
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomineralized metal–organic framework nanoparticles enable a primer exchange reaction-based DNA machine to work in living cells for imaging and gene therapy†
Juan
Zhang
,
Mengyun
He
,
Cunpeng
Nie
,
Manman
He
,
Qingshan
Pan
,
Chang
Liu
,
Yanlei
Hu
,
Tingting
Chen
 and
Xia
Chu
*
and
Xia
Chu
*
State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: xiachu@hnu.edu.cn
First published on 16th June 2020
Abstract
Sensitive tumor imaging and precise tumor therapy play critical roles in the cancer combat. Herein, we build a DNA machine based on a primer exchange reaction (PER) for mRNA imaging and gene therapy. By using zeolitic imidazolate framework-8 nanoparticles (ZIF-8 NPs) to co-deliver the components including a primer, hairpin and strand displacing polymerase to the living cells, the PER-based DNA machine can be initiated by intracellular survivin mRNA and continuously produce Bcl-2 antisense DNA (ASD), which enables the DNA machine not only to image survivin mRNA but also to implement gene therapy. The results demonstrate that ZIF-8 NPs can protect the polymerases and nucleic acid probes from protease attack and nuclease degradation. After internalization, pH-responsive ZIF-8 NPs can efficiently release cargos from endo-lysosomes due to the protonation effect. The intracellular PER-based DNA machine has been demonstrated to be able to sensitively image survivin mRNA expression levels and selectively kill the cancer cells and has no effect on the normal cells. The PER-based DNA machine may provide a promising platform for early stage tumor diagnosis and more precise tumor therapy.
Introduction
Antisense DNA (ASD) is a DNA single strand with a length of 18–25 nucleotides. It can hybridize with the mRNA of the target gene and inhibit the translation of the target protein.1,2 So, ASDs are a promising therapy as a gene drug.3–5 However, free ASDs are negatively charged, which makes it difficult for them to cross the cell membrane, accordingly limiting their extensive clinical applications.6,7 In recent years, many kinds of nanocarriers have been constructed for ASD delivery, such as DNA nanostructures,8,9 liposomes10–12 and inorganic nanoparticles.13–15 They can transport ASDs to the cytoplasm. However, these nanoparticles commonly tend to accumulate in the liver, kidneys and spleen when they target the tumor site, which results in the damage to healthy cells and causes serious side effects. Therefore, development of strategies that can deliver or produce ASDs exclusively in the cancer cells remains a great challenge. In addition, delivery or production of adequate amounts of ASDs in cancer cells is also significantly important for the efficacy of the tumor therapy.A recent study reported a primer exchange reaction (PER) to program the autonomous synthesis of single-stranded DNA (ssDNA) from a short (7–9 nt) DNA primer in vitro.16–18 Using a single PER hairpin as a catalytic template, the PER pathway starts with a prescribed short DNA primer, then repeatedly appends a nascent single-stranded sequence to the short primer with the aid of a strand displacing polymerase (for example, Klenow (exo-)). By the random walk process of three-way branch migration, the extended primer can spontaneously dissociate from the PER hairpin, and the hairpin becomes free and reacts with another primer to induce the next cycle of primer exchange. Inspired by this specific reaction, we want to employ the PER system to build a DNA machine, which is driven by a strand displacing polymerase and fueled by dNTPs, to continuously produce ASDs if the nucleotide sequences of the ASDs are programmed to design the PER hairpin. More importantly, if the region of the primer hybridization is first blocked by a DNA strand, the machine can be designed to be triggered by the over-expressed target molecules in cancer cells, such as over-expressed mRNAs and miRNAs. Accordingly, the DNA machine triggered by specific molecules in cancer cells can selectively kill cancer cells and has no effect on normal cells. However, how to deliver efficiently the components of the DNA machine including the primer, hairpin and especially strand displacing polymerase to living cells is a major challenge we face.
Recently, nanoscale metal–organic framework nanoparticles (MOF NPs) have been used for loading therapeutic or imaging agents due to their good biocompatibility, simple preparation, porous structure and multifunctional features.19–23 Among these MOF NPs, zeolitic imidazolate framework-8 nanoparticles (ZIF-8 NPs) constructed using zinc ions (Zn2+) and 2-methylimidazole (2-MIM) possess unique pH-sensitive biodegradability, which makes them an ideal nanocarrier for the delivery of various cargos into living cells including nucleic acid probes24,25 and chemical drugs.26–29 In addition, the green biomimetic mineralization synthesis process in aqueous media is favourable for the maintenance of the activities of bio-macromolecules and improvement of the bio-macromolecular stability against various denaturing conditions. To date, several enzymes30,31 and antibodies32 have been loaded in the ZIF-8 NPs for catalysis and disease treatment. Moreover, our group has demonstrated that ZIF-8 NPs can efficiently deliver and release native active proteins in living cells,33 and has realized a rolling circle amplification (RCA) reaction in living cells by using ZIF-8 NPs to deliver DNA polymerase and nucleic acids.34 So, the biomineralized ZIF-8 NPs should be suited for the delivery of components including a primer, hairpin and strand displacing polymerase to build a PER-based DNA machine working in living cells.
Herein, we build a tumor-associated biomarker initiated PER-based DNA machine for mRNA imaging and gene therapy. Using nanoscale ZIF-8 NPs to co-deliver Klenow (exo-)fragment polymerase (KFP) and nucleic acid probes into living cells, we implement an intracellular PER-based polymerase reaction. By programming the nucleotide sequences of the ASD to design the PER hairpin, the machine can continuously produce Bcl-2 ASD, which enables the DNA machine not only to image survivin mRNA but also to perform gene therapy. The results demonstrate that ZIF-8 NPs can protect the polymerases and nucleic acid probes from protease attack and nuclease degradation. After internalization into cells, pH-responsive ZIF-8 NPs enable efficient release and escape of cargos from endo-lysosomes due to the protonation effect. The intracellular PER-based DNA machine has been demonstrated to be able to sensitively image survivin mRNA expression levels and selectively kill the cancer cells and has no effect on the normal cells. The PER-based DNA machine may provide a potential platform for early stage diagnosis, and more precise and efficient therapy of tumors.
Results and discussion
Mechanism and design of the PER-based DNA machine
The mechanism and design of the mRNA-triggered primer exchange reaction (PER)-based DNA machine are depicted in Scheme 1. A dumbbell shaped hairpin with two loops was first designed. One loop (domain c, blue) was programmed to be complementary to the sequences of survivin mRNA and could hybridize with survivin mRNA and expose the complementary domain of the primer a*. Primer a with a length of 8 nucleotides is subsequently bound with domain a* (step 1) and extended with the aid of a strand displacing polymerase (such as KFP) to append a nascent single-stranded sequence b (termed the copied b domain) to the 3′-end of the short primer until the strand displacing elongation reaction was halted at the stop sequence on the PER hairpin (step 2). The b domain on the hairpin was then able to compete with the copied b domain via the random walk process of three-way branch migration (step 3).35 Finally, the extended primer was spontaneously dissociated from the PER hairpin once the copied b domain was displaced (step 4). The PER hairpin became free and bound with another primer to induce the next cycle of the primer exchange reaction. Therefore, a DNA single strand with an arbitrary sequence could be appended to the 3′-end of the short primer through the PER-based machine. Importantly, the machine could be designed to be triggered by a specific biomarker such as tumor-associated mRNA or miRNA through programming domain c. | ||
| Scheme 1 Schematic illustration of a mRNA-triggered primer exchange reaction (PER)-based DNA machine. In step 1, a dumbbell shaped hairpin with two loops (PER hairpin) was first designed. One loop (domain c, blue) was programmed to be complementary to the sequences of the survivin mRNA, and could hybridize with the survivin mRNA and expose the complementary domain of the primer a* (green). In step 2, primer a with a length of 8 nucleotides subsequently bound with domain a* and was extended using domain b* as the template such as in the PCR with the aid of Klenow (exo-)fragment polymerase to append a nascent single-stranded sequence b (termed the copied b domain, cb) to the 3′-end of the short primer until the strand displacing elongation reaction was halted at the stop sequence, two pairs of synthetic nucleotides, iso-dG (2′-O-methyl G) and iso-dC (2′-O-methyl C), on the PER hairpin. In step 3, the b domain on the hairpin was then able to compete with the copied b domain (cb) via the random walk process of three-way branch migration, and the b domain on the hairpin again hybridized with domain b*. In step 4, the copied b domain was displaced and a part of the 8 nucleotide long primer also was away from the PER hairpin due to the lower hybridization temperature. The extended primer was spontaneously dissociated from the PER hairpin. The PER hairpin became free and bound with another primer to induce the next cycle of the primer exchange reaction. | ||
Here, we chose survivin mRNA, a mRNA over-expressed in most cancers,36,37 as the model target to initiate the DNA machine. The B-cell lymphoma 2 (Bcl-2) protein is an anti-apoptotic protein and is commonly associated with therapy resistance in various human cancers.38,39 So, Bcl-2 antisense DNA (Bcl-2 ASD) was chosen as the gene drug. A dumbbell shaped hairpin was first designed, whose domain c (blue) was complementary to the survivin mRNA and domain b sequence (red) was the same as that of Bcl-2 ASD. The principle of the intracellular PER-based DNA machine for mRNA imaging and gene therapy is illustrated in Scheme 2. The ZIF-8 NPs encapsulated BSA and KFP were first prepared. Then, the nucleic acid including the hairpin, primer and molecular beacon (MB) was adsorbed on the surface of ZIF-8 NPs by electrostatic interaction. After cellular uptake, the ZIF-8 NPs were biodegraded in acidic endo-lysosomes, releasing the KFP, hairpin, primer and MB. The intracellular survivin mRNA caused the DNA machine to produce Bcl-2 ASD continuously in the presence of KFP and dNTPs through the mechanism in Scheme 1. For survivin mRNA imaging, the MB complementary to Bcl-2 ASD was delivered into the cells, the produced Bcl-2 ASD hybridized with the MB and resulted in the fluorescence recovery of Cy5. When the MB was not delivered, the Bcl-2 ASD bound with a key region of Bcl-2 mRNA, accordingly inhibiting the translation of the Bcl-2 protein and resulting in cell apoptosis. So, this PER-based DNA machine provided a promising strategy for sensitive tumor imaging and precise gene therapy.
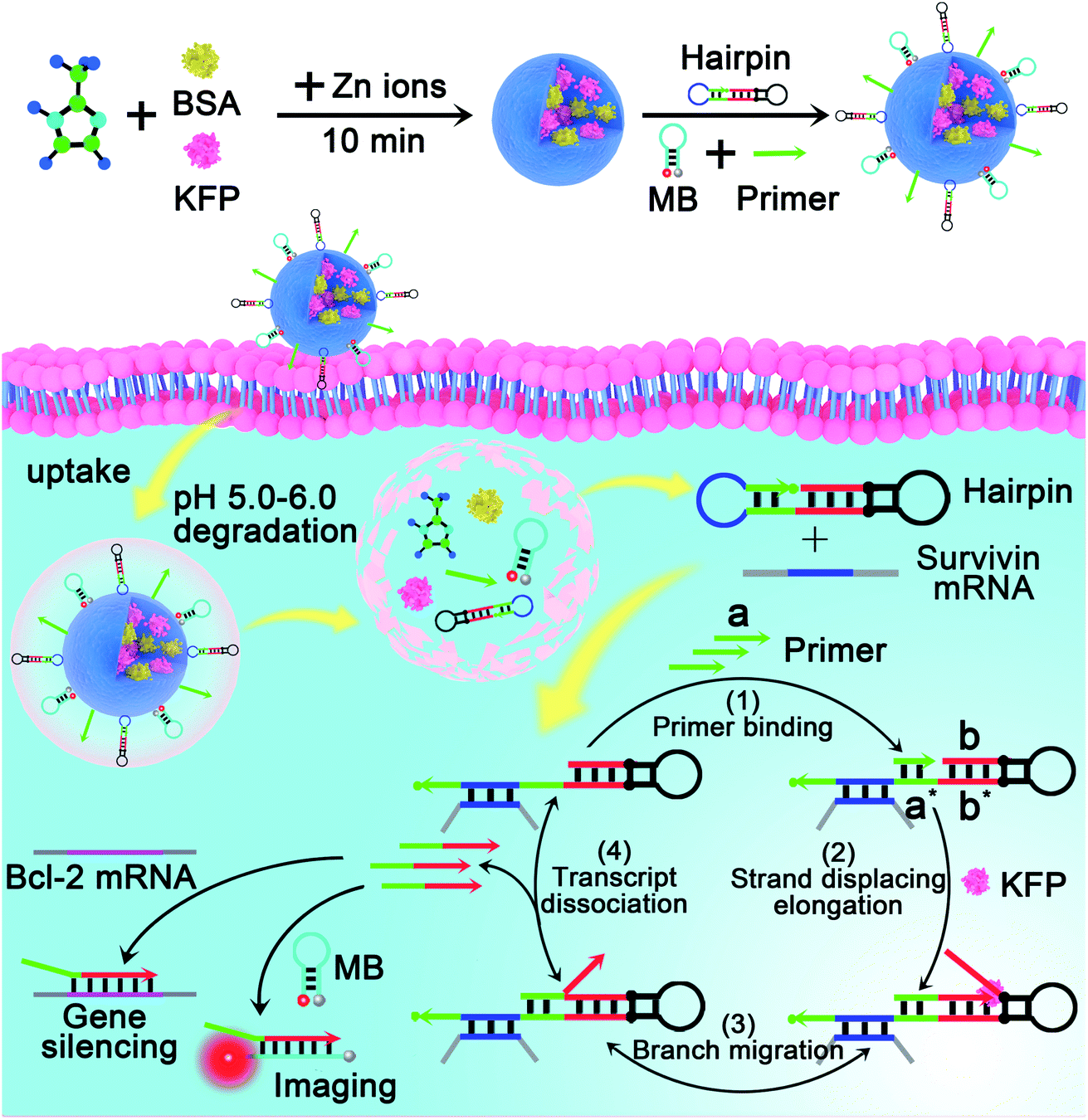 | ||
| Scheme 2 Illustration of the intracellular survivin mRNA-triggered primer exchange reaction (PER)-based DNA machine for mRNA imaging and gene silencing. By using zeolitic imidazolate framework-8 nanoparticles (ZIF-8 NPs) to co-deliver the components including the primer, hairpin and strand displacing polymerase to the living cells, the PER-based DNA machine can be initiated by intracellular survivin mRNA and continuously produce Bcl-2 ASD through the mechanism in Scheme 1, which enables the DNA machine not only to image survivin mRNA but also to implement gene therapy. | ||
Synthesis and characterization of ZIF-8 NPs
The ZIF-8 NPs encapsulated bovine serum albumin (BSA), a model protein, were first prepared according to the method previously reported by our group.32,33 The transmission electron microscopy (TEM) image showed that the monodisperse BSA@ZIF-8 NPs were obtained with an average size of 87 ± 6.8 nm, which is a desirable size for intracellular delivery (Fig. 1A). The nucleic acid probes were then adsorbed on the surface of the BSA@ZIF-8 NPs through electrostatic interaction. The dynamic light scattering (DLS) analysis indicated that the average hydrodynamic diameters of the ZIF-8 NPs, BSA@ZIF-8 NPs and BSA@ZIF-8/DNA NPs were 80.47 ± 19.25, 108.6 ± 18.59 and 161.9 ± 25.69 nm, respectively (Fig. S1†). Moreover, zeta potential measurements suggested that the surface charges of ZIF-8 NPs and BSA@ZIF-8 NPs were positive due to the abundant Zn2+ on the surface of ZIF-8 (Fig. 1B), which enabled negatively charged nucleic acid probes to adsorb on the surface of BSA@ZIF-8 NPs through electrostatic interaction. Therefore, the zeta potential of the BSA@ZIF-8 NPs changed from positive to negative after adsorption of nucleic acid probes (Fig. 1B). The crystalline form of BSA@ZIF-8 and BSA@ZIF-8/DNA NPs was confirmed by powder X-ray diffraction (PXRD), which was the same as that of the ZIF-8 NPs (Fig. 1C) and showed that proteins and nucleic acid probes did not affect the crystalline structure of ZIF-8 NPs. Fourier transform infrared spectroscopy (FTIR) analysis also demonstrated the successful incorporation of BSA and adsorption of nucleic acid probes. As seen in Fig. 1D, the characteristic absorption peaks of BSA corresponding to amide I in the range from 1640 to 1660 cm−1 were obtained in the spectrum of BSA@ZIF-8 and absorption peaks of DNA corresponding to the phosphodiester bond and stretching vibrations of PO43− about 1236 cm−1 and 1040 cm−1 were observed in the FTIR spectrum of the BSA@ZIF-8/DNA NPs. However, the FTIR spectrum of the ZIF-8 NPs did not show these characteristic peaks. All the above data showed that the protein was indeed encapsulated in the ZIF-8 NPs and nucleic acid probes were also successfully adsorbed on the surface of the BSA@ZIF-8 NPs. | ||
| Fig. 1 Characterization of the prepared nanoparticles. (A) TEM image of the BSA@ZIF-8 NPs. (B) Zeta potential distribution of ZIF-8 NPs, BSA@ZIF-8 NPs, and BSA@ZIF-8/DNA NPs. (C) PXRD patterns of simulated ZIF-8 NPs, synthetic ZIF-8 NPs, BSA@ZIF-8 NPs, and BSA@ZIF-8/DNA NPs. (D) FTIR spectral characterization of the model protein (BSA), ZIF-8 NPs, BSA@ZIF-8 NPs, and BSA@ZIF-8/DNA NPs. (E) UV-vis absorption spectra of native BSA provided for preparation of the BSA@ZIF-8 NPs (red) and BSA released from BSA@ZIF-8 NPs through acidic decomposition (green). (F) The pH-responsive release kinetics of the guest protein BSA from BSA@ZIF-8 NPs at pH 7.4 and 5.5. | ||
The protein encapsulation efficiency was determined using UV-vis absorption analysis as shown in Fig. 1E. The protein encapsulation efficiency (the percentage of the protein encapsulated inside BSA@ZIF-8 NPs over the protein amount initially added in the reaction mixture) was calculated to be ∼93.2%, corresponding to an estimate of the loading capacity of ∼77.6 μg BSA encapsulated in 1 mg BSA@ZIF-8 NPs. In addition, the nucleic acid probe's loading efficiency was also investigated by fluorescence spectroscopy. As shown in Fig. S2,† the loading efficiencies of the FAM-labeled HP, the Cy3-labeled primer and the Cy5-labeled MB were calculated to be 73.5 ± 2.5%, 90.5 ± 3.5% and 92.4 ± 4.8%, respectively, corresponding to the loading capacity of ∼2.46 × 10−10 mol FAM-HP, ∼3.02 × 10−9 mol Cy3-primer and ∼4.62 × 10−10 mol Cy5-MB probe per mg BSA@ZIF-8/DNA NPs.
Moreover, the ZIF-8 NPs undergo pH-sensitive biodegradation for stimulating the release of guest cargoes. The release kinetics of FITC-BSA from FITC-BSA@ZIF-8 NPs was investigated under acidic (pH 5.5) and physiological (pH 7.4) conditions. The FITC-BSA@ZIF-8 NPs were stable under physiological conditions (pH 7.4), whereas they rapidly degraded and released encapsulated FITC-BSA in 2 hours under acidic conditions (pH 5.5) (Fig. 1F). The pH-sensitive biodegradation may be associated with the fact that the metal–ligand bonds of 2-methylimidazole with Zn2+ were easily hydrolyzed to yield the protonated ligand under acidic conditions. This unique property makes the ZIF-8 NPs especially suited for intracellular delivery of cargoes because the ZIF-8 NPs can release the cargoes in acidic endosomes/lysosomes. In addition, the ZIF-8 NPs could prevent the proteases from accessing the caged proteins, thereby protecting the encapsulated proteins from degradation in the biological environment. As can be seen from Fig. S3,† the KFP released from BSA + KFP@ZIF-8 NPs with protease pre-treatment still could trigger a primer exchange reaction (PER). However, the protease-treated KFP could not initiate a PER, and a faint signal the same as that in the absence of KFP was obtained. Similarly, ZIF-8 NPs can also protect the adsorbed nucleic acid probes from digestion by DNase I. As is seen in Fig. S4,† the fluorescence signal of the MB labeled with Cy5 and BHQ2 increased rapidly with time after treatment with DNase I, whereas no obvious fluorescence change was shown when the MB was adsorbed on the surface of BSA@ZIF-8 NPs after incubation with the same concentration of DNase I. As a result, the protein@ZIF-8/DNA NP represents a satisfactory nanocarrier for transporting proteins and nucleic acid probes into the cytosol in living cells.
The biocompatibility and cytotoxicity of the BSA + KFP@ZIF-8 NPs were evaluated by using CCK-8 assays. The different concentrations of BSA + KFP@ZIF-8 NPs (60, 80 and 100 μg mL−1) in a culture medium were added to HeLa cells and incubated for different periods. As shown in Fig. S5A,† 60 μg mL−1 BSA + KFP@ZIF-8 NPs showed only low cytotoxicity, and more than 85.1% of the cells remained alive after incubation for 24 h, whereas higher concentrations of BSA + KFP@ZIF-8 NPs caused substantial toxicity to the cells. Also, another cytotoxicity experiment was performed by using CCK-8 assay. The HeLa cells were treated with BSA + KFP@ZIF-8 NPs (60, 80 and 100 μg mL−1) at 37 °C for 3 h followed by replacement of another fresh culture medium for 48 hours. As shown in Fig. S5B,† BSA + KFP@ZIF-8 NPs (60 μg mL−1) had lower cytotoxicity than other concentrations. In addition, comet assay (Fig. S6†) and ROS assay (Fig. S7†) were also carried out to investigate the effect of the BSA + KFP@ZIF-8 NPs on the biological events such as DNA damage and ROS formation. The results indicated that the 60 μg mL−1 BSA + KFP@ZIF-8 working concentration did not induce obvious DNA damage and ROS formation.
in vitro performance of the PER-based DNA machine
The in vitro performance of the survivin mRNA triggered PER-based DNA machine was first investigated. We used in vitro survivin DNA, which had the same sequences as survivin mRNA except for T instead of U, to hybridize with the hairpin to initiate the PER. The produced extended primer strand was monitored using fluorescence recovery resulting from the hybridization with the molecular beacon (MB) labeled with Cy5 and BHQ2. As can be seen from Fig. 2A, a significantly increased fluorescence signal could be obtained, indicating that the survivin DNA indeed could initiate a PER in the presence of the hairpin (HP), primer, dNTPs and KFP (curve e). In contrast, no obvious fluorescence signal was observed when either the hairpin (curve a), primer (curve b), dNTPs (curve c) or KFP (curve d) was omitted. In addition, PAGE assay was also carried out. As can be seen from Fig. 2B, survivin DNA could hybridize with the hairpin to form a double-stranded complex (lane 4). However, in the absence of the dNTPs or KFP, this double-stranded complex could not initiate a PER even if the primer is present (lane 6 and 7). An obvious new band corresponding to the extended primer strand could be obtained in the presence of survivin DNA, hairpin, primer, KFP and dNTPs (lane 8), indicating that a PER was performed. These results were consistent with those obtained by the fluorescence experiment, strongly demonstrating that the survivin DNA indeed could trigger a PER-based DNA machine and the latter could produce a great number of extended primer strands.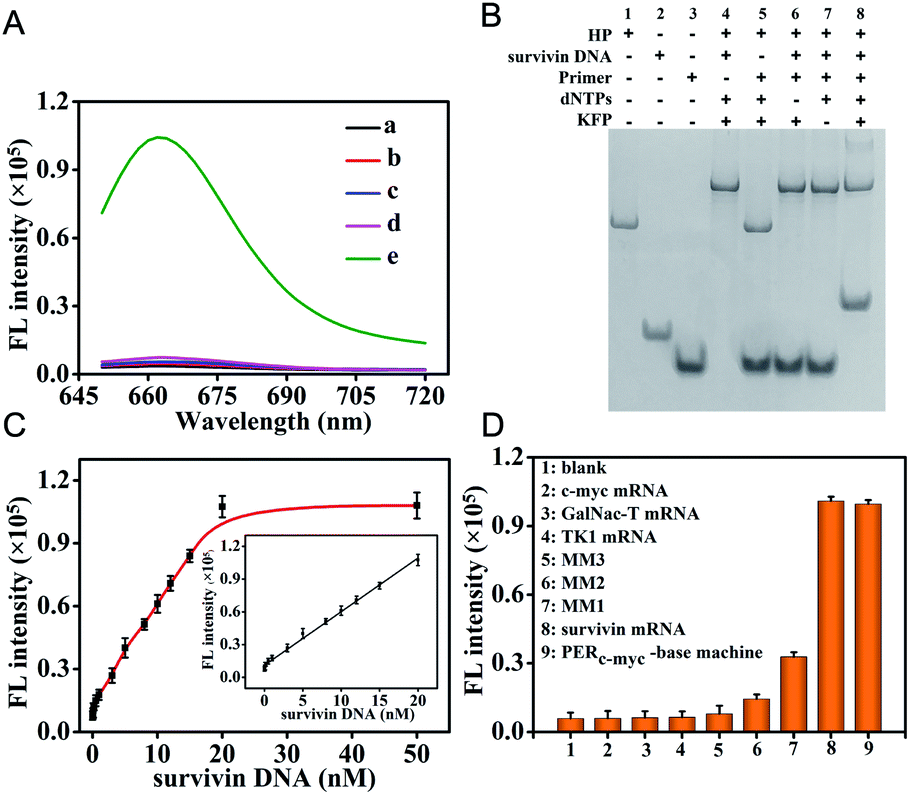 | ||
| Fig. 2 (A) Fluorescence responses of the activable PER based-DNA machine to target mRNA. (a) 200 nM primers + 20 nM survivin DNA + 200 μM dNTPs + 25 U mL−1 KFP + 30 nM MB; (b) 20 nM HP + 20 nM survivin DNA + 200 μM dNTPs + 25 U mL−1 KFP + 30 nM MB; (c) 20 nM HP + 200 nM primers + 20 nM survivin DNA + 25 U mL−1 KFP + 30 nM MB; (d) 20 nM HP + 200 nM primers + 20 nM survivin DNA + 200 μM dNTPs + 30 nM MB; and (e) 20 nM HP + 200 nM primers + 20 nM survivin DNA + 200 μM dNTPs + 25 U mL−1 KFP + 30 nM MB. (B) Corresponding to polyacrylamide gel electrophoresis (PAGE) of the PER based-DNA machine as mentioned above. (C) Relationship between the fluorescence intensities at 665 nm and the concentrations of the survivin DNA target from 0 to 50 nM. Insert: linear correlation between fluorescence intensities and survivin DNA concentrations in the range of 0.005–20 nM. (D) Specificity of the survivin DNA initiated PER-based DNA machine. | ||
Next, to obtain an optimal PER-based DNA machine reaction efficiency, we optimized the experimental conditions including the KFP concentration and PER reaction time (Fig. S8†). The effect of the KFP concentration on the PER reaction was first investigated. The result showed that the fluorescence intensity gradually increased with the KFP concentration from 0 to 20 U mL−1, and reached saturation at about 25 U mL−1. So, 25 U mL−1 KFP was employed in the subsequent experiments. The effect of the PER reaction time was also studied. The fluorescence intensity increased rapidly with the reaction time and reached a maximal signal in 2 h. Therefore, 2 h was selected as the optimal reaction time.
Under the optimized conditions, we investigated the changes of fluorescence with the target survivin DNA concentrations. The fluorescence signals increased gradually with the increase of the survivin DNA concentration from 0 nM to 50 nM (Fig. S9†). The fluorescence intensities at 665 nm were correlated linearly with the survivin DNA concentrations in the range of 5 pM to 20 nM (Fig. 2C), and the detection limit was calculated to be 2.3 pM (in terms of the 3σ/slope), indicating the high sensitivity of our strategy. This high sensitivity should be attributed to the powerful ability of the PER-based DNA machine to produce extended primer strands continuously after the activation by the target survivin DNA.
To investigate the specificity of PER-based DNA machine activated by the target survivin DNA, some other mRNAs including c-myc mRNA, GalNac-T mRNA, TK1 mRNA and three variants including single base-mismatched (MM1), double base-mismatched (MM2), and three base-mismatched (MM3) survivin mRNA were examined instead of the survivin DNA target. As can be seen from Fig. S10† and 2D, only single base-mismatched (MM1) survivin mRNA showed relatively high fluorescence signals, and the other five mRNA molecules all gave negligible fluorescence signals. The HP was replaced by the HPc-myc hybridized with the c-myc mRNA and the primer was replaced by the primerc-myc. When c-myc mRNA reacted with the HPc-myc in the presence of the primerc-myc, dNTPs and KFP, c-myc mRNA also gave relatively high fluorescence signals. This result indicated that the PER-based DNA machine could be triggered exclusively by the target mRNA and other mRNAs could not initiate the DNA machine to produce antisense oligonucleotides.
Imaging of survivin mRNA in living cells
Having demonstrated the feasibility of the survivin mRNA initiated PER-based DNA machine in vitro, we then attempted to carry out the PER in living cells by using BSA + KFP@ZIF-8/HP + primer + MB NPs to co-deliver the KFP and nucleic acid probes. Before survivin mRNA imaging, the time-dependent fluorescence imaging experiments were performed by incubating BSA + KFP@ZIF-8/HP + primer + MB NPs with HeLa cells for different time periods to monitor the PER-based DNA machine work. As can be seen from Fig. S11,† no apparent red fluorescence can be observed for 1 h. This was probably due to the insufficient delivery of ZIF-8 nanoparticles and the relatively short PER time. Further extension of the incubation time to 3 h resulted in the delivery of more nanoparticles into the cells for the survivin mRNA-triggered PER-based DNA machine work, and increasing red fluorescence intensity can be obtained. A longer treatment time would not increase the red fluorescence signal, suggesting that the PER-based DNA machine had reached the maximal signal in 3 h. Therefore, a 3 h treatment time was selected for living cells.The ability of the ZIF-8 NPs to co-deliver the polymerase and nucleic acid probes to living cells was first inspected. HeLa cells were incubated with the ZIF-8 NPs containing FITC-labeled BSA inside and Cy5-labeled MB adsorbed to the outside (denoted as FITC-BSA@ZIF-8/Cy5-MB) for 3 h. Significant green and red fluorescence in the whole cytoplasm was observed from the confocal laser scanning microscopy (CLSM) images (Fig. S12†), implying that the ZIF-8 NPs could successfully deliver the protein and nucleic acid probes into the living cells. The intracellular distribution of the probes was also investigated. The colocalization coefficients were analyzed using NIS-Elements AR software (Nikon) by calculating Pearson's correlation coefficient. The colocalization coefficient is essentially a statistics of linear correlation that can quantitatively measure the degree of colocalization between the organelle and the delivered probe in our assay. After incubating with BSA + KFP@ZIF-8/Cy5-MB for 3 h, the organelle endo-lysosomes of HeLa cells were localized with green Lyso@tracker (green fluorophore), and the delivered probe MB was marked using Cy5 (Cy5-labeled MB, red fluorophore) and Hoechst 33342 for nuclei. The fluorescence of individual channel was recorded and counted using confocal images (Fig. S13†). A scatter plot of colocalization of the green channel (Lyso@tracker) and red channel (Cy5) was obtained using NIS-Elements AR software (Nikon), which calculated the correlation coefficient. Therefore, a smaller correlation coefficient (0.29) for the Cy5-labeled MB and endo-lysosomes means that a smaller portion of the MB was retained in lysosomes, suggesting efficient escape of the MB from endo-lysosomes. The result demonstrated that the MB probes escaped successfully from lysosomes to the cytoplasm. Previous work by our group32 also gave plain evidence for efficient and successful release of ZIF-8 NP caged proteins to the cytosol. The efficient endo-lysosomal release and escape might be ascribed to the “proton sponge” effect. Due to the acid-labile noncovalent bond, the decomposition of the pH-sensitive ZIF-8 NPs in acidic endosomes and lysosomes could produce positively charged imidazole ligands, which would disrupt the endosomal membrane and release guest molecules through the “proton-sponge” effect.40–42
Next, we tested the feasibility of the PER-based DNA machine for survivin mRNA imaging in living cells. The HeLa cells with high survivin mRNA expression levels were incubated with the BSA + KFP@ZIF-8/HP + primer + MB NPs. The confocal laser scanning microscopy (CLSM) image showed bright red fluorescence in the cytoplasm (Fig. 3B(d)), suggesting that the intracellular survivin mRNA initiated a PER in the presence of the KFP, HP and primer and produced a large number of extended primer strands. The resulting extended primer strands hybridized with the MB labeled with Cy5 and BHQ2 and resulted in red fluorescence signal recovery (Fig. 3A). In contrast, no obvious red fluorescence signal could be observed when either the HP, primer or KFP was not delivered into HeLa cells (Fig. 3B(a–c)). Furthermore, the corresponding flow cytometric experiments were also performed (Fig. S14†), whose results were consistent with those obtained by CLSM. Taken together, these results demonstrated that a survivin mRNA initiated PER indeed took place in living cells by using ZIF-8 NPs to co-deliver the KFP, HP and primer to living cells, which could be used for sensitive imaging of intracellular survivin mRNA. In addition, because the KFP activity was dependent on the Mg2+ concentration, the influence of the Mg2+ concentration on the fluorescence imaging was also investigated. As can be seen from Fi. S15,† the significantly enhanced red fluorescence signal was observed in HeLa cells pretreated with 10 mM Mg2+ and 5 μM calcimycin,43 compared with the fluorescence signal in HeLa cells without pretreatment, suggesting that the concentration of endogenous Mg2+ in living cells was not enough to maintain the biological activity of the KFP. Hence, the pretreatment with 10 mM Mg2+ and 5 μM calcimycin was applied in the following imaging experiments.
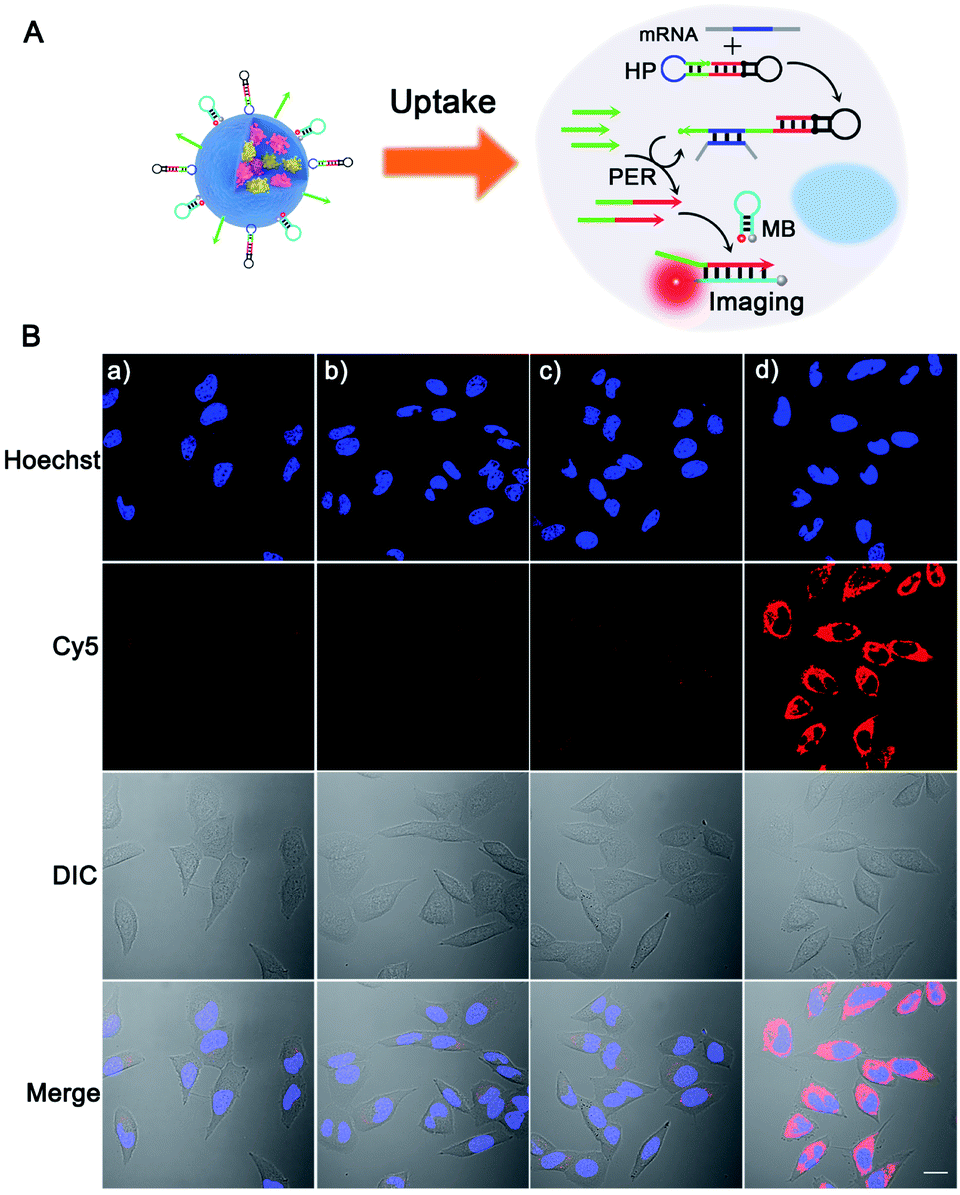 | ||
| Fig. 3 (A) Schematic illustration of the intracellular survivin mRNA triggered PER based-DNA machine system for sensitive imaging of survivin mRNA. (B) Confocal laser scanning microscopy (CLSM) images of HeLa cells. The HeLa cells were pretreated with 10 mM Mg2+ and 5 μM calcimycin for 30 min and then incubated for 3 h with different nanoparticles: (a) BSA + KFP@ZIF-8/primer + MB NPs; (b) BSA + KFP@ZIF-8/HP + MB NPs; (c) BSA@ZIF-8/HP + primer + MB NPs; and (d) BSA + KFP@ZIF-8/HP + primer + MB NPs. Scale bar: 20 μm. | ||
We further investigated the PER-based DNA machine for survivin mRNA imaging in different types of cells. HeLa and MCF-7 cells, which were reported to over-express survivin mRNA,35 were tested. The normal liver L02 cells with low expression levels of survivin mRNA44 were used as the negative control. After pretreatment with 10 mM Mg2+ and 5 μM calcimycin for 30 min, the cells were incubated with BSA + KFP@ZIF-8/HP + primer + MB NPs for 3 h. Bright red fluorescence could be observed in HeLa and MCF-7 cells, whereas no appreciable fluorescence signal was seen in L02 cells (Fig. 4A). To investigate the fluorescence signal of the entire cell population, the corresponding flow cytometric experiments were also carried out (Fig. 4C). The results were consistent with those obtained by cellular CLSM imaging. These results clearly verified that the intracellular survivin mRNA could initiate a PER and this PER-based DNA machine could be used for sensitive imaging of the survivin mRNA in living cells. In addition, the quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of the expression levels of survivin mRNA in these three cells was performed (Fig. S16†) by Sangon Biotechnology Co., Ltd. and the results were also consistent with those determined from the fluorescence images.
 | ||
| Fig. 4 (A) Cellular CLSM images of the survivin mRNA in different cells including HeLa cells, MCF-7 cells and L02 cells. These cells were pretreated with 10 mM Mg2+ and 5 μM calcimycin for 30 min and then incubated with 60 μg mL−1 BSA + KFP@ZIF-8/HP + primer + MB NPs for 3 h, followed by staining with Hoechst 33342 for 10 min. Scale bar: 20 μm. (B) Fluorescence intensity of different cells. (C) Flow cytometry analysis of different cell lines before (HeLa-control, MCF-7-control, and L02-control) and after (HeLa, MCF-7, and L02) incubation with 60 μg mL−1 BSA + KFP@ZIF-8/HP + primer + MB NPs. | ||
The PER-based DNA machine showed no fluorescent signal in L02 cells when the MB was included, which could be attributed to the absence of survivin mRNA, less efficient delivery or inhibition of the PER in L02 cells. To rule out the two last possibilities, we chose β-actin mRNA that is present in L02 cells as activator mRNA, and an additional control experiment has been completed. As shown in Fig. S17,† the L02 cells treated with the BSA + KFP@ZIF-8/HPβ-actin + primerβ-actin + MB NPs showed bright red fluorescence in the cytoplasm upon CLSM imaging (Fig. S17d†), suggesting that the intracellular β-actin mRNA initiated a PER. In contrast, no obvious red fluorescence signal could be observed when either the HPβ-actin, primerβ-actin or KFP was omitted from ZIF-8 NPs (Fig. S17a–c†). These data verified that the reason that the PER-based DNA machine could not be triggered and the result in L02 cells were because of the absence of survivin mRNA rather than less efficient delivery or inhibition of the PER.
The fluctuation of the survivin mRNA expression levels in HeLa cells was also monitored. Sepantronium bromide (YM155) was used as the survivin mRNA suppressant to decrease the expression levels of survivin mRNA in living cells.45 It can be seen from Fig. S18A† that the red fluorescence in HeLa cells gradually became faint with the increase in the suppressant YM155 concentrations, suggesting the down-regulation of survivin mRNA expression levels after YM155 treatment. These observations were further confirmed by the corresponding flow cytometry (Fig. S18C†). In addition, the survivin mRNA expression levels of HeLa cells pretreated with different suppressant YM155 concentrations were also determined by qRT-PCR experiments (Fig. S19†), whose results were consistent with those obtained by CLSM imaging and flow cytometry. Therefore, the survivin mRNA initiated PER could also be used to monitor the changes in the survivin mRNA expression levels in living cells.
Gene therapy efficacy of the PER-based DNA machine in living cells
Having demonstrated the ability of the PER-based machine to sensitively detect intracellular survivin mRNA, we next explored the gene therapy efficacy through survivin mRNA-initiated Bcl-2 protein silencing. As shown in Fig. 5A, a special PER hairpin containing domain b (red), the sequence of Bcl-2 antisense DNA (Bcl-2 ASD), was designed. When the target survivin mRNA was present, the hairpin was hybridized with the survivin mRNA and exposed the segment that was complementary to the primer. Then, the primer directed the synthesis of domain b at the 3′ end of the primer strand through the PER pathway via the mechanism in Scheme 1. This process produced a large number of Bcl-2 ASDs, which bound to a key region of the Bcl-2 mRNA and inhibited the translation of the Bcl-2 protein, accordingly resulting in cell apoptosis.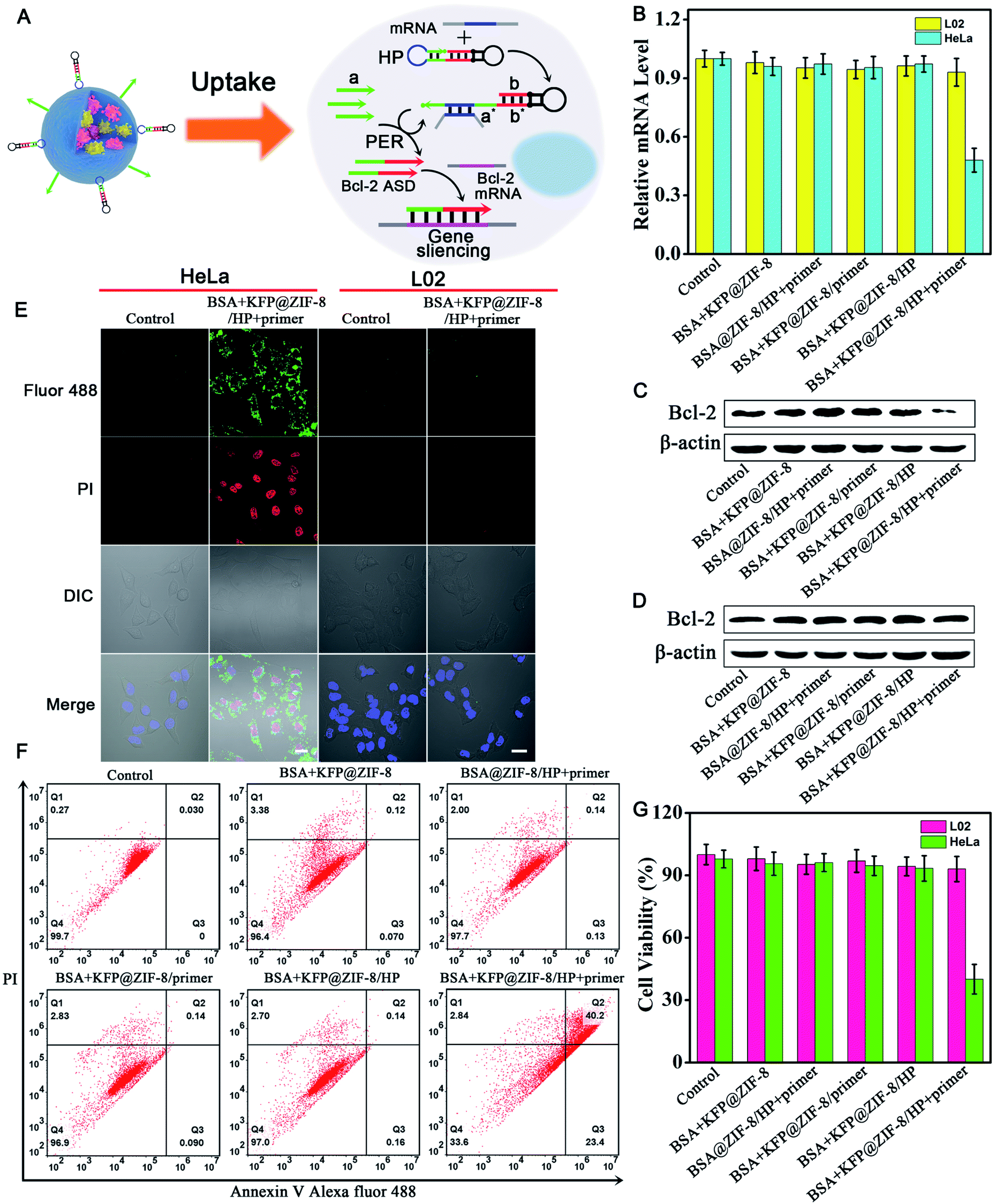 | ||
| Fig. 5 Gene silencing efficacy of the survivin mRNA triggered PER-based DNA machine in living cells. (A) Schematic illustration of gene silencing based on the PER DNA machine triggered by survivin mRNA. (B) Reverse transcription quantitative PCR (qRT-PCR) analysis of the relative Bcl-2 mRNA expression levels in total RNA extracted from HeLa cells and L02 cells treated with 60 μg mL−1 different nanoparticles. Western blot analysis of the Bcl-2 protein and β-actin protein in cell lysates from HeLa cells (C) and L02 cells (D). (E) Confocal images of annexin V Alexa Fluor 488 (AV 488)/PI stained apoptotic HeLa and L02 cells without treatment and after treatment with BSA + KFP@ZIF-8/HP + primer NPs. (F) Flow cytometry assay of apoptotic HeLa cells after treatment with the samples: BSA + KFP@ZIF-8 NPs; BSA@ZIF-8/HP + primer NPs; BSA + KFP@ZIF-8/primer NPs; BSA + KFP@ZIF-8/HP NPs; and BSA + KFP@ZIF-8/HP + primer NPs. (G) Cell viability of HeLa and L02 cells after incubation with different nanoparticles. Cells without treatment served as the control. Scale bar: 20 μm. | ||
The relative expression levels of Bcl-2 mRNA in HeLa cells were first measured using quantitative reverse transcription-PCR (qRT-PCR) analysis (Fig. 5B). As can be seen, the Bcl-2 mRNA expression levels of HeLa cells treated with BSA + KFP@ZIF-8/HP + primer NPs for 3 h followed by replacement with another fresh culture medium for 48 h decreased by approximately 50% compared with those of untreated cells, whereas no obvious changes in the Bcl-2 mRNA expression levels could be observed for the cells treated with other nanoparticles, in which either the KFP, HP or primer was omitted. This result indicated that the Bcl-2 ASD could be produced in the HeLa cells using the PER-based machine only when the KFP, HP and primer were present and no Bcl-2 ASD could be produced when either the KFP, HP and primer or absent, further demonstrating the reliability of the PER-based machine. It was noteworthy that the Bcl-2 mRNA expression levels in L02 cells showed no significant change even when the cells were treated with BSA + KFP@ZIF-8/HP + primer NPs. This was due to the lack of survivin mRNA in the L02 cells, leading to the failure to initiate the PER-based machine. This result implied that the PER-based machine had the potential to selectively kill cancer cells and has no effect on normal cells. The Bcl-2 protein expression levels were also studied by western blotting analysis. It could be clearly seen from Fig. 5C that Bcl-2 protein expression levels decreased remarkably in HeLa cells treated with BSA + KFP@ZIF-8/HP + primer NPs compared with those of untreated cells or the cells treated with other nanoparticles, in which either the KFP, HP or primer was omitted. In contrast, no appreciable decrease in the Bcl-2 protein expression levels could be observed in the L02 cells treated with BSA + KFP@ZIF-8/HP + primer NPs (Fig. 5D), suggesting that the PER-based machine could be triggered only by the survivin mRNA in the HeLa cells. In addition, the Bcl-2 protein expression levels were also investigated by immunofluorescence assays. HeLa cells treated with BSA + KFP@ZIF-8/HP + primer NPs showed weaker green fluorescence in the cytoplasm than the untreated cells or the cells treated with other nanoparticles (Fig. S20†). Likewise, L02 cells treated with BSA + KFP@ZIF-8/HP + primer NPs still exhibited bright green fluorescence (Fig. S21†). The results confirmed strongly that the intracellular survivin mRNA indeed could initiate the PER-based machine to produce Bcl-2 ASD to decrease the Bcl-2 protein expression levels, providing a powerful gene-silencing approach.
To investigate the gene therapy efficacy, CLSM imaging after annexin V/PI co-staining was performed. As is seen in Fig. S22,† the Bcl-2 ASD bound with the a key region of Bcl-2 mRNA, accordingly inhibiting the translation of the Bcl-2 protein and resulting in cell apoptosis through Bcl-2 anti-apoptosis-related signaling pathway. Phosphatidylserine (PS), a lipid, is normally restricted to the inner leaflet of the plasma membrane and is only exposed to the cell cytoplasm. However, PS will be exposed on the outer leaflet of the plasma membrane during apoptosis. Annexin V, a calcium-binding protein, can bind to PS. Therefore, fluorescently labeled annexin V is used to detect PS that is exposed on the outside of apoptotic cells. In addition, apoptotic cells can be distinguished by co-staining with propidium iodide (PI) because PI enters apoptotic cells but is excluded from living cells. So, bright green and red fluorescence was observed in HeLa cells incubated with BSA + KFP@ZIF-8/HP + primer NPs (Fig. 5E and S23†), indicating that the PER-based system could serve as an efficient gene drug machine for continuous production of Bcl-2 ASD to induce cell apoptosis. In contrast, the L02 cells gave no obvious green and red fluorescence after incubation with BSA + KFP@ZIF-8/HP + primer NPs (Fig. 5E and S24†), indicating that the survivin mRNA initiated PER-based machine provided an efficient strategy to selectively kill the cancer cells. In addition, the flow cytometry analysis of annexin V/PI co-staining was also carried out (Fig. 5F). The total apoptotic cell percent (including early and late apoptosis) induced by BSA + KFP@ZIF-8 NPs, BSA@ZIF-8/HP + primer NPs, BSA + KFP@ZIF-8/primer NPs, BSA + KFP@ZIF-8/HP NPs and BSA + KFP@ZIF-8/HP + primer NPs was determined to be 3.6%, 2.3%, 3.1%, 3.0% and 63.6%, respectively. Nevertheless, no significant increase in the total apoptotic cell percent was obtained in L02 cells even when the cells were incubated with BSA + KFP@ZIF-8/HP + primer NPs (Fig. S25†). This result was consistent with that obtained by the CLSM imaging. Finally, the cell viability of HeLa cells, L02 cells (Fig. 5G and S26†) and MCF-7 cells (Fig. S27†) was evaluated after incubation with different nanoparticles. The cell viability of HeLa cells and MCF-7 incubated with BSA + KFP@ZIF-8/HP + primer NPs significantly decreased compared with that of untreated cells or the cells treated with other nanoparticles. In contrast, the cell viability of L02 cells was nearly unchanged even when the cells were treated with BSA + KFP@ZIF-8/HP + primer NPs. Otherwise, these results strongly demonstrated that the survivin mRNA initiated PER-based machine could efficiently produce a gene drug to induce cell apoptosis and provide a promising strategy to selectively kill the cancer cells.
Conclusions
In summary, we successfully built an intracellular survivin mRNA initiated PER-based DNA machine by using ZIF-8 NPs to co-deliver the components including the hairpin, primer and KFP to living cells. The machine was driven by the strand displacing polymerase KFP and fueled by dNTPs and could continuously produce Bcl-2 ASD after being triggered by the intracellular survivin mRNA. The results demonstrated that the machine could sensitively image survivin mRNA expression levels and selectively kill the cancer cells and has no effect on the normal cells. The PER-based DNA machine holds great potential for early stage tumor diagnosis and precise tumor therapy. In addition, by reprogramming the hairpin stem domain and loop domain, the DNA machine could be retrofitted to produce the desired DNA single strand with arbitrary sequences after being triggered by other biomarkers such as mRNA, miRNA, small molecules and even proteins (when reprogramming the loop domain with the aptamer). Moreover, the ability of ZIF-8 NPs to co-deliver proteins and nucleic acids also provides an excellent platform to design various protein enzyme-based nucleic acid amplification reactions in living cells for disease diagnosis and therapy.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21525522, 21991080 and 21705039), Hunan Provincial Science and Technology Department (No. 2019RS3011), the Foundation for Innovative Research Groups of NSFC (Grant 21521063), and the Fundamental Research Funds for the Central University.Notes and references
- O. Nakagawa, X. Ming, L. Huang and R. L. Juliano, J. Am. Chem. Soc., 2010, 132, 8848–8849 CrossRef CAS PubMed.
- Q. B. Mou, Y. Ma, F. Ding, X. H. Gao, D. Y. Yan, X. Y. Zhu and C. Zhang, J. Am. Chem. Soc., 2019, 141, 6955–6966 CrossRef CAS PubMed.
- D. R. Corey, Nat. Neurosci., 2017, 20, 497–499 CrossRef CAS PubMed.
- M. A. Havens and M. L. Hastings, Nucleic Acids Res., 2016, 44, 6549–6563 CrossRef PubMed.
- X. J. Ren, R. J. Deng, L. D. Wang, K. X. Zhang and J. H. Li, Chem. Sci., 2017, 8, 5692–5698 RSC.
- C. A. Stein and Y. C. Cheng, Science, 1993, 261, 1004–1012 CrossRef CAS PubMed.
- A. Astriab-Fisher, D. Sergueev, M. Fisher, B. R. Shaw and R. L. Juliano, Pharm. Res., 2002, 19, 744–754 CrossRef CAS PubMed.
- J. J. Yang, Q. Jiang, L. He, P. F. Zhen, Q. Liu, S. L. Liu, M. F. Fu, J. B. Liu, C. Li and B. Q. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 23693–23699 CrossRef CAS PubMed.
- J. B. Liu, T. T. Wu, X. H. Lu, X. H. Wu, S. L. Liu, S. Zhao, X. H. Xu and B. Q. Ding, J. Am. Chem. Soc., 2019, 141, 19032–19037 CrossRef CAS PubMed.
- Y. Wang, L. Miao, A. Satterlee and L. Huang, Adv. Drug Delivery Rev., 2015, 87, 68–80 CrossRef CAS PubMed.
- O. B. Garbuzenko, M. Saad, V. P. Pozharov, K. R. Reuhl, G. Mainelis and T. Minko, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10737–10742 CrossRef CAS PubMed.
- O. Nakagawa, X. Ming, L. Huang and R. L. Juliano, J. Am. Chem. Soc., 2010, 132, 8848–8849 CrossRef CAS PubMed.
- Z. J. Wang, Y. Fu, Z. Z. Kang, X. G. Liu, N. Chen, Q. Wang, Y. Q. Tu, L. H. Wang, S. P. Song, D. S. Ling, H. Y. Song, X. Q. Kong and C. H. Fan, J. Am. Chem. Soc., 2017, 139, 15784–15791 CrossRef CAS PubMed.
- R. Huschka, A. Barhoumi, Q. Liu, J. A. Roth, L. Ji and N. J. Halas, ACS Nano, 2012, 6, 7681–7691 CrossRef CAS PubMed.
- M. Z. Alyami, S. K. Alsaiari, Y. Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715–1720 CrossRef CAS PubMed.
- T. E. Schaus, S. W. Woo, F. Xuan, X. Chen and P. Yin, Nat. Commun., 2017, 8, 696–704 CrossRef PubMed.
- J. Y. Kishi, T. E. Schaus, N. Gopalkrishnan, F. Xuan and P. Yin, Nat. Chem., 2018, 10, 155–164 CrossRef CAS PubMed.
- J. Y. Kishi, B. J. Beliveau, S. W. Lapan, E. R. West, A. Zhu, H. M. Sasaki, S. K. Saka, Y. Wang, C. L. Cepko and P. Yin, Nat. Methods, 2019, 16, 533–544 CrossRef CAS PubMed.
- J. T. Liu, T. R. Liu, P. Du, L. Zhang and J. P. Lei, Angew. Chem., Int. Ed., 2019, 131, 7890–7894 CrossRef.
- D. W. Zhang, Q. Lei, J. Y. Zhu, J. X. Fan, C. X. Li, C. Li, Z. S. Xu, S. X. Cheng and X. Z. Zhang, Nano Lett., 2017, 17, 284–291 CrossRef PubMed.
- X. T. Yang, Q. Tang, Y. Jiang, M. N. Zhang, M. Wang and L. Q. Mao, J. Am. Chem. Soc., 2019, 141, 3782–3786 CrossRef CAS PubMed.
- C. Liu, J. Xing, O. U. Akakuru, L. J. Luo, S. Sun, R. F. Zou, Z. S. Yu, Q. L. Fang and A. G. Wu, Nano Lett., 2019, 19, 5674–5682 CrossRef CAS PubMed.
- F. J. Lyu, Y. F. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- J. T. Yi, T. T. Chen, J. Huo and X. Chu, Anal. Chem., 2017, 89, 12351–12359 CrossRef CAS PubMed.
- H. M. Wang, Y. Q. Chen, H. Wang, X. Q. Liu, X. Zhou and F. N. Wang, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- M. Zheng, S. Liu, X. G. Guan and Z. G. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 22181–22187 CrossRef CAS PubMed.
- C. Y. Sun, C. Qin, X. L. Wang, G. S. Yang, K. Z. Shao, Y. Q. Lan, Z. M. Su, P. Huang, C. G. Wang and E. B. Wang, Dalton Trans., 2012, 41, 6906–6909 RSC.
- H. Y. Zhang, Q. Li, R. L. Liu, X. K. Zhang, Z. H. Li and Y. X. Luan, Adv. Funct. Mater., 2018, 28, 1802830–1802840 CrossRef.
- Y. Zhang, F. M. Wang, E. G. Ju, Z. Liu, Z. W. Chen, J. S. Ren and X. G. Qu, Adv. Funct. Mater., 2016, 26, 6454–6461 CrossRef CAS.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS PubMed.
- G. Cheng, W. Q. Li, L. Ha, X. H. Han, S. J. Hao, Y. Wan, Z. G. Wang, F. P. Dong, X. Zou, Y. W. Mao and S. Zheng, J. Am. Chem. Soc., 2018, 140, 7282–7291 CrossRef CAS PubMed.
- Y. F. Feng, H. R. Wang, S. N. Zhang, Y. Zhao, J. Gao, Y. Y. Zheng, P. Zhao, Z. J. Zhang, M. J. Zaworotko, P. Cheng, S. Q. Ma and Y. Chen, Adv. Mater., 2018, 31, 1805148–1805154 CrossRef PubMed.
- T. T. Chen, J. T. Yi, Y. Y. Zhao and X. Chu, J. Am. Chem. Soc., 2018, 140, 9912–9920 CrossRef CAS PubMed.
- J. Zhang, M. Y. He, C. P. Nie, M. M. He, Q. S. Pan, C. Liu, Y. L. Hu, J. T. Yi, T. T. Chen and X. Chu, Anal. Chem., 2019, 91, 9049–9057 CrossRef CAS PubMed.
- C. S. Lee, R. W. Davis and N. Davidson, J. Mol. Biol., 1970, 48, 1–22 CrossRef CAS PubMed.
- Z. Wu, G. Q. Liu, X. L. Yang and J. H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef CAS PubMed.
- W. Pan, T. T. Zhang, H. J. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581–10588 CrossRef CAS PubMed.
- A. Webb, D. Cunningham, F. Cotter, P. A. Clarke, F. Stefano, P. Ross, M. Corbo and Z. Dziewanowska, Lancet, 1997, 349, 1137–1141 CrossRef CAS.
- X. J. Yang, C. G. Koh, S. J. Liu, X. G. Pan, R. Santhanam, B. Yu, Y. Peng, J. X. Pang, S. Golan, Y. Talmon, Y. Jin, N. Muthusamy, J. C. Byrd, K. K. Chan, L. J. Lee, G. Marcucci and R. J. Lee, Mol. Pharm., 2008, 6, 221–230 CrossRef PubMed.
- Y. W. Li, N. Xu, W. H. Zhu, L. Wang, B. Liu, J. X. Zhang and Z. G. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 22974–22984 CrossRef CAS PubMed.
- H. Ren, L. Zhang, J. An, T. Wang, L. Li, X. Si, L. He, X. Wu, C. Wang and Z. Su, Chem. Commun., 2014, 50, 1000–1002 RSC.
- C. Moreira, H. Oliveira, L. R. Pires, S. Simões, M. A. Barbosa and A. P. Pego, Acta Biomater., 2009, 5, 2995–3006 CrossRef CAS PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, K. Quan, L. Wang, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2016, 88, 5981–5987 CrossRef CAS PubMed.
- D. G. He, X. He, X. Yang and H. W. Li, Chem. Sci., 2017, 8, 2832–2840 RSC.
- L. Liu, J. W. Liu, H. Wu, X. N. Wang, R. Q. Yu and J. H. Jiang, Anal. Chem., 2018, 90, 1502–1505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00339e |
| This journal is © The Royal Society of Chemistry 2020 |