
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric allylic substitution–isomerization to axially chiral enamides via hydrogen-bonding assisted central-to-axial chirality transfer†
Chao
Sun‡
a,
Xiaotian
Qi‡
b,
Xiao-Long
Min
a,
Xue-Dan
Bai
a,
Peng
Liu
 *bc and
Ying
He
*a
*bc and
Ying
He
*a
aSchool of Chemical Engineering, Nanjing University of Science & Technology, Nanjing 210094, China. E-mail: yhe@njust.edu.cn
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: pengliu@pitt.edu
cDepartment of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, USA
First published on 7th September 2020
Abstract
Axially chiral enamides bearing a N–C axis have been recently studied and were proposed to be valuable chiral building blocks, but a stereoselective synthesis has not been achieved. Here, we report the first enantioselective synthesis of axially chiral enamides via a highly efficient, catalytic approach. In this approach, C(sp2)–N bond formation is achieved through an iridium-catalyzed asymmetric allylation, and then in situ isomerization of the initial products through an organic base promoted 1,3-H transfer, leading to the enamide products with excellent central-to-axial transfer of chirality. Computational and experimental studies revealed that the 1,3-H transfer occurs via a stepwise deprotonation/re-protonation pathway with a chiral ion-pair intermediate. Hydrogen bonding interactions with the enamide carbonyl play a significant role in promoting both the reactivity and stereospecificity of the stepwise 1,3-H transfer. The mild and operationally simple formal N-vinylation reaction delivered a series of configurationally stable axially chiral enamides with good to excellent yields and enantioselectivities.
Introduction
Asymmetric isomerization of allylic compounds has attracted great attention from synthetic chemists due to its 100% atom economy.1–3 Typical approaches for this transformation are achieved through the transition metal-mediated [1,3]-hydride transfer catalysis. With the development of the area, organic molecule catalysis has enabled efficient enantioselective [1,3]-allylic rearrangements since 2010.4–13 In this case, [1,3]-H (proton) transfer would occur in the presence of an organic base catalyst. Very recently, the detailed mechanistic studies of stereospecific [1,3]-allylic rearrangements showing chirality transfer were reported by Paton and co-workers.14 Exemplified by base-catalyzed [1,3]-rearrangement of arylindenols, they proposed that a suprafacial prototopic shift is mediated by the amine base; and the chirality transfer is achieved through a resilient contact ion-pair held together by electrostatic attraction and an NH⋯π interaction (Fig. 1a). Alternatively, cognizant of significant role of hydrogen-bonding interactions in asymmetric olefin isomerization,15 we were intrigued by the effect of hydrogen-bonding interactions of stereospecific [1,3]-allylic rearrangements.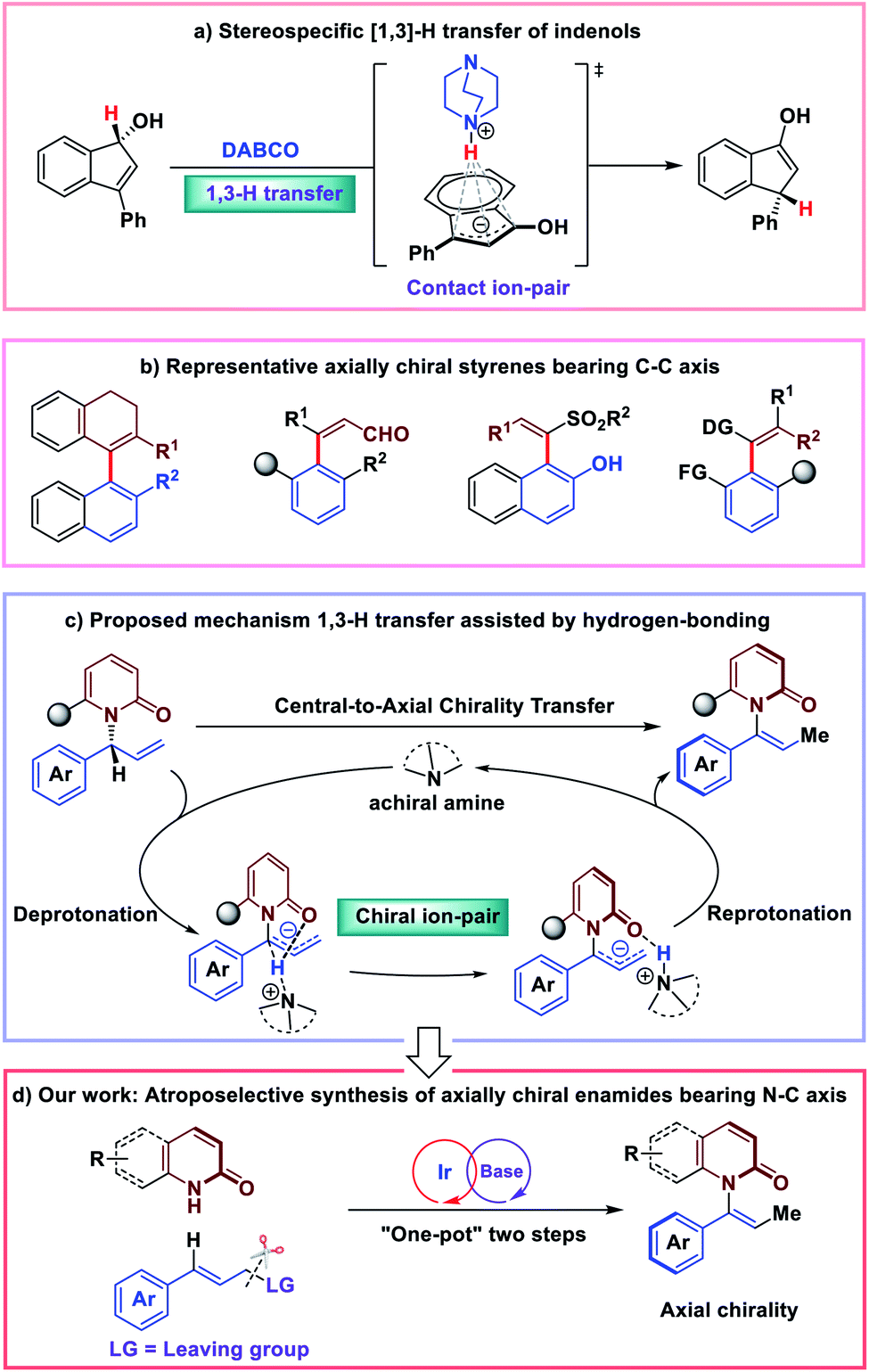 | ||
| Fig. 1 Central-to-axial chirality transfer inspired by [1,]-H transfer strategy. | ||
Recently, we became interested in the construction of axially chiral styrenes. While axially chiral styrenes were proposed as intermediates of interest in the study of chirality transfer decades ago,16 only recently have strategies for their enantioselective synthesis been disclosed (Fig. 1b).17–23 Inspired by stereospecific [1,3]-H transfer strategy, we envisioned the preparation of axially chiral styrenes from enantioenriched allylic compounds. N-substituted bulky anilide derivatives have attracted much attention as important atropoisomeric compounds with a N–C chiral axis.24–32 In 2016, Curran and co-workers prepared a collection of axially chiral enamides in racemic form and measured the rate of racemization for chromatographically-separated enantiomers. The authors concluded that, in many cases, enantioenriched enamides are configurationally-stable for periods of days to weeks at room temperature and may serve as building blocks of interest for asymmetric synthesis and construction of molecular gears and machines.33 Despite their potential applications and inherent interest, access to these compounds remained limited to resolution by chiral chromatography. Catalytic enantioselective synthesis of this new class of axially chiral compounds would constitute a highly desirable synthetic process.
In this scenario, we wondered whether axially chiral enamides could be synthesized via stereospecific [1,3]-H transfer. In this case, [1,3]-H transfer would occur via a stepwise deprotonation/re-protonation pathway with a chiral ion-pair intermediate. Hydrogen bonding interactions with the enamide carbonyl would play a significant role in promoting both the reactivity and stereospecificity of the stepwise [1,3]-H transfer (Fig. 1c). More importantly, enantioenriched allylic amides could be easily synthesized by classic iridium-catalyzed asymmetric allylation.34–42 Due to the basic conditions of the reaction system, we expected that in situ isomerization43,44 of the enantioenriched allylic products by an organic base would lead to the axially chiral enamides via “one-pot” two steps process (Fig. 1d).
Nevertheless, at the outset of our studies we anticipated several challenges of finding conditions to achieve our goal of developing a catalytic, enantioselective synthesis of axially-chiral enamides: (1) the products must be obtained with high enantioselectivity, regioselectivity and geometrical (Z/E) selectivity; (2) the weakly acidic intermediate (more weakly acidic than previously reported substrates for base-mediated chirality transfer) must be deprotonated and re-protonated with high stereochemical fidelity via chiral ion pair; and (3) high temperatures and other harsh conditions must be avoided to suppress racemization of the enantioenriched product. Here we report the first catalytic synthesis of axially chiral enamides using a combination of iridium-catalyzed allylation and base-mediated central-to-axial chirality transfer.
Results and discussion
Optimized conditions
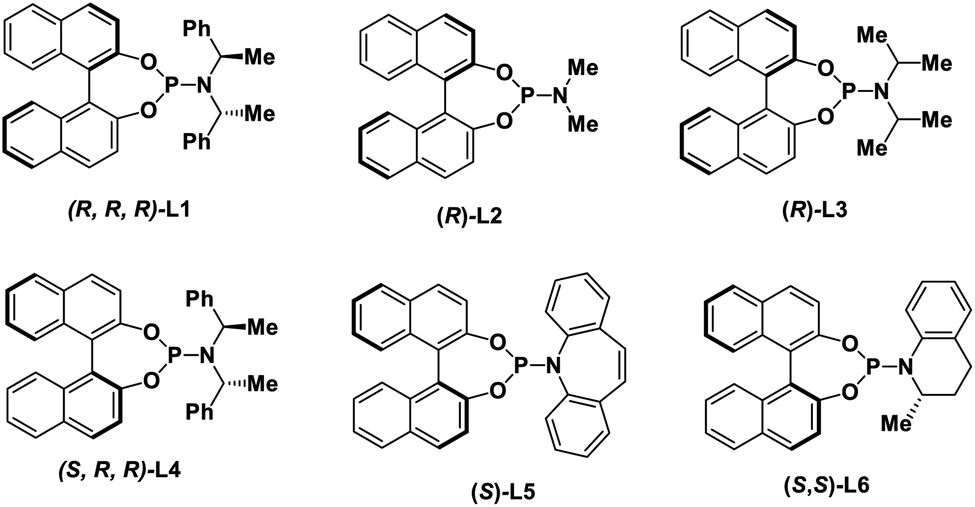
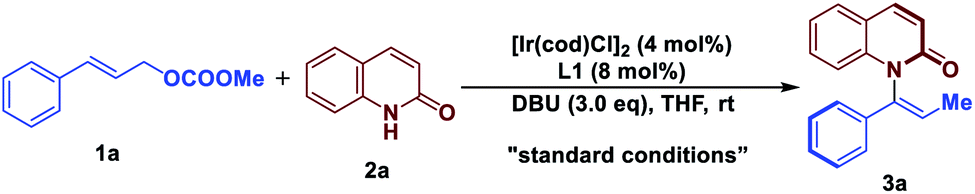
With these considerations in mind, we initiated our studies by examining the reaction of cinnamyl carbonate (1a) and 2-quinolinol (2a) using in situ generated iridacycle catalyst (Table 1, see ESI for details†). To our delight, the reaction proceeded smoothly under the “standard conditions”, affording the desired product 3a in 88% yield with 92% ee using L1 as a ligand (Table 1, entry 1). Upon switching the ligand to L2–L6 did not yield the product 3a efficiently (entries 2–6). The use of different bases such as Et3N, DABCO and TBD led to diminished enantioselectivities and yields (entries 7–9). Examination of solvent effect revealed that compatible yield and enantioselectivity of 3a was obtained (entries 10 and 11). The leaving groups of 1a such as –OBz and –Boc led to good enantioselectivities but low yields of 38% and 34%, respectively (entries 12 and 15). A dramatically low ee of −3% and 50% were obtained using –Cl and –OPO(OEt)2 as the leaving group in substrate 1a.|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | eeb (%) | Yieldc (%) |
| a Reaction conditions: all reactions were run on 0.1 mmol scale with respect to 1. b ee determined by chiral HPLC. c Isolated yield. DABCO = 1,4-diazabicyclo[2.2.2]octane, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene. | |||
| 1 | None | 92 | 88 |
| 2 | L2 instead of L1 as ligand | — | Trace |
| 3 | L3 instead of L1 as ligand | 13 | <5% |
| 4 | L4 instead of L1 as ligand | — | Trace |
| 5 | L5 instead of L1 as ligand | −80 | — |
| 6 | L6 instead of L1 as ligand | 72 | 33 |
| 7 | Et3N instead of DBU as base | 89 | 33 |
| 8 | DABCO instead of DBU as base | 88 | 35 |
| 9 | TBD instead of DBU as base | 86 | 37 |
| 10 | 1,4-dioxane instead of THF as solvent | 91 | 86 |
| 11 | Toluene instead of THF as solvent | 87 | 64 |
| 12 | –OBz instead of –OCOOMe | 93 | 38 |
| 13 | –Cl instead of –OCOOMe | –3 | 46 |
| 14 | –OPO(OEt)2 instead of –OCOOMe | 50 | 42 |
| 15 | –OBoc instead of –OCOOMe | 89 | 34 |
|
|||
Substrate Scope of Reaction
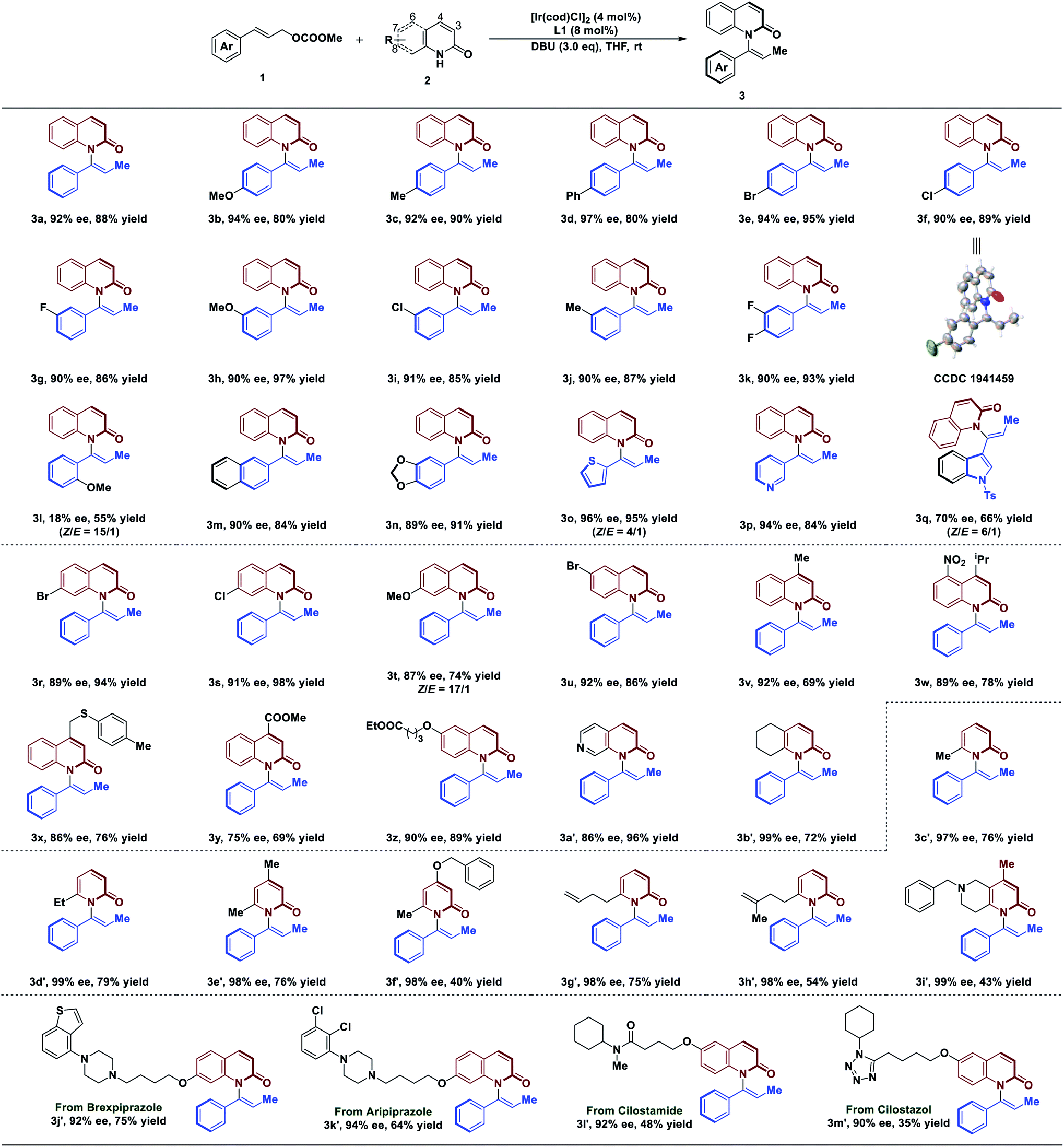
With the optimized conditions in hand, we next explored the substrate scope of the reaction (Table 2). In general, C–H amination of a variety of substituted cinnamyl carbonates are well tolerated under optimized conditions. For example, reactions with a range of para-substituted cinnamyl carbonates bearing electron-neutral, electron-donating, and electron-withdrawing groups proceeded efficiently to afford the corresponding products in 90–97% ee with 80–95% yield (3a–3f). Cinnamyl carbonates containing the meta-substituted, electron-poor, and electron-rich substituents also gave us high ee of 90–91% with 85–97% yield (3g–3j). Difunctionalized cinnamyl carbonate was compatible with the reaction, leading to the desired product in 90% ee with 93% yield (3k). To our disappointment, a lower ee of 18% was obtained using substrate 1 bearing ortho-substituent of aryl group (3l). Remarkably, reaction tolerated bulky substituent in 1, affording the desired products in 89–90% ee (3m and 3n). Notably, the reaction was not limited to aryl moieties of substrate 1; it was also amenable to the heterocyclic rings that afforded the desired products in 94–96% ee with 84–95% yield (3o and 3p). However, the product 3q was obtained in slightly lower ee (70%) with 66% yield. In addition, the absolute configuration of 3f was determined by single X-ray crystallographic analysis, and others were assigned by analogy to 3f.
a Reaction conditions: all reactions were run on 0.1 mmol scale with respect to 1a. ee determined by chiral HPLC. Isolated yield. Unless noted, products were obtained with more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of Z/E. :1 of Z/E.
|
|---|
|
With respect to 2-quinolinol scope, we investigated the generality of this reaction. As depicted in Table 2, the electronic properties of substituents on 2-quinolinol have a significant effect on the enantioselectivity. Substrates 2 bearing substituents at C8, C7, C6 and C4 positions were well tolerated, affording products in 86–92% ee with 69–98% yield (3r–3x and 3z). However, the use of substrate 2 processing electron-withdrawing group at C4 position gave the product in 75% ee (3y). Several 2-quinolinol analogues were then investigated in our catalysis system. Expectedly, the reaction of 2-quinoxalinol with 1a proceeded readily to afford 3a′ in 86% ee with 96% yield. In addition, the reaction of cinnamyl carbonate with 5,6,7,8-tetrahydroquinolin-2(1H)-one resulted in the desired product in 99% ee with 72% yield (3b′). Moreover, the reaction is not limited to 2-quinolinol derivatives; substrates such as pyridin-2(1H)-one analogues were tolerated, affording the axially chiral enamides 3c′–3f′ in up to 99% ee. It's worth noting that the reaction tolerates the functionalized substrates affording the products 3g′–3i′ in 98–99% ee.
Since quinolinols represent a vital class of heterocyclic units that are extensively utilized in natural products and pharmaceuticals,45–47 decoration of bioactive molecules has been carried out. For example, brexpiprazole and aripiprazole, as antipsychotic medications, are used to treat the symptoms of schizophrenia. Under our conditions, they are readily functionalized to afford 3j′ and 3k′ in 92% ee and 94% ee, respectively. A PDE3 inhibitor, cilostamide, could also undergo atroposelective vinylation to generate axially chiral enamide 3l′ in 92% ee, albeit with 48% yield. The reaction also tolerates a cilostazol derivative which afforded 3m′ in 90% ee. Having established the atroposelective vinylation process for the construction of axially chiral enamides, we next explored the preparative-scale synthesis of product 3a. As a result, the product was obtained in 91% ee with 73% yield, suggesting that this method has the potential for large-scale chemical production (see ESI for details†).
Mechanistic studies
In an effort to gain more insight into the reaction mechanism, deuterium labeling and control experiments were carried out. To our delight, allylic product 4a could be obtained by omitting DBU and conducting the reaction using in situ generated iridacycle catalyst under near-neutral conditions (Fig. 2, eqn (1)).48 The subjection of 4a, isolated in 95% ee, to DBU resulted in efficient and stereospecific isomerization (>99% es) to deliver 3a in 95% ee with full conversion (eqn (2)); and no isomerization product was obtained in the absence of base. Determination of the absolute stereochemistry of 4a49 and 3a indicated that re-protonation occurred on the face from which deprotonation initially occurred, consistent with a [1,3]-H transfer mechanism involving a contact ion pair (eqn (2), see ESI for details†).14 High levels of deuterium transfer (>90% D of 6 from 99% D of 5, eqn (3)) were also consistent with reactions previously proposed that take place through an ion pair intermediate (see ESI for details†).14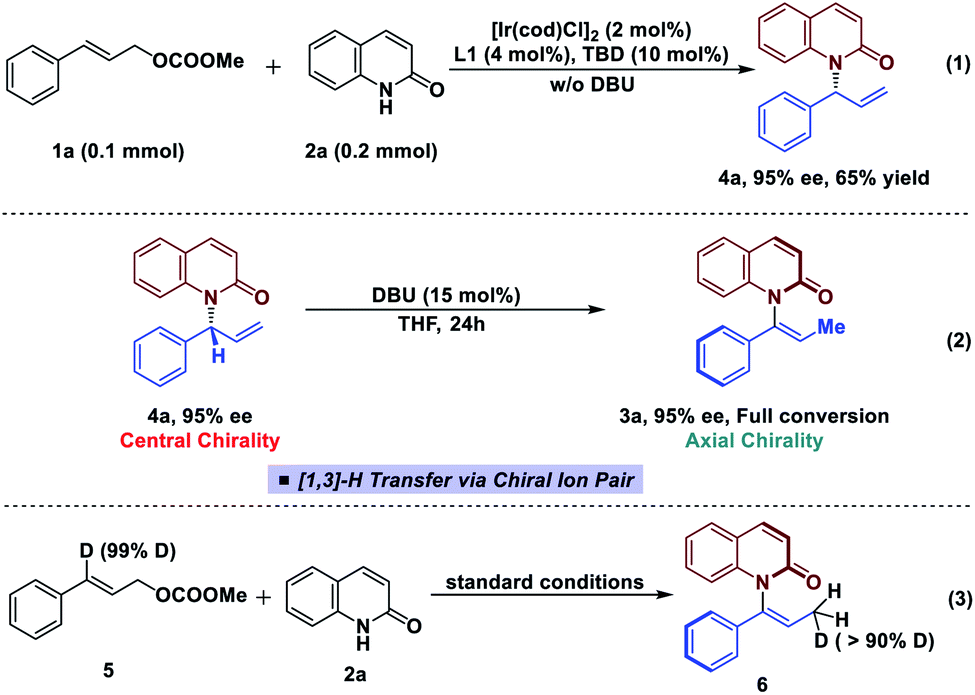 | ||
| Fig. 2 Control experiments and deuterium labeled experiment. | ||
To further clarify the origin of the high levels of stereospecificity during the central-to-axial chirality transfer, density functional theory (DFT) calculations were performed to study the DBU-promoted [1,3]-H transfer of allylation product 4a which bears an (R)-stereogenic center. As shown in Fig. 3 and 4, [1,3]-H transfer pathways from several conformers of 4a were considered. The most favorable benzylic C–H deprotonation occurs from the most stable conformer of 4avia transition state TS-1a with an activation free energy of 21.8 kcal mol−1. This pathway requires a much lower kinetic barrier than the deprotonation of conformer 4b, which requires 24.5 kcal mol−1 with respect to 4a. Intrinsic reaction coordinate (IRC) calculation and Born–Oppenheimer molecular dynamics (BOMD) simulation confirmed that this deprotonation transition state leads to a chiral ion-pair intermediate 7a, which involves strong hydrogen bonding between the iminium N–H and the amide carbonyl group. This N–H⋯O hydrogen bonding is also observed in the deprotonation transition state (TS-1a), as evidenced by a relatively short H⋯O distance of 2.31 Å. In the deprotonation pathway from conformer 4b (Fig. 3, dashed lines), such hydrogen bonding interaction is absent because the benzylic C–H bond and the carbonyl are anti-periplanar. Moreover, the short H⋯H distance of 1.95 Å highlighted in TS-1b implies the steric repulsion between benzylic and phenyl C–H bonds. Therefore, both TS-1b and 7b are significantly less stable than TS-1a and 7a, respectively. Furthermore, the comparison between 7a and 7b implies that the prominent hydrogen bonding interaction could promote the stabilization of 7a by 5.3 kcal mol−1. Because of the hydrogen bonding and electrostatic attraction (see Fig. S1† for the NCI plot of 7a), the dissociation of chiral ion-pair 7a is endergonic by 9.1 kcal mol−1 (see Fig. S3† for details), which is consistent with the deuterium labelling experiment (Fig. 2, eqn (3)). From 7a, racemization via C–N bond rotation (TS-3) requires a very high barrier. Instead, protonation of the terminal allylic carbon takes place viaTS-2a with an energy barrier of 8.3 kcal mol−1, generating the (P)-enantiomer of the enamide product 3a. This preferred stereochemistry is consistent with the experimental mechanistic studies (Fig. 2). Once (P)-3a is formed, the racemization to form (M)-3a requires a high barrier of 27.4 kcal mol−1 (viaTS-4), which is consistent with the configurational stability of the product under room temperature.
 | ||
| Fig. 3 Free energy profiles of central-to-axial chirality transfer through DBU-promoted [1,]-H transfer. All energies are calculated at M06-2X/6-311++G(d,p)/SMD(THF)//M06-2X/6-31G(d)/SMD(THF) level of theory. | ||
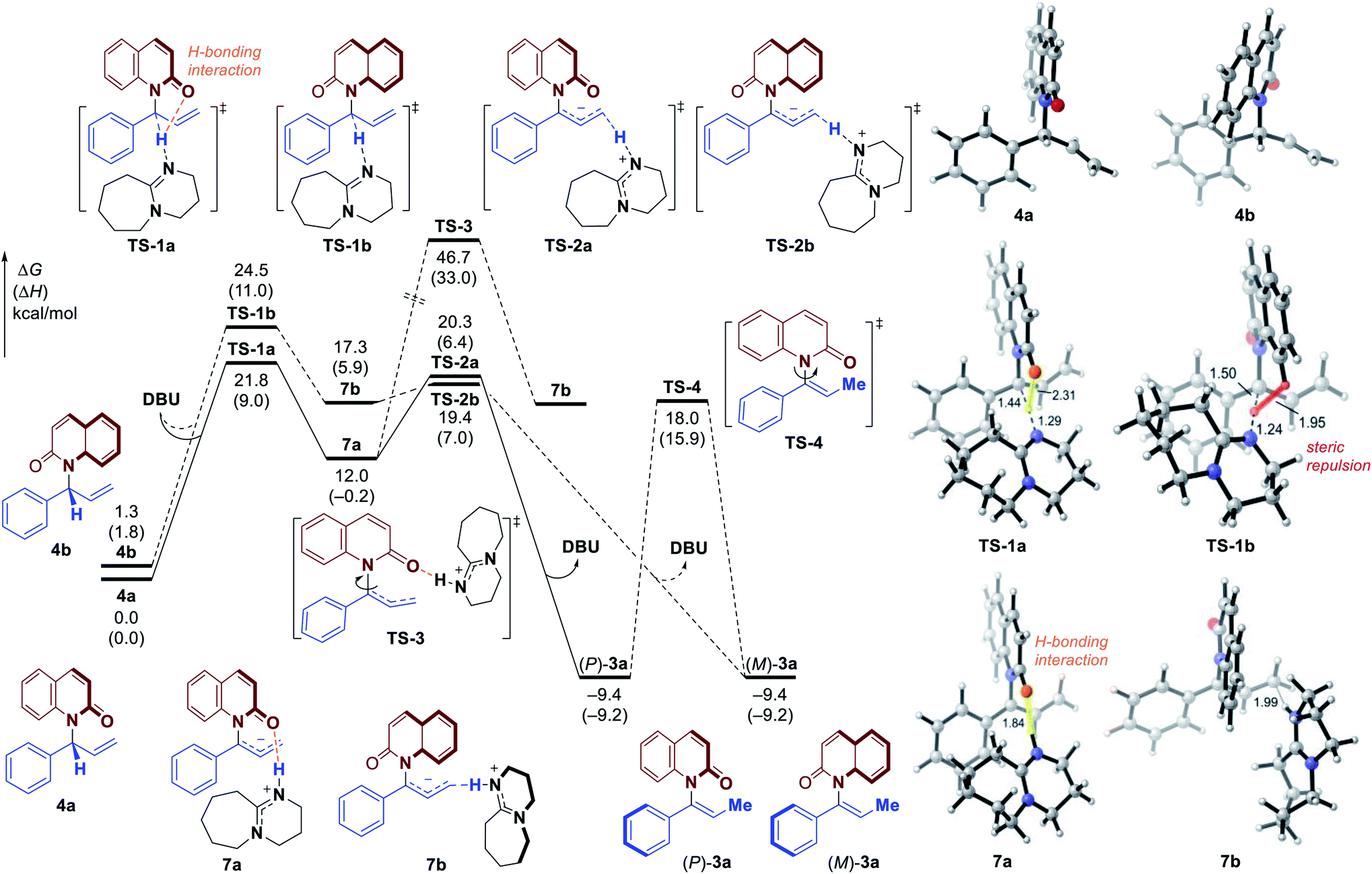
 | ||
| Fig. 4 Free energy profile for the formation of E-alkene (E)-3a through 1,3-H transfer. All energies are calculated at M06-2X/6–311++G(d,p)–SMD(THF)//M06-2X/6-31G(d)–SMD(THF) level of theory. | ||
It should be noted that the high levels of stereospecificity is kinetically controlled, because the less stable conformer 4b, which would lead to the unobserved (M)-enantiomer of 3a, is less stable than 4a by only 1.3 kcal mol−1 with an isomerization barrier of 12.0 kcal mol−1 (see Fig. S4† for details), indicating a noticeable population of 4b in the ground state (∼10%). Because deprotonation from 4b is kinetically disfavored, this pathway to form (M)-3a is completely suppressed.
Based on the experimental results, the atroposelective vinylation products were obtained in high Z/E ratio (usually >20/1). Our computational studies indicate that the deprotonation of conformer 4c to form E-alkene (E)-3a has an activation free energy of 24.7 kcal mol−1 (TS-1c, Fig. 4), which is 2.9 kcal mol−1 higher than that of TS-1a. The isomerization of 7a to 7c (viaTS-5) is found to be endergonic and requires a high barrier. Therefore, the two possible pathways to form 7c and eventually E-alkene (E)-3a are both less favorable. Optimized structures show that the TS-1c and 7c are both destabilized by 1,3-allylic strain with the phenyl group – the highlighted H⋯H distances in TS-1c and 7c (Fig. 4) are only 2.07 and 1.94 Å, respectively.
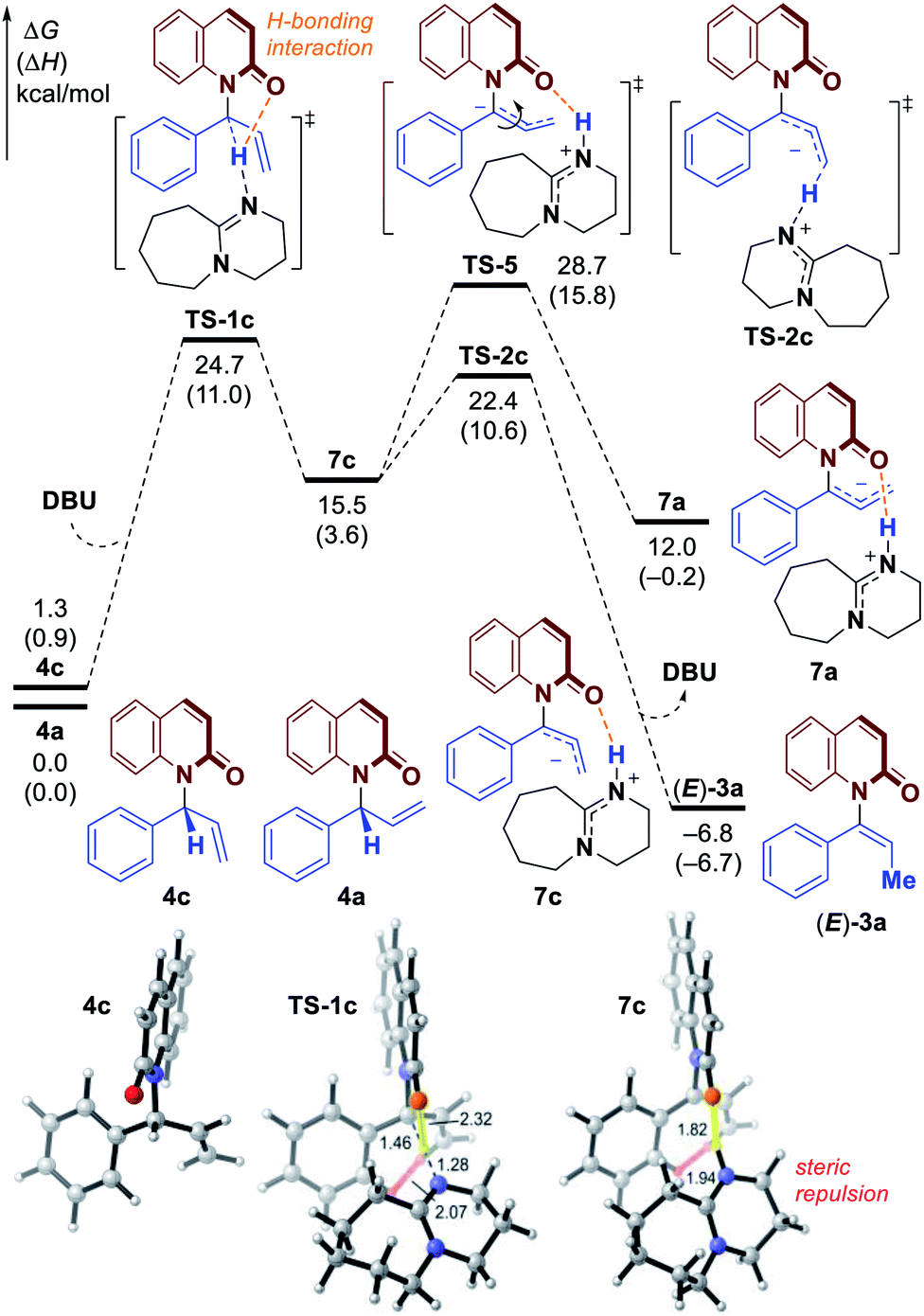
Based on these studies above, the mechanism and origin of stereoselectivity of [1,3]-H transfer are summarized in Fig. 5. After the enantioenriched allylation product I is formed under the classic iridium-catalyzed asymmetric allylic substitution (Fig. 2), the DBU promoted deprotonation of I at the benzylic position would occur to generate a chiral ion-pair intermediate III through transition state II-TS. Both II-TS and III are stabilized by the H-bonding interaction between the iminium N–H and the amide carbonyl group. As deprotonation is the rate-determining step, subsequent re-protonation of the terminal allylic carbon from III could accomplish the [1,3]-H transfer and generate the axially chiral product (P)-IV. In contrast, the disfavored (M)-enantiomer (M)-IV is generated through the same mechanism from I-b, which is a conformer of I. The corresponding deprotonation transition state IIb-TS is less stable due to the steric repulsion and the absence of H-bonding interaction. Thus, the deprotonation of I-b is much slower. Moreover, the racemization of chiral ion-pair III and III-b is difficult to occur compared with the re-protonation step, which suggests that the deprotonation is also the stereoselectivity-determining step. Therefore, we could draw the conclusion that the stabilizing H-bonding interaction not only facilitates the [1,3]-H transfer but also ensure the high levels of stereospecificity during the central-to-axial chirality transfer.
 | ||
| Fig. 5 Proposed mechanism of H-bonding assisted stereospecific [1,]-H transfer process. | ||
Axial to central chirality transfer
Catalytic asymmetric epoxidation is a powerful transformation in organic synthesis because the generated optically active epoxides are common motifs in biologically active compounds and biosynthetic intermediates.50–54 Finally, to demonstrate the utility of the strategy, epoxidations of products 3 were investigated for their stereospecificity. To our delight, treatment of 3 with m-CPBA afforded the epoxide scaffolds in good yield, while maintaining the enantiomeric excess (Table 3). The protocol established here represents a novel strategy of asymmetric epoxidation via axial-to-central chirality transfer.| a Reaction conditions: all reactions were run on 0.1 mmol scale with respect to 3. ee determined by chiral HPLC. Isolated yield. |
|---|
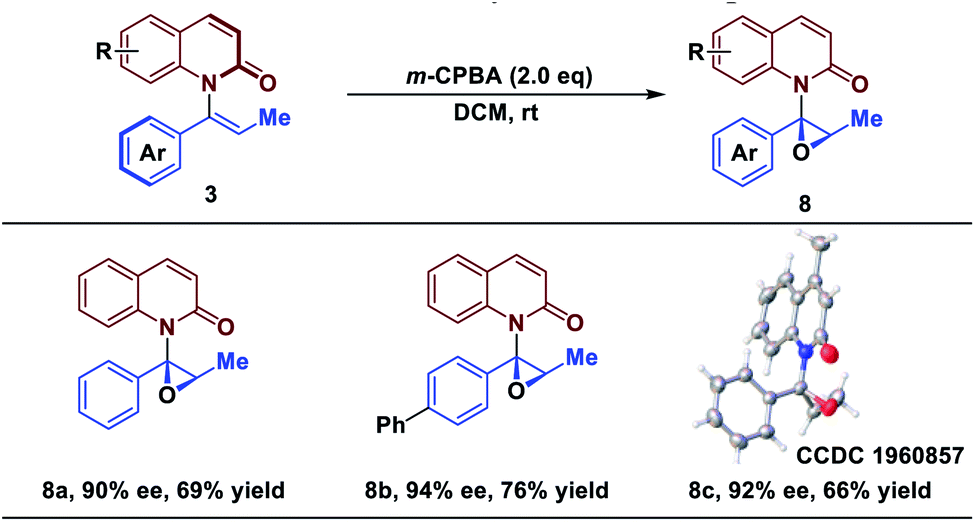
|
Conclusion
In conclusion, we have demonstrated the first example of an atroposelective synthesis of axially chiral enamides bearing the N–C axis. The reaction provides entry to a wide range of quinolone- and pyridone-derived axially chiral enamides in excellent yields with high enantioselectivities. Preliminary mechanistic studies revealed that the reaction proceeds via a pathway in which isomerization is the terminal reaction, occurring after the asymmetric allylic amination. Theoretical calculation combined with experimental studies revealed that the isomerization occurs through a hydrogen bonding interaction assisted [1,3]-H transfer, which enantioselectively generates the enamide products. Moreover, the assistance of hydrogen bonding interaction is crucial for the reactivity and stereospecificity during central-to-axial chirality transfer. On one hand, it plays a vital role in stabilizing the deprotonation transition state and the chiral ion-pair intermediate; on the other hand, the lack of hydrogen bonding interaction plus the steric repulsion greatly suppressed the formation of the undesired enantiomer. Furthermore, the epoxidation of axially chiral enamides promises to serve as a useful approach for the preparation of other chiral derivatives by central-to-axial-to-central chirality transfer sequence. Extension of this methodology to other axially chiral styrenes as well as detailed mechanistic studies is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Natural Science Foundation of Jiangsu Province (BK20180447), the Fundamental Research Funds for the Central Universities (30918011313, 30919011404), the NSF (CHE1707490). P. L and X. Q. thank the University of Pittsburgh and the National Science Foundation (CHE-1654122) for financial support of the computational work. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh, the Texas Advanced Computing Center (TACC) at The University of Texas at Austin, and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation grant number ACI-1548562. We especially Prof. Yi-Ming Wang (University of Pittsburgh) for helpful suggestions in the preparation of the manuscript. We also thank Prof. F. Dean Toste (UC Berkeley) and Prof. Hiroshi Naka (Nagoya University) for helpful discussions.References
- D. Cahard, S. Gaillard and J.-L. Renaud, Tetrahedron Lett., 2015, 56, 6159 CrossRef CAS.
- A. H. Hoveyda, S. J. Malcolmson, S. J. Meek and A. R. Zhugralin, Angew. Chem., Int. Ed., 2010, 49, 34 CrossRef CAS.
- E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816 RSC.
- Y. Saga, R. Motoki, S. Makino, Y. Shimizu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 7905 CrossRef CAS.
- Y. Wu, R. P. Singh and L. Deng, J. Am. Chem. Soc., 2011, 133, 12458 CrossRef CAS.
- Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334 CrossRef CAS.
- X. Zhou, Y. Wu and L. Deng, J. Am. Chem. Soc., 2016, 138, 12297 CrossRef CAS.
- S. Martinez-Erro, A. Sanz-Marco, A. B. Gómez, A. Vázquez-Romero, M. S. G. Ahlquist and B. Martín-Matute, J. Am. Chem. Soc., 2016, 138, 13408 CrossRef CAS.
- S. Martinez-Erro, V. García-Vázquez, A. Sanz-Marco and B. Martín-Matute, Org. Lett., 2020, 22, 4123 CrossRef CAS.
- N. Molleti, S. Martinez-Erro, A. C. Cerdán, A. Sanz-Marco, E. Gomez-Bengoa and B. Martín-Matute, ACS Catal., 2019, 9, 9134 CrossRef CAS.
- J. A. Dabrowski, F. Haeffner and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 7694 CrossRef CAS.
- V. Bizet, X. Pannecoucke, J.-L. Renaud and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 6467 CrossRef CAS.
- J. C. Golec, E. M. Carter, J. W. Ward, W. G. Whittingham, L. Simón, R. S. Paton and D. J. Dixon, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006202.
- D. M. H. Ascough, F. Duarte and R. S. Paton, J. Am. Chem. Soc., 2018, 140, 16740 CrossRef CAS.
- X.-S. Xue, X. Li, A. Yu, C. Yang, C. Song and J.-P. Cheng, J. Am. Chem. Soc., 2013, 135, 7462 CrossRef CAS.
- T. Kawabata, K. Yahiro and K. Fuji, J. Am. Chem. Soc., 1991, 113, 9694 CrossRef CAS.
- S.-C. Zheng, S. Wu, Q. Zhou, L. W. Chung, L. Ye and B. Tan, Nat. Commun., 2017, 8, 15238 CrossRef.
- J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558 CrossRef CAS.
- Y. Tan, S. Jia, F. Hu, Y. Liu, L. Peng, D. Li and H. Yan, J. Am. Chem. Soc., 2018, 140, 16893 CrossRef CAS.
- S. Jia, Z. Chen, N. Zhang, Y. Tan, Y. Liu, J. Deng and H. Yan, J. Am. Chem. Soc., 2018, 140, 7056 CrossRef CAS.
- J. Feng, B. Li, Y. He and Z. Gu, Angew. Chem., Int. Ed., 2016, 55, 2186 CrossRef CAS.
- Y. Liang, J. Ji, X. Zhang, Q. Jiang, J. Luo and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 4959 CrossRef CAS.
- L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497 CAS.
- S.-L. Li, C. Yang, Q. Wu, H.-L. Zheng, X. Li and J.-P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836 CrossRef CAS.
- J.-W. Zhang, J.-H. Xu, D.-J. Cheng, C. Shi, X.-Y. Liu and B. Tan, Nature Commun, 2016, 7, 10677 CrossRef.
- S. Shirakawa, K. Liu and K. Maruoka, J. Am. Chem. Soc., 2012, 134, 916 CrossRef CAS.
- Y. Kikuchi, C. Nakamura, M. Matsuoka, R. Asami and O. Kitagawa, J. Org. Chem., 2019, 84, 8112 CrossRef CAS.
- S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147 CrossRef CAS.
- F. Eudier, P. Righi, A. Mazzanti, A. Ciogli and G. Bencivenni, Org. Lett., 2015, 17, 1728 CrossRef CAS.
- J. Zhang, Y. Zhang, L. Lin, Q. Yao, X. Liu and X. Feng, Chem. Commun., 2015, 51, 10554 RSC.
- N. Di lorio, P. Righi, A. Mazzanti, M. Mancinelli, A. Ciogli and G. Bencivenni, J. Am. Chem. Soc., 2014, 136, 10250 CrossRef.
- H.-Y. Bai, F.-X. Tan, T.-Q. Liu, G.-D. Zhu, J.-M. Tian, T.-M. Ding, Z.-M. Chen and S.-Y. Zhang, Nature Commun, 2019, 10, 3063 CrossRef.
- A. J. Clark, D. P. Curran, D. J. Fox, F. Ghelfi, C. S. Guy, B. Hay, N. James, J. M. Phillips, F. Roncaglia, P. B. Sellars, P. Wilson and H. Zhang, J. Org. Chem., 2016, 81, 5547 CrossRef CAS.
- R. Takeuchi and S. Kezuka, Synthesis, 2006, 3349 CrossRef CAS.
- J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS.
- P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147 RSC.
- X.-J. Liu, C. Zheng, Y.-H. Yang, S. Jin and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 10493 CrossRef CAS.
- J. C. Hethcox, S. E. Shockley and B. M. Stoltz, ACS Catal., 2016, 6, 6207 CrossRef CAS.
- J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539 CrossRef CAS.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855 CrossRef CAS.
- S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657 CrossRef.
- X. Zhang, Z.-P. Yang, L. Huang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 1873 CrossRef CAS.
- X.-d. Bai, J. Wang and Y. He, Adv. Synth. Catal., 2019, 361, 496 CrossRef CAS.
- For selected example on central-to-axial chirality transfer via in stiu isomerization, see: C. Xu, H. Zheng, B. Hu, X. Liu, L. Lin and X. Feng, Chem. Commun., 2017, 53, 9741 RSC.
- S. Heeb, M. P. Fletcher, S. R. Chhabra, S. P. Diggle, P. Williams and M. Cámara, FEMS Microbiol. Rev., 2011, 35, 247 CrossRef CAS.
- J. P. Michael, Nat. Prod. Rep., 2007, 24, 223 RSC.
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS.
- It was speculated that catalytic amount TBD play a role of base for construction of 4. See ESI for more substrate scope.†.
- Absolute configuration of 4a was assigned by analogy to that of 4f, which was determined by comparation of literature (ESI for details†). A. Rodrigues, E. E. Lee and R. A. Batey, Org. Lett., 2010, 12, 260 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS.
- Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS.
- R. L. Davis, J. Stiller, T. Naicker, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 7406 CrossRef CAS.
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603 CrossRef CAS.
- G. D. Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1941459 and 1960857. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02828b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |