
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe-catalyzed three-component dicarbofunctionalization of unactivated alkenes with alkyl halides and Grignard reagents†
Lei
Liu
 ,
Wes
Lee
,
Cassandra R.
Youshaw
,
Mingbin
Yuan
,
Michael B.
Geherty
,
Peter Y.
Zavalij
and
Osvaldo
Gutierrez
*
,
Wes
Lee
,
Cassandra R.
Youshaw
,
Mingbin
Yuan
,
Michael B.
Geherty
,
Peter Y.
Zavalij
and
Osvaldo
Gutierrez
*
Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, USA. E-mail: ogs@umd.edu
First published on 24th July 2020
Abstract
A highly chemoselective iron-catalyzed three-component dicarbofunctionalization of unactivated olefins with alkyl halides (iodides and bromides) and sp2-hybridized Grignard reagents is reported. The reaction operates under fast turnover frequency and tolerates a diverse range of sp2-hybridized nucleophiles (electron-rich and electron-deficient (hetero)aryl and alkenyl Grignard reagents), alkyl halides (tertiary alkyl iodides/bromides and perfluorinated bromides), and unactivated olefins bearing diverse functional groups including tethered alkenes, ethers, protected alcohols, aldehydes, and amines to yield the desired 1,2-alkylarylated products with high regiocontrol. Further, we demonstrate that this protocol is amenable for the synthesis of new (hetero)carbocycles including tetrahydrofurans and pyrrolidines via a three-component radical cascade cyclization/arylation that forges three new C–C bonds.
Introduction
Olefins are ubiquitous in natural products and bioactive compounds and serve as versatile commodity feedstocks. 1,2-Difunctionalization of olefins represents one of the most widely used strategies to build synthetic complexity in organic synthesis and serves as a platform to introduce concepts of chemo-, regio-, and stereoselectivity.1 Recently, there has been a surge in the development of three-component transition metal-catalyzed difunctionalization that employs olefins because of its potential to rapidly increase diversity in a single step (Scheme 1a).2–4 However, selective transition metal-catalyzed three-component alkylarylation of unactivated alkenes without electronically biased substrates or directing groups is rare.5 Moreover, despite the inherent attractive features of iron as a catalyst (Earth abundant, less toxic, inexpensive, and environmentally benign in comparison to Pd or Ni) in pharmaceutical settings, there are no general methods for iron-catalyzed three-component 1,2-dicarbofunctionalization of olefins.6–13 Recently, our group reported the use of a strained vinyl cyclopropanes to promote a three-component Fe-catalyzed reaction leading to 1,5-alkylarylation products (Scheme 1b).14,15 Unfortunately, despite numerous attempts, the 1,2-difunctionalization products were not observed, presumably due to much more rapid ring-opening of the incipient alkyl radical followed by C–C bond formation. Herein, we report the first iron-catalyzed 3-component dicarbofunctionalization of unactivated alkenes with both alkyl iodides and bromides with sp2-hybridized Grignard nucleophiles leading to 1,2-alkylarylation or 1,2-alkylvinylation of alkenes with broad scope and excellent regio- and chemoselectivity (Scheme 1c). Further, we applied this concept to develop a three-component radical alkylation/cyclization/arylation cascade leading to diverse (hetero)cyclic compounds. We anticipate that this report will lead to greater application of Fe as a catalyst in three-component difunctionalization of olefins.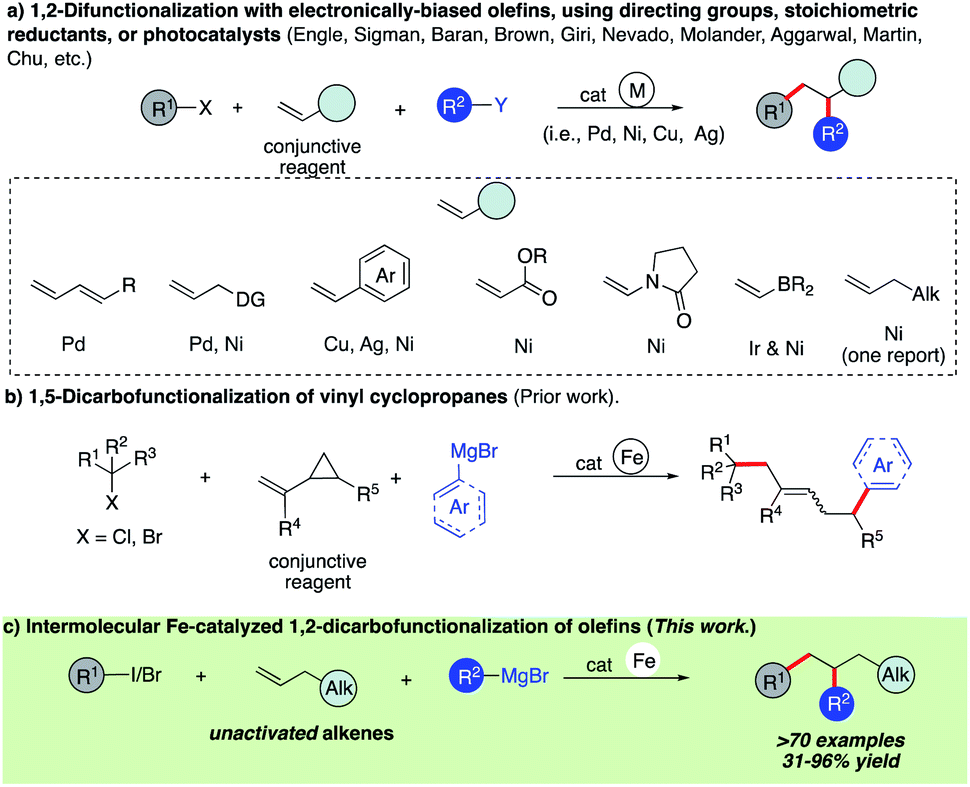 | ||
| Scheme 1 Transition metal-catalyzed three-component difunctionalization of olefins. | ||
As shown in Scheme 2, we hypothesize that alkyl halide 1 would react with Fe species A to form the alkyl radical int-1 and B.12,13 Due to the high barrier associated with sterically hindered alkyl radicals and aryl iron B to undergo direct cross-coupling, we anticipate that the tertiary radical int-1 (or a fast reacting alkyl radical) would favor regioselective Giese addition to olefin 2 to form, in the absence of cyclopropyl groups, a transient secondary alkyl radical int-2.16 Then the longer lived (persistent) aryl iron species B can trap the less sterically hindered 2° alkyl radical int-2, and undergo reductive elimination from C to form the desired 1,2-dicarbofunctionalization product and D. Finally, facile transmetallation with aryl Grignard 3 restarts the catalytic cycle.17 Recognizing that the success of the 3-component dicarbofunctionalization hinges on driving the equilibrium towards formation of int-2, presumably by favoring Giese addition over addition to aryl iron B, we initiated our studies under solvent-free conditions and at high concentrations of alkenes.
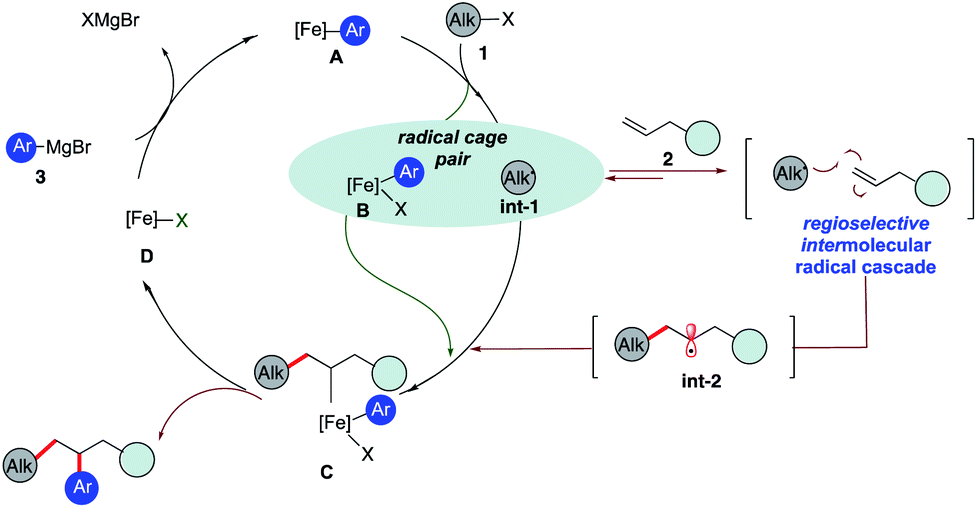 | ||
| Scheme 2 Proposed pathway to realize the 1,2-dicarbofunctionalization of alkenes using iron catalysis. | ||
The challenge remains whether (a) we can drive the kinetics towards the Giese addition to 2, (b) int-2 is sufficiently long-lived to be intercepted by the persistent iron species B, and (c) C will undergo reductive elimination to form the desired 1,2- dicarbofunctionalization product.
Results and discussion
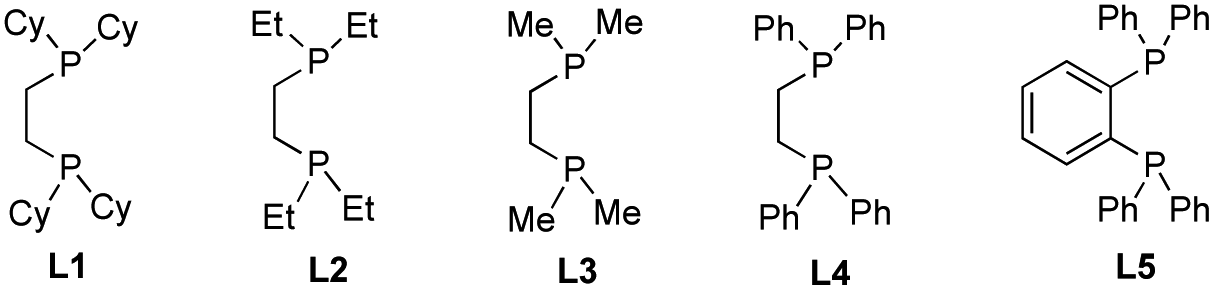
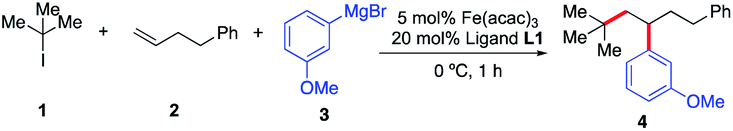
Initially, we elected to use tert-butyl iodide 1, 4-phenyl-1-butene 2, and meta-methoxy phenyl Grignard 3 as model substrates (Table 1). Gratifyingly, under our modified conditions for radical cross-coupling with vinyl cyclopropanes (i.e., using Fe(acac)3 as a precatalyst and 1,2-bis(dicyclohexylphosphino)ethane as a ligand),14a we observed the formation of the desired 1,2-alkylaryl product 4 in 86% yield and complete regioselectivity with unactivated olefin 2 (Table 1, entry 1). Notably, other bisphosphine ligands commonly employed in direct Fe-catalyzed cross-coupling reactions with alkyl halides10 significantly decrease the yield (entries 2–5). Further, the use of the iron precatalyst bearing strongly coordinating ligands inhibits the reaction (entry 6) while other precatalysts were less efficient (entries 7 and 8). Moreover, the use of THF as solvent had a minor effect on the overall efficiency of the 3-component 1,2-dicarbofunctionalization (entry 9). Finally, we could also perform the reaction in high yield under lower catalytic loading (entry 10). Control experiments show that the Fe and ligand are both critical for the reaction (entries 11 and 12). For full details of reaction optimization and screening conditions, see the ESI.†|
|
||
|---|---|---|
| Entry | Deviations from above | Yieldb [%] |
| a The reaction was performed with tert-butyl iodide1 (0.1 mmol, 1.0 equiv.), 4-phenyl-1-butene 2 (14 equiv.; 1–1.3 equiv. based on the recovered starting material; see the ESI) and meta-methoxy phenyl Grignard 3 (1.4 equiv.) without any additional solvent. Aryl Grignard3 was added dropwise via a syringe pump over 1 h. b The yield was determined by 1H NMR using dibromomethane as the internal standard. In parentheses is given the isolated yield after column chromatography. c 1.5 equiv. of 3. d 0.20 mmol scale. | ||
| 1 | None | 86 |
| 2 | L2 (20 mol%) | 0 |
| 3 | L3 (20 mol%) | 0 |
| 4 | L4 (20 mol%) | 2 |
| 5 | L5 (20 mol%) | 14 |
| 6 | Using Fe(OAc)2 (5 mol%) | <5 |
| 7 | Using FeBr2 (5 mol%) | 80 |
| 8 | Using Fe(OTf)2 (5 mol%) | 41 |
| 9c | In THF (0.2 mL) | 83 |
| 10c,d | Using Fe(acac)3 (3 mol%) and L1 (12 mol%) | 90 (85) |
| 11c | No L1 | <5 |
| 12c | No Fe(acac)3 and no L1 | 0 |
|
||
With a set of optimized reaction conditions in hand, an exploration of the reaction scope and limitations of this bisphosphine iron-catalyzed 3-component dicarbofunctionalization was undertaken. As shown in Scheme 3, the reaction tolerated a wide range of electron-rich (e.g., 4, 6, 7, 9, 12, 13, 15, and 16) and electron-deficient aryl Grignard nucleophiles (e.g., 5, 8, 11, 14, and 17) forming the desired 1,2-alkylaryl products. Further, various substituent positions on the aryl nucleophiles were tolerated including meta and para mono- and disubstituted aryl Grignard nucleophiles. Importantly, vinyl Grignard reagents are also competent nucleophilic partners forming the regioselective 1,2-alkylvinyl product 18 in 41% yield. This represents the first example of transition-metal catalyzed 1,2-alkylvinyl functionalization of unactivated olefins. Unfortunately, sterically hindered Grignard reagents are not compatible reagents in this transformation, presumably due to the high energy required to undergo inner-sphere reductive elimination.11,12
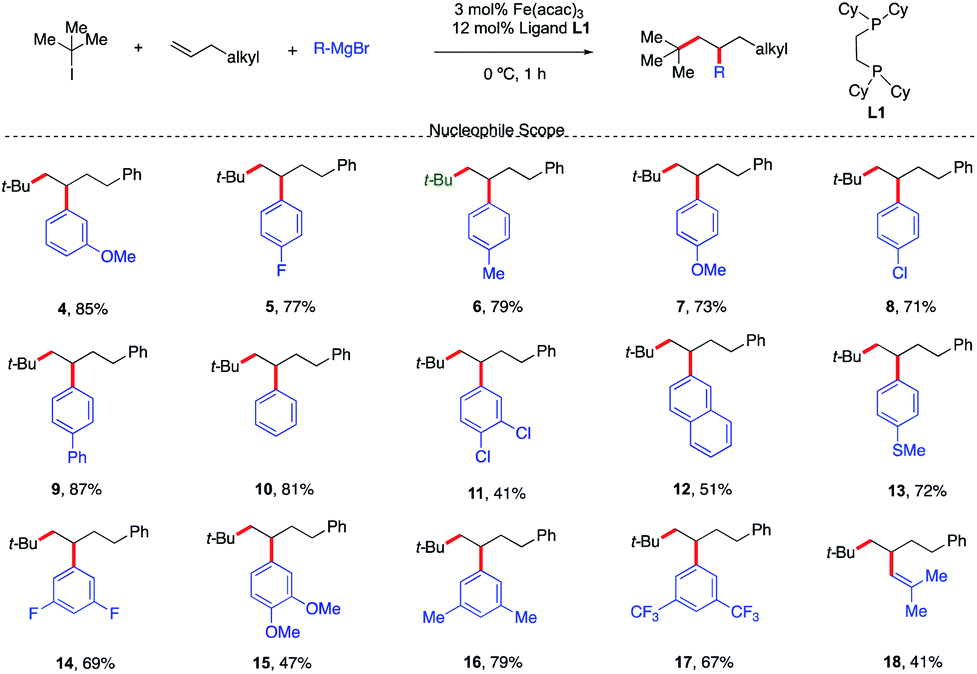 | ||
| Scheme 3 Scope of the Grignard nucleophile in the 3-component dicarbofunctionalization with unactivated alkenes. Unless otherwise stated, all reactions were performed under the optimized conditions (Table 1, entry 10). Isolated yields. | ||
Next, we explored the olefin scope using tert-butyl iodide 1 and meta-methoxy phenyl Grignard 3 as dicarbofunctionalization partners (Scheme 4). In general, a wide range of unactivated olefinic partners were tolerated. Compatible partners include olefins with tethered aliphatic chains, alkenes, alkoxy, protected alcohols, aldehydes and amines, esters, and even pyridine and furan moieties producing the desired products in 32–83% yield (19–34). However, alkenes bearing O- and S-heteroatoms were not compatible with this transformation (see the ESI†). Importantly, this Fe-catalyzed three-component method provides unique reactivity with dienes. In particular, we found that the method is highly chemo- and regioselective for monofunctionalization of less substituted alkenes (23–25) even at lower concentrations of alkenes (see the ESI†). To showcase the practical application of this method, we also scaled up the reaction that formed the monofunctionalized product 22 in 83% yield (1.38 g). Furthermore, we also found that the perfluororated n-alkyl bromides were competent partners with unactivated cyclic alkenes (35 and 36) yielding the desired products as single diastereoisomers in 53–74% yield. For aliphatic chain internal alkene (37) using the perfluororated n-alkyl radical, we obtained the desired products as a mixture of diastereomers (dr = 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; see the ESI†) in 46% yield.
:1; see the ESI†) in 46% yield.
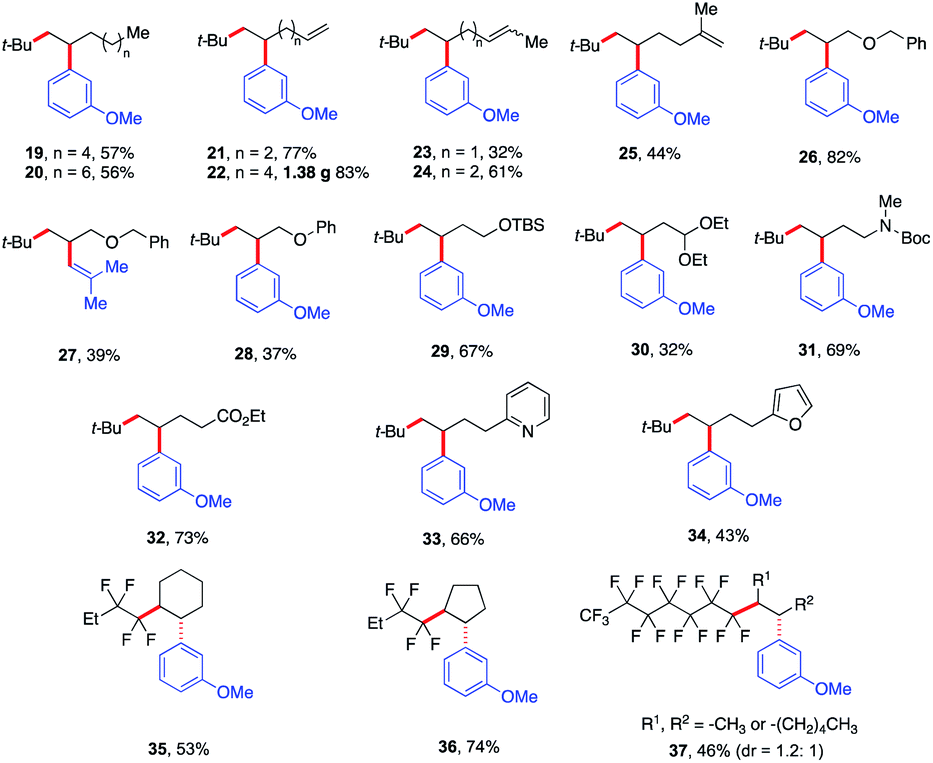 | ||
| Scheme 4 Scope of alkenes in the reaction. Unless otherwise stated, all reactions were performed under the optimized conditions (Table 1, entry 10) in THF (0.2 mL). Isolated yields. | ||
As shown in Scheme 5, contrary to current state-of-the-art TM-catalyzed three-component dicarbofunctionalization, this method tolerates a range of diverse radical precursors and operates under short reaction times and at low temperatures. Specifically, tertiary alkyl bromides also form the desired 1,2-alkylaryl products 38–50 with similar efficiency to alkyl iodides. These results represent the first examples of using alkyl bromides in a transition metal-catalyzed 3-component intermolecular 1,2-alkylarylation of unactivated olefins and can complement existing methods using reductive cross-couplings as reported by Nevado.5 Furthermore, other tertiary alkyl iodides/bromides are compatible in this transformation yielding the desired products 51–55 in 31–63% yield. Finally, consistent with our hypothesis (Scheme 2), we also found that perfluororated n-alkyl radicals (much more reactive towards Giese addition to alkenes)18 were competent in this Fe-catalyzed three-component dicarbofunctionalization reaction yielding the desired products 56–57 in 77–87% yield. Unfortunately, other primary and secondary alkyl halides are not compatible in this transformation due to the competing direct cross-coupling formation (see the ESI†).
 | ||
| Scheme 5 Scope of alkenes in the reaction. Unless otherwise stated, all reactions were performed under the optimized conditions (Table 1, entry 10). Isolated yields. (a) THF (0.2 mL). | ||
To expand the synthetic utility of this Fe-catalyzed three-component dicarbofunctionalization, we next explored the possibility of performing a radical cascade cyclization/arylation with a series of 1,6-dienes leading to the formation of three carbon–carbon bonds in one synthetic step (Scheme 6a). We hypothesize that regioselective Giese addition to the olefin will form the secondary alkyl radical intermediate G•. If the rates of Fe-arylation are slower than the rate of ring-closure, then we should only observe the ring-closed arylated product (i.e., 58). However, if the rate for Fe-arylation of G• is faster than the rate for Fe-arylation of radical 5-exo-trig, then we should observe only the uncyclized product (i.e., 59). As shown in Scheme 6a, we found that this method delivered the desired carbocycle 58 in good yield (71%). We also observed the uncyclized product 59, presumably from direct arylation of G•, albeit in low yield (9%).
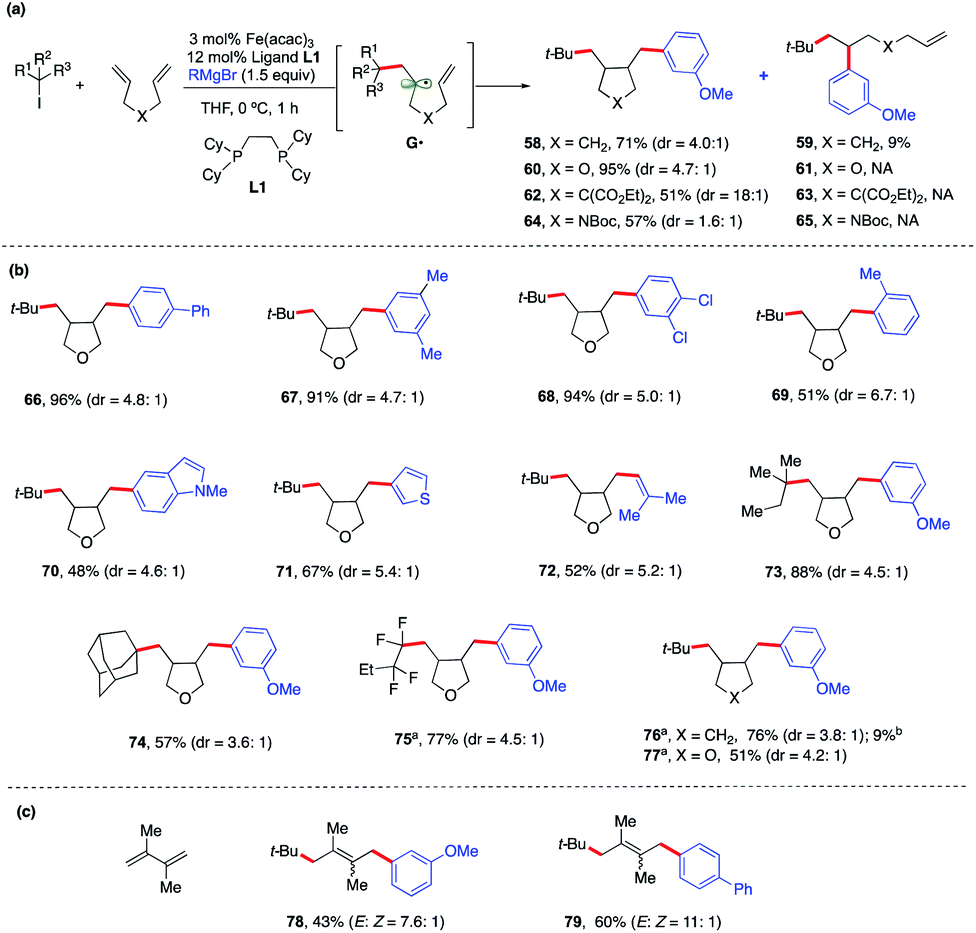 | ||
| Scheme 6 Scope and energetics for radical cascade cyclization/arylation. Unless otherwise stated, all reactions were performed under the optimized conditions (Table 1, entry 10) in THF (0.2 mL). Isolated yield. (a) Alkyl bromides. (b) Yield of the acyclic/arylation product. | ||
Notably, incorporation of heteroatoms (O or N) or addition of diester linkage results in exclusive formation of the cyclic product. Specifically, we found the desired formation of alkylaryl tetrahydrofuran 60, di-ester substituted carbocycle 62, and pyrrolidine 64 in good to excellent yield (51–95%) and without the formation of the uncyclized product. DFT calculations [UPBEPBE-D3/6-311+G(d,p)-CPCM(THF)//UB3LYP/6-31G(d)] using the tBu radical and 1,6-heptadiene predict a barrier of 13.2 kcal mol−1 for irreversible Giese addition leading to G•, 5.2 kcal mol−1 downhill in energy. In agreement with the experiment, G• preferentially favors radical cyclization leading to a cis isomer, while (irreversible) radical cyclization leading to a trans isomer is only 1.2 kcal mol−1 higher in energy. However, consistent with the experiment, the rates for radical cyclization for X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O substituted diene are faster and the energy difference between cis and trans radical cyclization is much higher (1.7 kcal mol; see the ESI†). However, at this stage, we cannot rule out alternative mechanistic pathways such as olefin coordination to the metal center preceding alkyl radical addition or 1,2-migratory insertion of the iron-aryl into the alkene. Future work on elucidating the mechanism of this transformation is on-going and will be reported in due course. Given the prevalence of saturated heterocyclic compounds (tetrahydrofurans and pyrrolidines) in pharmaceuticals, we used an oxygen-substituted diene as a model compound to explore the reaction scope of this Fe-catalyzed three-component radical cascade cyclization/arylation (Scheme 6b). As shown in Scheme 6b, this reaction is very robust with aryl Grignard nucleophiles forming the desired products in excellent yields, and the cis-isomer is the major product (as determined by 1H NMR and via crystal structure determination of 66; see the ESI†). The use of sterically hindered, heteroaryl or vinyl nucleophiles was also tolerated (69–72). Moreover, other tertiary alkyl iodides and perfluorinated alkyl and tertiary bromides also work in this transformation forming the radical cascade cyclization/arylation products 73–77 in 51–88% yield. Finally, the method is regioselective for addition to conjugated 1,3-diene to form 1,4-alkylaryl products 78–79 in good yield (up to 11:1 E:Z, Scheme 6c).
O substituted diene are faster and the energy difference between cis and trans radical cyclization is much higher (1.7 kcal mol; see the ESI†). However, at this stage, we cannot rule out alternative mechanistic pathways such as olefin coordination to the metal center preceding alkyl radical addition or 1,2-migratory insertion of the iron-aryl into the alkene. Future work on elucidating the mechanism of this transformation is on-going and will be reported in due course. Given the prevalence of saturated heterocyclic compounds (tetrahydrofurans and pyrrolidines) in pharmaceuticals, we used an oxygen-substituted diene as a model compound to explore the reaction scope of this Fe-catalyzed three-component radical cascade cyclization/arylation (Scheme 6b). As shown in Scheme 6b, this reaction is very robust with aryl Grignard nucleophiles forming the desired products in excellent yields, and the cis-isomer is the major product (as determined by 1H NMR and via crystal structure determination of 66; see the ESI†). The use of sterically hindered, heteroaryl or vinyl nucleophiles was also tolerated (69–72). Moreover, other tertiary alkyl iodides and perfluorinated alkyl and tertiary bromides also work in this transformation forming the radical cascade cyclization/arylation products 73–77 in 51–88% yield. Finally, the method is regioselective for addition to conjugated 1,3-diene to form 1,4-alkylaryl products 78–79 in good yield (up to 11:1 E:Z, Scheme 6c).
Conclusions
In summary, we have developed a three-component 1,2-alkylarylation of unactivated olefins using bisphosphine iron as the catalyst. Further, we demonstrated that this protocol can forge three carbon–carbon bonds in one synthetic step leading to a diverse set of carbo- and heterocyclic compounds. We expect that this method will be adapted by the pharmaceutical community for the synthesis of bioactive products, fine chemicals, and late-stage diversification of promising leads. Although this method is currently limited to the use of a large excess of olefins, preliminary experiments show that the use of activated alkenes could circumvent the need for excess alkenes, and this will be reported in due course. Future work is ongoing to elucidate the mechanism of this transformation using computational, experimental, and spectroscopic tools. We are actively pursuing other three-component Fe-catalyzed reactions with other π-acceptors, nucleophiles, and electrophiles including asymmetric variants and will report in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the NSF (CAREER 1751568) and by the NIGMS of the NIH (R35GM137797). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. A provisional patent application has been submitted by the University of Maryland for this reaction, with L.L., W.L., and O.G. as inventors. We are grateful to the University of Maryland College Park for start-up funds and computational resources from UMD Deepthought2 and MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053).References
- For representative reviews on alkene functionalization, see: (a) V. Saini, B. J. Stokes and M. S. Sigman, Angew. Chem., Int. Ed., 2013, 52, 11206 CrossRef CAS PubMed; (b) J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636 CrossRef CAS PubMed.
- For representative reviews on dicarbofunctionalization of alkenes, see: (a) J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21 CAS; (b) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013 CrossRef CAS PubMed.
- For some examples leading to dicarbofunctionalization of alkenes, see: (a) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (c) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (d) S. Kc, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801 CrossRef CAS PubMed; (e) P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653 CrossRef CAS; (f) M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 14245 CrossRef CAS; (g) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS; (h) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375 CrossRef CAS PubMed; (i) S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 CrossRef CAS PubMed; (j) S. KC, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047 CrossRef CAS PubMed.
- For recent examples using directing groups, see: (a) J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 1201 CrossRef CAS PubMed; (b) Y. Tao, X. Chen, W. Rao and M. J. Koh, Chem, 2020, 6, 738 CrossRef.
- W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 CrossRef CAS PubMed.
- For a recent review, see: A. Piontek, E. Bisz and M. Szostak, Angew. Chem., Int. Ed., 2018, 57, 11116 CrossRef CAS.
- (a) C.-L. Sun, H. Krause and A. Fürstner, Adv. Synth. Catal., 2014, 356, 1281 CrossRef CAS; (b) T. Hatakeyama, Y. Okada, Y. Yoshimoto and M. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 10973 CrossRef CAS; (c) K. G. Dongol, H. Koh, M. Sau and C. L. L. Chai, Adv. Synth. Catal., 2007, 349, 1015 CrossRef CAS.
- (a) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed; (b) J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484 CrossRef CAS PubMed; (c) R. B. Bedford, E. Carter, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, D. M. Murphy, E. C. Neeve and J. Nunn, Angew. Chem., Int. Ed., 2013, 52, 1285 CrossRef CAS PubMed; (d) C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. A. Cartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333 CrossRef CAS PubMed; (e) T. Hatakeyama, Y. Kondo, Y. Fujiwara, H. Takaya, S. Ito, E. Nakamura and M. Nakamura, Chem. Commun., 2009, 1216 RSC; (f) R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC.
- (a) N. Nakagawa, T. Hatakeyama and M. Nakamura, Chem. Lett., 2015, 44, 486 CrossRef CAS; (b) T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834 CrossRef CAS PubMed; (c) T. Hashimoto, T. Hatakeyama and M. Nakamura, J. Org. Chem., 2011, 77, 1168 CrossRef PubMed; (d) T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674 CrossRef CAS.
- T. L. Mako and J. A. Byers, Inorg. Chem. Front., 2016, 3, 766 RSC.
- M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed.
- W. Lee, J. Zhou and O. Gutierrez, J. Am. Chem. Soc., 2017, 139, 16126 CrossRef CAS PubMed.
- A. K. Sharma, W. M. C. Sameera, M. Jin, L. Adak, C. Okuzono, T. Iwamoto, M. Kato, M. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2017, 139, 16117 CrossRef CAS PubMed.
- (a) L. Liu, W. Lee, M. Yuan, C. Acha, M. B. Geherty, B. Williams, O. Gutierrez, Chem. Sci., 2020, 11, 3146. A previous version of this manuscript was deposited on a preprint server ChemRxiv, DOI:10.26434/chemrxiv.9858500.v1; (b) L. Liu, W. Lee, J. Zhou, S. Bandyopadhyay and O. Gutierrez, Tetrahedron, 2019, 75, 129 CrossRef CAS.
- Z.-Q. Zhang, X.-Y. Meng, J. Sheng, Q. Lan and X.-S. Wang, Org. Lett., 2019, 21, 8256 CrossRef CAS.
- D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74 CrossRef CAS PubMed.
- (a) M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischaeur, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2019, 52, 140 CrossRef CAS PubMed; (b) J. D. Sears, P. G. N. Neate and M. L. Neidig, J. Am. Chem. Soc., 2018, 140, 11872 CrossRef CAS PubMed; (c) L. Liu, W. Lee, M. Yuan and O. Gutierrez, Comments Inorg. Chem., 2018, 38, 210 CrossRef CAS.
- D. V. Avila, K. U. Ingold, J. Lusztyk, W. R. Dolbier and H. Q. Pan, J. Am. Chem. Soc., 1993, 115, 1577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization of all starting materials and products, spectroscopic data, and computational details. CCDC 1996911. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02127j |
| This journal is © The Royal Society of Chemistry 2020 |