1995 RSC.
- T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- D. Kroiss, G. Ashkenasy, A. B. Braunschweig, T. Tuttle and R. V. Ulijn, Chem, 2019, 5, 1917–1920 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, organic synthesis, NMR, MS, TEM, AFM, HPLC, UV-Vis, fluorescence emission spectroscopy and 1O2 calculations. See DOI: 10.1039/c9sc06481h |
| This journal is © The Royal Society of Chemistry 2020 |