
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitchable dynasore analogs to control endocytosis with light†
Núria
Camarero‡
 a,
Ana
Trapero‡
ab,
Ariadna
Pérez-Jiménez
a,
Eric
Macia
c,
Alexandre
Gomila-Juaneda
a,
Andrés
Martín-Quirós
a,
Laura
Nevola§
d,
Artur
Llobet
e,
Amadeu
Llebaria
b,
Jordi
Hernando
f,
Ernest
Giralt
dg and
Pau
Gorostiza
*ahi
a,
Ana
Trapero‡
ab,
Ariadna
Pérez-Jiménez
a,
Eric
Macia
c,
Alexandre
Gomila-Juaneda
a,
Andrés
Martín-Quirós
a,
Laura
Nevola§
d,
Artur
Llobet
e,
Amadeu
Llebaria
b,
Jordi
Hernando
f,
Ernest
Giralt
dg and
Pau
Gorostiza
*ahi
aInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology (BIST), Spain. E-mail: pau@icrea.cat
bInstitute for Advanced Chemistry of Catalonia (IQAC-CSIC), Spain
cInstitut de Pharmacologie Moléculaire et Cellulaire (IPMC), Université Nice Sophia Antipolis, France
dInstitute for Research in Biomedicine (IRB Barcelona), Spain
eBellvitge Biomedical Research Institute (IDIBELL), Spain
fDepartament de Química, Universitat Autònoma de Barcelona (UAB), Spain
gUniversitat de Barcelona (UB), Spain
hCIBER-BBN, Spain
iICREA, Spain
First published on 17th August 2020
Abstract
The spatiotemporal control of cellular dynamic processes has great fundamental interest but lacks versatile molecular tools. Dynamin is a key protein in endocytosis and an appealing target to manipulate cell trafficking using patterns of light. We have developed the first photoswitchable small-molecule inhibitors of endocytosis (dynazos), by a stepwise design of the photochromic and pharmacological properties of dynasore, a dynamin inhibitor. We have characterized their photochromism with UV-visible and transient absorption spectroscopy and their biological activity using fluorescence microscopies and flow cytometry. Dynazos are water-soluble, cell permeable, and photostable, and enable fast, single-wavelength photoswitchable inhibition of clathrin-mediated endocytosis at micromolar concentration.
Introduction
Most of the proteins in the cell require the interaction with other molecules or proteins to play their biological function. These interactions are crucial for the proper control of a wide variety of cellular processes, such as cell division, membrane trafficking, signaling, etc. A way to understand how these cellular processes work is by perturbing the activity of one key component in the system and observing how the process is affected by this perturbation. Small molecules can be used to interfere with the biological function of a protein, by inhibiting or activating their function. These compounds are a powerful tool to probe the molecular mechanisms involved in the transduction of specific biological signals and lead to the development of novel therapeutic strategies.1 Potency and selectivity to a target protein subtype are desirable qualities to avoid off-target effects, since the stronger the binding and its consequences, the longer they last and the more difficult they are to revert. Compared to genetic approaches to interfere with the action of a protein, such as RNAi or the overexpression of dominant negative proteins, the use of small molecules offers several advantages in terms of controlling the dynamics and reversibility of the response. The effects of overexpressing a protein can take hours or days to appear and could be irreversible, whereas small molecules are often fast acting and can be readily removed. Acute stimulation with these compounds can also prevent the development of homeostatic or compensatory mechanisms by the cell, which plague the outcomes and interpretation of genetic manipulations.2Local administration of compounds can further improve their efficacy and the reversibility of the elicited responses, e.g. when studying the pharmacological profile of an ion channel with electrophysiological recordings and fast perfusion of selective inhibitors.3 However, the use of diffusible molecules poses limitations on the temporal and spatial precision that can be achieved. Once the molecule is administered, it can be diluted or distributed in tissue, and the control on spatial location and precise timing of its activity is lost.
This is important when investigating transient or irreversible events (e.g. the cellular processes involved in membrane trafficking) or when disturbing the activity of proteins essential for cell survival (whose consequences could be extended to other cells or tissues).4,5 These limitations can also affect the efficacy and specificity of drugs used in pharmacotherapy. As an example, the cytotoxic anti-neoplastic agents used in cancer treatment result in severe side effects due to the lack of specificity towards healthy tissue.6
These problems can be addressed with compounds whose biological activity can be turned on and off remotely and reversibly, like light-regulated drugs used in photopharmacology. These molecules can be obtained by the introduction of a light-sensitive group (photoswitch) in the structure of a biologically active ligand.5,7 Careful molecular design and extensive testing allow in some cases to use light as the external trigger to control the drug's pharmacologic activity with high spatiotemporal resolution.8 The azobenzene is one of the best-studied photoswitchable groups for this purpose and has been extensively used in photopharmacology to regulate the activity of ion channels,3,9–11 G protein-coupled receptors,12,13 and enzymes.14,15 It undergoes a trans/cis isomerization process upon irradiation with ultraviolet (UV) light, which can be reverted by illumination with visible light or by thermal relaxation of the less stable cis isomer.16 This results in light-induced changes of the molecular length, geometry and dipolar moment, which can be exploited to alter the pharmacological properties of an appended bioactive moiety.
Among all fundamental biological processes, endocytosis greatly exemplifies both the interest and challenges to manipulate the cellular spatiotemporal dynamics. It is used by all eukaryotic cells to internalize extracellular material along with portions of the plasma membrane through the formation of vesicles. It regulates many physiological processes, including the uptake of nutrients, growth factors and transmembrane receptors and it also constitutes the entry pathway of a variety of pathogens.17 There are many different routes of endocytosis operating at the same time at the cell surface,18 which are critical in controlling the membrane turnover and plasticity in essential cellular functions such as mitosis, cell differentiation, adhesion, migration, and transmembrane receptor signaling. Endocytic processes involve the concerted action of many proteins, complexes, cell membranes and compartments, and constitute potential targets to prevent infections and to kill cells.
Although the main molecular players of endocytosis are well characterized, and several experimental methods have been developed to observe their dynamics, the molecular mechanisms are far from being understood due in part to the lack of acute manipulation tools.19,20 One of the key molecular players involved in most endocytic pathways is dynamin, a GTPase required for membrane scission during the endocytic vesicle formation.21,22
Dynamin is essential not only for clathrin-mediated endocytosis (CME), one of the best-characterized endocytic pathways which is dependent on the recruitment of clathrin cages to the membrane, but also for other clathrin-independent routes for cellular endocytosis.23,24 The GTPase activity of dynamin represents a highly efficacious target to control endocytosis and a growing number of small molecules that specifically inhibit dynamin activity have been described.25–29 Nowadays these inhibitors constitute not only useful research tools for the study of endocytosis, but also a promising strategy for the treatment of viral and bacterial infections. Thus, dynamin is an appealing target to manipulate endocytosis in spatiotemporally designated patterns with light.
To photocontrol dynamin activity, we combined the pharmacological properties of the dynamin inhibitor dynasore28 (Fig. 1a) and the photochromic properties of an azobenzene group, to yield a series of photoswitchable analogues of dynamin inhibitors that we called dynazos (compounds dynazo-2, dynazo-3 and dynazo-4 in Fig. 1a). Here we present their design strategy, synthesis, and their photochromic and biological characterization.
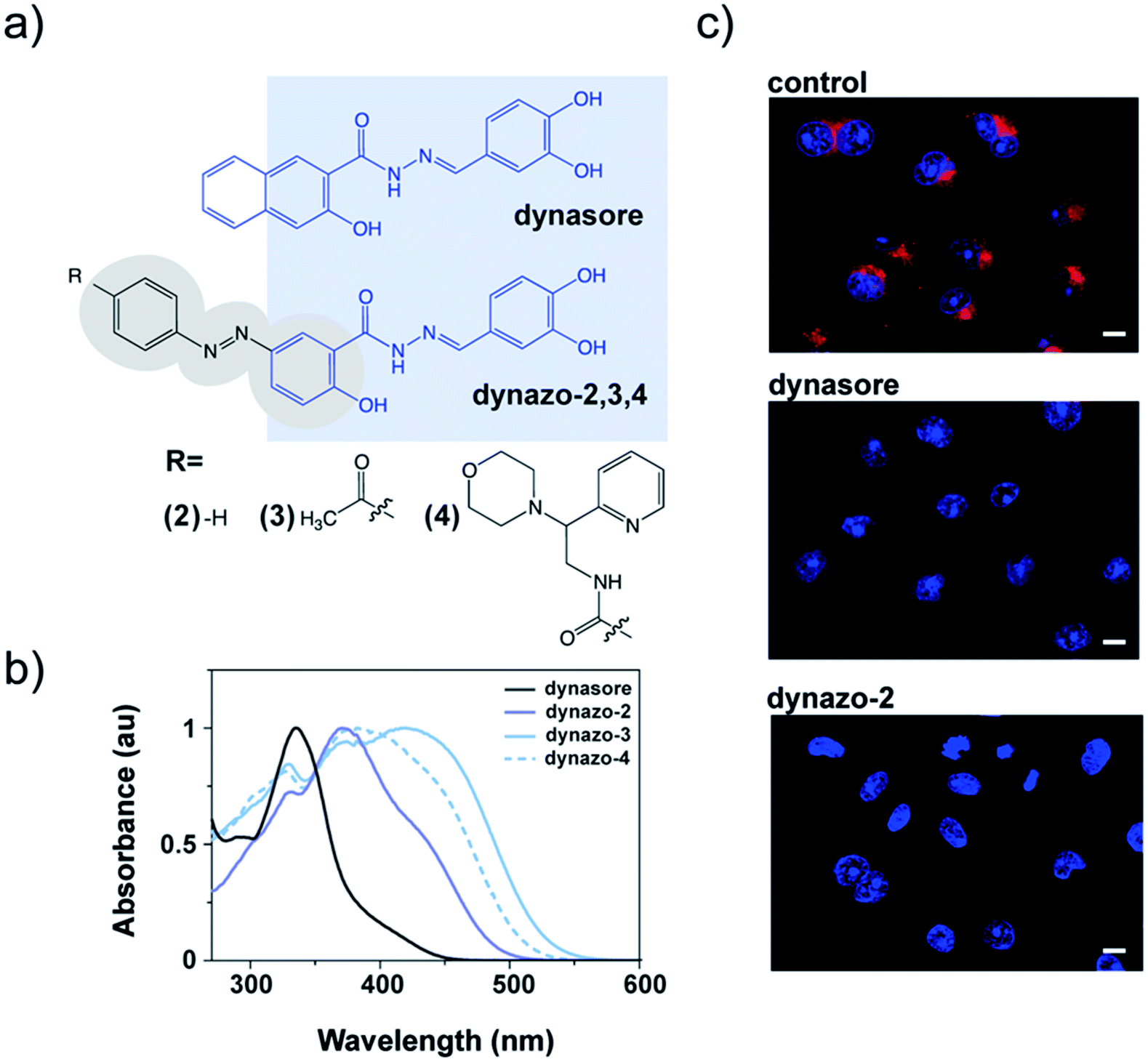 | ||
| Fig. 1 Molecular design and biological activity of dynazos, photochromic dynasore analogs. (a) Chemical structures of dynasore, a well-known dynamin inhibitor,28 and dynazos. Highlighted in blue, the structure of dynasore that has been conserved in the design of dynazos-2, 3 and 4. There are also shown the electron-withdrawing substituents introduced on the azobenzene core of dynazos-3 and 4 (gray ovals) to improve their photochromic and biological properties with respect to dynazo-2. (b) UV-visible absorption spectra in PBS with 1% DMSO showing the red-shifted azobenzene absorption of dynazos-2, 3 and 4 compared to dynasore upon incorporation of strong electron-donating and electron-withdrawing substituents. (c) Confocal microscopy images of transferrin (Tf) uptake assays in COS-7 cells. The uptake of Alexa-568-Tf (red) is inhibited in cells treated with dynazo-2 (100 μM) to a similar extent as in cells treated with dynasore (80 μM) compared to control vehicle-treated cells. DRAQ5 staining shows nuclei (blue) (scale bar: 10 μm). | ||
Results and discussion
We decided to partially integrate azobenzene into the structure of dynasore as a constitutive part of the inhibitor pharmacophore (shaded in light blue in Fig. 1a). We expected that this might allow preservation of the inhibitory activity to an extent that was sensitive to the photoisomer configuration, thereby enabling control of inhibition with light. The number and the relative position of the hydroxyl groups in the catechol moiety of dynasore are important for its biological activity,25,30 and for this reason the azobenzene unit was introduced at the position of the naphthyl fragment.We had to take into account an additional constraint as an integral part of the design, namely that dynasore absorbs in the same spectral region as regular trans-azobenzenes16 (∼330 nm, Fig. 1b, black plot) and, as a result, it photodegrades upon continuous direct excitation with the UV light required for photoisomerization (e.g. 355 nm, ESI Fig. S1†). In order to decouple the absorption spectra of dynasore and the azobenzene moiety of the dyad, we selected azobenzene groups with red-shifted photoisomerization wavelengths, which have the additional advantages of deeper penetration than UV light and reduced cellular damage upon prolonged exposure.31,32 It is reported that azobenzenes with strong electron-donating substituents at 2- or 4-position (e.g. –NH2 or –OH) absorb at longer wavelengths and display fast thermal back isomerization to the trans configuration in the dark.16,33
We thus took advantage of the hydroxyl group present in the naphthyl moiety of the pharmacophore, which was preserved in the azobenzene core of the dynazo compounds designed in order to achieve the desired spectral red shift (Fig. 1a).
With these design principles in mind, we first synthesized dynazo-2, the simplest of the compounds prepared. Compared to dynasore, the UV-visible absorption spectrum of dynazo-2 shows an additional absorption peak at 372 nm in phosphate buffer saline (PBS) that confirms successful red shift of the absorption spectrum of its azobenzene photochrome (violet solid plot in Fig. 1b). This bathochromic displacement is even more pronounced in DMSO, the lowest energy azobenzene absorption band appearing at 447 nm (dark blue solid plot in ESI Fig. S2†). Based on previous reports on 4-hydroxyazobenzenes, the larger spectral effect found in DMSO might be ascribed to intramolecular hydrogen bonding between the nearby hydroxyl and carbonyl substituents of the azo chromophore, which will not be favored in aqueous media.34 Next, we tested if the biological activity of dynasore was retained in dynazo-2. A well-established method to study dynamin inhibitors on endocytosis is to measure receptor-mediated transferrin (Tf) uptake.35 Fluorescently labeled Alexa 563-Tf was allowed to bind to its endogenous receptor on the surface of COS-7 intact cells by incubation at low temperature to slow down endocytosis. After removal of the Tf excess, cells were transferred to 37 °C for 15 minutes to allow internalization of the Tf-receptor complex and cell surface-bound Tf was removed by an acidic wash. After nuclei staining and cell fixation, internalized Tf-receptor complexes accumulated at the perinuclear-recycling late endosomes and could be observed by fluorescence confocal microscopy (Fig. 1c). Incubation with dynazo-2 (100 μM) reduced Tf internalization to a similar extent than dynasore (80 μM) compared to control vehicle-treated cells (Fig. 1c). These results confirmed that our approach for the integration of the azobenzene moiety into the dynasore structure does not compromise its biological activity and prompted us to undertake further chemical modifications of the azobenzene core away from the pharmacophore to improve its photochromic response and activity modulation.
Two derivatives of dynazo-2 were thus designed and synthesized. Dynazo-3 has an acetyl group at the 4′-position of the azobenzene core (Fig. 1a). We introduced this electron-withdrawing group to generate a push–pull azobenzene derivative with an even larger absorption bathochromic shift with respect to dynazo-2. In addition, the incorporation of this group was also intended to reduce the affinity of the cis form towards dynamin by increasing the steric hindrance around the biologically active fragment of the compound upon photoisomerization. This design concept was further exploited in dynazo-4, where a much bulkier electron-withdrawing amide group was introduced. To facilitate chemical access to a variety of potential candidates, the ketone group was replaced by an amide to which several commercially available N-substituents could be attached. Importantly, this should not detrimentally affect the optical properties of the resulting compound (i.e. absorption red-shift), as proven by our spectroscopic measurements. The N-(2-morpholino-2-(pyridin-2-yl)ethyl)amide substituent chosen for this purpose trades off steric hindrance and aqueous solubility, compared to other options. For instance, for an N-(2-diphenylethyl)amide group, we observed a strong reduction in aqueous solubility that essentially prevented biological experiments (data not shown in the manuscript). The photophysical properties of the new dynazos were again analyzed by UV-visible spectroscopy (Fig. 1b). Compared to parent compound dynazo-2, the substituents introduced in dynazo-3 and dynazo-4 caused an additional red shift of the absorption spectra, especially for dynazo-3 (∼45 nm and ∼40 nm in PBS and DMSO respectively, Fig. 1d and ESI Fig. S2†).
The synthesis of dynazo-4 was achieved as shown in Scheme 1. Diazotization of 4-aminobenzoic acid and coupling of the resulting diazonium salt with methyl 2-hydroxybenzoate yielded azobenzene 5. The reaction of the obtained intermediated 5 with hydrazine hydrate led to the formation of acid hydrazide 6, which underwent condensation with 3,4-dihydroxybenzaldehye affording hydrazine 7. Finally, amide coupling of acid 7 and 2-morpholino-2-(pyridin-2-yl)ethanamine afforded dynazo-4.
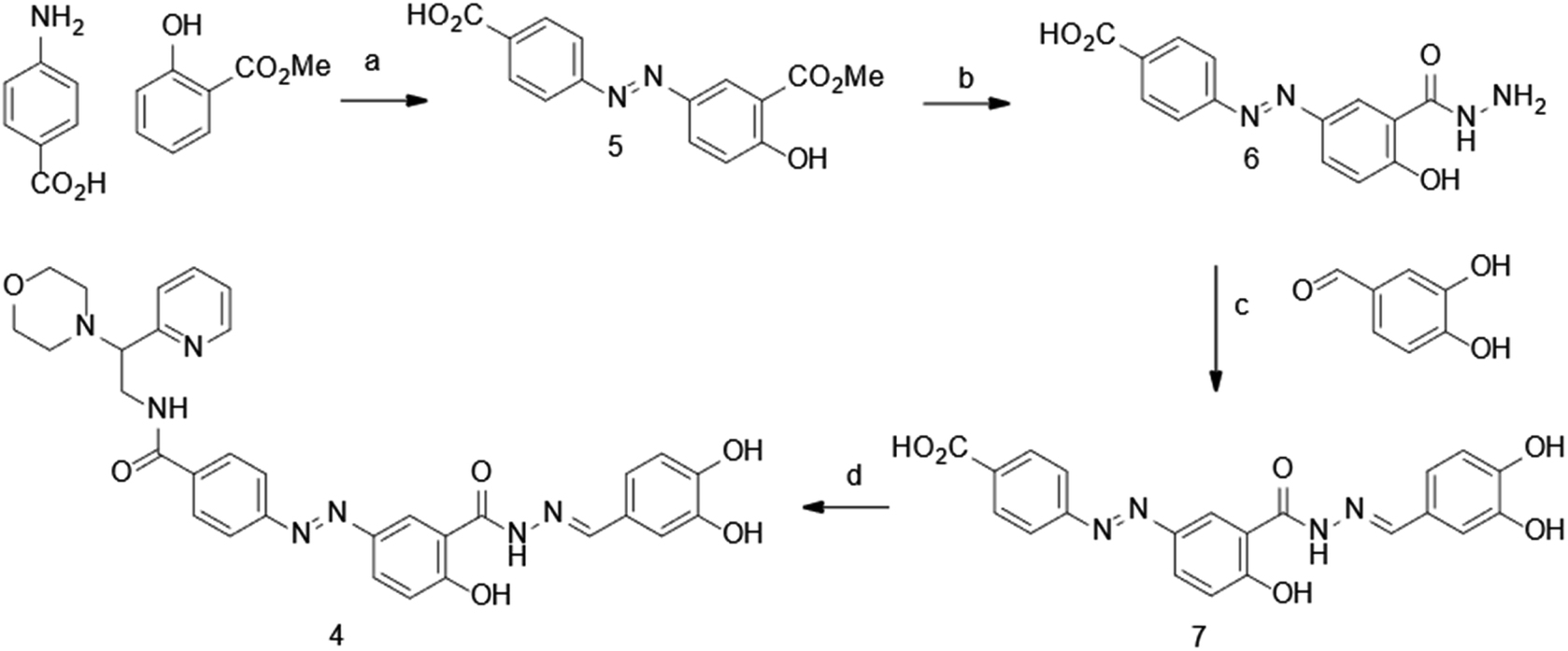 | ||
Scheme 1 Synthesis of dynazo-4. Reagents and conditions: (a) (i) NaNO2 (1.1 eq.), HCl/H2O (v/v = 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :92), −5 °C; (ii) NaOH (2 eq.), Na2CO3 (2 eq.), H2O, −5 °C, 61% yield; (b) NH2NH2·H2O (4 eq.), EtOH, reflux, 62% yield; (c) EtOH, reflux, 83% yield; (d) 2-morpholino-2-(pyridin-2-yl)ethanamine (1.2 eq.), EDC (1.5 eq.), HOBt (1.5 eq.), TEA (2.6 eq.), DMF, RT, 57% yield. :92), −5 °C; (ii) NaOH (2 eq.), Na2CO3 (2 eq.), H2O, −5 °C, 61% yield; (b) NH2NH2·H2O (4 eq.), EtOH, reflux, 62% yield; (c) EtOH, reflux, 83% yield; (d) 2-morpholino-2-(pyridin-2-yl)ethanamine (1.2 eq.), EDC (1.5 eq.), HOBt (1.5 eq.), TEA (2.6 eq.), DMF, RT, 57% yield. | ||
As reported for azobenzene derivatives with strong mesomeric electro-donating and/or electron-withdrawing groups33 dynazo-2, 3, 4 displayed very fast cis to trans thermal isomerization rates, which prevented observation of the photoconversion of these switches using steady state UV-visible absorption spectroscopy. Instead, time-resolved absorption spectroscopy allowed recording with nanosecond accuracy the time evolution of the cis states formed after pulsed irradiation of these compounds.
Fig. 2 shows the transient absorption spectra measured for dynazo-2, 3, 4 in PBS right after photoexcitation (ΔAbs at t = 0), which display negative values in all cases that must be attributed to the light-induced conversion of their initial trans isomers into less absorbing species. This, together with the fast time decay of the negative ΔAbs signal to recover the initial situation, prove reversible trans/cis azobenzene photoisomerization in dynazo-2, 3, 4 and allow estimating the most efficient wavelengths to induce this process:36ca. 390 nm (dynazo-2) and 405 nm (dynazo-3, 4) in PBS, which are in good agreement with the red-shifted absorption bands shown by these compounds with respect to dynasore in Fig. 1b. As such, photoexcitation of the azobenzene and dynasore units in dynazo-2, 3, 4 could be decoupled, which should enable trans/cis photoisomerization without degrading their pharmacophore. This was demonstrated by illumination with violet light at 405 nm, which should lead to efficient photoisomerization. Remarkably, no photodegradation of dynasore or of any of the dynazos was observed under these excitation conditions (ESI Fig. S3†), thus confirming the success of our design approach.
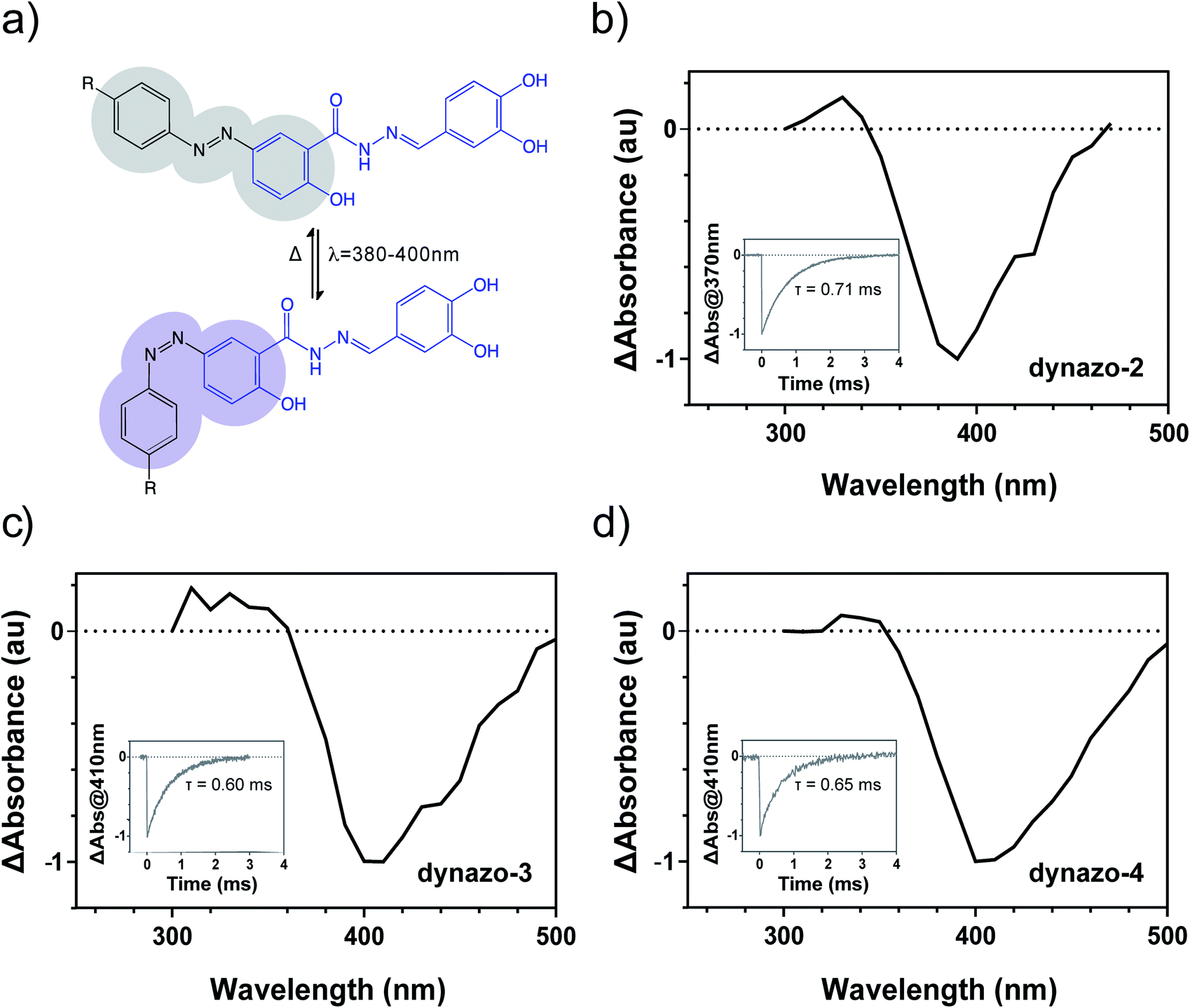 | ||
| Fig. 2 Characterization of dynazos' photochromism. (a) Photoisomerization of dynazos occurs upon illumination of the thermally stable trans isomer (gray ovals) with a wavelength between ca. 350–450 nm. The trans isomer is recovered by thermal back isomerization of the cis isomer (violet ovals). Transient absorption spectra of (b) dynazo-2, (c) dynazo-3 and (d) dynazo-4 after nanosecond pulsed laser excitation (λ = 355 nm, t = 0) confirms efficient photoisomerization and bathochromic shift of the most efficient irradiation wavelength from 390 nm in dynazo-2 to 405 nm in dynazo-3, 4. The transient absorption decays and the thermal lifetimes of the cis isomers (τ) retrieved from monoexponential fits are shown in the insets of (b) dynazo-2, (c) dynazo-3 and (d) dynazo-4. Measurements were conducted in PBS with 1% DMSO. | ||
In agreement with the behavior expected owing to their azobenzene substitution pattern, fast thermal back isomerization of the cis isomers of dynazo-2, 3, 4 was clearly observed by monitoring the time evolution of transient absorption and the corresponding cis state lifetimes (τ) were determined by fitting monoexponential functions to the recorded data. As it is shown in the insets of Fig. 2b–d, dynazos' relaxation times are between 0.6 and 0.7 ms, confirming that these compounds back isomerize to their trans configuration as soon as illumination is turned off. Continuous irradiation is thus required to maintain a significant fraction of cis isomers, which is advantageous to spatiotemporally constrain dynamin photoresponses regardless of compound diffusion, in contrast to the behavior of slow-relaxing diffusible compounds. In addition, fast-relaxing compounds also allow single-wavelength operation to control the target protein activity, thus simplifying the experimental resources needed.
To study the effect of dynazos on CME, we quantified the uptake of Alexa 488-labeled Tf by large populations of cells using flow cytometry. We used the same protocol than in the experiments of Fig. 1c, with cell resuspension in complete medium after the acidic wash, and propidium iodide staining to exclude dead cells from the analysis. The fluorescence of cells incubated in increasing amounts of each compound (ranging from 0.5 to 10 μM) was measured and plotted as a percentage of the mean fluorescence of vehicle-treated cells (Fig. 3a–c, black solid plots corresponding to dark conditions). These results confirm that photoisomerizable dynazos inhibit Tf uptake in a dose-dependent manner. To assess if the inhibitory activity of dynazos on CME could be remotely controlled with light, we repeated Tf uptake assays incubating dynazo-2, 3, 4 in separate cell plates in the dark and under optimal illumination conditions in each case, i.e. 380 nm for dynazo-2, and 405 nm for dynazo-3, 4 (Fig. 3a–c). Control experiments showed no effect of light on Tf uptake assays performed in untreated cells (ESI Fig. S4 and Methods/ESI for further details†). A Tf uptake time-course in untreated cells is also shown in ESI Fig. S5† to show the progression of Tf internalization without addition of dynazos. The inhibitory activities of all dynazos displayed significant differences between dark and illuminated conditions, at concentrations of 1 μM for dynazo-2 and between 0.5 μM and 5 μM for dynazo-3 and dynazo-4. The greatest relative differences in the inhibitory activities between the two conditions are ∼20% for 1 μM dynazo-2, 3 and ∼35% for 1 μM dynazo-4. It must be noted that flow cytometry measurements are performed in thousands of cells and that the significance of the observed differences is therefore very high (dynazo-2, P < 0.005, dynazo-3, P < 0.0001 and dynazo-4, P < 0.0005; 1 μM concentration, two-tailed t-test).
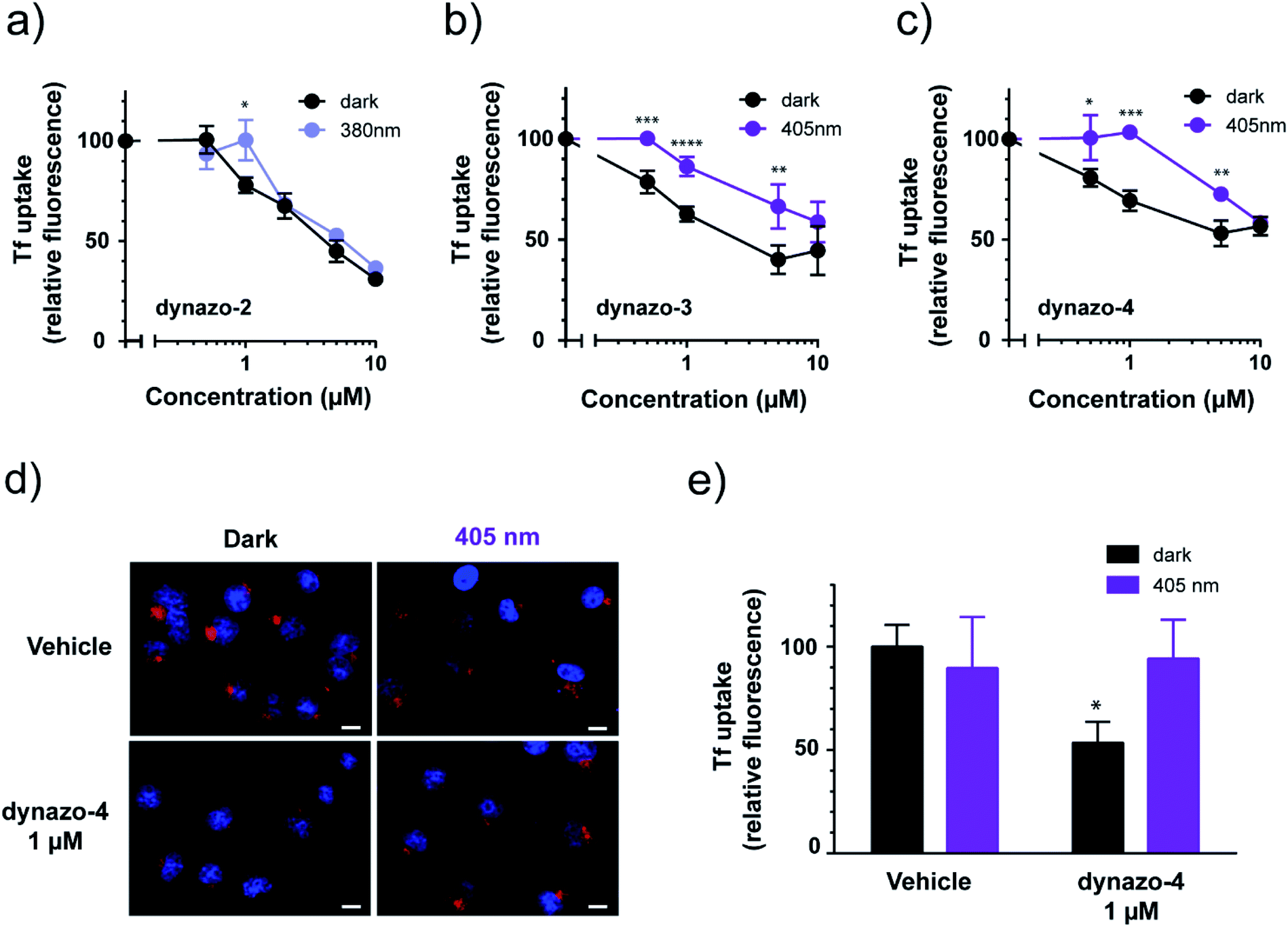 | ||
| Fig. 3 Characterization of dynazos' biological activity. (a) Dose–response curve of Tf uptake by flow cytometry in the dark and under illumination at 380 nm for dynazo-2 shows dose-dependent inhibition of Alexa-488-Tf uptake and significant differences in activity with light at 1 μM (*P < 0.05). (b) Dynazo-3 and (c) dynazo-4 dose–response curves in the dark and under illumination at 405 nm show dose-dependent inhibition and significant differences in activity with light between 0.5–5 μM (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). In all these cases, dose–response curves were normalized to those measured for vehicle-treated samples (100%). Data are representative of three independent experiments performed in duplicate and values are expressed in mean ± SEM. (d) Confocal microscopy images of Tf uptake assays in the dark and at 405 nm irradiation shows a significant reduction in Alexa-568-Tf uptake only in cells treated with dynazo-4 (1 μM) in the dark condition. Internalized Tf is shown in red and DRAQ5 blue staining shows nuclei (scale bar: 10 μm). Quantification of Tf uptake assay from (d) [normalized to vehicle-treated in the dark (100%)] is shown in (e) (*P < 0.05, n = 112 cells for vehicle/dark, n = 122 cells for vehicle/405 nm, n = 151 for dynazo-4 1 μM/dark and n = 153 cells for dynazo-4 1 μM/405 nm). All data are expressed as the mean ± SEM. | ||
Having determined the useful operating conditions, we further characterized the effect of illumination in Tf uptake using confocal microscopy. We performed Alexa-563-Tf uptake assays as in Fig. 1c but separately incubating cells in the dark and under illumination (405 nm) in the presence of 1 μM dynazo-4. Representative confocal microscopy images of this experiment are shown in Fig. 3d. In Fig. 3e, the average fluorescence intensities of 112 to 153 cells for each condition are plotted relative to the control dark condition (100% internalization). Dynazo-4 significantly reduced Tf uptake by 47% (P < 0.0163) in the dark compared to the 405 nm illumination conditions or to vehicle-incubated controls, in good agreement with the cytometry experiments of Fig. 3c. These results demonstrate that CME can be remotely controlled in intact, untransfected cells by means of small molecules (photochromic inhibitors of dynamin) that are directly applied to the bath. Dynazos are active at 1 μM concentration in the dark and can be deactivated to different extents with violet illumination, which may produce significantly lower toxicity and less unspecific effects than dynasore at the usual 80 μM concentration. In particular, at 1 μM concentration dynazo-4 can inhibit CME up to 47% and be photoswitched to a fully inactive state using an illumination wavelength available in most commercial laser scanning microscopes.
We also assessed the capacity of dynazo-4 to photocontrol the dynamics of clathrin-coated pit (CCP) formation during endocytosis. To this end, we used live-cell total internal reflection fluorescence microscopy (TIRFM) to monitor the CCP lifetimes by tracking the fluorescently labeled AP-2 protein in BSC-1 cells, using customized particle-tracking software.37 Together with clathrin, AP-2 is the most abundant coat protein in the CCP and it is also present during the entire lifetime of a CPP.38,39 We recorded time lapse movies of BSC-1 cells expressing the eGFP-tagged adaptor protein AP-2 before and after the incubation with 25 μM dynazo-4 for 40 minutes. Following, we recorded the CCP dynamics under continuous 405 nm illumination in the presence of dynazo-4 (ESI Fig. S6–S8†). Dynazo-4 treatment significantly increases the time duration of the CCP at the plasma membrane, indicating a slowdown of the endocytic process in full agreement with transferrin uptake assays by flow cytometry and fluorescence microscopy (Fig. 3). This effect is reversible under 405 nm illumination in the case of the CCP with a time duration of ≥120 seconds, but not at shorter time durations, probably due to the fast relaxation lifetime of dynazo-4, which limits the effective percentage of (inactive) cis isomer that can be achieved under illumination (ESI Fig. S7c†). These differences could also be due to the absence of dynamin recruitment in some CCP pools, which accelerates their disappearance from the plasma membrane.40 Control experiments in untreated cells showed no effect of 405 nm illumination on CCP lifetime (ESI Fig. S7d†).
Compared to traffic light peptides,37 dynazo-4 targets a protein bearing wider impact in endocytosis, it achieves comparable photocontrol of CME at 20-fold lower concentration, it has 4.6-fold smaller molecular weight (609.63 Da compared to 2830.8 Da of TL-2) and simpler synthetic accessibility.
Due to the rapid cis to trans relaxation of dynazos, we could not estimate the content of the cis isomer in the illuminated sample. According to the results in Fig. 2, it is likely that the cis/trans mixture in the PSS of illuminated dynazos contains a residual fraction of the more active isomer (trans) that is sufficiently high to limit the range of concentrations displaying light-dependent inhibition of Tf uptake. Reducing the trans isomer content in the PSS of illuminated dynazos (i.e. photoconverting more efficiently to the cis state) would likely enhance the differences in activity between the cis/trans forms, thereby expanding the useful concentration range of dynazos.
In addition, an enhancement of the photoswitchable steric hindrance around the pharmacophore with even bulkier substituents could further reduce the activity of cis isomers and increase cis/trans differential biological activity. It is worth noting that the inhibition of endocytosis by 1 μM dynazo-4 can be completely shut off under illumination, despite partial photoconversion (Fig. 3c). However, it would also be useful for certain experiments that the inactive isomer was most abundant in the dark, and that endocytosis could be inhibited with light (e.g. at focalized spots). This could in principle be achieved by means of a “reverse switch” with a bridged azobenzene which adopts a more stable cis configuration in the dark,41 although in turn, the resulting compound may display partial (basal) inhibition in the relaxed form instead of the complete inactivity of cis-dynazo-4. On the other hand, the large separation of the cis and trans absorbance bands in bridged azobenzenes should enable complete bidirectional switching with violet/green light.
Conclusions
In summary, we have used rational design to obtain the first light-regulated small-molecule inhibitor of endocytosis. Integration of substituted azobenzenes in the structure of dynasore while preserving the integrity of its pharmacophore is a successful strategy to endow photosensitivity to a dynamin inhibitor. In a series of stepwise modifications of the azobenzene core, first we have red-shifted the photoisomerization wavelength of dynazos in order to uncouple it from dynasore absorption, prevent photodegradation, and allow the use of visible light to switch off their activity. And second, we have blocked the activity of the cis isomer and enhanced functional photoswitching by increasing the steric hindrance around the pharmacophore and reducing the affinity of the cis isomer for its target. All compounds in the series are active in living cells and can be photomodulated to different extents. Remarkably, the activity of dynazos in living cells indicates that these dynasore derivatives are also cell-permeable compounds. These results constitute the proof of principle for the development of optimized photoswitchable dynasore analogs to manipulate dynamin-dependent endocytosis. These photopharmacological tools can be useful to dissect the role of endocytosis in cellular functions where it exerts a crucial role, such as cell growth, differentiation, cell motility and invasiveness, modulation of transmembrane receptor signalling and synaptic transmission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Ris3CAT-CECH project (001-P-001682, co-financed by the European Union Regional Development Fund within the framework of the ERDF Operational Program of Catalonia 2014-2020), from the CERCA Programme and the 2017-SGR-1442 grant (Government of Catalonia), from the European Union’s Horizon 2020 Framework Programme for Research and Innovation under the specific grant agreement no. 945539 (Human Brain Project SGA3), and from grant PID2019-111493RB-I00 (MINECO). A. G.-J., was supported by fellowship BES-2017-083025. We are grateful to Anna Lladó and Sebastian Tosi from the Advanced Digital Microscopy Core Facility of Institute for Research in Biomedicine (IRB) Barcelona for technical support in TIRF imaging and analysis, to Benjamín Torrejón from the Advanced Optical Microscopy Unit for technical assistance and the Cytometry unit of the CCiT-UB. We also thank T. Kirchhausen for the AP-2 BSC-1 cell line.Notes and references
- S. L. Schreiber, J. D. Kotz, M. Li, J. Aubé, C. P. Austin, J. C. Reed, H. Rosen, E. L. White, L. A. Sklar, C. W. Lindsley, B. R. Alexander, J. A. Bittker, P. A. Clemons, A. De Souza, M. A. Foley, M. Palmer, A. F. Shamji, M. J. Wawer, O. McManus, M. Wu, B. Zou, H. Yu, J. E. Golden, F. J. Schoenen, A. Simeonov, A. Jadhav, M. R. Jackson, A. B. Pinkerton, T. D. Y. Chung, P. R. Griffin, B. F. Cravatt, P. S. Hodder, W. R. Roush, E. Roberts, D. H. Chung, C. B. Jonsson, J. W. Noah, W. E. Severson, S. Ananthan, B. Edwards, T. I. Oprea, P. J. Conn, C. R. Hopkins, M. R. Wood, S. R. Stauffer and K. A. Emmitte, Cell, 2015, 161, 1252–1265 CrossRef CAS PubMed.
- T. G. Iversen, G. Skretting, B. Van Deurs and K. Sandvig, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5175–5180 CrossRef CAS PubMed.
- M. Izquierdo-Serra, A. Bautista-Barrufet, A. Trapero, A. Garrido-Charles, A. Diaz-Tahoces, N. Camarero, S. Pittolo, S. Valbuena, A. Perez-Jimenez, M. Gay, A. Garcia-Moll, C. Rodriguez-Escrich, J. Lerma, P. De La Villa, E. Fernandez, M. A. Pericas, A. Llebaria and P. Gorostiza, Nat. Commun., 2016, 7, 12221 CrossRef CAS PubMed.
- Y. Posor, M. Eichhorn-Gruenig, D. Puchkov, J. Schöneberg, A. Ullrich, A. Lampe, R. Müller, S. Zarbakhsh, F. Gulluni, E. Hirsch, M. Krauss, C. Schultz, J. Schmoranzer, F. Noé and V. Haucke, Nature, 2013, 499, 233–237 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- N. Carelle, E. Piotto, A. Bellanger, J. Germanaud, A. Thuillier and D. Khayat, Cancer, 2002, 95, 155–163 CrossRef PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48(7), 1947–1960 CrossRef CAS PubMed.
- X. Gómez-Santacana, S. Pittolo, X. Rovira, M. Lopez, C. Zussy, J. A. R. Dalton, A. Faucherre, C. Jopling, J. P. Pin, F. Ciruela, C. Goudet, J. Giraldo, P. Gorostiza and A. Llebaria, ACS Cent. Sci., 2017, 3, 81–91 CrossRef PubMed.
- P. Gorostiza, M. Volgraf, R. Numano, S. Szobota, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10865–10870 CrossRef CAS PubMed.
- R. H. Kramer, A. Mourot and H. Adesnik, Nat. Neurosci., 2013, 16, 816–823 CrossRef PubMed.
- J. Broichhagen, M. Schönberger, S. C. Cork, J. A. Frank, P. Marchetti, M. Bugliani, A. M. J. Shapiro, S. Trapp, G. A. Rutter, D. J. Hodson and D. Trauner, Nat. Commun., 2014, 5, 5116 CrossRef CAS PubMed.
- S. Pittolo, X. Gómez-Santacana, K. Eckelt, X. Rovira, J. Dalton, C. Goudet, J. P. Pin, A. Llobet, J. Giraldo, A. Llebaria and P. Gorostiza, Nat. Chem. Biol., 2014, 10, 813–815 CrossRef CAS PubMed.
- M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2014, 53(12), 3264–3267 CrossRef PubMed.
- B. Schierling, A. J. Noel, W. Wende, T. Hien le, E. Volkov, E. Kubareva, T. Oretskaya, M. Kokkinidis, A. Rompp, B. Spengler and A. Pingoud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1361–1366 CrossRef CAS PubMed.
- W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Chem.–Eur. J., 2015, 21, 16517–16524 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- C. B. Harper, M. R. Popoff, A. McCluskey, P. J. Robinson and F. A. Meunier, Trends Cell Biol., 2013, 23, 90–101 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS.
- M. Mettlen, P.-H. Chen, S. Srinivasan, G. Danuser and S. L. Schmid, Annu. Rev. Biochem., 2018, 87, 871–896 CrossRef CAS.
- A. I. Ivanov, Methods Mol. Biol. DOI:2014, 1174, 3-18.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2019, 78, 857–902 CrossRef PubMed.
- S. M. Ferguson and P. De Camilli, Nat. Rev. Mol. Cell Biol., 2012, 13, 75–88 CrossRef CAS PubMed.
- S. Mayor, R. G. Parton and J. G. Donaldson, Cold Spring Harbor Perspect. Biol., 2014, 6, a016758 CrossRef PubMed.
- H. T. McMahon and E. Boucrot, Nat. Rev. Mol. Cell Biol., 2011, 12, 517–533 CrossRef CAS PubMed.
- A. Mccluskey, J. A. Daniel, G. Hadzic, N. Chau, E. L. Clayton, A. Mariana, A. Whiting, N. N. Gorgani, J. Lloyd, A. Quan, L. Moshkanbaryans, S. Krishnan, S. Perera, M. Chircop, L. von Kleist, A. B. Mcgeachie, M. T. Howes, R. G. Parton, M. Campbell, J. A. Sakoff, X. Wang, J. Y. Sun, M. J. Robertson, F. M. Deane, T. H. Nguyen, F. A. Meunier, M. A. Cousin and P. J. Robinson, Traffic, 2013, 14, 1272–1289 CrossRef CAS PubMed.
- M. J. Robertson, G. Hadzic, J. Ambrus, D. Y. Pomè, E. Hyde, A. Whiting, A. Mariana, L. Von Kleist, N. Chau, V. Haucke, P. J. Robinson and A. McCluskey, ACS Med. Chem. Lett., 2012, 3(5), 352–356 CrossRef CAS PubMed.
- A. Quan, A. B. McGeachie, D. J. Keating, E. M. Van Dam, J. Rusak, N. Chau, C. S. Malladi, C. Chen, A. McCluskey, M. A. Cousin and P. J. Robinson, Mol. Pharmacol., 2007, 72, 1425–1439 CrossRef CAS PubMed.
- E. Macia, M. Ehrlich, R. Massol, E. Boucrot, C. Brunner and T. Kirchhausen, Dev. Cell, 2006, 10, 839–850 CrossRef CAS PubMed.
- T. Hill, L. R. Odell, J. K. Edwards, M. E. Graham, A. B. McGeachie, J. Rusak, A. Quan, R. Abagyan, J. L. Scott, P. J. Robinson and A. McCluskey, J. Med. Chem., 2005, 48, 7781–7788 CrossRef CAS PubMed.
- S. Lee, K. Y. Jung, J. Park, J. H. Cho, Y. C. Kim and S. Chang, Bioorg. Med. Chem. Lett., 2010, 20, 4858–4864 CrossRef CAS PubMed.
- J. Forman, M. Dietrich and W. Todd Monroe, Photochem. Photobiol. Sci., 2007, 6, 649–658 RSC.
- Q. Dong, K. Svoboda, T. R. Tiersch and W. Todd Monroe, J. Photochem. Photobiol., B, 2007, 88, 137–146 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- N. J. Dunn, W. H. Humphries IV, A. R. Offenbacher, T. L. King and J. A. Gray, J. Phys. Chem. A, 2009, 113(47), 13144–13151 CrossRef CAS PubMed.
- T. Kirchhausen, E. Macia and H. E. Pelish, Methods Enzymol., 2008, 438, 77–93 CAS.
- M. Izquierdo-Serra, M. Gascón-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, È. Ferrer, R. Alibés, F. Busqué, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136(24), 8693–8701 CrossRef CAS.
- L. Nevola, A. Martín-Quirõs, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704–7708 CrossRef CAS PubMed.
- M. Mettlen, P.-H. Chen, S. Srinivasan, G. Danuser and S. L. Schmid, Annu. Rev. Biochem., 2018, 87, 871–896 CrossRef CAS PubMed.
- M. Mettlen and G. Danuser, Cold Spring Harbor Perspect. Biol., 2014, 6, a017038 CrossRef PubMed.
- F. Aguet, C. N. Antonescu, M. Mettlen, S. L. Schmid and G. Danuser, Dev. Cell, 2013, 26(3), 279–291 CrossRef CAS PubMed.
- S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03820b |
| ‡ NC and AT contributed equally. |
| § Present address: IDP Discovery Pharma, Parc Científic de Barcelona (PCB), Spain. |
| This journal is © The Royal Society of Chemistry 2020 |