
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElusive structural changes of New Delhi metallo-β-lactamase revealed by ultraviolet photodissociation mass spectrometry†
M. Rachel
Mehaffey
a,
Yeong-Chan
Ahn
b,
Dann D.
Rivera
b,
Pei W.
Thomas
b,
Zishuo
Cheng
c,
Michael W.
Crowder
 c,
R. F.
Pratt
d,
Walter
Fast
*b and
Jennifer S.
Brodbelt
*a
c,
R. F.
Pratt
d,
Walter
Fast
*b and
Jennifer S.
Brodbelt
*a
aDepartment of Chemistry, University of Texas at Austin, Austin, TX 78712, USA. E-mail: jbrodbelt@cm.utexas.edu
bDivision of Chemical Biology and Medicinal Chemistry, College of Pharmacy, University of Texas at Austin, Austin, TX 78712, USA. E-mail: walt.fast@austin.utexas.edu
cDepartment of Chemistry and Biochemistry, Miami University, Oxford, OH 45056, USA
dDepartment of Chemistry, Wesleyan University, Middletown, CT 06459, USA
First published on 11th August 2020
Abstract
We use mass spectrometry (MS), under denaturing and non-denaturing solution conditions, along with ultraviolet photodissociation (UVPD) to characterize structural variations in New Delhi metallo-β-lactamase (NDM) upon perturbation by ligands or mutation. Mapping changes in the abundances and distributions of fragment ions enables sensitive detection of structural alterations throughout the protein. Binding of three covalent inhibitors was characterized: a pentafluorphenyl ester, an O-aryloxycarbonyl hydroxamate, and ebselen. The first two inhibitors modify Lys211 and maintain dizinc binding, although the pentafluorophenyl ester is not selective (Lys214 and Lys216 are also modified). Ebselen reacts with the sole Cys (Cys208) and ejects Zn2 from the active site. For each inhibitor, native UVPD-MS enabled simultaneous detection of the closing of a substrate-binding beta-hairpin loop, identification of covalently-modified residue(s), reporting of the metalation state of the enzyme, and in the case of ebselen, observation of the induction of partial disorder in the C-terminus of the protein. Owing to the ability of native UVPD-MS to track structural changes and metalation state with high sensitivity, we further used this method to evaluate the impact of mutations found in NDM clinical variants. Changes introduced by NDM-4 (M154L) and NDM-6 (A233V) are revealed to propagate through separate networks of interactions to direct zinc ligands, and the combination of these two mutations in NDM-15 (M154L, A233V) results in additive as well as additional structural changes. Insight from UVPD-MS helps to elucidate how distant mutations impact zinc affinity in the evolution of this antibiotic resistance determinant. UVPD-MS is a powerful tool capable of simultaneous reporting of ligand binding, conformational changes and metalation state of NDM, revealing structural aspects of ligand recognition and clinical variants that have proven difficult to probe.
Introduction
Infection by antibiotic resistant organisms remains a serious health threat. A 2019 report from the Centers for Disease Control and Prevention (CDC) indicates that in the U.S., bacteria and fungi cause over 2.8 million antibiotic resistant infections each year and that 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die due to these infections.1 The CDC ranks carbapenem-resistant Enterobacteriaceae in the top tier of “Urgent Threats”. Resistance against carbapenems is particularly dangerous because these β-lactam drugs are often held in reserve as life-saving “drugs of last resort” for patients with complicated infections.2 Some carbapenems can serve two purposes: dual inhibition of peptidoglycan biosynthesis and activity of some serine-β-lactamases. However, these drugs do not inhibit metallo-β-lactamases (MBLs), which instead use active-site zinc ion(s) to catalyze efficient hydrolysis and inactivation of carbapenems. For example, New Delhi metallo-β-lactamase (NDM) readily catalyzes hydrolysis of meropenem and imipenem with specificity constants (kcat/KM) > 106 M−1 s−1.3 Currently, there are no FDA approved drugs that counter the activity of NDM, or any other MBL, so NDM inhibitors are an unmet clinical need.4
000 people die due to these infections.1 The CDC ranks carbapenem-resistant Enterobacteriaceae in the top tier of “Urgent Threats”. Resistance against carbapenems is particularly dangerous because these β-lactam drugs are often held in reserve as life-saving “drugs of last resort” for patients with complicated infections.2 Some carbapenems can serve two purposes: dual inhibition of peptidoglycan biosynthesis and activity of some serine-β-lactamases. However, these drugs do not inhibit metallo-β-lactamases (MBLs), which instead use active-site zinc ion(s) to catalyze efficient hydrolysis and inactivation of carbapenems. For example, New Delhi metallo-β-lactamase (NDM) readily catalyzes hydrolysis of meropenem and imipenem with specificity constants (kcat/KM) > 106 M−1 s−1.3 Currently, there are no FDA approved drugs that counter the activity of NDM, or any other MBL, so NDM inhibitors are an unmet clinical need.4
Despite more than 100 structural models of NDM deposited in the protein data bank, there are still significant gaps in our knowledge about NDM structure. NDM drug development efforts would greatly benefit from specific information detailing how ligands bind to active site loops and how clinical variants of the enzyme impact structure, but aspects of these structural differences often remain elusive. For example, MBLs similar to NDM have a flexible beta-hairpin loop containing a hydrophobic amino acid at the apex that closes down over a ligand upon inhibitor binding or during catalysis, making important binding interactions with the ligand as revealed by mutagenesis, kinetic analysis, X-ray crystallography, and protein NMR studies.5–9 However, the conformation of the homologous beta-hairpin loop in NDM is often obscured or artifactually constrained in X-ray studies owing to interactions with a neighboring NDM monomer during crystal formation.10 Mutagenesis and kinetic studies indicate an important role for this NDM loop in ligand binding and substrate turnover that may differ somewhat from its role in homologous MBLs, underscoring the need to better characterize structural changes upon ligand binding.11,12 Two spectroscopic methods (19F NMR and RFQ-DEER) have been used along with covalent incorporation of labels into the NDM loop to detect conformational changes upon ligand binding.13,14 The conformations that this loop adopts appear to be ligand dependent and ligand binding may even trigger loop opening, although the covalent incorporation of these labels may perturb the system and complicate interpretation.13,14 Alternative methods are needed to better understand the structural implications of ligand-binding, preferably using unlabeled proteins.
A second example of inadequate structural information relates to clinical variants of NDM. Almost 30 different protein sequences have been reported for NDM clinical variants (NDM-1 through NDM-29). Many of these variants improve thermostability and the affinity of Zn2 (some variants have 10-fold lower Kd values) and appear to be emerging due to the combined selective pressures of antibiotic treatment and zinc scarcity at infection sites brought on by the host innate immune response.3,15–18 The structural models of seven different NDM variants (NDM-1, 3, 4, 5, 8, 9, 12) are deposited in the protein data bank, but the structural differences observed among these variants by X-ray crystallography are small and the mechanisms whereby mutations lead to improvements in zinc affinity are difficult to discern.15 Alternative strategies that can better detect the impact of mutation on NDM structure would help elucidate how clinical variants achieve improved resistance and aid in predicting the impact of newly sequenced variants.
The development of native mass spectrometry (MS) represents an alternative technique to probe protein structure by enabling the transfer of intact protein complexes with bound ligands into the gas phase using electrospray ionization (ESI) of high ionic strength solutions.19,20 While traditional collisional activation provides some sequence information on native protein complexes in the gas phase, typically this MS/MS approach disrupts non-covalent interactions and ejects ligands and individual protein constituents. As such, alternative MS/MS methods are necessary to probe the structure of intact protein complexes.21,22 The ability of electron-based activation methods, including electron transfer dissociation (ETD),23 electron capture dissociation (ECD),24–26 and electron ionization dissociation (EID),27 to allow retention of noncovalent ligands and metal cofactors on fragment ions, referred to as holo ions, has been used to identify structural differences between ligand-bound (holo) and ligand-free (apo) ions. The propensity of these electron-based strategies for fragmenting a given region correlates with protein flexibility (i.e. B-factors), enabling aspects of tertiary structure to be determined.23,28 In many ways mirroring the scope of electron-based activation methods, ultraviolet photodissociation (UVPD) at 193 nm has proven capable of providing sequence information, localizing ligand binding sites, and probing conformational changes of ligand:protein complexes.29–31 Retention of noncovalent ligands during photoactivation yields ligand-bound “holo” fragment ions that can be mapped along the sequence to elucidate binding sites.30 Tracking enhancement or suppression of backbone cleavages upon UVPD highlights regions where there are changes in stabilizing noncovalent interactions and in protein flexibility (i.e. enhancement of backbone fragmentation indicates fewer interactions, more flexibility, and typically leads to greater production of sequence ions; whereas suppression signifies more extensive interactions that limit the separation and detection of fragment ions).31 Additionally, UVPD affords unsurpassed sequence coverage29,30 and retention of labile covalent moieties32 with little dependence on precursor charge state.33 This native UVPD-MS approach has previously been applied for detecting conformational changes induced by ligand binding to dihydrofolate reductase (DHFR),34 sequence variants of rat sarcoma GTPase K-Ras35,36 and DHFR,37 and structural re-organization of the phosphotransferase enzyme adenylate kinase during its catalytic cycle.38
Here, we use native UVPD-MS to track changes in fragmentation patterns as a means to infer changes in the active site loop conformation, zinc binding, and conformations of surrounding residues in NDM-1 upon binding to three different small molecule inhibitors known to covalently modify the enzyme. Combining a native MS strategy with UVPD allows us to simultaneously define changes in protein conformation and zinc binding arising from interaction with inhibitors. We also compare four representative clinical NDM variants: NDM-1 (the reference sequence), NDM-4 (M154L), NDM-6 (A233V), and NDM-15 (M154L, A233V), specifically focusing on variations in backbone fragmentation adjacent to the six zinc-coordinating residues. Application of this method reveals structural differences not easily monitored by other approaches and provides information useful for NDM inhibitor development and better understanding how clinical variants lead to increased zinc affinity and enhanced drug resistance.
Experimental
Sample preparation
The reference sequence (NDM-1) and three clinical variants (NDM-4, NDM-6, and NDM-15) of recombinant NDM were expressed and purified as previously described, all of which include an N-terminal truncation to remove a lipidation signal sequence to make soluble versions of each protein.3,17 The expressed protein sequence with mutated sites highlighted and structures of the small molecule inhibitors are shown in Fig. S1.† The numbering scheme includes the initial 35 residues although the coding region for this sequence is omitted in the expression constructs. The three covalent inhibitors (1–3) were synthesized or purchased.A covalent inhibitor of imipenemase-1 (IMP-1), the pentafluorophenyl ester of 3-mercaptopropionic acid (1), was synthesized as described elsewhere39 and reconstituted in dimethyl sulfoxide stock solutions immediately prior to incubation with NDM-1 [1H NMR (400 MHz, CDCl3): δ 3.01 (2H, t), 2.91 (2H, dt), 1.75 (1H, t); ESI-MS (m/z): 273.0014 (M + H)+]. Inhibitor 1 and NDM-1 were combined at various stoichiometric ratios (1:1, 5:1, and 100:1 inhibitor:NDM-1) in 20 mM ammonium acetate (pH 6.8) and incubated at 25 °C for 1 h. The synthesis of a covalent inhibitor of NDM-1, N-(benzyloxycarbonyl)-O-[(phenyoxycarbonyl)]hydroxylamine, which is an O-aryloxycarbonyl hydroxamate (2), was previously described,40,41 and a 125:1 stoichiometric ratio (inhibitor:NDM-1) was incubated with NDM-1 at 2 °C for 18 h in 50 mM HEPES (pH 7.0). The covalent NDM-1 inhibitor ebselen,42 a benzisoselenazol (3), was purchased from Sigma-Aldrich (St. Louis, MO) and incubated at a 1:1 stoichiometric ratio (inhibitor:NDM-1) at 25 °C for 30 min in 20 mM ammonium acetate (pH 6.8). Prior to use in MS experiments, these incubation solutions, as well as stock solutions of the variants NDM-1, -4, -6, and -15, were desalted and exchanged into water or 20 mM ammonium acetate using 10 kDa molecular weight cutoff filter devices (MilliporeSigma, Burlington, MA). Samples were subsequently diluted for MS analysis to 10 μM protein concentration in 50/49.5/0.5 (v/v/v) acetonitrile/water/formic acid for denaturing experiments or 20 mM ammonium acetate (pH 6.8) for native conditions.
Mass spectrometry
An offline nano-ESI source with borosilicate emitters fabricated in-house and coated in Au/Pd was used to ionize proteins and protein complexes. The source was operated at applied voltages of 1.0–1.1 kV and set at a temperature of 200 °C to transfer proteins into a Thermo Scientific Orbitrap Elite mass spectrometer (Bremen, Germany). This instrument was modified previously by aligning an Excistar 193 nm ArF excimer laser (Coherent, Santa Cruz, CA) with the HCD cell to perform UV photodissociation.29 MS/MS experiments involved ion trap isolation of a single charge state of the protein species of interest using isolation widths of 10–20 m/z and subsequent collisional activation using 15–20% NCE or a single 3 mJ pulse for UVPD. MS1 spectra represent sixty scans, while MS/MS spectra contain 500 transients with a scan range of m/z 220–4000. Using a resolving power of 240 K at 400 m/z and a maximum ion time of 2 s, the automatic gain control was set at 1 × 106 for MS1 and 5 × 105 for MS/MS spectra. All MS/MS spectra were collected in triplicate.Data analysis
The Thermo Xtract algorithm was used to de-charge and de-isotope all ESI mass spectra and HCD or UVPD mass spectra (S/N ratio of 3, fit factor of 44%, remainder of 25%). ProSight Lite v1.4 assigned monoisotopic fragment ions from the MS/MS spectra as apo sequence ions by searching against the NDM sequence. For HCD mass spectra, only b- and y-type ions were considered, while for UVPD mass spectra all nine ion types were considered (a, a + 1, b, c, x, x + 1, y, y − 1, z). Holo fragment ions bound to zinc(II) resulting from photodissociation were also identified for the clinical variants and protein–inhibitor complexes by including the corresponding mass shifts at the termini: 61.913–63.929 Da for one zinc(II) or 123.827–127.858 Da for two zinc(II) ions. Covalently attached inhibitors were considered static modifications and included in searches using the expected mass shifts listed in Fig. S1.†The relative efficiencies of backbone cleavages induced upon UVPD were calculated for clinical variants and protein–inhibitor complexes using the fragment abundance tab of the Web-based utility UV-POSIT.43 Briefly, in a protein with R residues (numbered 1 to R from N-terminus to C-terminus), this program sums the abundances of all the fragment ions resulting from backbone cleavages adjacent to each individual amino acid and calculates a backbone cleavage yield for each amino acid position. In essence, the total backbone cleavage yield of amino acid n is the sum of all N-terminal sequence ions (an, bn, cn) resulting from cleavage C-terminal to the nth residue and all C-terminal product ions (xR−n+1, yR−n+1, zR−n+1) produced by backbone cleavage N-terminal to the nth residue. The summed values for each backbone position are then normalized to the total ion current of the spectrum and reported as the cleavage “efficiencies” (i.e. relative propensities) adjacent to each amino acid.43 This method provides a semi-quantitative way to evaluate variations in fragmentation throughout the protein sequence. Two protein states are compared (e.g. NDM-1 versus clinical variant, or unmodified versus inhibitor-bound) by subtraction of corresponding backbone cleavage efficiencies and represented as difference plots or heatmaps. Statistical significance of observed differences is established by pooling standard deviations and calculating p-values using a two-tailed Student's t-test. The log of these values is plotted for each protein:inhibitor complex compared to the corresponding unmodified protein (Fig. S2A and B†) or for each clinical variant compared to the reference NDM-1 protein (Fig. S2C(1–3)†). A histogram of p-values for the entire clinical variant data set is included in Fig. S2C(4).† The ∼25% of residues with p-values of 1.00 correspond to those with no adjacent backbone cleavages (i.e. no sequence coverage). The p-values less than 1.00 (∼75% of the values which also correlates to the observed 75% sequence coverage) correspond to those residues for which bracketing backbone cleavages were observed for both NDM-1 and an NDM-1:inhibitor complex or for both NDM-1 and a variant, thus allowing comparison of the abundances and calculation of a statistical significance. All differences in backbone fragmentation efficiencies discussed below are significant at a confidence threshold of 99% (i.e. p-value < 0.01), with this cutoff represented as a black line for each of the five difference graphs in Fig. S2† and collectively represented by the left-most blue bar in Fig. S2C(4)† (highest significance). A crystal structure of NDM-1 (PDB ID 3SPU)44 with important regions labelled is included to aid in visualization and detail the residues involved in the defined active site loops (ASLs) (Fig. S3†).
Results and discussion
Inhibitor selection
We applied a native UVPD-MS approach to simultaneously detect changes in protein structure and zinc content upon inhibitor binding to NDM-1, using two distinct types of MBL covalent inhibitors as examples: lysine 211-modifying inhibitors that retain binding of both zinc ions, and a cysteine 208-modifying inhibitor that ejects one zinc from the active-site. The first Lys-modifying inhibitor, 3-mercaptopropionic acid pentafluorophenyl ester (1), contains a zinc-binding thiol group tethered to a reactive ester and was previously shown to serve as an affinity label that covalently modifies the Lys244 in IMP-1 (KI = 3.45 μM; kinact = 4.6 min−1), and retains both zinc ions at the active site after inhibition.39 Because of the homology between IMP and NDM, we reasoned that 1 would also serve as an affinity label for Lys211 in NDM-1 (Fig. 1). Due to an unexpected lack of selectivity for 1 (vide infra), we also investigated a second Lys-modifying affinity label, an O-aryloxycarbonyl hydroxamate (2), that we previously determined to modify Lys211 in NDM-1 (KI = 140 μM; kinact = 0.045 min−1) and to leave the dizinc active site intact (Fig. 1).41 For a Cys-modifying inhibitor, we chose a non-selective thiol-modifying reagent ebselen (3) that has been previously shown to covalently modify the sole Cys in soluble NDM-1 constructs (Cys208) as an affinity label (KI = 0.38 μM; kinact = 0.034 min−1), and to eject one equivalent of zinc from the dinuclear zinc cluster (Fig. 1).42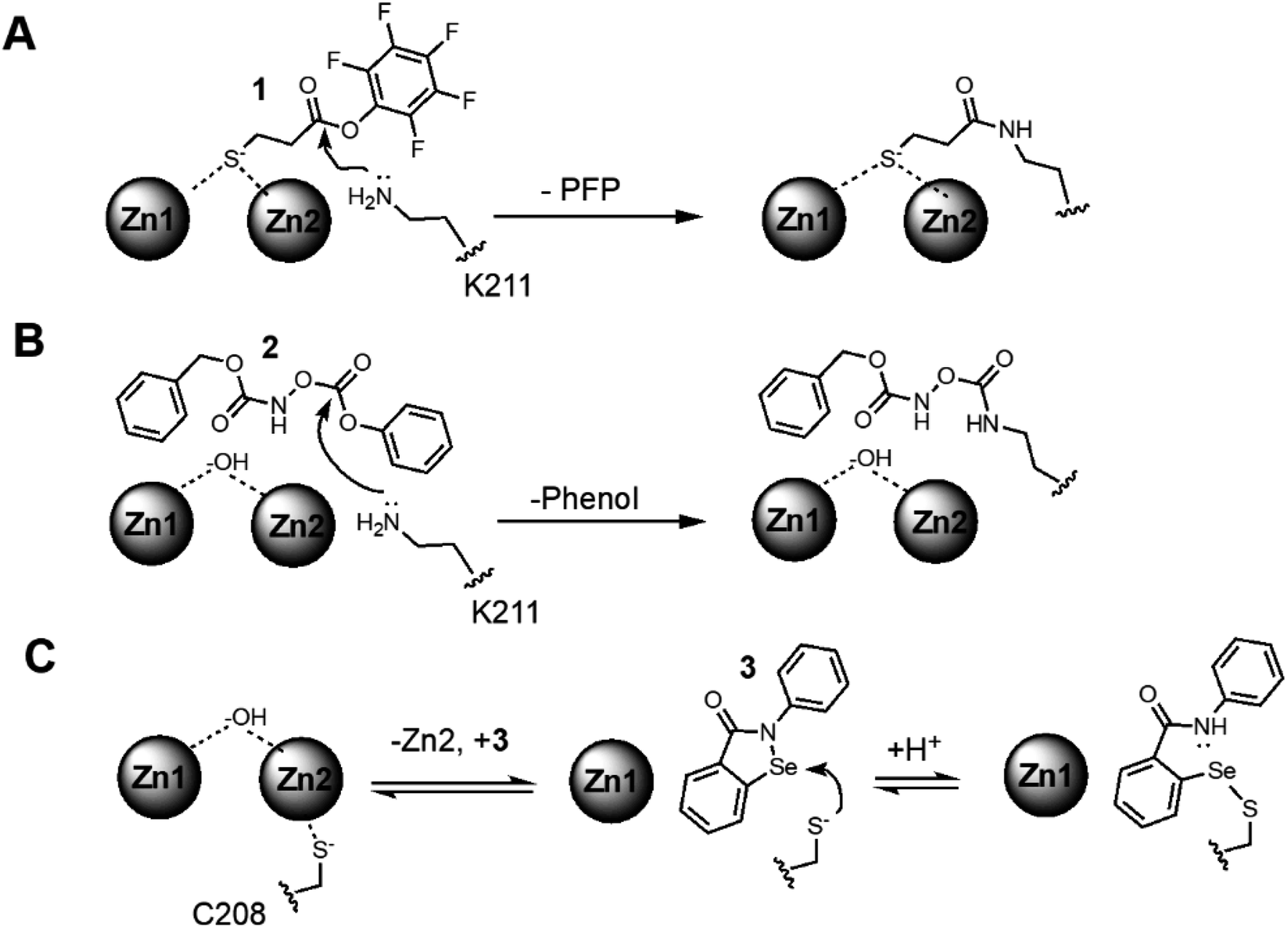 | ||
| Fig. 1 Proposed covalent NDM-1 inactivation mechanisms: (A) a reactive pentafluorophenol (PFP) ester (1) is bound in proximity to active-site K211 to facilitate reaction and loss of pentafluorophenol (PFP). (B) A reactive O-aryloxycarbonyl hydroxamate (2) is bound in proximity to active-site K211 to facilitate reaction. (C) Because the C208 thiol is not solvent accessible in the dinuclear zinc form of NDM-1, and Zn2 is bound relatively weakly,3 a dynamic equilibrium is depicted between dizinc and monozinc metalloforms, enabling binding of the thiol-reactive ebselen (3) in proximity to Cys208 to facilitate reaction and loss of Zn2 affinity. | ||
UVPD-MS to localize a lysine-selective covalent inhibitor of NDM-1
We found that a Lys-targeted pentafluorophenyl ester affinity label reported for IMP-1 (1) can also readily inactivate the homologous NDM-1 enzyme in a manner that is irreversible to dilution into excess substrate (Fig. S4†). A full kinetic characterization was not completed owing to unexpected lack of selectivity noted below. ESI-MS data were collected for denaturing and non-denaturing (high ionic strength) solutions containing NDM-1 without or with inhibitor 1 (Fig. S5,† inhibitor:protein ratios 1:1, 5:1, 100:1). Even at a 1:1 inhibitor:protein ratio, up to two inhibitors were observed to bind covalently to NDM-1 with up to three inhibitors per protein for solutions containing higher inhibitor:protein ratios. This outcome indicates that inhibitor 1 is less specific for one particular Lys in NDM-1 as compared to IMP-1.39 Similar results were observed for spectra collected after various incubation time-points. Using non-denaturing ESI conditions yields similar results with addition of up to three inhibitor molecules to NDM-1 and also confirms that neither of the active site Zn(II) ions are displaced due to the reaction (Fig. S5B†).
To localize the reaction sites of inhibitor 1, the 25+ charge states of the singly, doubly, and triply modified NDM-1 were individually isolated and characterized using HCD and UVPD (Fig. S6†). As opposed to the diverse array of sequence ions produced by UVPD, HCD results in a relatively small number of b/y ions dominated by preferential Pro cleavages (e.g., y100 and b114). Maps of the backbone cleavage sites corresponding to the observed fragment ions shown in Fig. S7† highlight the higher sequence coverage afforded by UVPD (74–84%) compared to HCD (16–26%). The mass shifts of the y10011+ ions observed for both HCD and UVPD in Fig. S6† corresponds to addition of one, two, or three equivalents of 1. In this context, the specific positions of the each adduct can be determined by accounting for the mass shift(s) at each of the eight possible Lys residues of NDM-1. Covalent attachment of inhibitor 1 through disulfide bridging of the thiol end (opposite the expected reactive moiety) to C208 was considered but the retention of both Zn(II) ions (Fig. S5B†) after modification as well as the similar HCD and UVPD fragmentation patterns observed even after addition of a reducing agent provide strong evidence against this possibility. Both HCD and UVPD methods indicate that the modification sites are all located along the same loop that borders the active-site (ASL4: G207-H228). Localization of binding sites is summarized in Fig. 2 for NDM-1 containing one, two, or three equivalents of inhibitor 1. Briefly, for the singly-bound species, the UVPD data indicates that reaction occurs at either K211 (expected site) or K214. When two equivalents of 1 are bound, UVPD confidently localizes them to K211 and K214. The UVPD results indicate that the third equivalent is added at K216. There is no evidence for covalent modification by 1 at K125 (buried), or at other solvent accessible Lys residues (K106, K181, K242, K268). The only Lys residues that are modified are contained within a single loop consisting of residues 209–224 that neighbor the active site (ASL4), and none of the other Lys residues are targeted. In comparison, the homologous loop in IMP-1 is considerably shorter and lacks the two Lys residues in NDM-1 that account for additional modifications. Therefore, sequence differences likely underlie the apparent difference in Lys modification selectivity between IMP-1 and NDM-1.
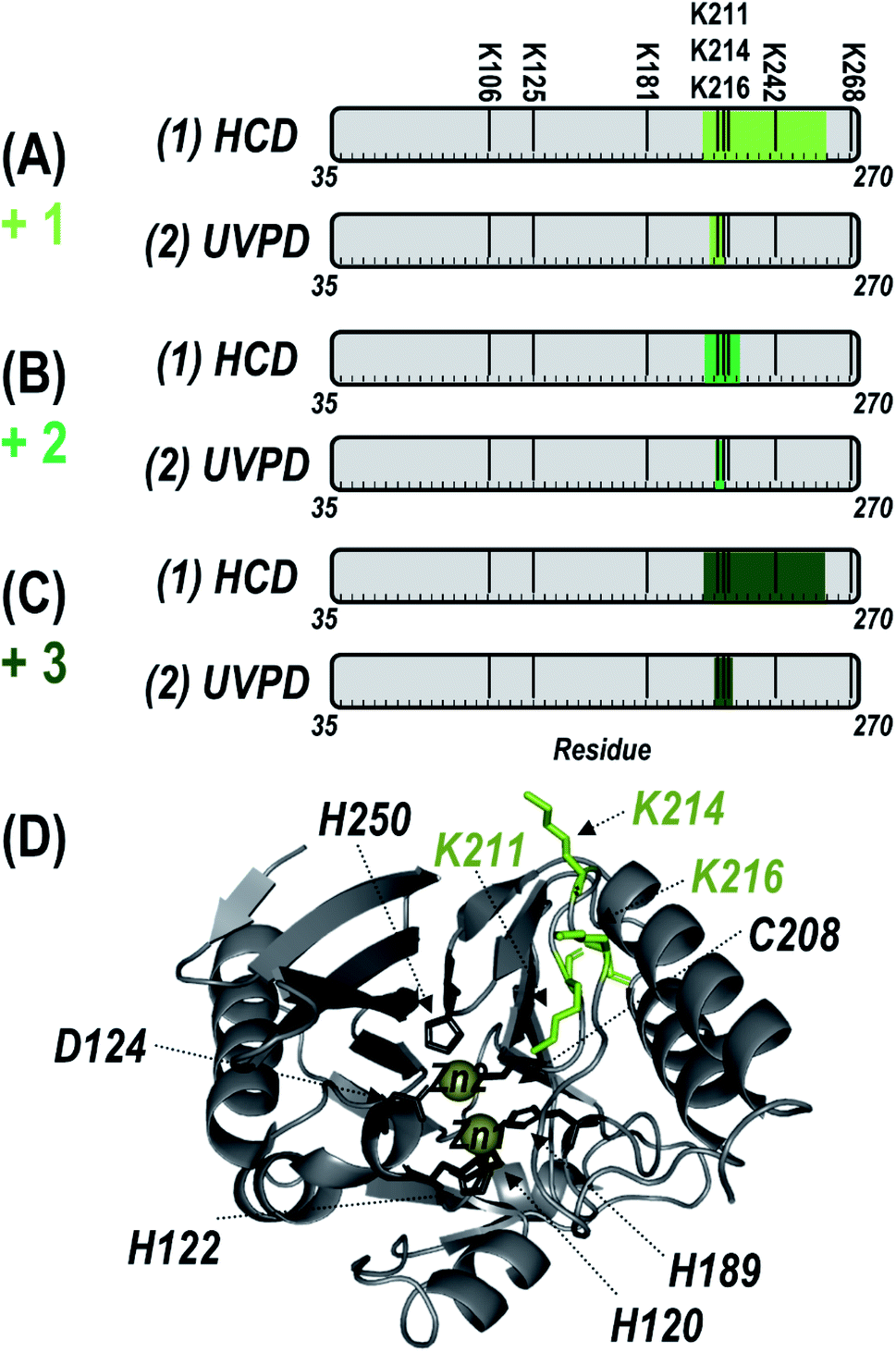 | ||
| Fig. 2 Summary of the localization of (A) one, (B) two, or (C) three inhibitor (1) moieties (with loss of pentafluorophenol) covalently attached to Lys residues of NDM-1 by using (1) HCD and (2) UVPD. Green shaded regions indicate localization according to the MS/MS spectra in Fig. S6† and corresponding sequence coverage maps of identified fragment ions in Fig. S7.† The eight possible Lys sites are labelled above the gray residue bar in (A). (D) Sites at which the inhibitor was localized (K211, K214, K216) are labelled and shown as green sticks on the crystal structure of NDM-1 (PDB ID 3SPU). The six Zn(II) binding residues are also labelled and shown as black sticks. | ||
Tracking closure of an active site loop over a lysine-modifying covalent NDM-1 inhibitor
To characterize conformational changes resulting from a more well-defined binding event, we substituted a different affinity label, an O-aryloxycarbonyl hydroxamate (2), that selectively modifies Lys211 in NDM-1 (although minor amounts of an undefined secondary modification site were reported when excess 2 was used for labeling).41 In a previous study both X-ray crystallography and MS were used to confirm covalent modification of K211, but the positioning of the substrate-binding beta-hairpin loop was perturbed artifactually by interaction with a second monomer found in the crystal lattice, and by non-enzymatic degradation of the adduct during crystallization.41 Here, we used native ESI-MS to better characterize structural changes that occur upon incubation of 2 with NDM-1. Even with treatment of excess 2, only one equivalent of expected adduct was detected on NDM-1 (Fig. S8†), with retention of both zinc ions after modification. The degraded adduct (a carbamoylated K211) observed earlier by X-ray crystallography was not observed under these conditions.We then used UVPD-MS to characterize the structural impact on NDM-1 caused by inhibition with 2via changes in observed fragmentation. Specifically, we have consistently found that regions of a protein that exhibit increased flexibility and/or fewer stabilizing interactions frequently result in enhanced fragmentation and thus yield more abundant sequence ions. In contrast, engagement of a region in stabilizing noncovalent interactions may prevent separation of fragment ions, thus hindering their detection and leading to an apparent suppression in the backbone cleavages.31 UVPD of the 9+ charge state yielded a wide array of sequence ions, including those retaining both the covalently-bound inhibitor and one or two non-covalently bound Zn(II) ions (Fig. S9†). The binding site of the inhibitor was localized to the expected residue, K211 (Fig. S9B and S9C†), based on backbone cleavages that confidently bracketed the mass shift of the inhibitor. Summation of all the fragment ions arising from backbone cleavages as described in the Experimental section yielded the graphical displays shown in Fig. S10† for unmodified NDM-1 and the inhibitor 2:NDM-1 complex. A plot of calculated p-values per residue in Fig. S2A† assigns statistical significance to observed differences from t-test calculations. Conversion of the two displays to a difference plot in Fig. 3A or as a heatmap in Fig. 3B facilitates visualization of the variations in fragmentation of NDM-1 after reaction with this affinity label.
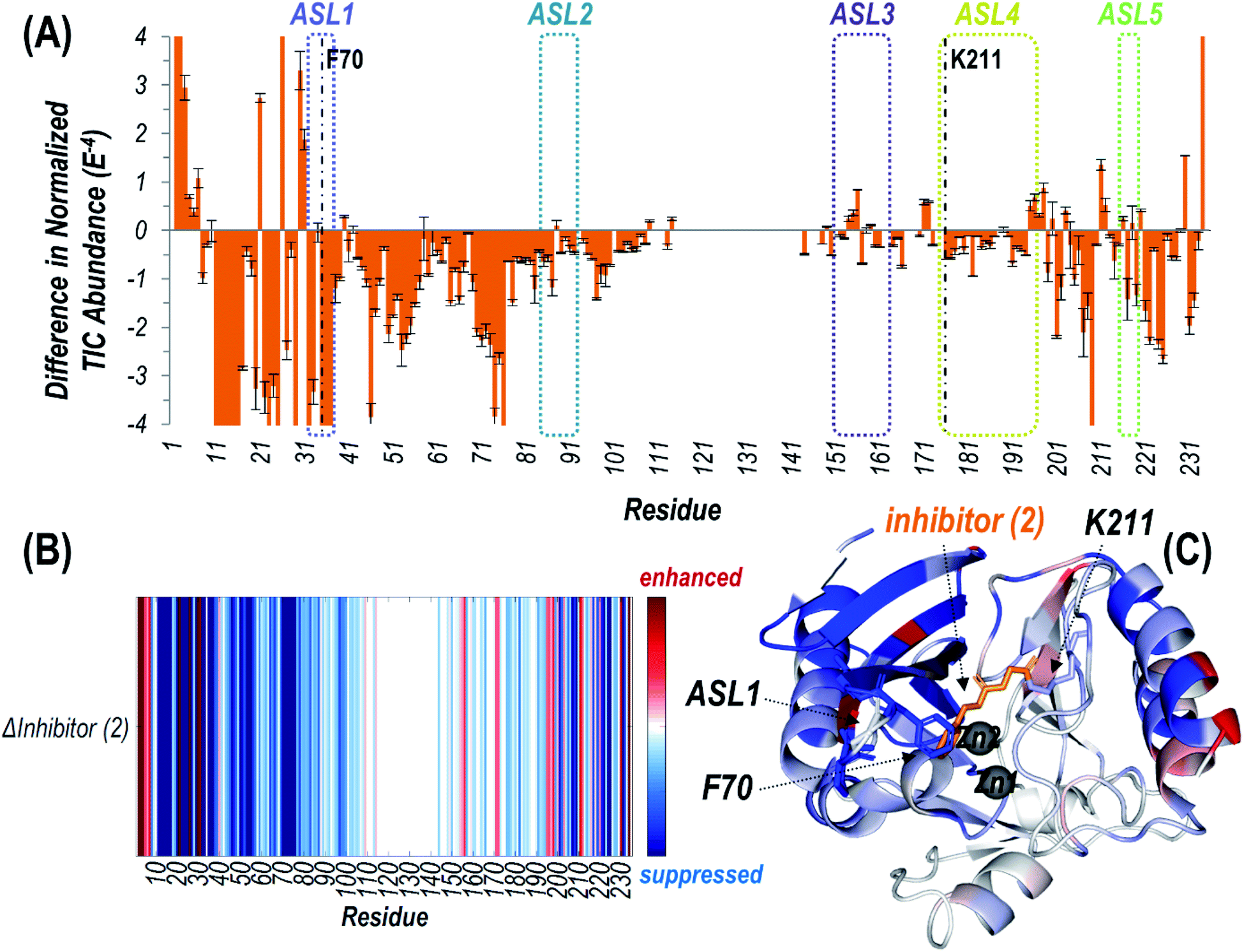 | ||
| Fig. 3 (A) Difference plot showing the change in summed abundances of Zn(II) bound holo and apo fragment ions produced upon UVPD of NDM-1 covalently bound to inhibitor (2) (with loss of phenol) compared to uninhibited NDM-1. The UVPD fragmentation plots for both samples are shown in Fig. S10.† The five active site loops are outlined with dotted lines while the residue at which the inhibitor attaches (Lys211) is denoted with a dashed line. Heatmaps of these differences are represented (B) linearly along the protein sequence or (C) mapped on the crystal structure of NDM-1 bound to inhibitor (2) (PDB ID 6OVZ). Red regions correspond to enhancement of backbone cleavage efficiency for the inhibitor bound sample compared to the uninhibited while blue regions indicate suppression. Active site loop 1 (including F70), Lys211, and inhibitor (2) are displayed as sticks, while the Zn(II) ions are shown as spheres in (C). | ||
Suppression of backbone fragmentation (colored blue in the heatmap) is observed throughout large stretches of the protein for inhibitor-bound NDM-1, particularly encompassing the N-terminal half of the protein that includes two of the active site loops (ASL1: M67-G71 and ASL2: V117-D124) as well as the three other active site loops in the C-terminal region (Fig. 3). ASL3 (F183-T195) and ASL5 (M248-S251) are short loops that form a deep cavity in which the two Zn(II) ions reside. ASL4 (G207-H228) is significantly longer and creates the floor, while ASL2 acts as the ceiling. Notably, ASL1 is the beta-hairpin loop proposed to play an important role in substrate binding (Fig. S3†). The suppression of fragmentation indicates a general loss of flexibility in all five active site loops that frame the active site. This observation is consistent with other studies that show a general increase in overall thermostability of NDM-1 upon binding inhibitors.45 More specifically, our use of native UVPD-MS reveals that fragmentation of the backbone spanning the ASL1 region shows the greatest suppression, specifically bracketing the residue F70, which is positioned at the apex of this substrate-binding beta-hairpin loop (Fig. 3). This observation is a clear indication that the beta-hairpin loop neighboring the active site NDM-1 closes and rigidifies over the bound ligand. The use of UVPD-MS avoids several artifacts in the crystallography of ligand:NDM-1 complexes: commonly observed ASL1 intertwining between neighboring monomers in the crystal lattice and degradation of the covalent adduct made by 2. Additionally, this MS approach avoids the requirement to covalently install reporter labels in the beta-hairpin loop that could perturb function, and clearly favors a loop closing event upon inhibitor binding. Our result is more consistent with prior 19F NMR results indicating loop closure upon ligand binding and implies that the loop opening previously suggested by RFQ-DEER studies may instead be due to perturbation by the spin label or loop twisting resulting in an apparent distance increase.13,14
Detecting displacement of Zn2 by a cysteine-modifying covalent NDM-1 inhibitor
Determining the impact of inhibitors on the metalation state of NDM-1 is an important yet often overlooked aspect of developing inhibitors suitable for therapeutic use.4 Compounds that inhibit by stripping both metal ions from the dinuclear zinc NDM-1 and sequestering them as small molecule complexes are likely to have significant off-target effects making them less suitable as potential therapeutics. Native MS has previously been used to detect changes in the metalation state of NDM-1 upon inhibitor binding.42,45 Here we apply UVPD-MS and an established Zn2 ejector to more extensively elucidate structural changes that occur throughout the NDM-1 protein upon inhibitor binding.The non-selective thiol reagent ebselen (3) has been examined as a ligand for a wide variety of cysteine-containing proteins by using mass spectrometry.46 In particular, ebselen is a potent inhibitor of NDM-1 that functions by covalently modifying the sole Cys residue found in soluble NDM-1 constructs (Cys208).42 Cys208 is a direct ligand to Zn2 (coordinated by D124, C208, H250) and treatment with ebselen (3) induces the loss of one zinc ion, previously presumed to be Zn2, from NDM-1 as detected by native MS. Based on using denaturing conditions for initial collection of ESI mass spectra in the present study, we confirmed addition of one ebselen equivalent to NDM-1 (Fig. S11A†). Using non-denaturing conditions, a narrow envelope of low charge states (8+ to 11+) is observed for uninhibited NDM-1 with the retention of two Zn(II) ions (Fig. S11B†). After reaction with ebselen, NDM-1 yields a much wider distribution of charge states (8+ to 18+) and the net mass shift corresponds to the attachment of a single ebselen along with the loss of one Zn(II) ion (Fig. S11B†). These results confirm removal of a metal cofactor after modification by ebselen, and also indicate unfolding of a portion of the NDM-1 protein as evidenced by the extended tail of the charge state distribution.
UVPD of the most abundant charge state (24+ for denaturing conditions; 9+ for non-denaturing conditions) allows the structure of the modified protein to be characterized in more detail. High sequence coverages were obtained for the protein in both the high (denatured) and low (non-denaturing) charge states (78% and 79%, respectively) and allowed confident localization of the inhibitor adduct to Cys208 based on bracketing the mass shift via backbone cleavages (Fig. S12 and S13†). Under non-denaturing conditions, zinc-bound holo fragment ions are observed in addition to apo (zinc-free) fragment ions (Fig. S13B†). Owing to the high energy deposited during UV photoactivation, backbone bonds are cleaved preferentially over the disruption of non-covalent interactions, resulting in production of holo (containing Zn) fragment ions as well as apo (without Zn) fragment ions that elucidate binding sites.30,34–36,38 For uninhibited NDM-1, holo fragment ions can contain up to two zinc(II) ions, whereas the ebselen-modified protein only has one remaining Zn(II) that can be retained in fragment ions (in addition to the covalently-bound ebselen moiety). Zn-bound holo ions resulting from UVPD were mapped along the protein sequence and upon the crystal structure for uninhibited NDM-1 (Fig. 4). Corresponding structures for inhibited NDM-1 in which holo ions containing one or two Zn(II) were considered separately are shown in Fig. S14.† Based on this analysis, it is evident that it is exclusively Zn2, and not Zn1, that is lost upon binding of ebselen to NDM-1. Specifically, Zn-bound holo fragment ions are no longer observed from backbone cleavages adjacent to the three residues responsible for coordinating Zn2 (D124, C208, H250). Also, N-terminal holo ions are only produced C-terminal to the first Zn1 binding residue (H120) and C-terminal holo ions N-terminal to H189 (Fig. 4). This assessment of the appearance and disappearance of key Zn-containing fragment ions allows the identity of the lost Zn ion to be directly observed as Zn2.
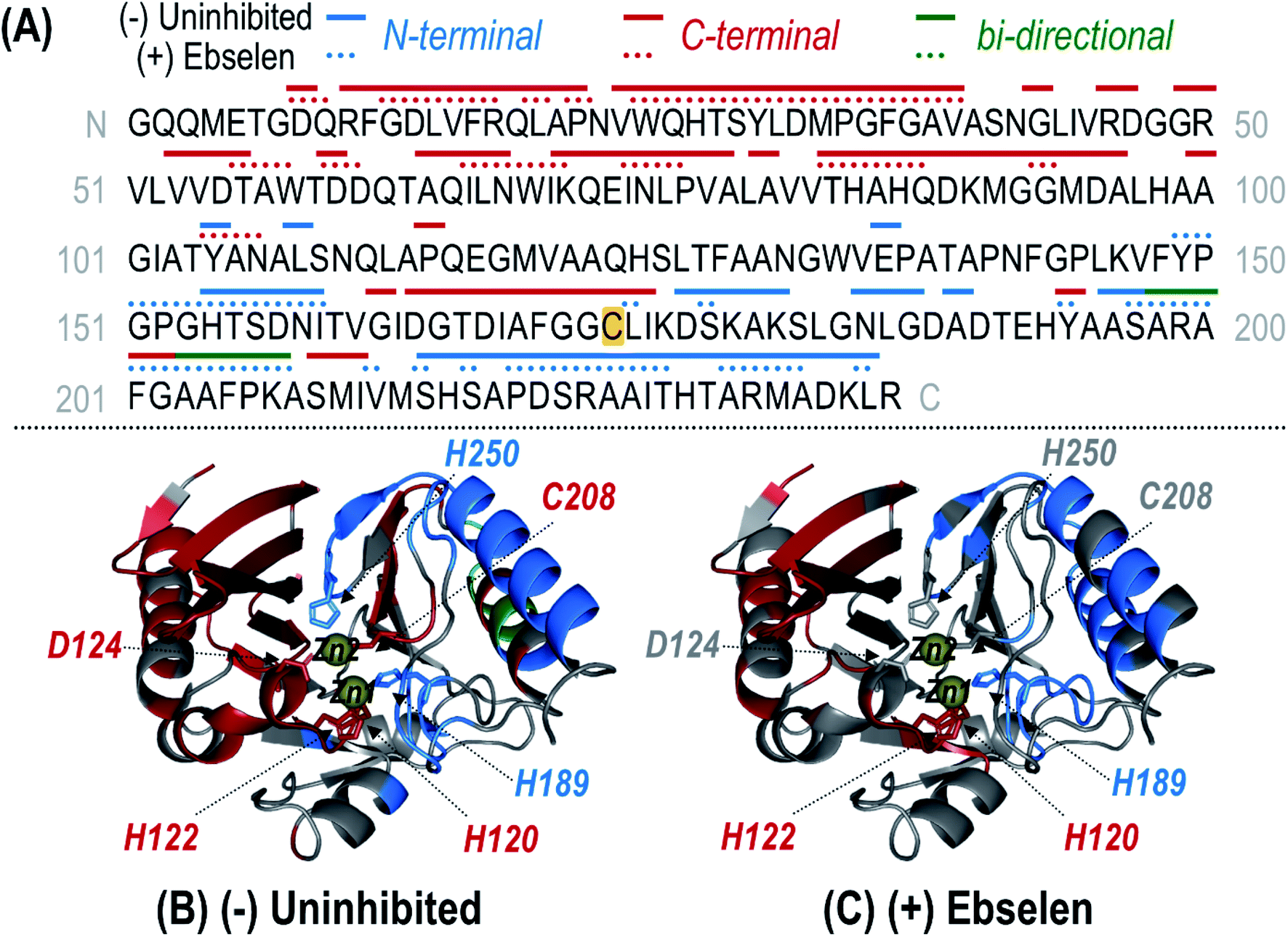 | ||
| Fig. 4 (A) Sequence of NDM-1 with Zn(II)-containing holo ions observed during UVPD of the uninhibited (solid line) and ebselen (3) reacted (dotted line) samples mapped above as linear segments. Coloring of the lines corresponds to N-terminal (blue), C-terminal (red), or bi-directional (green) Zn(II)-containing holo fragment ions identified from the UVPD spectra of the corresponding 9+ species shown in Fig. S13.† Complementary N- and C-terminal holo ions arising from the same backbone position indicate bi-directional fragmentation. For the uninhibited protein, one or two bound Zn(II) ions were considered while for the reacted protein, only holo ions containing a single Zn(II) were identified. The residue at which ebselen covalently attaches (Cys208) is highlighted in yellow. This information is mapped along the crystal structure of NDM-1 (PDB ID 3SPU) for the uninhibited (B) and (+) ebselen (C) proteins. Corresponding crystal structure maps for the uninhibited protein separately identifying singly and doubly Zn(II) bound holo ions are given in Fig. S14.† Zn(II) binding residues are shown as sticks and the two Zn(II) ions are represented as yellow spheres. | ||
Lastly, considering both apo and holo fragment ions, variations in the backbone fragmentation efficiencies for ebselen-modified NDM-1 versus uninhibited NDM-1 were charted (Fig. S15†) and shown as a difference plot in Fig. S16.† Suppression of backbone cleavage is observed for the ASL1 region (the substrate-binding beta-hairpin loop) as well as for the three residues coordinating Zn1 (H120, H122, H189), suggesting a reorganization that curbs fragmentation throughout the N-terminal region of the protein after modification by ebselen. Interestingly, there is slight suppression of UVPD at D124 suggesting that this residue, no longer coordinated to Zn2, may engage in new interactions with the bound inhibitor, which is expected to be located nearby due to the proximity of Cys208.
Here we demonstrate the application of UVPD-MS to characterize inhibitor binding, and to simultaneously report on the site of covalent reaction (Cys208), the metalation state of NDM-1 (specifically the loss of Zn2), the closure of the substrate-binding beta-hairpin loop (ASL1), and the partial denaturation of C-terminal domain of the protein. These results are consistent with prior work that shows loss of one zinc equivalent upon addition of one equivalent of ebselen, and additionally provide direct evidence that Cys208 is adducted and that Zn2 is ejected.42 Our results are also consistent with prior studies that show an overall decrease in thermostability upon loss of Zn2 and the formation of putative inclusion bodies upon ebselen treatment, but provide more specific structural details about how the structure of Zn2 ligands, surrounding residues, and the C-terminal domain of the protein are impacted by Zn2 ejection.15,47
Examining structural changes in NDM clinical variants associated with Zn(II) binding residues
Given the capability of UVPD-MS to detect structural changes in NDM-1 upon Zn2 ejection by ebselen (3), we reasoned that this MS technique may also be useful to detect and better understand the structural implications of sequence differences introduced by clinical variants of NDM. Many of the NDM variants (currently NDM-1 through NDM-29) have increased thermostability and increased affinity for Zn2, presumably indicating that the blaNDM gene is evolving in response to the dual selective pressures of antibiotic treatment and zinc depravation by host innate immune responses.3,15–18 Here, we compare NDM-1 (the reference sequence) and the double mutant NDM-15 (M154L, A233V), which has a ∼10-fold increase in Zn2 Kd value and ∼7 °C increase in thermostability, as well as the individual single mutants NDM-4 (M154L) and NDM-6 (A233V) (Fig. S3†).17 X-ray crystallography has revealed only minor structural differences between NDM-1 and NDM-4 (or other variants), and the mechanism whereby these mutations communicate to the Zn2 site to impact affinity is not obvious. Here, we apply UVPD-MS to interrogate structural differences among representative clinical NDM variants.Upon electrospray ionization, each of the variants was observed to produce intact protein ions in low charge states (9+, 10+, 11+) bound to two Zn(II) (Fig. S17†). Isolation and UV photoactivation of the 9+ charge state yielded the fragmentation patterns in Fig. S18.† Deconvolution allows the isotopically-resolved fragments to be readily assigned as sequence ions (Fig. S19†). In addition to apo (Zn-free) fragment ions, holo (Zn-bound) fragment ions were identified, the latter readily assigned based on inclusion of a mass shift of one or two Zn(II) ions relative to the metal-free fragment ions. Combining the observed metal-free and holo fragment ions accounts for coverage of 69–78% of the protein sequence as shown in the maps in Fig. S19.†
Differences in relative abundances of some of the fragment ions between variants are apparent upon visual inspection of the UVPD mass spectra in Fig. S18.† Regions of the protein in which noncovalent interactions or flexibility are impacted by single point mutations can be elucidated by examining the enhancement or suppression of UVPD backbone cleavages.35–37 For each NDM variant, the backbone cleavage efficiency upon UVPD was determined and is represented graphically in Fig. S20† as a function of each residue of the protein. The fragmentation behavior of three clinical variants (NDM-4, NDM-6, NDM-15) is compared to that of the reference sequence of NDM-1 by subtraction of the backbone cleavage values at each residue and visualized as difference plots (Fig. S21†). Negative values are indicative of suppression in UVPD backbone cleavages for each clinical variant compared to NDM-1, whereas positive values designate enhancement in backbone cleavages for the clinical variant compared to NDM-1. Statistical significance of the observed differences is determined from t-test calculations and represented as p-values plotted per residue in Fig. S2C.† To aid in visualization, the changes are represented as a heatmap spanning blue (suppression of fragmentation of the clinical variant) to red (enhancement of fragmentation of the clinical variant) along the protein sequence from N-terminus to C-terminus (Fig. S22A†). Imprinting the heatmap on the crystal structure allows differences in fragmentation to be related to structural features and specific regions of the protein, as illustrated Fig. S22B–D.†
Overall, the variations in backbone fragmentation (enhancement of some regions and suppression of others) observed for NDM-15 (Fig. S22D†) generally mirror the collective differences for NDM-4 and NDM-6 (Fig. S22B and C†). Specifically, for NDM-15 suppression in UVPD fragmentation is observed along the loop containing H120-D124 (ASL2) and the loop containing C208 (ASL4), with the same modest degree of suppression occurring for both NDM-4 and NDM-6. These two loop regions are shaded in blue on the crystal structure in Fig. S22D† to highlight the suppression of backbone fragmentation of NDM-15. The suppression in fragmentation likely represents a rigidification of these domains that is qualitatively consistent with the observed changes in thermostability. Reported melting temperatures (Tm) for NDM-1, -4, -6, and -15 (54.5, 58.4, 56.3, and 61.5 °C, respectively) show increases in stability and the gain for NDM-15 (∼7 °C) is only slightly more than the sum of the two individual mutants, which is also consistent with the mostly additive nature of the observed structural changes.17
We then focused particularly on the six residues that anchor the dinuclear zinc ion cluster at the active site. The higher affinity Zn1 site consists of H120, H122, and H189 ligands, and the lower-affinity Zn2 site consists of D124, C208, and H250 (Fig. S3†).48 The relative degrees of backbone fragmentation bracketing these six key residues of NDM-1 and each of the other variants are highlighted in Fig. 5. For NDM-4 (M154L) compared to NDM-1, the only significant differences in fragmentation related to the six targeted Zn-binding residues are suppression of backbone cleavages at H120 and H122. This observation aligns well with the location of M154 near the α-carbon of H122, and with a prior proposal that substitution of Met to Leu shifts the position of H122 to enable more stabilizing interactions with Zn1.15 Here, we clearly observe the stronger H122:Zn1 interactions experimentally as a suppression in the efficiency of backbone cleavages at H122. This same type of suppression of fragmentation at H122 is not noted for NDM-6 (A233V), indicating a different stabilization mechanism is used for these two variants.
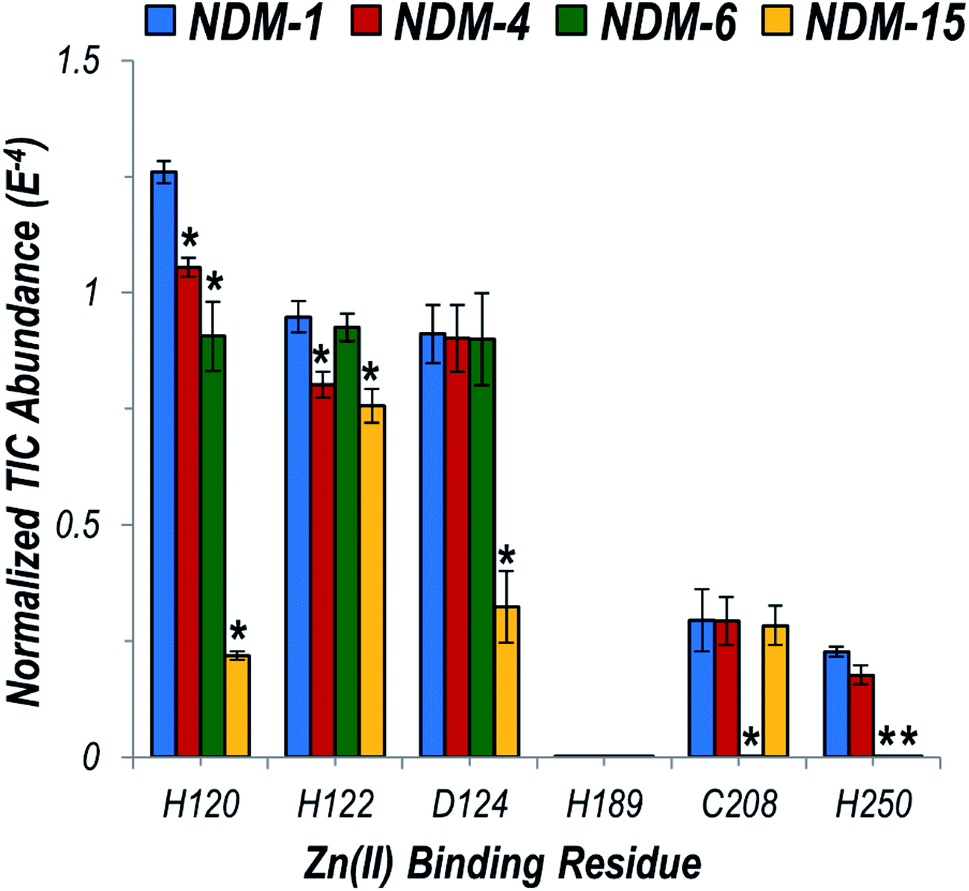 | ||
| Fig. 5 Graph displaying summed UVPD fragment ion abundances normalized to the total ion current (TIC) corresponding to fragment ions originating from cleavages occurring proximal to each of the six Zn(II) binding residues for NDM-1 (blue), NDM-4 (red), NDM-6 (green), and NDM-15 (yellow). Corresponding values for the entire protein sequence are given in the UVPD abundance plots in Fig. S20.† Asterisks indicate residues at which the difference in backbone cleavages for the clinical variant compared to the reference NDM-1 protein is statistically significant (99% confidence threshold). | ||
For NDM-6 (A233V), the backbone cleavage efficiency is substantially suppressed at a different Zn1-ligand, H120, as well as at two Zn2-ligands, C208 and H250. The shift of the flexible loop containing C208 owing to the introduction of a bulkier Val for Ala would place C208, H120 and H250 (on neighboring loops) in more ideal positions for stabilizing Zn2. Lastly, for NDM-15 (M154L, A233V), suppression of backbone fragmentation was observed at four of the six zinc-binding residues, representing mostly a sum of the changes observed in individual single mutations. However, backbone fragmentation at C208 was not suppressed for NDM-15, suggesting that the M154L substitution has a greater impact on enhancing Zn(II) affinity compared to A233V when both mutations are present. The use of native UVPD-MS reveals in increased detail how distant structural changes located on each side of the dizinc site are propagated and combine to increase metal ion affinity, helping to elucidate the mechanisms used by clinical variants to overcome zinc scarcity at infection sites.
Conclusion
Structural information detailing ligand:NDM interactions is important for design and optimization of selective and potent inhibitors to serve as probes for chemical biology experiments and as lead compounds for therapeutic development. However, interactions between ligand and the essential substrate-binding beta-hairpin loop are obscured due to method-dependent artifacts. Additionally, structure–activity relationships are often difficult to extract from libraries if the mechanisms of inhibition are not determined. Attributes that lead to more effective metal stripping and attributes that lead to more effective ternary complex formation (inhibitor:dizinc cluster:NDM) can both show increased inhibition, but only the latter category is suitable for developing NDM-selective probes and therapeutic leads. Here, we demonstrate a powerful strategy utilizing native MS along with UVPD relying on observed changes in fragmentation to interrogate structural differences between uninhibited and inhibited NDM-1, simultaneously revealing conformational changes in the substrate-binding beta-hairpin loop, reporting on the metalation state of the enzyme, identifying modified residues (if any), and tracking other changes throughout the protein structure. We characterized two structurally divergent inhibitors, a K211-selective covalent inhibitor and a C208 covalent modifier and found that each induced a similar structural change - closure of the beta-hairpin loop. These cases were characterized using covalent inhibitors, but native UVPD-MS can also be applied to the similar characterization of non-covalent ligands.31,34–36,38 Previous solution phase studies of NDM led to conflicting conclusions, that ligand binding either induces loop closure or opens the cavity even wider.13,14 However, the prior studies required mutation and chemical modification of the loop which likely impact both structure and dynamics, and one approach used a non-native metalloform. Here, native UVPD-MS was completed using wild type dizinc NDM to conclusively show loop closing upon ligand binding. This result is particularly significant because optimization of loop:inhibitor interactions is required for NDM drug design, and native UVPD-MS now provides a novel strategy to probe this interaction with minimal perturbation that was not possible with previous methods.
Another advantage of native UVPD-MS as showcased here for NDM in the present study is the ability to simultaneously report on modification sites (for covalent inhibitors), metalation state, and structural perturbations in addition to loop closure. Several notable conclusions can be drawn from the study of inactivators 1–3. The pentafluorophenyl ester (1) was previously shown to be specific for one Lys residue in IMP-1, but we find it readily modifies three Lys residues neighboring the active site (although no other surface Lys residues). This finding reveals two additional Lys residues as alternative available targets for design of more effective covalent NDM inhibitors. Simultaneous targeting of multiple Lys residues could be useful in overcoming resistance-causing mutations. Previous characterization of the O-aryloxycarbonyl hydroxyamate inactivator (2) by X-ray crystallography revealed two covalent adducts, and one was proposed to be a degradation product of the other.41 Here, native UVPD-MS revealed only the parent modification and not the degradation product, supporting the proposed inactivation mechanism and suggesting that the MS method may give less ambiguous results. Finally, native UVPD-MS of ebselen (3)-treated NDM-1 confirms a prior report that one zinc ion is ejected,15,47 and now reveals the identity of the ejected zinc as Zn2 and provides direct evidence that Cys208 is the modified residue. Unexpectedly, we also detected partial denaturation of the C-terminus of the protein, which provides structural information that helps explain decreased thermostability and inclusion body formation upon Zn2 loss and ebselen treatment. We note that ebselen is a useful probe to show the structural and mechanistic implications of Cys modification and Zn2 loss in NDM-1 that may be more broadly applicable. NDM-1 appears to be less effective than other carbapenemases in some infection models and patients.49 One possibility is that the weak affinity for Zn2 allows NDM-1 to be more easily inhibited by host zinc sequestering proteins such as calprotectin or Cys oxidation by ROS during inflammation, which would be expected to result in deleterious structural changes similar to those we observed with ebselen using native UVPD-MS.
The ability of native UVPD-MS to report on structural changes related to metalation state prompted us to use this method to characterize select NDM clinical variants. Notably, we were able to track the structural impact of distant mutations to direct zinc ligands. Specifically, the increased Zn2 affinity (with respect to NDM-1) previously measured for NDM-4 (M154L) and NDM-6 (A233V) appear to be due to structural changes largely propagated through different networks of residues emanating from the distant mutated sites toward the dizinc cluster. Many of the structural changes induced by the individual mutations are conserved when combined in the NDM-15 variant (M154L, A233V), but additional structural changes are also observed, highlighting the structural basis of the small synergism observed in this double mutant. Identification of these networks of residues may enable prediction of how newly emerging clinical variants perturb structure, function and zinc affinity. These structural changes were not easily observed using X-ray crystallography, but native UVPD-MS provides an innovative approach to determine the impact of NDM clinical variants on structure and dynamics and elucidates in greater mechanistic detail how structural changes at distant mutation sites are propagated to the zinc binding site.
In summary, native UVPD-MS is a powerful analytical tool that enables simultaneous detection of inhibitor binding, covalent modification sites, metalation state, and structural changes to NDM, including closing of the active-site beta-hairpin loop and other perturbations owing to ligand binding or mutation, all using unlabeled wild-type dizinc protein. This tool should help to easily distinguish metal stripping agents from ternary complex forming inhibitors, which are preferred for drug design,4 while providing additional information about inhibitor interactions with the substrate-binding beta-hairpin loop that were not easily obtained previously. Moreover, new insight into the structure and dynamics of emerging NDM clinical variants has been uncovered.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported in part by the National Institutes of Health (R01GM121714 to JSB and GM111926 to WF) and the Robert A. Welch Foundation (F-1572 to WF and F-1155 to JSB).References
- Centers for Disease Control and Prevention (CDC), Antibiotic resistance threats in the United States, 2019, Centers for Disease Control and Prevention (U.S.), Atlanta, 2019 Search PubMed.
- K. M. Papp-Wallace, A. Endimiani, M. A. Taracila and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 4943–4960 CrossRef CAS PubMed.
- P. W. Thomas, M. Zheng, S. Wu, H. Guo, D. Liu, D. Xu and W. Fast, Biochemistry, 2011, 50, 10102–10113 CrossRef CAS PubMed.
- L.-C. Ju, Z. Cheng, W. Fast, R. A. Bonomo and M. W. Crowder, Trends Pharmacol. Sci., 2018, 39, 635–647 CrossRef CAS PubMed.
- J. H. Toney, P. M. D. Fitzgerald, N. Grover-Sharma, S. H. Olson, W. J. May, J. G. Sundelof, D. E. Vanderwall, K. A. Cleary, S. K. Grant, J. K. Wu, J. W. Kozarich, D. L. Pompliano and G. G. Hammond, Chem. Biol., 1998, 5, 185–196 CrossRef CAS PubMed.
- C. Moali, C. Anne, J. Lamotte-Brasseur, S. Groslambert, B. Devreese, J. Van Beeumen, M. Galleni and J.-M. Frère, Chem. Biol., 2003, 10, 319–329 CrossRef CAS PubMed.
- S. D. B. Scrofani, J. Chung, J. J. A. Huntley, S. J. Benkovic, P. E. Wright and H. J. Dyson, Biochemistry, 1999, 38, 14507–14514 CrossRef CAS PubMed.
- J. J. A. Huntley, S. D. B. Scrofani, M. J. Osborne, P. E. Wright and H. J. Dyson, Biochemistry, 2000, 39, 13356–13364 CrossRef CAS PubMed.
- J. J. A. Huntley, W. Fast, S. J. Benkovic, P. E. Wright and H. J. Dyson, Protein Sci., 2003, 12, 1368–1375 CrossRef CAS PubMed.
- W. Fast and L. D. Sutton, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 1648–1659 CrossRef CAS PubMed.
- J. Chiou, T. Y.-C. Leung and S. Chen, Antimicrob. Agents Chemother., 2014, 58, 5372–5378 CrossRef PubMed.
- A. R. Palacios, M. F. Mojica, E. Giannini, M. A. Taracila, C. R. Bethel, P. M. Alzari, L. H. Otero, S. Klinke, L. I. Llarrull, R. A. Bonomo and A. J. Vila, Antimicrob. Agents Chemother., 2019, 63, e01754-18 CrossRef PubMed.
- M. Aitha, L. Moritz, I. D. Sahu, O. Sanyurah, Z. Roche, R. McCarrick, G. A. Lorigan, B. Bennett and M. W. Crowder, J. Biol. Inorg Chem., 2015, 20, 585–594 CrossRef CAS PubMed.
- A. M. Rydzik, J. Brem, S. S. van Berkel, I. Pfeffer, A. Makena, T. D. W. Claridge and C. J. Schofield, Angew. Chem., Int. Ed., 2014, 53, 3129–3133 CrossRef CAS PubMed.
- A. C. Stewart, C. R. Bethel, J. VanPelt, A. Bergstrom, Z. Cheng, C. G. Miller, C. Williams, R. Poth, M. Morris, O. Lahey, J. C. Nix, D. L. Tierney, R. C. Page, M. W. Crowder, R. A. Bonomo and W. Fast, ACS Infect. Dis., 2017, 3, 927–940 CrossRef CAS PubMed.
- G. Bahr, L. Vitor-Horen, C. R. Bethel, R. A. Bonomo, L. J. González and A. J. Vila, Antimicrob. Agents Chemother., 2018, 62, e01849–17 Search PubMed.
- Z. Cheng, P. W. Thomas, L. Ju, A. Bergstrom, K. Mason, D. Clayton, C. Miller, C. R. Bethel, J. VanPelt, D. L. Tierney, R. C. Page, R. A. Bonomo, W. Fast and M. W. Crowder, J. Biol. Chem., 2018, 293, 12606–12618 CrossRef CAS PubMed.
- A. Makena, J. Brem, I. Pfeffer, R. E. J. Geffen, S. E. Wilkins, H. Tarhonskaya, E. Flashman, L. M. Phee, D. W. Wareham and C. J. Schofield, J. Antimicrob. Chemother., 2015, 70, 463–469 CrossRef CAS PubMed.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem., 2013, 85, 2–9 CrossRef CAS PubMed.
- A. C. Leney and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef CAS PubMed.
- T. M. Allison and C. Bechara, Biochem. Soc. Trans., 2019, 47, 317–327 CrossRef CAS PubMed.
- L. A. Macias, I. C. Santos and J. S. Brodbelt, Anal. Chem., 2020, 92, 227–251 CrossRef CAS PubMed.
- F. Lermyte and F. Sobott, Proteomics, 2015, 15, 2813–2822 CrossRef CAS PubMed.
- H. Zhang, W. Cui, J. Wen, R. E. Blankenship and M. L. Gross, Anal. Chem., 2011, 83, 5598–5606 CrossRef CAS PubMed.
- J. Zhang, G. R. Malmirchegini, R. T. Clubb and J. A. Loo, Eur. J. Mass Spectrom., 2015, 21, 221–231 CrossRef CAS PubMed.
- P. Wongkongkathep, J. Y. Han, T. S. Choi, S. Yin, H. I. Kim and J. A. Loo, J. Am. Soc. Mass Spectrom., 2018, 29, 1870–1880 CrossRef CAS PubMed.
- H. Li, Y. Sheng, W. McGee, M. Cammarata, D. Holden and J. A. Loo, Anal. Chem., 2017, 89, 2731–2738 CrossRef CAS PubMed.
- H. Li, H. H. Nguyen, R. R. Ogorzalek Loo, I. D. G. Campuzano and J. A. Loo, Nat. Chem., 2018, 10, 139–148 CrossRef CAS PubMed.
- J. B. Shaw, W. Li, D. D. Holden, Y. Zhang, J. Griep-Raming, R. T. Fellers, B. P. Early, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, J. Am. Chem. Soc., 2013, 135, 12646–12651 CrossRef CAS PubMed.
- J. P. O'Brien, W. Li, Y. Zhang and J. S. Brodbelt, J. Am. Chem. Soc., 2014, 136, 12920–12928 CrossRef PubMed.
- M. B. Cammarata and J. S. Brodbelt, Chem. Sci., 2015, 6, 1324–1333 RSC.
- M. R. Robinson, J. M. Taliaferro, K. N. Dalby and J. S. Brodbelt, J. Proteome Res., 2016, 15, 2739–2748 CrossRef CAS PubMed.
- S. N. Sipe and J. S. Brodbelt, Phys. Chem. Chem. Phys., 2019, 21, 9265–9276 RSC.
- M. B. Cammarata, R. Thyer, J. Rosenberg, A. Ellington and J. S. Brodbelt, J. Am. Chem. Soc., 2015, 137, 9128–9135 CrossRef CAS PubMed.
- M. B. Cammarata, C. L. Schardon, M. R. Mehaffey, J. Rosenberg, J. Singleton, W. Fast and J. S. Brodbelt, J. Am. Chem. Soc., 2016, 138, 13187–13196 CrossRef CAS PubMed.
- M. R. Mehaffey, C. L. Schardon, E. T. Novelli, M. B. Cammarata, L. J. Webb, W. Fast and J. S. Brodbelt, Chem. Sci., 2019, 10, 8025–8034 RSC.
- M. Cammarata, R. Thyer, M. Lombardo, A. Anderson, D. Wright, A. Ellington and J. S. Brodbelt, Chem. Sci., 2017, 8, 4062–4072 RSC.
- M. R. Mehaffey, M. B. Cammarata and J. S. Brodbelt, Anal. Chem., 2018, 90, 839–846 CrossRef CAS PubMed.
- H. Kurosaki, Y. Yamaguchi, T. Higashi, K. Soga, S. Matsueda, H. Yumoto, S. Misumi, Y. Yamagata, Y. Arakawa and M. Goto, Angew. Chem., Int. Ed., 2005, 44, 3861–3864 CrossRef CAS PubMed.
- R. Tilvawala, M. Cammarata, S. A. Adediran, J. S. Brodbelt and R. F. Pratt, Biochemistry, 2015, 54, 7375–7384 CrossRef CAS PubMed.
- P. W. Thomas, M. Cammarata, J. S. Brodbelt, A. F. Monzingo, R. F. Pratt and W. Fast, Biochemistry, 2019, 58, 2834–2843 CrossRef CAS PubMed.
- J. Chiou, S. Wan, K.-F. Chan, P.-K. So, D. He, E. W. Chan, T. Chan, K. Wong, J. Tao and S. Chen, Chem. Commun., 2015, 51, 9543–9546 RSC.
- J. Rosenberg, W. R. Parker, M. B. Cammarata and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2018, 29, 1323–1326 CrossRef CAS PubMed.
- D. King and N. Strynadka, Protein Sci., 2011, 20, 1484–1491 CrossRef CAS PubMed.
- A. Y. Chen, C. Thomas, P. W. Thomas, K. Yang, Z. Cheng, W. Fast, M. W. Crowder and S. M. Cohen, ChemMedChem, 2020, 15, 1272–1282 CrossRef CAS PubMed.
- K. Xu, Y. Zhang, B. Tang, J. Laskin, P. J. Roach and H. Chen, Anal. Chem., 2010, 82, 6926–6932 CrossRef CAS PubMed.
- C. Chen, Y. Xiang, K.-W. Yang, Y. Zhang, W.-M. Wang, J.-P. Su, Y. Ge and Y. Liu, Chem. Commun., 2018, 54, 4802–4805 RSC.
- Y. Guo, J. Wang, G. Niu, W. Shui, Y. Sun, H. Zhou, Y. Zhang, C. Yang, Z. Lou and Z. Rao, Protein Cell, 2011, 2, 384–394 CrossRef CAS PubMed.
- S. E. Boyd, D. M. Livermore, D. C. Hooper and W. W. Hope, Antimicrob. Agents Chemother., 2020 DOI:10.1128/AAC.00397-20.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S22 which include the expressed protein sequence of NDM, structures of the covalent inhibitors, crystal structure of NDM-1 with relevant regions labelled, NDM-1 inactivation data, p-value plots highlighting statistical significance of measured differences, ESI-MS, HCD or UVPD MS/MS, holo ion plots, UVPD intensity plots, and difference plots or heatmaps of uninhibited NDM-1, NDM-1 bound to each of the three inhibitors, and three other clinical variants (NDM-4, NDM-6, NDM-15). See DOI: 10.1039/d0sc02503h |
| This journal is © The Royal Society of Chemistry 2020 |