
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rationally designed peptoid for the selective chelation of Zn2+ over Cu2+†
Pritam
Ghosh
 and
Galia
Maayan
*
and
Galia
Maayan
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 3200008, Israel. E-mail: gm92@technion.ac.il
First published on 28th August 2020
Abstract
The selective removal of Zn2+ from proteins by using a synthetic chelator is a promising therapeutic approach for the treatment of various diseases including cancer. Although the chelation of Zn2+ is well known, its removal from a protein in the presence of potential competing biologically relevant ions such as Cu2+ is hardly explored. Herein we present a peptoid – N-substituted glycine trimer – incorporating a picolyl group at the N-terminus, a non-coordinating but structurally directing benzyl group at the C-terminus and a 2,2':6′,2′′-terpyridine group in the second position, that selectively binds Zn2+ ions in the presence of excess Cu2+ ions in water. We further demonstrate that this chelator can selectively bind Zn2+ from a pool of excess biologically relevant and competitive ions (Cu2+, Fe3+, Ca2+, Mg2+, Na+, and K+) in a simulated body fluid (SBF), and also its ability to remove Zn2+ from a natural zinc protein domain (PYKCPECGKSFSQKSDLVKHQRTHTG) in a SBF.
Introduction
The metal ion Zn2+ plays an important role in several enzymes and transcription factors within the human body, where it acts as a structural co-factor by allowing the protein to fold into a stable conformation, thus enabling a specific function.1 Zinc finger proteins (ZFPs), 3% in abundance among the entire human genome, are a common DNA binding motif in transcription factors.2 Recent reports indicate extensive participation of ZFPs in several diseases, such as parasitic diseases,3 cancer,4 viral contamination5 or even neurological problems.6 Therefore ZFPs are increasingly recognized as therapeutic targets.2 The approach for treating diseases by targeting ZFPs is mainly associated with the replacement and/or removal of Zn2+ from the protein scaffold; proteins lacking Zn2+ would become apo-proteins (Scheme 1), will not be able to bind the nucleotide, and eventually would become inactive.7 One approach to obtain an apo-protein is by replacing Zn2+ with other metal ions.2 Although this strategy is well explored, it can lead to possible side effects from the substituting metal ions. Thus, removal of Zn2+ from ZFPs (unstructured protein, shown in blue in Scheme 1) by using an unbound chelator specific to zinc (shown in cyan in Scheme 1) to form an un-structured apo-protein (shown in grey, Scheme 1) would be advantageous. The major challenge related to Zn2+ extraction is the development of water-soluble small molecular probe-based chelators that could bind Zn2+ in competition with a strong opponent such as Cu2+; even molecules that are routinely used to chelate zinc, e.g. ethylenediaminetetraacetic acid (EDTA) and N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) are not selective to Zn, and this limits their utilization.8a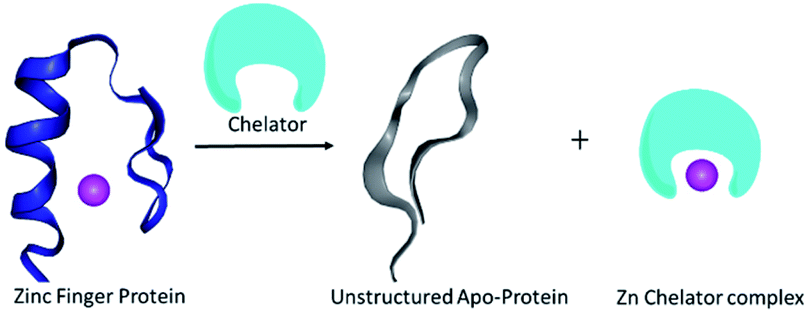 | ||
| Scheme 1 A schematic representation of Zn2+ chelation from a structured ZFP domain (shown in blue) by using a suitable chelator resulting in an unstructured apo-protein (shown in grey). | ||
Only a handful of examples of peptidomimetics are known for Zn2+ chelation.8b–d Most of these examples, however, do not consider the binding of Zn2+ in the presence of other biologically relevant ions including Ca2+, Mg2+, Na+, K+ and especially Cu2+, which is the binding competitor of Zn2+. It was previously reported that a cytochrome-based assembly can bind Zn2+ in the presence of Ni2+, Co2+ or Cu2+; however, this assembly was not selective to Zn2+, as it was also binding the other metal ions.8d Moreover, selective binding to Zn2+ was tested neither in a mixture of different metal ion competitors nor in an environment similar to body fluids such as blood plasma (i.e.; in the presence of other ions such as Cl−, HCO3−, HPO42−etc.).9 In such a fluid, the Cu2+/Zn2+ concentration ratio can range between 1.06 (ref. 9c) to 1.39.(ref. 9d) Therefore, only the exclusive selectivity to Zn2+ over Cu2+ chelation within such ratios would be adequate for therapeutic applications.
Peptoids, N-substituted glycine oligomers, are peptidomimetics akin to peptides that are composed of primary amines instead of amino acids. Due to their ease of synthesis on a solid support,10 sequence versatility and high stability, they exhibit great potential for application in catalysis,11 self-assembly,12 metal binding13 and recognition,14 and medicine.15 Compared to small molecule chelators, peptoid chelators are advantageous because various metal-binding ligands and structural elements of choice can be incorporated within their scaffold with high sequence specificity, leading to control over a desired coordination geometry and complex stability. They include different N-donor ligands, many of which are the basis for systems used for Zn2+ chelation and/or sensing.16 Indeed, peptoids bearing the N-donor ligands 8-hydroxyquinoline (HQ) and imidazole, can bind Zn2+ (among other metal ions).13d,8e Moreover, peptoids having imidazole groups were shown to bind Zn2+ more efficiently than peptoids having thiols as ligands.8e The N-donor ligand 2,2′:6′,2′′-terpyridine (terpy) was incorporated together with HQ within a helical peptoid hexamer in a sequence-specific manner, resulting in selective intermolecular binding of Zn2+ by two terpy ligands.14b Other N-donor ligands that were successfully incorporated within peptoids are 2,2′-bipyridine,13a and 1,10-phenanthroline,13c but they were not used as Zn2+ chelators. Based on our achievements in the area of metallopeptoids, we prepared a series of water-soluble peptoid trimers, and found that one of these trimers can bind Zn2+ selectively from a mixture containing metal ion competitors including Cu2+ in a Cu2+/Zn2+ concentration ratio that exists in human body fluids. Significantly, we demonstrated successful chelation and extraction from a zinc finger protein.
Results and discussion
Design and synthesis of a Zn2+-peptoid chelator
In general, quinolone-based Zn2+ probes suffer from poor solubility and/or low cell permeability.17 Thus, although HQ-based peptoids were shown to bind Zn2+, HQ might not be a good choice for our current peptoid design. On the other hand, fluorescence studies with terpy-based ligands, which were carried out in a solution containing either Zn2+ or Cu2+, demonstrated the ability of terpy to bind Zn2+ but not Cu2+. Significantly, it was shown that in an aqueous solution, the association constant of terpy for Zn2+ is much higher than that for Cu2+, suggesting that terpy has a higher affinity for Zn2+ in comparison with Cu2+.18 Additionally, the role that terpy plays in various metallo-intercalating drugs (where terpy helps to increase the DNA-binding ability),19 stands for its utility as a bio-compatible compound, making it the preferred choice as a chelating unit in the design of biomimetic chelators. As terpy is a tridentate ligand, and Zn can be stabilized via a tetra, penta or hexa-coordination, it is indeed required to incorporate a co-ligand that will stabilize an intramolecular 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Zn:peptoid complex. This is because intramolecular metallopeptoid complexes were shown to have greater stability than their equivalent intermolecular metallopeptoid complexes, as evident from their higher association constants.20 As the most common coordination number of Zn2+ complexes is four,21 we chose to incorporate as the co-ligand a picolyl group (Npam), another N-donor metal-binding moiety22 preferable for Zn2+ chelation.8e We previously showed that a non-coordinating bulky side chain incorporated next to an active site within the peptoid scaffold has an important role as a structure-directing group, and facilitates the peptoid's activity.11 As the terpy and picolyl groups are anticipated to form a distinct coordination site, we sought to incorporate a non-coordinating group next to it and explore its effect on Zn2+ binding. To this aim, we incorporated one additional aromatic23 or aliphatic24 non-coordinating side chain at the C-terminus of the peptoid. Thus, peptoids PT-1 to PT-7 bearing a benzyl, naphthyl, cyclohexyl, (R)-(−)-3,3-dimethyl-2-butyl, pentyl, allyl and 2-methoxyethyl group, respectively (Fig. 1 and S2†), were synthesized and characterized. In addition, the peptoid dimer PD-1, which lacks the non-coordinating group, was synthesized as a control (Fig. 1). All the peptoids were synthesized by the sub-monomer approach on resin,10 cleaved, purified by HPLC and lyophilized to obtain the pure peptoid products (>95% purity). The mass of the peptoids as determined by ESI-MS was consistent with their sequences (Fig. S3–S12† for HPLC and Fig. S13–S22 for ESI-MS, ESI†).
:1 Zn:peptoid complex. This is because intramolecular metallopeptoid complexes were shown to have greater stability than their equivalent intermolecular metallopeptoid complexes, as evident from their higher association constants.20 As the most common coordination number of Zn2+ complexes is four,21 we chose to incorporate as the co-ligand a picolyl group (Npam), another N-donor metal-binding moiety22 preferable for Zn2+ chelation.8e We previously showed that a non-coordinating bulky side chain incorporated next to an active site within the peptoid scaffold has an important role as a structure-directing group, and facilitates the peptoid's activity.11 As the terpy and picolyl groups are anticipated to form a distinct coordination site, we sought to incorporate a non-coordinating group next to it and explore its effect on Zn2+ binding. To this aim, we incorporated one additional aromatic23 or aliphatic24 non-coordinating side chain at the C-terminus of the peptoid. Thus, peptoids PT-1 to PT-7 bearing a benzyl, naphthyl, cyclohexyl, (R)-(−)-3,3-dimethyl-2-butyl, pentyl, allyl and 2-methoxyethyl group, respectively (Fig. 1 and S2†), were synthesized and characterized. In addition, the peptoid dimer PD-1, which lacks the non-coordinating group, was synthesized as a control (Fig. 1). All the peptoids were synthesized by the sub-monomer approach on resin,10 cleaved, purified by HPLC and lyophilized to obtain the pure peptoid products (>95% purity). The mass of the peptoids as determined by ESI-MS was consistent with their sequences (Fig. S3–S12† for HPLC and Fig. S13–S22 for ESI-MS, ESI†).
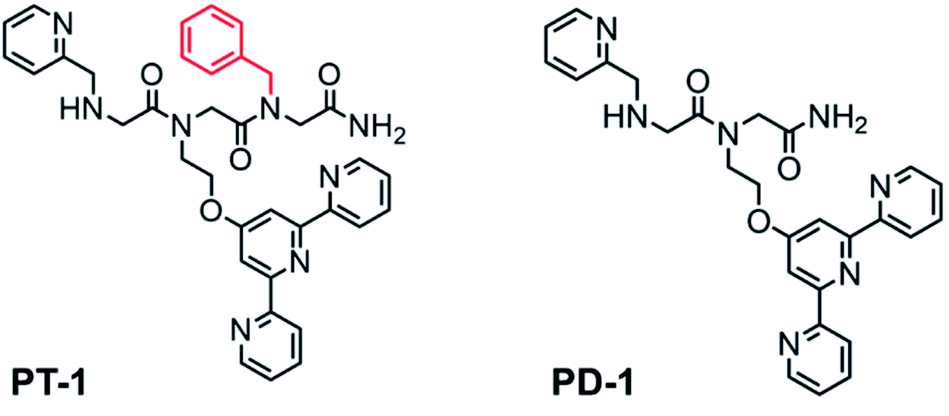 | ||
| Fig. 1 Chemical structures of peptoids PT-1 and PD-1. | ||
Binding of Zn2+ and Cu2+ to the peptoids PT1–PT7 and PD-1 in water
The peptoids PT1–PT7 and PD-1 are soluble in water and thus UV-Vis characterization has been carried out in a non-buffered water solution. Metal free PT-1 exhibits absorbance maxima at 235 and 275 (with a small hump at 262) nm in a water medium arising from the ligands terpy and picolyl, respectively. Upon addition of zinc acetate, the bands near 235 and 275 nm shifted to 244 and 274 nm respectively, and new absorption bands near λ = 310 and 322 nm were produced (Fig. 2a). A metal-to-peptoid ratio plot constructed from these UV-Vis titrations was consistent with the 1:1 peptoid:Zn2+ ratio, demonstrating the formation of the intramolecular complex ZnPT-1 (Fig. 2a, inset). Addition of 1 equiv. of copper(II) acetate to PT-1 produced new bands near λ = 315 and 328 nm (Fig. 2b and S23b†). The difference between the UV spectra of the two complexes ZnPT-1 and CuPT-1 enables the detection of the coordination of one of these ions over the other in competition experiments followed by UV-Vis analysis. Indeed, the UV-Vis spectrum of PT-1 with a mixture of Zn2+ and Cu2+ in a 1:4 ratio was obtained, revealing an expected selective chelation of Cu2+ over Zn2+ (see Fig. S24a† for UV-Vis and Fig. S26b† for ESI-MS).
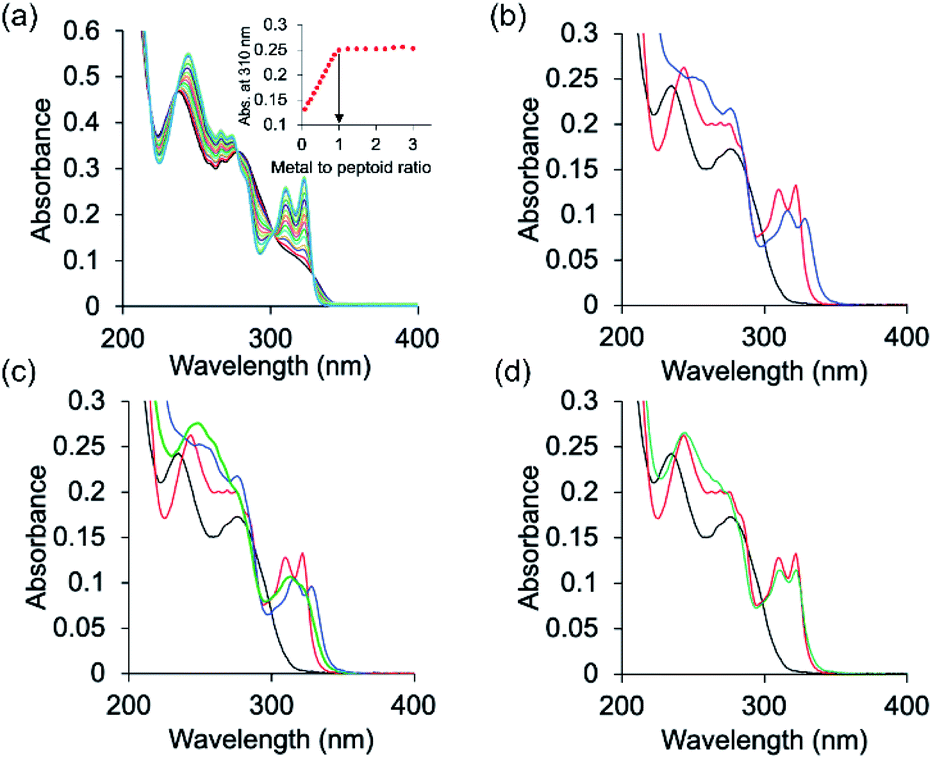 | ||
| Fig. 2 UV-Vis spectra of (a) PT-1 titration with Zn2+. PT-1 (17 μM) in water was titrated with 2 μL aliquots of metal ions (2 mM in H2O) in multiple steps (black = free peptoid PT-1 and blue = metal complex). Inset: metal-to-peptoid ratio plot for Zn2+ binding to PT-1. (b) Zn2+ added to PT-1 (red line) and Cu2+ added to PT-1 (blue line), (c) competition experiment between Zn2+ and Cu2+ with PT-1 in a 1:3 Zn2+:Cu2+ ratio (black: PT-1; red: PT-1 with Zn2+; blue: PT-1 with Cu2+; green: PT-1 with 1:3 Zn2+:Cu2+) and (d) competition experiment between Zn2+ and Cu2+ with PT-1 in a 1:2 Zn2+:Cu2+ ratio (black: PT-1; red: PT-1 with Zn2+; green: PT-1 with 1:2 Zn2+:Cu2+), (8 μM in b–d, solvent: water). | ||
This selectivity decreases in a 1:3 Zn2+:Cu2+ ratio (Fig. 2d and S26a† for ESI-MS) and is, remarkably, completely lost and actually turns to selectivity towards Zn2+ in a 1:2 Zn2+:Cu2+ ratio (Fig. 2c and S25a† for ESI-MS). A sample taken from this UV-Vis solution was subjected to ESI-MS analysis and the results supported chelation of zinc only, as evident from the well-matched ESI-MS pattern of simulated and experimental data (Fig. S25†). In Fig. 2a, which shows the titration of PT-1 with Zn2+, we could identify five isosbestic points, i.e.; 218, 236, 278, 300 and 329 nm. The points near 218 and 329 nm do not show much change in the absorbance value before and after metal binding, and the isosbestic point near 278 nm is common for both metal ions. Therefore, we considered the points near 236 and 300 nm for explaining the metal binding. In a 1:4 (Zn2+:Cu2+) ratio, the UV signal is similar to that of Cu2+ and there is no similarity in isosbestic points to those of the Zn2+ signal. In a 1:3 (Zn2+:Cu2+) ratio, the UV signal passes through the 300 nm isosbestic point only and other points are not similar to those of both Zn2+ and Cu2+, which explains the binding of both Zn2+ and Cu2+ to PT-1. However in a 1:2 (Zn2+:Cu2+) ratio, the UV signal passes through 235 and 300 nm isosbestic points which explains the binding of Zn2+ only with PT-1 (supported by mass spectrometric analysis).
The role of the non-coordinating group that was incorporated at the C-terminus was examined by conducting competition experiments similar to the experiment that was carried out with PT-1. Thus, each of the peptoids PT-2 to PT-7 and PD-1 was mixed with a 1:2 Zn2+:Cu2+ solution in water, and the solutions were analyzed by UV-Vis spectroscopy. Interestingly, the spectra of these solutions indicated that all these peptoids do not bind Zn2+ but rather bind Cu2+ (Fig. S27 and S33, ESI†). To understand this result, the dissociation constants of peptoids PT-1 to PT-3 with Zn2+ were measured by a competition experiment with EDTA at pH 7. The dissociation constant of PT-1 was found to be 1.85 × 10−13 M (Fig. S31†), 1.4–2.4 times lower than that of PT-2 (2.65 × 10−13 M, Fig. S31†) and PT-3 (4.5 × 10−13 M, Fig. S31†), and about 10 times lower than its dissociation constant with Cu2+ (2.2 × 10−12 M, Fig. S31†). The results from all the above experiments suggest that a non-coordinating structure-directing group is required to achieve selective binding of Zn2+ to the terpy/picolyl coordination site, but that such a group should not be too bulky to hinder the binding. Among the seven non-coordinating groups studied here, the benzyl side chain is the best one to function as a non-hindering structure-directing group and assists the coordinating ligands to selectively bind Zn2+ over Cu2+. Hence, this non-coordinating group provides the advantages of a second coordination sphere around the binding site, similar to the one afforded by the folds within the active site of functional proteins.
We have previously showed that the N terminal –NH group of metal-binding peptoids can coordinate Cu2+.12,13a We therefore anticipated that acetylating the N-terminal of PT-1 will enable the minimization of the possibility of Cu2+ binding, and increase the selectivity towards Zn2+ even with Zn2+:Cu2+ ratios higher than 1:3. Thus, PT-1 was re-synthesized and its N-terminus has been acetylated to obtain PT-1Ac (Fig. 3). This peptoid was mixed with a 1:2 Zn2+:Cu2+ solution in water, followed by UV-Vis spectroscopy. Despite our expectations, the resulting spectrum indicated that acetylation at the N-terminal cannot improve the selectivity, as PT-1Ac failed to bind Zn2+ in the presence of two equivalents of Cu2+. These results also suggest that the N-terminal secondary amine has a role in the selectivity of PT-1 for Zn2+.
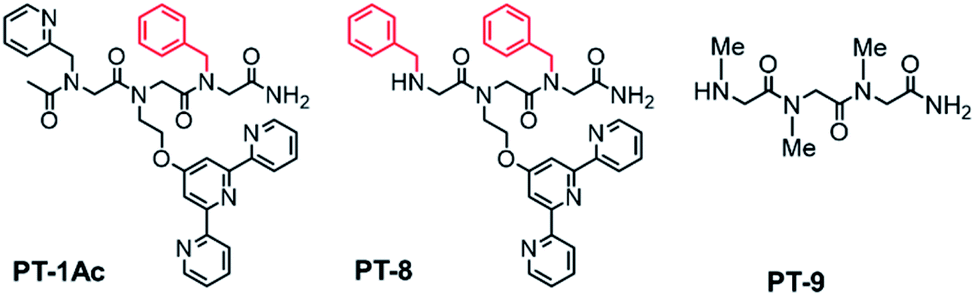 | ||
| Fig. 3 Additional peptoid oligomers designed and synthesized for this study. | ||
To establish the role of the picolyl group in the selective binding to Zn2+, we have also synthesized PT-8, which incorporates a benzyl group instead of the picolyl side chain (Fig. 3). A similar competition experiment performed with a 1:2 Zn2+:Cu2+ solution followed by UV-Vis spectroscopy revealed that PT-8 binds Cu2+ rather than Zn2+, indicating that the picolyl side chain is also required for selectivity to Zn2+ in the presence of excess Cu2+ in solution (Fig. 4). A similar competition experiment performed with a 1:1 mixture of terpy and picolyl amine in acetonitrile solution also demonstrated selectivity to Cu2+ only. Overall, the competition experiments with peptoids PT-2 to PT-8, PD-1 and PT-1Ac, as well as with a mixture of the two ligands, with 1:2 Zn2+:Cu2+ (Fig. S38, ESI†), collectively established PT-1 as a unique chelator for Zn2+, which can bind it selectively from a solution containing an excess of up to 3 equiv. of Cu2+.
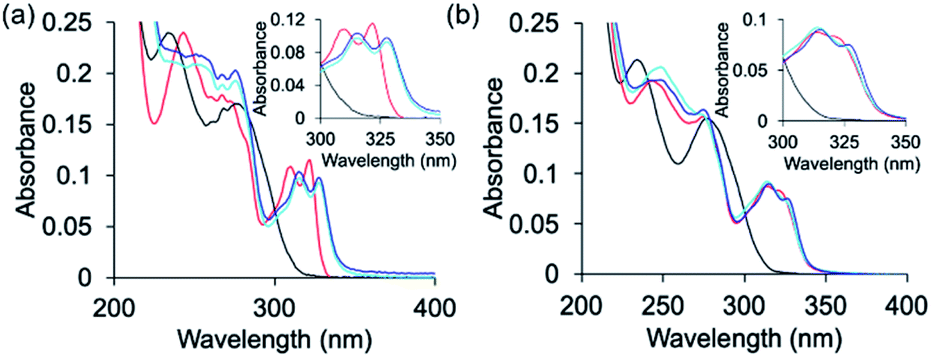 | ||
| Fig. 4 UV-Vis spectra describing the competition between Zn2+ and Cu2+ in peptoid binding. The spectra were recorded upon addition of a 1:2 Zn2+:Cu2+ mixture (8 μM in water) to (a) PT-1Ac and (b) PT-8 (black: Peptoid, red: Zn2+ complex, blue: Cu2+ complex and cyan: mixture). | ||
Exploring the selectivity of PT-1 towards Zn2+ in solutions containing various other metal ions
To explore the ability of PT-1 to bind Zn2+ selectively from a pool of other biologically relevant metal ions, PT-1 was titrated with 1 equiv. of Ca2+, K+, Mg2+, Na+ or Fe3+ in a non-buffered water solution followed by UV-Vis measurements. The results suggested no interaction between the examined metal ions and PT-1, as no shift in the absorbance bands was recorded and the UV-Vis spectra were mostly identical before and after the titration (Fig. S57, ESI†). The chelation of biologically relevant metal ions, however, does not take place in water or buffered solutions but rather in a human body fluid. Thus, we have prepared a simulated body fluid (SBF) according to a previously reported method25 and tested the chelation of Zn2+ and Cu2+ by PT-1 in this SBF solution. The complexation of both Zn2+ and Cu2+ was successful in the SBF (Fig. S58†), indicating that PT-1 is a good chelator of these ions in the SBF also. The stability of ZnPT-1 was monitored by a 24 hour UV-Vis measurement in the SBF at 37 °C and the obtained spectrum matched the one measured in water, suggesting that ZnPT-1 maintains its stability in a SBF. To further explore the selectivity of PT-1 towards Zn2+ in the SBF, competition experiments between Zn2+, Cu2+, Fe3+, Ca2+, Mg2+, Na+, and K+ in varying ratios were performed. Initially, two mixture solutions were prepared, in which the ratio between Zn2+ and Cu2+ was varied between 1:2 and 1:3, while keeping the other metal ions in excess owing to their abundance in the human body with respect to Zn2+.26 Both PT-1 and the two mixtures (A with the Zn2+:Cu2+:Fe3+:Ca2+:Mg2+:Na+:K+ ratio 1:2:5:10:10:10:10 and B with the ratio 1:3:5:10:10:10:10) were prepared in the SBF. Each mixture was added to PT-1 in two different UV-Vis experiments and the spectra were recorded. Absolute selectivity towards Zn2+ was observed upon addition of mixture A (Fig. S59, ESI†), consistent with the results obtained in the competition experiment between Zn2+ and Cu2+ in binding to PT-1 (Fig. 2d).
Characterization of the ZnPT-1 and CuPT-1 complexes
To understand the ability of PT-1 to selectively bind Zn2+ over Cu2+ we have characterized both complexes ZnPT-1 and CuPT-1 and analyzed the structural differences between them, specifically the coordination geometries about their metal center. To this aim, ZnPT-1 and CuPT-1 complexes were prepared under different reaction conditions and characterized by UV-Vis spectroscopy, ESI-MS, IR spectroscopy, EPR (CuPT-1) and 1H NMR (ZnPT-1), and their possible coordination geometries were computed by density-functional theory (DFT) calculations. Initially, the two complexes were prepared by mixing PT-1 in water with the acetate salt of either Zn2+ or Cu2+ in an exact 1:1 peptoid:metal ratio, for 4 hours in a non-buffered water solution at room temperature. After 4 hours, the solutions were analyzed by UV-Vis spectroscopy and ESI-MS. The UV-Vis spectra were identical to those obtained from the titration experiments and the MS analysis confirmed the formation of the ZnPT-1 and CuPT-1 complexes, suggesting coordination to the acetate counter ion (Fig. S43 and 44,† ESI†). The solutions were than lyophilized to obtain the solid complexes ZnPT-1 and CuPT-1 (92% and 88% yield, respectively). To test the stability of these complexes with time, the same reactions were performed but the reaction mixtures were allowed to stir for 24 hours at room temperature. The samples from these solutions were analyzed by UV-Vis spectroscopy every several hours and the spectra in all the cases were almost identical to the ones recorded after 4 hours, indicating high complex stability over time. Additionally, the same reactions were also performed in water at 35 °C and 50 °C for 24 hours. The samples taken from these mixtures after 24 hours were analyzed by UV-Vis spectroscopy. These spectra also showed a similar absorbance signal with almost no change in the UV-Vis signal intensity compared to that of the spectra obtained from the solutions that were mixed at room temperature (Fig. S45–46, ESI†). These results imply that the formed complexes are both kinetic and thermodynamic products. Finally, the absorbance signal of the complexes was measured when the reactions were performed at room temperature for four hours in methanol or acetonitrile as the solvent, showing UV-Vis spectra nearly similar to those obtained when the reactions were performed in water. These findings suggest that Zn2+ and Cu2+ complex formation does not depend on one coordinating solvent or another.
For further insight, 1H-NMR of the PT-1 and its Zn2+ complex was carried out in solution. We note here, that assigning the 1H-NMR chemical shift of the peptoids is not straightforward because peptoids adopt multiple conformations in solution caused by cis–trans interconversions at the backbone amides, and because there are no intramolecular CO–HN hydrogen bonds, which collectively result in highly flexible scaffolds.27 Indeed, the NMR spectra of PT-1 and ZnPT-1 in water were too broad to analyze, and possible interaction of the N-terminal –NH group with the metal center could not be detected due to proton exchange with the solvent. Thus, in order to be able to assign the 1H-NMR chemical shift of PT-1 and its zinc complex, we synthesized the complex by mixing 1 equiv. of PT-1 with one equiv. of zinc acetate dihydrate in deuterated acetonitrile and performed both measurements of PT-1 and ZnPT-1 in this solvent. In addition, we have designed the peptoid trimer PT-9 (Fig. 3), having three methyl side chains, as a simple model that can represent the scaffold of PT-1 in 1H-NMR analysis. PT-9 was synthesized on a solid support via the submonomer method, cleaved and purified by HPLC (>95% purity), and its identity was confirmed by ESI-MS. The 1H-NMR spectrum of PT-9 in acetonitrile exhibits four groups of signals; among them a small broad signal at 9 ppm that can be assigned to the proton of the N-terminal –NH group, a multiplet in the 5.8–7 ppm region assigned to the two protons of the –NH2 group at the C-terminal, and a multiplet in the region between 3.7 and 4.4 ppm corresponding to the prochiral geminal protons in the glycine-like backbone are observed (Fig. S49†). In addition, the 1H-NMR spectra of benzylamine, 2,2′:6′,2′′-terpyridine and 2-picolyl amine were measured in acetonitrile and the signals were fully assigned (Fig. S50–52, ESI†).
With these spectra in hand, we performed 1H-NMR measurements on PT-1 and its corresponding 1:1 Zn2+ complex in acetonitrile and analyzed the obtained spectra based on the 1H-NMR data of PT-9 and the three side chains (Fig. 5). In general, the NMR signals of both the peptoid and its zinc complex appear as groups of multiplets between 7.75 ppm and 9 ppm, between 7.2 and 7.6 ppm, in the range of 5.8–6.7 ppm, between 4.3 and 4.6 ppm and about 3.8–4.2 ppm (Fig. 5a and b). The NMR spectrum of PT-1 exhibits: (1) a broad signal at 9.2 ppm that we assigned to the N-terminal –NH proton, together with a multiplet near 9.18 ppm that is assigned to three α-protons adjacent to pyridine nitrogen, (2) multiplets about 7.75–9 ppm ascribed to the protons of terpy and picolyl groups, (3) signals in the 7.2–7.6 ppm range assigned to the five protons of the benzyl group together with other less acidic aromatic protons from the pyridine-based ligands, (4) a multiplet in the 5.8–6.7 ppm region assigned to the two protons of the C-terminal –NH2 group, and (5) signals in the 4.3–4.6 ppm and 3.8–4.2 ppm range that we ascribed to the protons of the skeletal methylene groups, and to the protons of the side-chain methylene linkers, respectively, a total of fourteen protons (Fig. S53, ESI† for a more detailed assignment). The NMR spectrum of ZnPT-1 exhibits a downfield shift of the aromatic protons from the pyridine-based ligands (e.g.; proton signal near 7.9 ppm shifted to 8 ppm, 8 to 8.1 ppm, 8.25 ppm to 8.3 ppm, 9.18 to 9.20 ppm etc., Fig. 5c), indicating the participation of terpy and picolyl in the zinc binding.28 The lack of the terminal –NH signal could be attributed to either its coordination to Zn2+ or to some interactions with either water molecules or acetate ions from the zinc acetate dihydrate salt used for the complex formation.
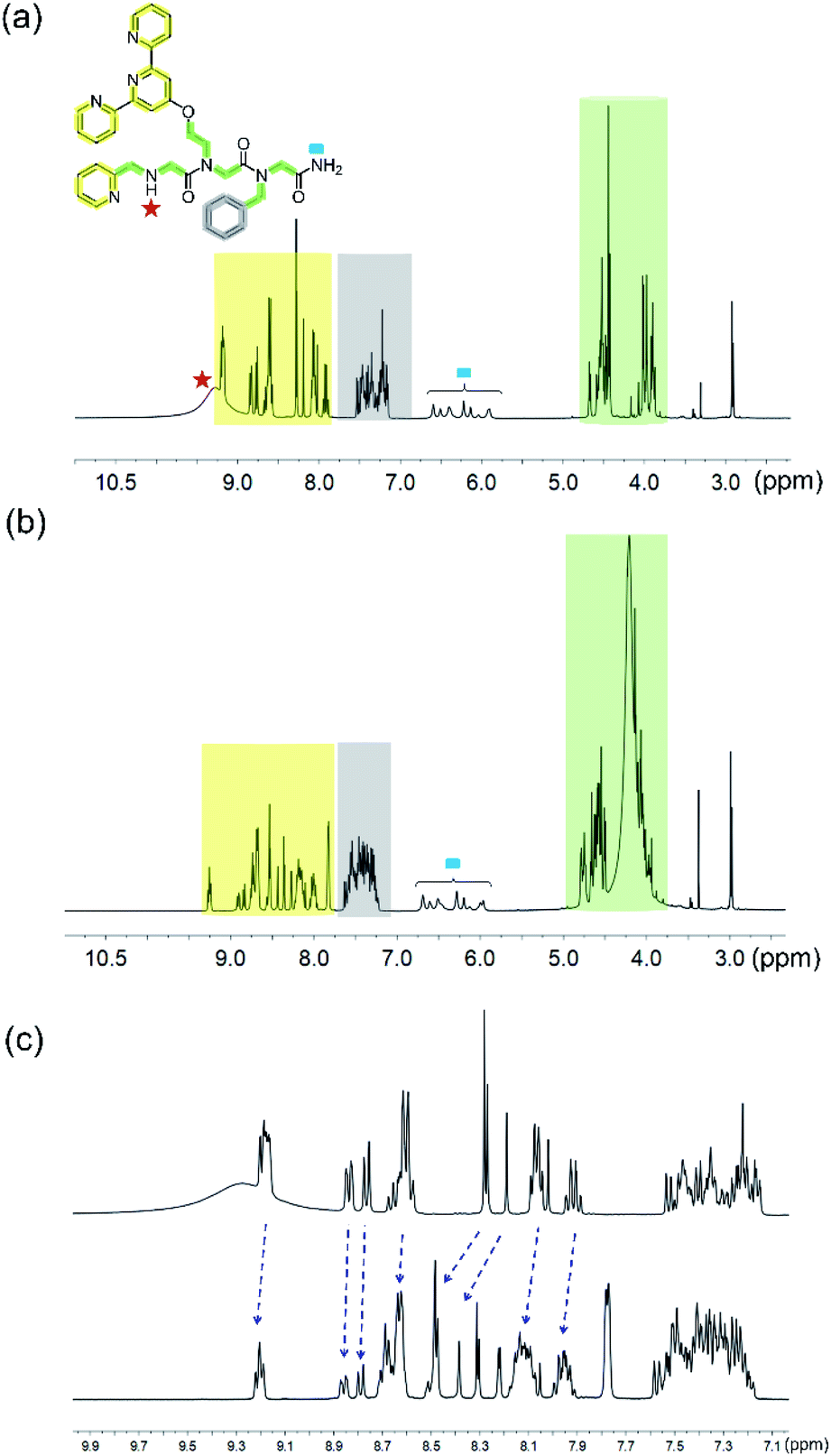 | ||
| Fig. 5 1H-NMR (in 400 MHz) spectra of (a) PT-1 and (b) ZnPT-1. (c) Expanded aromatic region of PT-1 (top) and ZnPT-1 (down) in CD3CN; the changes are marked with blue arrows. | ||
To prove this point we have performed an FT-IR measurement on the zinc complex, which we have isolated from the acetonitrile solution, as well as on PT-1, in the solid state. The FT-IR data revealed that the bands corresponding to the –NH stretching in the range between 2700 and 3400 cm−1 are present in both spectra, indicating that the terminal –NH group is not coordinated to zinc (Fig. S54†).13a,29a Interestingly, the FT-IR spectrum of ZnPT-1 exhibits bands near 1535 cm−1 and 1440 cm−1, both assigned to COO− stretching, with a Δυ(COO−) = 95 cm−1, suggesting a bidentate coordination of the acetate ligand to Zn2+ (Fig. S54†).29b This FT-IR spectrum was almost identical to the FT-IR spectrum of ZnPT-1 formed in water.
Based on our structural experimental data, we could suggest that within ZnPT-1, Zn2+ binds to the three nitrogen atoms of terpy, the nitrogen atom of picolyl, and the two oxygen atoms from one acetate molecule in a bidentate mode, forming a hexa-coordination geometry. To support this assumption, we computed this structure using DFT-D4.30 Unrestricted DFT calculations resulted in a geometry-optimized conformation with a hexa-coordinated zinc center, including a bidentate coordination to the acetate ion in a distorted octahedral geometry (Fig. 6a and the ESI† for the coordinates).
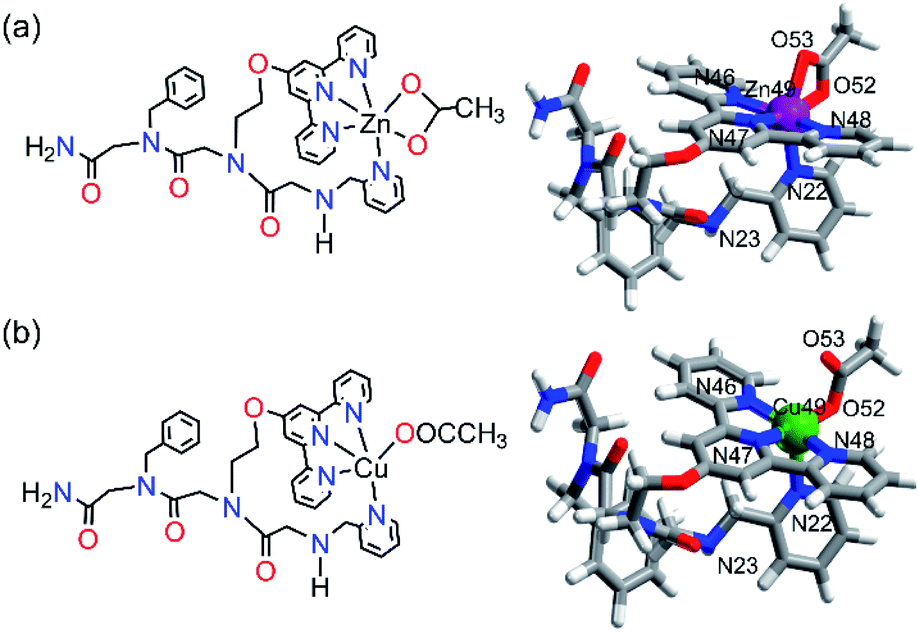 | ||
| Fig. 6 Chemical structure representation and DFT-D4 energy optimized configurations of (a) ZnPT-1 and (b) CuPT-1 (color code: grey: carbon, white: hydrogen, red: oxygen, blue: nitrogen, purple: zinc, and green: copper). | ||
To understand the selectivity of PT-1 to Zn2+ in the presence of Cu2+, we first characterized CuPT-1via FT-IR and EPR spectroscopies. The FT-IR spectrum obtained from measuring a solid sample of CuPT-1 suggested that similar to ZnPT-1, there is no coordination of the terminal –NH group to copper. On the other hand, the FT-IR spectrum of CuPT-1 showed bands corresponding to υasym(COO−) and υsym(COO−) at 1593 cm−1 and 1420 cm−1, respectively, leading to a Δυ(COO−) of 173 cm−1, which stands for a pseudo bridged uni-dentate arrangement of Cu2+ (Fig. S54b†).31 The X-band EPR measurements were performed in a frozen solution, and the resulting experimental spectrum was simulated to obtain the Hamiltonian parameter as g_II = 2.22; g┴ = 2.06 and A_II = 165 G. These parameters suggest that CuPT-1 adopts a square pyramidal geometry32 about the copper center (Fig. S55†). Overall, these data suggest that within CuPT-1, Cu2+ binds to the three nitrogen atoms of terpy, the nitrogen atom of picolyl, and one oxygen atom from one acetate molecule, forming a penta-coordination geometry. DFT-D4 calculations resulted in a geometry-optimized conformation with a square pyramidal copper center, including a mono-dentate linkage from the acetate ion (Fig. 6b and the ESI† for the coordinates). The calculated structures of ZnPT-1 and CuPT-1 were utilized to investigate their relative stability based on the HOMO–LUMO gap, as the larger the energy difference between the HOMO and LUMO, the more stable is the complex.33 Indeed, the HOMO–LUMO gap in ZnPT-1 (4.313 eV) is higher than that in CuPT-1 (3.671 eV) suggesting the higher kinetic stability of ZnPT-1 over CuPT-1, which can explain the selectivity to Zn2+ (see the ESI†).
Utility of the peptoid chelator
Following our results showing high selectivity in the binding of Zn2+ by PT-1, we wished to explore the ability of PT-1 to bind and remove Zn2+ from a Zn finger protein. To this aim, we chose the apo-zinc finger protein domain PYKCPECGKSFSQKSDLVKHQRTHTG (ZF), because it has canonical two cysteines and two histidines, which form a CCHH type metal coordination site (Fig. 7a).34 Initially the circular dichroism spectrum of the apo-ZF was measured in acetate buffer (pH 6.5) in order to compare it to a previously reported spectrum (Fig. S62†).35 The CD spectrum of the unstructured apo protein has a minimum near 200 nm, demonstrating its random coil conformation. The CD spectrum of the zinc complex of this protein exhibits a maximum at 190 nm and double minima near 205 and 218 nm, suggesting the formation of a structured α-helical conformation. Upon addition of PT-1 in acetate buffer at pH 6.5, the minima shift back to 200 nm, suggesting the successful chelation of Zn2+ from the domain of the ZF. ESI-MS measurements of the zinc-bound protein before and after the addition of PT-1 support the extraction of Zn2+ from the protein by PT-1 (see the ESI†). This chelation experiment was repeated in a SBF followed by CD measurements. The spectrum corresponding to the apo-ZF exhibits a minimum near 200 nm, which changes after the addition of Zn2+, showing a maximum at 190 nm and double minima near 205 and 218 nm. This spectrum changes again after the addition of PT-1, exhibiting only one minimum near 200 nm (Fig. 7b). These results establish the ability of the peptoid chelator PT-1 to bind Zn2+ from the zinc finger protein in a biological-like medium that contains ions such as Cl−, HCO3−, SO42−, HPO42−, Na+, Ca2+, K+ and Mg2+ at a concentration similar to that of our body fluid.25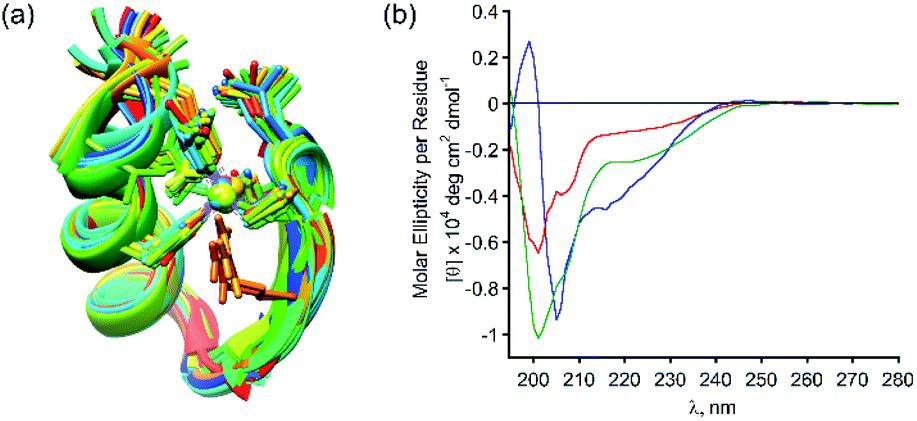 | ||
| Fig. 7 (a) A 3D model graphic representation of a zinc finger protein (PDB accession code 1ZNF);36 (b) CD spectra (at 100 μM SBF) of the apo-ZF (red line), Zn-ZF (blue line) and Zn2+ removed ZF (green line). | ||
Conclusions
Controlling chelation processes for the development of therapeutics that target specific metal ion(s) remains a great challenge in chemistry and chemical biology. Although small molecular probes incorporating bidentate and/or tridentate ligands, as well as peptidomimetic oligomers with tunable sequences were developed for Zn2+ chelation, competitive binding in the presence of other biologically relevant ions such as Cu2+ in an aqueous medium is scarce. Herein we combined the advantages from both systems, and by capitalizing on the water solubility of short peptoids, we rationally designed for the first time a water-soluble peptoid trimer chelator, PT-1, bearing the ligands terpy and picolyl, that can selectively bind Zn2+ in the presence of other biologically relevant and competitive ions in water, as well as in a simulated body fluid (SBF). The chelation of Zn2+ ions and their main metal ion competitor Cu2+ by PT-1 was investigated using several spectroscopic studies and the coordination geometry of both complexes ZnPT-1 and CuPT-1 was further studied by DFT-D4 calculations. The selectivity of PT-1 towards Zn2+ was rationalized based on the theoretical analysis. Finally we could show that PT-1 can extract Zn2+ from a Zn-finger protein domain in a SBF. These results open up a new avenue for the design of zinc chelators as a therapeutic approach targeting zinc finger proteins, towards multidirectional research in chemistry, biology and medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Boris Tumanskii from the Schulich Faculty of Chemistry for the EPR measurements and analysis and Mrs Larisa Panz from the Schulich Faculty of Chemistry for the MS measurements and analysis. PG thanks the Lady Davis Trust for his postdoctoral fellowship. This work was funded by the Israel Science Foundations, grant # 395/16 awarded to Prof. Galia Maayan.Notes and references
- (a) A. I. Anzellotti and N. P. Farrell, Chem. Soc. Rev., 2008, 37, 1629–1651 RSC; (b) M. Cassandri, A. Smirnov, F. Novelli, C. Pitolli, M. Agostini, M. Malewicz, G. Melino and G. Raschellà, Cell Death Discovery, 2017, 3, 17071, DOI:10.1038/cddiscovery.2017.71; (c) T. V. O'Halloran, Science, 1993, 261, 715–725 CrossRef.
- C. Abbehausen, Metallomics, 2019, 11, 15–28 RSC.
- N. G. Kolev, E. Ullu and C. Tschudi, Cell. Microbiol., 2014, 16, 482–489 CrossRef CAS.
- S. Vyas and P. Chang, Nat. Rev. Cancer, 2014, 14(7), 502–509 CrossRef CAS.
- N. Iraci, O. Tabarrini, C. Santi and L. Sancineto, Drug Discovery Today, 2018, 23(3), 687–695 CrossRef CAS.
- Y. Sun, D. Hu, J. Liang, Y. P. Bao, S. Q. Meng, L. Lu and J. Shi, Schizophr. Res., 2015, 162(1–3), 124–137 CrossRef.
- (a) Y. M. Lee, Y. Duh, S. T. Wang, M. M. C. Lai, H. S. Yuan and C. Lim, J. Am. Chem. Soc., 2016, 138(11), 3856–3862 CrossRef CAS; (b) Y. M. Lee, Y.-T. Wang, Y. Duh, H. S. Yuan and C. Lim, J. Am. Chem. Soc., 2013, 135, 14028–14031 CrossRef CAS.
- (a) E. Kawabata, K. Kikuchi, Y. Urano, H. Kojima, A. Odani and T. Nagano, J. Am. Chem. Soc., 2005, 127(3), 818–819 CrossRef CAS; (b) A. Balliu and L. Baltzer, ChemBioChem, 2017, 18(14), 1408–1414 CrossRef CAS; (c) E. M. Nolan, J. W. Ryu, J. Jaworski, R. P. Feazell, M. Sheng and S. J. Lippard, J. Am. Chem. Soc., 2006, 128(48), 15517–15528 CrossRef CAS; (d) J. D. Brodin, A. M. Morales, T. Ni, E. N. Salgado, X. I. Ambroggio and F. A. Tezcan, J. Am. Chem. Soc., 2010, 132, 8610–8617 CrossRef CAS; (e) B. C. Lee, T. K. Chu, K. A. Dill and R. N. Zuckermann, J. Am. Chem. Soc., 2008, 130, 8847–8855 CrossRef CAS.
- (a) N. F. Zakharchuk, G. S. Soldatova, T. V. Novikova and N. S. Borisova, Chem. Sustainable Dev., 2006, 231–240 Search PubMed; (b) W. I. Mortada, I. M. M. Kenawy, A. M. Abdelghany, A. M. Ismail, A. F. Donia and K. A. Nabieh, Mater. Sci. Eng. Carbon, 2015, 52, 288–296 CrossRef CAS; (c) J. Weissgarten, S. Berman, R. Bilchinsky, D. Modai and Z. Averbukh, Metabolism, 2001, 50, 270–276 CrossRef CAS; (d) L. J. Hinks, B. E. Clayton and R. S. Lloyd, J. Clin. Pathol., 1983, 36, 1016–1021 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, W. H. Moosf and S. B. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- (a) K. J. Prathap and G. Maayan, Chem. Commun., 2015, 51(55), 11096–11099 RSC; (b) D. C. Mohan, A. Sadhukha and G. Maayan, J. Catal., 2017, 355, 139–144 CrossRef; (c) T. Ghosh, P. Ghosh and G. Maayan, ACS Catal., 2018, 8(11), 10631–10640 CrossRef CAS.
- T. Ghosh, N. Fridman, M. Kosa and G. Maayan, Angew. Chem., Int. Ed., 2018, 57(26), 7703–7708 CrossRef CAS.
- (a) M. Baskin and G. Maayan, Dalton Trans., 2018, 47(31), 10767–10774 RSC; (b) M. Baskin and G. Maayan, Biopolym, 2015, 104(5), 577–584 CrossRef CAS; (c) H. Tigger-Zaborov and G. Maayan, J. Colloid Interface Sci., 2017, 508, 56–64 CrossRef CAS; (d) L. Zborovsky, A. Smolyakova, M. Baskin and G. Maayan, Chem. – Eur. J., 2018, 24(5), 1159–1167 CrossRef CAS; (e) L. Zborovsky, H. Tigger-Zaborov and G. Maayan, Chem. – Eur. J., 2019, 25(38), 9098–9107 CrossRef CAS.
- (a) M. Baskin, H. Zhu, Z. W. Qu, J. H. Chill, S. Grimme and G. Maayan, Chem. Sci., 2019, 10(2), 620–632 RSC; (b) M. Baskin and G. Maayan, Chem. Sci., 2016, 7(4), 2809–2820 RSC; (c) M. Baskin, L. Panz and G. Maayan, Chem. Commun., 2016, 52, 10350–10353 RSC; (d) A. D. ' Amato, P. Ghosh, C. Costabile, G. Della Sala, I. Izzo, G. Maayan and F. De Riccardis, Dalton Trans., 2020, 49(18), 6020–6029 RSC.
- J. A. Schneider, T. W. Craven, A. C. Kasper, C. Yun, M. Haugbro, E. M. Briggs, V. Svetlov, E. Nudler, H. Knaut and R. Bonneau, Nat. Commun., 2018, 9(1), 1–10, DOI:10.1038/s41467-018-06845-3.
- (a) F. Qian, C. Zhang, Y. Zhang, W. He, X. Gao, P. Hu and Z. Guo, J. Am. Chem. Soc., 2009, 131(4), 1460–1468 CrossRef CAS; (b) H. W. Rhee, H. Y. Choi, K. Han and J. I. Hong, J. Am. Chem. Soc., 2007, 129(15), 4524–4525 CrossRef CAS; (c) K. Komatsu, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2005, 127(29), 10197–10204 CrossRef CAS; (d) X. A. Zhang, D. Hayes, S. J. Smith, S. Friedle and S. J. Lippard, J. Am. Chem. Soc., 2008, 130(47), 15788–15789 CrossRef CAS; (e) E. Kawabata, K. Kikuchi, Y. Urano, H. Kojima, A. Odani and T. Nagano, J. Am. Chem. Soc., 2005, 127(3), 818–819 CrossRef CAS; (f) Z. Xu, K. H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132(2), 601–610 CrossRef CAS; (g) Y. Zhang, X. Guo, W. Si, L. Jia and X. Qian, Org. Lett., 2008, 10(3), 473–476 CrossRef CAS; (h) P. Du and S. J. Lippard, Inorg. Chem., 2010, 49(23), 10753–10755 CrossRef CAS; (i) W. Lin, D. Buccella and S. J. Lippard, J. Am. Chem. Soc., 2013, 135(36), 13512–13520 CrossRef CAS.
- X. Wang, and X. Chen, Novel Nanomaterials for Biomedical, Environmental, and Energy Applications, Elsevier, 2018 Search PubMed.
- (a) A. Mandal, A. Maity, S. Bag, P. Bhattacharya, A. K. Das and A. Basak, RSC Adv., 2017, 7(12), 7163–7169 RSC; (b) Z. Huang, J. Du, J. Zhang, X. Q. Yu and L. Pu, Anal. Methods, 2012, 4(7), 1909–1912 RSC.
- (a) P. Collery, Proceedings of the Fourth International Symposium on Metal Ions in Biology and Medicine, ed. P. Collery, J. Corbella, J. L. Domingo, J-C. Etienne and J. M. Llobet, John Libbey Eurotext, 1996, vol. 4, pp. 703–1996 Search PubMed; (b) J. Gao, J. H. Reibenspies and A. E. Martell, J. Inorg. Biochem., 2003, 94, 272–278 CrossRef CAS.
- G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC.
- M. J. Lachenmann, J. E. Ladbury, J. Dong, K. Huang, P. Carey and M. A. Weiss, Biochem, 2004, 43(44), 13910–13925 CrossRef CAS.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44(15), 5200–5219 RSC.
- (a) J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133(39), 15559–15567 CrossRef CAS; (b) K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(8), 4303–4308, DOI:10.1073/pnas.95.8.4303; (c) C. M. Darapaneni, P. J. Kaniraj and G. Maayan, Org. Biomol. Chem., 2018, 16(9), 1480–1488 RSC.
- C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125(44), 13525–13530 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27(15), 2907–2915 CrossRef CAS.
- F. B. Abdallah and T. R. Giffune, Biochim. Biophys. Acta, 2016, 1860, 879–891 CrossRef.
- (a) J. Karn, N. J. Keen, M. J. Churcher, F. Aboul-ela, G. Varani, F. Hamy, E. R. Felder, G. Heizmann and T. Klimkait, Pharmacochem. Libr., 1998, 29, 121–132 CAS; (b) C. M. Drapaneni, P. Ghosh, T. Ghosh and G. Maayan, Chem. – Eur. J., 2020, 26, 9573–9579 CrossRef.
- E. Manandhar, J. H. Broome, J. Myrick, W. Lagrone, P. J. Cragg and K. J. Wallace, Chem. Commun., 2011, 47(31), 8796–8798 RSC.
- (a) A. M. Díez-Pascual, Polymers, 2019, 11(4), 625–630 CrossRef; (b) M. S. Ghazvini, G. Pulletikurthi, A. Lahiri and F. Endres, ChemElectroChem, 2016, 3(4), 598–604 CrossRef CAS.
- (a) TURBOMOLE, Version 7.3, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, available from http://www.turbomole.com Search PubMed; (b) E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147(3), 034112 CrossRef.
- S. K. Papageorgiou, E. P. Kouvelos, E. P. Favvas, A. A. Sapalidis, G. E. Romanos and F. K. Katsaros, Carbohydr. Res., 2010, 345(4), 469–473 CrossRef CAS.
- (a) D. Floner, O. Lavastre, M. Zaarour, N. M. Saleh, F. Geneste, F. Justaud, R. Marion and N. A. Qachachi, J. Inorg. Biochem., 2011, 105(11), 1391–1397 CrossRef; (b) S. Muthuramalingam, K. Anandababu, M. Velusamy and R. Mayilmurugan, Inorg. Chem., 2020, 59(9), 5918–5928 CrossRef CAS.
- (a) Z. Zhou and R. G. Parr, J. Am. Chem. Soc., 1990, 112(15), 5720–5724 CrossRef CAS; (b) J. I. Aihara, J. Phys. Chem. A, 1999, 103(37), 7487–7495 CrossRef CAS; (c) L. P. Ding, F. H. Zhang, Y. S. Zhu, C. Lu, X. Y. Kuang, J. Lv and P. Shao, Sci. Rep., 2015, 5, 1–12, DOI:10.1038/srep15951.
- The pdb file of the zinc finger protein with accession code 1ZNF was taken and modelled with a chimera software suite (ref. 36).
- M. A. Franzman and A. M. Barrios, Inorg. Chem., 2008, 47(10), 3928–3930 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25(13), 1605–1612 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures of the peptoid and Zn2+ and/or Cu2+ complexes, UV-Vis measurements, ESI-MS analysis, coordinates of the geometry optimized configuration of Zn2+ and Cu2+ complexes with the peptoid. See DOI: 10.1039/d0sc03391j |
| This journal is © The Royal Society of Chemistry 2020 |