
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pnictogen-bonding catalysis: brevetoxin-type polyether cyclizations†
Andrea
Gini‡
a,
Miguel
Paraja‡
a,
Bartomeu
Galmés
b,
Celine
Besnard
 a,
Amalia I.
Poblador-Bahamonde
a,
Naomi
Sakai
a,
Antonio
Frontera
b and
Stefan
Matile
*a
a,
Amalia I.
Poblador-Bahamonde
a,
Naomi
Sakai
a,
Antonio
Frontera
b and
Stefan
Matile
*a
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Tel: +41 22 379 6523
bDepartment de Química, Universitat de les Illes Balears, Palma de Mallorca, Spain
First published on 25th June 2020
Abstract
Pnictogen-bond donors are attractive for use in catalysis because of deep σ holes, high multivalency, rich hypervalency, and chiral binding pockets. We here report natural product inspired epoxide-opening polyether cyclizations catalyzed by fluoroarylated Sb(V) > Sb(III) > Bi > Sn > Ge. The distinctive characteristic found for pnictogen-bonding catalysis is the breaking of the Baldwin rules, that is selective endo cyclization into the trans-fused ladder oligomers known from the brevetoxins. Moreover, tris(3,4,5-trifluorophenyl)stibines and their hypervalent stiborane catecholates afford different anti-Baldwin stereoselectivity. Lewis (SbCl3), Brønsted (AcOH) and π acids fail to provide similar access to these forbidden rings. Like hydrogen-bonding catalysis differs from Brønsted acid catalysis, pnictogen-bonding catalysis thus emerges as the supramolecular counterpart of covalent Lewis acid catalysis.
Pnictogen and tetrel bonds refer to non-covalent interactions1–7 between electron-rich acceptors and σ holes on group V (15) and group IV (14) atoms, respectively (Fig. 1a–c).3,4 σ Holes are regions with positive electrostatic potential appearing at the side opposite to σ bonds to electron-withdrawing substituents R. Compared to better established halogen5 and chalcogen bonds,6 pnictogen- and, although less important in this study, also tetrel-bond donors are of higher valency and thus offer more σ holes. Moreover, pnictogen-bond donors can be interconversion-free8 stereogenic centers9 and at the origin of chiral axes.10 σ-Hole interactions are primarily electrostatic. They strengthen with the depth of the σ hole, which relates to polarizability, thus increases downward and toward the left in the periodic table.1,2
 | ||
| Fig. 1 (a) Region of interest in the periodic table. (b and c) General structure of tetrel and pnictogen-bond donors (D; DIII: trivalent; DV: pentavalent) interacting with their acceptors (A); blue circles, σ holes; red orbitals, lone pairs. (d) Pnictogen-bonding catalysis defined as a non-covalent counterpart of (e) Lewis acid catalysis (La), analogous to (f) hydrogen-bonding and (g) Brønsted acid catalysis (Ba, conjugate base: Bb−). S, substrate; P, product; etc.*: ligand (L) exchange, proton release from S upon addition to La, etc. | ||
Here, we suggest to define pnictogen-bonding catalysis as the non-covalent, supramolecular counterpart of classical covalent Lewis acid catalysis (Fig. 1d and e). This is analogous to hydrogen-bonding and Brønsted acid catalysis, with interactions that become too strong transfer their proton and form new covalent bonds (Fig. 1f and g). Similarly overachieving cation-π and anion-π interactions can continue into electrophilic and nucleophilic aromatic substitution, respectively.11 Group 15 Lewis acids, however, have been studied exhaustively as reagents and catalysts.2,12–14 Except for a few recent examples,2,12,13 possible contributions from pnictogen bonds to these activities were either ignored or alluded to from different points of view.14 The question thus arises whether or not pnictogen-bonding catalysis is just a weak form of Lewis acid catalysis and thus essentially trivial. The differences between hydrogen-bonding and Brønsted acid catalysis are understood. The differences in structure and charge distribution between non-covalent pnictogen bonding and covalent ligand addition/exchange (Fig. 1d and e) further support that pnictogen-bonding catalysis should exist and matter. In the following, we show that this is indeed the case.
Most catalyst candidates 1–13 were readily accessible in a few steps from commercially available substrates (Fig. 2a, Schemes S1–S3,† X-ray structures: Fig. 2b, S75–S86†).2 Only Bi 7 was too unstable in our hands.15 Stibine 1 was obtained by nucleophilic substitution of SbCl3 with aryl anions derived from bromobenzene 14 (Fig. 2a). Sb(III) 1 was oxidized with chloranil (Ch) 1512 to give stiborane 2. Molecular electrostatic potential surfaces (MEP, BP86-D3/def2-TZVP level) confirmed12 that this oxidation converts the three deep σ holes on Sb(III) 1 into one deep σ hole on Sb(V) 2 (Fig. 2b and c). Consistent with increasing polarizability,1,2 Sn(IV) 3 excelled with four deep σ holes, whereas the σ holes of the smaller Ge(IV) 4 were not accessible. In 1–4, the ortho fluorines of the original perfluorinated 52 were replaced by hydrogens because the crystal structure of 5 indicated the existence of Sb–F pnictogen bonds that weaken and obstruct all σ holes (Fig. 2b). The acidic ortho hydrogens in 1–4 should further assist σ-hole interactions with proximal C–H⋯A bonds (see below).
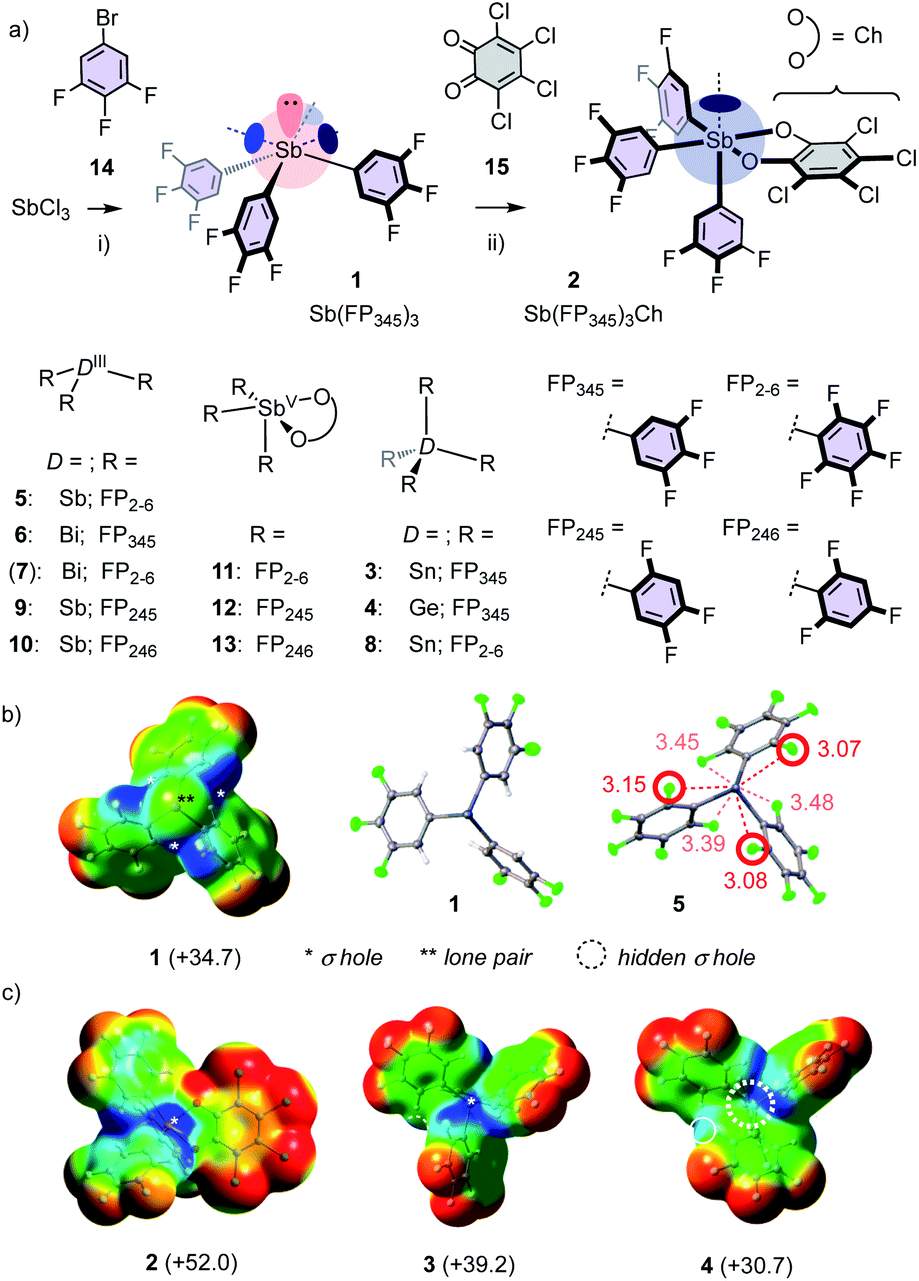 | ||
| Fig. 2 Catalyst synthesis and structures (a), with MEP and crystal structures (b and c); distances in Å; *: positive MEP maxima (kcal mol−1), corresponding to σ holes. Dashed circle: inaccessible σ hole. (i) nBuLi, Et2O, −78 °C to rt, 12 h, 45%; (ii) CH2Cl2, rt, 10 min, 78%. | ||
The structural complexity of epoxide-opening ether cyclizations16–18 was considered as ideal to identify possible differences between pnictogen-bonding and Lewis acid catalysis. Initial studies focused on monomers 16–19 (Fig. 3a). According to the Baldwin (B) rules, their 5-exo-tet cyclization into oxolanes 20–23 is preferred over 6-endo-tet anti-Baldwin (A) oxanes 24–27.16–18 After one day under standard conditions, Sb(FP345)31 converted 81% of cis epoxide 1717,18 into (B)-21 (Table 1, entry 1). Reactions were much slower with Bi 6, Sn 3 and Ge 4 (entries 2–5). However, tetrel-bonding Sn 3 remained operational as catalyst, as confirmed with high conversion at 20 mol% (entry 4). FP2-65 and 8 were unstable, supporting that the ortho hydrogens in FP345 minimize not only σ-hole obstruction but also catalyst decomposition.
 | ||
| Fig. 3 Cyclization of (a) monomers 16–19 and (b) dimers 28 with representative 1H NMR spectrum of a product mixture generated for 28 with 1. (c) NMR fingerprints for 28 cyclized with AcOH, SbCl3, 1 and 2. (d) 1H NMR spectrum and (e) crystal structure of trans,trans (AA)-33. | ||
| Cb | c C (mol%) | Sd | c S (M) | T (°C) | t (d) | η t (%) | B/Ai | |
|---|---|---|---|---|---|---|---|---|
| a For more data, see Tables S1–S11. b Catalysts. c Concentration (CD2Cl2). d Substrates. e Concentration. f Reaction temperature. g Time to reach the given. h Conversion; vf, very fast, <10 min. i Selectivity, B = Baldwin, A = anti-Baldwin products. | ||||||||
| 1 | 1 | 100 | 17 | 1.6 | 40 | 1 | 81 | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 :0 |
| 2 | 6 | 100 | 17 | 1.6 | 40 | 8 | 89 | 100:0 |
| 3 | 3 | 100 | 17 | 1.6 | 40 | 8 | 63 | 100:0 |
| 4 | 3 | 20 | 17 | 1.6 | 40 | 14 | 40 | 100:0 |
| 5 | 4 | 100 | 17 | 1.6 | 40 | 8 | 28 | 100:0 |
| 6 | 1 | 100 | 19 | 2.4 | 40 | 1 | 81 | 56:44 |
| 7 | 1 | 20 | 19 | 2.4 | rt | 9 | 71 | 59:41 |
| 8 | 2 | 10 | 19 | 1.0 | rt | vf | >99 | 46:54 |
| 9 | 2 | 0.1 | 19 | 1.0 | rt | vf | >99 | 25:75 |
| 10 | 11 | 1 | 19 | 1.0 | rt | vf | >99 | 68:32 |
| 11 | SbCl3 | 1 | 19 | 1.0 | rt | vf | >99 | 80:20 |
| 12 | AcOH | 100 | 19 | 2.1 | 40 | 1 | >99 | 92:8 |
With the permethylated monomer 19,17,18 stibine 1 produced significant amounts of anti-Baldwin product (B/A ≈ 6:4, entry 6, 7). Access to anti-Baldwin selectivity depended on substrate (18 > 19 > 17, 16) and catalyst structures (2 > 13 > 12 > 1–9 > 11 > 10, Tables 1, S2–S6†). Some small but significant irregularities in dependence of endo/exo selectivity on catalyst structure nicely illustrated the influence of the specific environment in the respective binding pockets with pnictogen-bonding catalysis. Proximity effects in binding pockets is a hallmark of supramolecular catalysis, much appreciated in hydrogen-bonding catalysis to access stereoselectivity, and absent in covalent “general” Brønsted acid catalysis, which is independent of the acid used. Very fast conversion on the “cyclopean” σ hole of Sb(V) stiborane 2 allowed for meaningful studies at lower temperature as well as lower catalyst loading, which caused the expected increase in anti-Baldwin selectivity (entry 8, 9). The ortho-fluorinated Gabbaï original 1112 failed to break the Baldwin rules, as did Lewis and Brønsted acid controls (entries 10–12).
Access to anti-Baldwin cascade cyclizations was of general interest also because, in nature, Baldwin oligomers such as the monensin-like ionophores are complemented by the rich family of brevetoxin-like ladder oligomers.16–18 Minimalist cascade cyclizations were explored with a cis–trans mixture of diepoxide 28 to maximize the number of constitutional and stereoisomers contributing to catalyst fingerprints (Fig. 3b and c). 1H NMR spectroscopy and X-ray analyses of at least partially purified products and comparison with literature data17 allowed us to assign NMR signals to isomers 29–33 (Fig. 3b–e, S10–S13†). The endo/exo selectivity was estimated from the ratio of characteristic peaks in the spectra of the product mixtures. Isolated, easy to integrate peaks of cis,cis-(BB)-30 and trans,trans-(AA)-33 were selected because they originate from the same substrate isomer, i.e., trans,syn-28 (Fig. S13†). The results are described as BB/AA ratios (Table S11†).
Brønsted acid catalysis with AcOH afforded (BB)-30 exclusively (Fig. 3c and Table S11†). With Lewis acid SbCl3, Baldwin selectivity persisted (BB/AA = 8:2). In contrast, pnictogen-bonding catalysts Sb(III) 1 (BB/AA = 3:7) and Sb(V) 2 (BB/AA = 1:9) both broke the Baldwin rules. The Gabbaï original 11, however, failed to do so (BB/AA = 6:4). The stereoselectivity of (AA)-33 produced by Sb(III) 1 and Sb(V) 2 differed. The according to the crystal structure (Fig. 3e and S87†) trans-fused trans epimer (AA)-33 was reasonably accessible only with Sb(III) 1 (tt/tc = 1:1), whereas the trans-fused cis epimer was the main product with the hypervalent Sb(V) 2 (tt/tc = 1:2; Fig. 3c, blue). The isolation of (A)-29 as a dominant intermediate supported that the cascades are directional.
The cyclizations of trimers 34 and tetramers 35 were characterized mainly by comparing their 1H NMR fingerprint to those of dimers (Fig. 4, S18 and S20†). The products obtained from 34 with AcOH showed a cluster of signals between 3.75 ∼ 4.10 ppm, characteristic of Baldwin products (Fig. 4b). With pnictogen-bonding catalysts 1 and 2, the appearance of up-field shifted peaks revealed anti-Baldwin selective cyclizations. NMR fingerprints of cascade cyclized tetramer 35 showed the same trends at increased complexity, containing up to 16 constitutional isomers, from (B4)-36 to (A4)-37 (Fig. S17†). In NMR fingerprints beyond dimers, differences between pnictogen-bonding and Lewis acid catalysis remained visible but became increasingly difficult to quantify. Gas chromatography (GC) proved more revealing, confirming the lessons learned on the dimer level: the reactivity of supramolecular pnictogen-bonding catalysts Sb(III) 1 and Sb(V) 2 differs from covalent Lewis acid catalysts like SbCl3, and the former excel with an almost complete suppression of all-Baldwin products (Fig. 4c, S19 and S21†).
 | ||
| Fig. 4 (a) Cascade cyclization of 34 and 35 with pure B and A oligomers shown as extreme products. (b) 1H NMR and (c) GC fingerprints of 34 converted with AcOH, SbCl3, 1 and 2 (c, Baldwin products: tR 6.5–6.9 min, anti-Baldwin: tR > 6.9 min, substrates: tR < 4.0 min). | ||
Computational studies were complicated by the high number of possible stereochemical and conformational isomers (Fig. S22–S28†). However, significant isolate observations could nevertheless be secured. Firstly, the binding of epoxide 19 to Sn 3 revealed a formal tetrel bond,1,4 shorter than the sum of vdW radii (3.69 Å) and longer than covalent bonds (2.03 Å, Fig. 5a). The smaller Ge 4 preserved the bidentate CH⋯O interactions but lost the tetrel bond (3.66 Å, vdW 3.62 Å). These findings were consistent with accessible σ holes on the MEP surface of Sn 3 but not Ge 4 (Fig. 2c). Although weak and presumably precedented in the Lewis acid literature,1,4,14 the cyclization of 17 with Sn 3 could thus be considered as one of the first examples of explicit tetrel-bonding catalysis (Table 1, entry 3, 4). Also worth noting were more than one tetrel bond with oligoepoxides (Fig. S27†), and four intermolecular tetrel bonds in the crystal structure of Sn 3 but not of Ge 4 (Fig. S77†).
 | ||
| Fig. 5 BP86-D3/def2-TZVP optimized intermediates with (a) 19 bound to 3, (b) 35 to 1, and (c) 34 to 2, with schematic drawings, distances in Å, polyepoxide foldamers in (b) carousel-like with parallel and (c) snake-like conformation with antiparallel epoxides. | ||
Most important were pnictogen bonds between epoxides of 35 to all three σ holes of 1 (Fig. 5b). This was not trivial because each pnictogen bond formed weakens the remaining σ holes.7 This finding thus supported contributions from multivalency, including entropy-driven substrate destabilization,17–19 to catalysis. The last epoxide is engaged in lonepair-π interactions, ready to occupy the σ hole liberated by the first ring formed. Finally, a single pnictogen bond to 34 confirmed the loss of multivalency of 2, which was compensated by CH⋯O and lonepair-π interactions. Affinity gradients in the resulting “triad” would be compatible with 2 crawling along the antiparallel epoxides in snake-like foldamer tracks (Fig. 5c).
In summary, with the hypersensitive epoxide-opening polyether cascade cyclizations, we show that pnictogen-bonding catalysts are more than just weak Lewis acids. Naturally slower and not autocatalytic like on π-acidic aromatic surfaces (Fig. S6–S8†),17,18 the distinctive characteristic of pnictogen-bonding catalysis is the breaking of the Baldwin rules. Important differences in regio- and stereoselectivity exist also between multivalent Sb(III) and hypervalent Sb(V) pnictogen-bonding catalysts. These initial results on pnictogen-bonding catalysis thus support the general expectation that the integration of unorthodox interactions20 will provide access to new reactivity. Attractive perspectives include antimony as stereogenic center9,10 combined with multivalency, and the integration into more advanced functional systems.11,21
The discussion about the difference between pnictogen-bonding and Lewis acid catalysis launched in this report will continue and spread into other, less affected σ-hole interactions. The need for such a distinction will have to be confirmed, and the tantalizing question where and how to draw the line will persist, particularly considering the underlying continuum and the dependence on the involved partner, either pnictogen-bond acceptor or Lewis base (i.e., every weak enough pnictogen-bond acceptor will turn also a strong Lewis acid like SbCl3 into a pnictogen-bond donor22). Differences in bond length, changes in geometry, charge distribution or deprotonation (Fig. 1) are all convincing but indirect measures to draw this line; direct functional differences as identified in this study will ultimately be needed. What remains for certain is that the IUPAC definition restricts Lewis acids to reactions and covalency,23 while extrapolation from halogen bonding24 defines pnictogen-bond donors as the supramolecular counterpart, i.e., electrophilic regions that interact non-covalently, rather than react covalently like Lewis acids. The comparison with non-covalent hydrogen-bonding catalysis and covalent Brønsted acids, presumably applicable to all σ-hole catalysis,25 could thus help in this situation because the same ambiguities exist but they are understood and appreciated. However, despite all compelling analogies, only the future will tell if σ-hole catalysis in general and pnictogen-bonding catalysis in particular will also become as important as the complementary hydrogen-bonding catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NMR and MS platforms for services, and the University of Geneva, the NCCR Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support. AF thanks the MINECO/AEI (project CTQ2017-85821-R, FEDER funds) for financial support.Notes and references
- (a) I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS; (b) J. Bamberger, F. Ostler and O. Garcia Mancheño, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed; (c) R. L. Sutar and M. S. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS; (d) M. S. Taylor, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS; (e) J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CrossRef CAS; (f) S. Scheiner and J. Lu, Chem.–Eur. J., 2018, 24, 8167–8177 CrossRef CAS PubMed.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- (a) P. Scilabra, G. Terraneo and G. Resnati, J. Fluorine Chem., 2017, 203, 62–74 CrossRef CAS; (b) S. Scheiner, Acc. Chem. Res., 2013, 46, 280–288 CrossRef CAS PubMed; (c) S. Moaven, J. Yu, M. Vega, D. K. Unruh and A. F. Cozzolino, Chem. Commun., 2018, 54, 8849–8852 RSC; (d) C. Leroy, R. Johannson and D. L. Bryce, J. Phys. Chem. A, 2019, 123, 1030–1043 CrossRef CAS PubMed; (e) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2016, 17, 1608–1614 CrossRef PubMed; (f) G. S. Girolami, J. Chem. Educ., 2009, 86, 1200 CrossRef CAS; (g) J. Starbuck, N. C. Norman and A. G. Orpen, New J. Chem., 1999, 23, 969–972 RSC.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed; (b) A. Karim, N. Schulz, H. Andersson, B. Nekoueishahraki, A.-C. C. Carlsson, D. Sarabi, A. Valkonen, K. Rissanen, J. Gräfenstein, S. Keller and M. Erdelyi, J. Am. Chem. Soc., 2018, 140, 17571–17579 CrossRef CAS PubMed; (c) S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824–1834 RSC; (d) M. Michalczyk, W. Zierkiewicz, R. Wysokiński and S. Scheiner, ChemPhysChem, 2019, 20, 959–966 CrossRef CAS PubMed; (e) S. Scheiner, J. Phys. Chem. A, 2018, 122, 2550–2562 CrossRef CAS PubMed; (f) H. Nakamura, K. Ishihara and H. Yamamoto, J. Org. Chem., 2002, 67, 5124–5137 CrossRef CAS PubMed; (g) R. Berger, K. Duff and J. L. Leighton, J. Am. Chem. Soc., 2004, 126, 5686–5687 CrossRef CAS PubMed; (h) R. Hrdina, C. E. Müller, R. C. Wende, K. M. Lippert, M. Benassi, B. Spengler and P. R. Schreiner, J. Am. Chem. Soc., 2011, 133, 7624–7627 CrossRef CAS PubMed.
- (a) X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400–404 CrossRef CAS PubMed; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- (a) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed; (b) P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CAS; (c) G. Garrett, E. Carrera, D. Seferos and M. Taylor, Chem. Commun., 2016, 52, 9881–9884 RSC; (d) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
- (a) N. Biot and D. Bonifazi, Chem.–Eur. J., 2018, 24, 5439–5443 CrossRef CAS PubMed; (b) V. Kumar, Y. Xu and D. L. Bryce, Chem.–Eur. J., 2020, 26, 3275–3286 CrossRef CAS PubMed; (c) S. Moaven, M. C. Andrews, T. J. Polaske, B. M. Karl, D. K. Unruh, E. Bosch, N. P. Bowling and A. F. Cozzolino, Inorg. Chem., 2019, 58, 16227–16235 CrossRef CAS PubMed; (d) K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef PubMed.
- P. Schwerdtfeger, L. J. Laakkonen and P. Pyykkö, J. Chem. Phys., 1992, 96, 6807–6819 CrossRef CAS.
- (a) K. Mislow, A. Zimmerman and J. T. Melillo, J. Am. Chem. Soc., 1963, 85, 594–597 CrossRef CAS; (b) S. Yasuike, Y. Kishi, S. Kawara, K. Yamaguchi and J. Kurita, J. Organomet. Chem., 2006, 691, 2213–2220 CrossRef CAS; (c) C. I. Raţ, C. Silvestru and H. J. Breunig, Coord. Chem. Rev., 2013, 257, 818–879 CrossRef.
- (a) S. Rizzo, T. Benincori, V. Bonometti, R. Cirilli, P. R. Mussini, M. Pierini, T. Pilati and F. Sannicolò, Chem.–Eur. J., 2013, 19, 182–194 CrossRef CAS PubMed; (b) B. A. Chalmers, C. B. E. Meigh, P. S. Nejman, M. Bühl, T. Lébl, J. D. Woollins, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2016, 55, 7117–7125 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- (a) M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed; (b) M. Yang, M. Hirai and F. P. Gabbaï, Dalton Trans., 2019, 48, 6685–6689 RSC.
- J. Schmauck and M. Breugst, Org. Biomol. Chem., 2017, 15, 8037–8045 RSC.
- (a) J. Lei, L. Peng, R. Qiu, Y. Liu, Y. Chen, C.-T. Au and S.-F. Yin, Dalton Trans., 2019, 48, 8478–8487 RSC; (b) K. Komeyama, N. Saigo, M. Miyagi and K. Takaki, Angew. Chem., Int. Ed., 2009, 48, 9875–9878 CrossRef CAS PubMed; (c) L. Liu, Y. Zhu, K. Huang, W. Chang and J. Li, Eur. J. Org. Chem., 2013, 2013, 2634–2645 CrossRef CAS; (d) O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS PubMed; (e) K. Ishihara, Y. Kuroki, N. Hanaki, S. Ohara and H. Yamamoto, J. Am. Chem. Soc., 1996, 118, 1569–1570 CrossRef CAS; (f) S. Kobayashi, T. Ogino, H. Shimizu, S. Ishikawa, T. Hamada and K. Manabe, Org. Lett., 2005, 7, 4729–4731 CrossRef CAS PubMed; (g) R. Nomura, A. Ninagawa and H. Matsuda, J. Org. Chem., 1980, 45, 3735–3738 CrossRef CAS; (h) P. Ondet, G. Lemière and E. Duñach, Eur. J. Org. Chem., 2017, 2017, 761–780 CrossRef CAS; (i) T. Harada and T. Mukaiyama, Chem. Lett., 1992, 21, 81–84 CrossRef; (j) G. A. Olah, Angew. Chem., Int. Ed., 1995, 34, 1393–1405 CrossRef CAS; (k) K. Ishihara, Lewis Acids in Organic Synthesis, John Wiley & Sons, Ltd, 2008, pp. 523–541 Search PubMed.
- A. Schmuck and K. Seppelt, Chem. Ber., 1989, 122, 803–808 CrossRef CAS.
- (a) H. Liu, S. Lin, K. M. Jacobsen and T. B. Poulsen, Angew. Chem., Int. Ed., 2019, 58, 13630–13642 CrossRef CAS PubMed; (b) S. Sittihan and T. F. Jamison, J. Am. Chem. Soc., 2019, 141, 11239–11244 CrossRef CAS PubMed; (c) K. Nakanishi, Toxicon, 1985, 23, 473–479 CrossRef CAS PubMed; (d) K. C. Nicolaou, C. V. C. Prasad, P. K. Somers and C. K. Hwang, J. Am. Chem. Soc., 1989, 111, 5335–5340 CrossRef CAS; (e) J. C. Valentine, F. E. McDonald, W. A. Neiwert and K. I. Hardcastle, J. Am. Chem. Soc., 2005, 127, 4586–4587 CrossRef CAS PubMed.
- (a) M. Paraja and S. Matile, Angew. Chem., Int. Ed., 2020, 59, 6273–6277 CrossRef CAS PubMed; (b) X. Zhang, X. Hao, L. Liu, A.-T. Pham, J. López-Andarias, A. Frontera, N. Sakai and S. Matile, J. Am. Chem. Soc., 2018, 140, 17867–17871 CrossRef CAS PubMed.
- M. Paraja, X. Hao and S. Matile, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202000681.
- (a) M. F. Menger and F. Nome, ACS Chem. Biol., 2019, 14, 1386–1392 CrossRef PubMed; (b) R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS PubMed.
- (a) L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS PubMed; (b) H. Wagner, K. Harms, U. Koert, S. Meder and G. Boheim, Angew. Chem., Int. Ed. Engl., 1996, 35, 2643–2646 CrossRef CAS; (c) B. Baumeister, N. Sakai and S. Matile, Org. Lett., 2001, 3, 4229–4232 CrossRef CAS PubMed.
- U. Müller and A. T. Mohammed, Z. Anorg. Allg. Chem., 1983, 506, 110–114 CrossRef.
- IUPAC. Compendium of Chemical Terminology, The Gold Book, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, Online version (2019-) created by S. J. Chalk, DOI:10.1351/goldbook.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. CCDC 1999316–1999326. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02551h |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |