
Small molecule-mediated self-assembly behaviors of Pluronic block copolymers in aqueous solution: impact of hydrogen bonding on the morphological transition of Pluronic micelles†
Haiyan
Luo
 ab,
Kun
Jiang
ab,
Xiangfeng
Liang
c,
Huizhou
Liu
*a and
Yingbo
Li
*a
ab,
Kun
Jiang
ab,
Xiangfeng
Liang
c,
Huizhou
Liu
*a and
Yingbo
Li
*a
aCAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: hzliu@ipe.ac.cn; ybli@ipe.ac.cn; Fax: +86 10 62554264; Tel: +86 10 62554264
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cCAS Key Laboratory of Bio-Based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266061, China
First published on 12th November 2019
Abstract
The influence of hydrogen bonding on the self-assembly behaviors of Pluronic P123 micelles is experimentally and theoretically investigated by introducing three small molecules, i.e. propyl benzoate (PB), propyl paraben (PP) and propyl gallate (PG) into the aqueous solution. It is discovered that the number of phenolic hydroxyl groups and concentration of the tested small molecules exhibit a profound impact on the micellar morphology. Although all the small molecules increase the size and polydispersity of Pluronic micelles in a concentration-dependent manner, the micellar morphologies induced by them vary considerably as demonstrated by DLS and cryo-TEM measurement. PB, without phenolic hydroxyl, cannot bring about the morphological change of P123 micelles, while PP induces a series of morphological transitions from spheres to long worm-like micelles and then to unilamellar vesicles by increasing the PP content. Upon increasing the number of phenolic hydroxyls in small molecules, i.e. PG, the fusion of the intermicellar core takes place, resulting in the formation of large micelles and micellar clusters. A qualitative study by NMR reveals that the different locations of small molecules within the micelles are attributed to the balance of hydrogen bonding and hydrophobic interaction between small molecules and copolymers. In addition, molecular dynamics simulations (MDS) are performed to further confirm the experimental results and provide quantitative information on intermolecular interaction strength. It is supposed that the mechanism of micellar morphological transition mediated by small molecules is ascribed to the hydrogen bonding interactions with varying strengths between the PEO blocks and their phenolic hydroxyls, which governs their locations in micelles, affecting the free energies from different regions of micelles, and consequently leads to the varying micellar morphologies. This study deepens our understanding of the role of hydrgen bonding in the self-assembly behaviors of Pluronic micelles and provides an alternative strategy for manipulating the nanostructure of Pluronic micelles.
1. Introduction
Amphiphilic block copolymers composed of both hydrophilic and hydrophobic moieties have attracted increasing interest in the recent decades due to their ability to self-assemble into versatile nanostructures with well-defined morphology and size in selective solvents.1 The potential applications of these nanostructures have been well documented in many fields including emulsions, nano-material preparation, electronics, catalysts, separation, etc.2–4 In particular, the employment of polymeric micelles as drug delivery nanocarriers is blooming. Meanwhile, recent findings demonstrated that the morphologies of polymeric micelles have a significant impact on the drug encapsulation capacity and delivery efficiency.5–7 Therefore, research studies on the regulation of morphology of block copolymer micelles is an important issue toward their application, which would not only enrich our knowledge in supramolecular science, but also provide fundamentals for fabricating novel functional materials.In principle, the formation of various equilibrium morphologies of block copolymer aggregates are determined by the free energy of the aggregates from the thermodynamic point of view, which consist of three components: the free energy of the core, which relates to the stretching degree of the core-forming blocks; the free energy of the interface, which relates to the surface tension between the core-forming block and the outside solvent; and the free energy of corona, to which the repulsive interactions among corona-forming blocks contribute.8,9 The morphologies can therefore be fine-tuned by factors affecting any of the three contributions. It has been reported that many factors can affect the above three terms. Generally, these factors can be divided into two categories: copolymer composition that includes the chemical structure of the repeating units of blocks, the size of each block, the overall molecular weight and the architecture.10,11 Another is solution factors that include the concentration of the copolymer, the nature of solvents, the temperature, the pH value and the additives such as salts and homopolymers.12–15
Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO) triblock copolymer, known as Pluronic or Poloxamer, is a typical amphiphilic surfactant that has received considerable attention from both scientific and industrial communities. The aggregation behaviors of this block copolymer in aqueous medium have been extensively studied in the past decades, and fascinating micellar morphologies including spheres, rods, vesicles, lamellae and tubular have been found depending on the solution temperature, composition and concentration of the block copolymers.16–19 More recently, it has been reported that small molecules can also have a profound impact on the morphologies of Pluronic micelles by several research groups. Kim and coworkers have discovered that the P85/5-methyl salicylic acid (5mS) mixture exhibits sphere–rod–vesicle morphological transition as the concentration of 5mS increases, based on which they have prepared unilamellar vesicles by mixing P85 and 5mS in aqueous solution.20 However, how the 5mS induced such a morphological change is not revealed. By the methods of SANS and viscosity, Khimani et al. reported that methyl paraben induces the sphere-to-rod transition of P104 and P105 micelles, while butyl paraben leads to the formation of micellar clusters.21 The formation of micelle clusters induced by butyl paraben is ascribed to the onset of attractive interactions between micelles. Nevertheless, the nature of the driving force of the attractive intermicellar interaction between micelles is not revealed in that work. Despite the underlying mechanism of small molecules influencing the aggregation behaviors remaining poorly understood, these examples inspire us to employ small molecules to tune the morphology of Pluronic micelles.
Indeed, small molecules with various functional groups could interact with block copolymers through various weak interactions, i.e. hydrogen bonding, hydrophobic interaction and electrostatic interaction, etc., which offer many possibilities to vary the microstructures of micelles. Among these weak interactions, hydrogen bonding interaction due to its high selectivity, directionality, saturation, and tunable bonding strength plays a very important role in the formation of specific structures of biomacromolecules such as nucleic acids, proteins, and polysaccharides with explicit functions. In the synthesized polymer field, hydrogen bonding interaction has also been extensively employed to coordinate the physical behaviors of the block copolymer in the bulk state. In comparison, the hydrogen bonding interaction is rarely used to manipulate the micellar morphologies of block copolymers in selective media. Zhu et al. reported that a variety of nanostructures of the triblock copolymer poly(4-vinylpyridine)43-b-polystyrene366-b-poly(4vinylpyridine)43 (P4VP43-b-PS366-b-P4VP43) could be obtained by introducing different amounts of pentadecylphenol (PDP) into a dilute solution.22 A recent study by Guo et al. demonstrated that different micellar morphologies of the diblock copolymer PS-b-P4VP were formed which were induced by octyl gallate (OG) in different common solvents.23 The nature of different common solvents affects the strength of the hydrogen bonding interaction between PS-b-P4VP and OG, resulting in the formation of various nanostructures. Until now, few studies on the regulation of Pluronic micellar morphologies by hydrogen bonding interactions in aqueous solution are reported.
In the present work, the effects of three small molecules, i.e. propyl benzoate (PB), propyl paraben (PP) and propyl gallate (PG) on the self-assembly behavior of the PEO–PPO–PEO triblock copolymer in aqueous solution were investigated by a combination of experimental methods and molecular simulations. These molecules possess different numbers of phenolic hydroxyl groups and are thus selected as model molecules to study how the hydrogen bonding and its strength affect the nanostructure of Pluronic micelles. It was found that PP and PG having phenolic hydroxyls induced the morphological transition of PEO–PPO–PEO block copolymer micelles, while PB without the phenolic hydroxyl did not. This implies that the hydrogen bonding interaction between small molecules and the PEO block of copolymers plays a significant role in switching morphologies. To explore the nature of these phenomena, qualitative and quantitative research on the interaction between these small molecules and block copolymers by NMR and molecular dynamic simulations (MDS) were performed in detail. On the basis of experimental findings and MDS results, the mechanism of micelle morphological transition of PEO–PPO–PEO triblock copolymers induced by the tested small molecules is discussed and suggested.
2. Experimental section
2.1 Materials
The triblock copolymer Pluronic P123 (EO20PO70EO20, Fig. 1), propyl benzoate (PB), propyl paraben (PP) and propyl gallate (PG) were purchased from Sigma-Aldrich and used as received. The chemical structure and octanol–water partition coefficients of PB, PP and PG are presented in Fig. 1. The reference 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS, ≥97%) and D2O (≥99.9%) was also purchased from Sigma-Aldrich.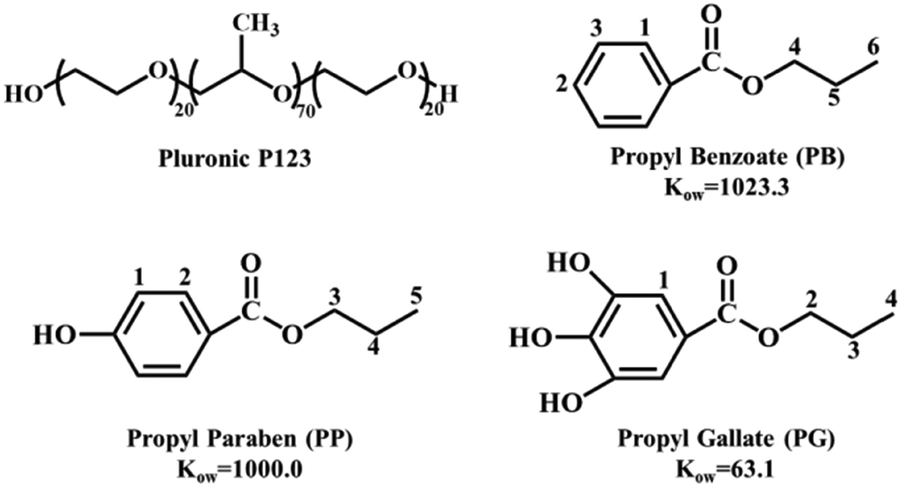 | ||
| Fig. 1 Chemical structures of Pluronic P123, propyl benzoate, propyl paraben and propyl gallate. | ||
2.2 Dynamic light scattering (DLS)
DLS measurements were performed using a commercial LLS spectrometer (ALV/CGS-3) equipped with a multi-digital time correlator (LSE-5004). The light source was a cylindrical He–Ne laser operated at 623.8 nm with a maximum output power of 22 mW. The samples were prepared with Milli-Q water followed by filtration through 0.22 μm Millipore filters into a dust-free light scattering cell. The scattering angle is 90°. All samples were allowed to equilibrate at 30 °C for at least 1 hour before measurement. Each measurement was repeated three times. The apparent hydrodynamic radii (Rh) of scatterers were calculated using the Stokes–Einstein equation Rh = kBT/6πηD, where kB is the Boltzmann constant, η is the viscosity of the solvent, T is the temperature and D is the diffusion coefficient.2.3 Nuclear magnetic resonance (NMR)
NMR experiments were conducted using a Bruker Avance 700 spectrometer with a 1H frequency of 700.13 MHz. The temperature was controlled by a Bruker BVT 3200 temperature control unit with an accuracy of 0.1 °C. All samples were prepared in D2O and DSS was used as an external reference standard to eliminate temperature-induced shifts. For all the experiments, the samples were equilibrated at 30 °C for at least 10 min before each measurement. The interaction sites of P123 with tested small molecules were determined by NOESY experiments. For each sample, 48 cans for each of the 2048 increments were recorded. The mixing time was 300 ms.2.4 Cryogenic transmission electron microscopy (cryo-TEM)
The vitrified specimens for cryo-TEM were prepared in a controlled environment vitrification system at 30 °C and 100% relative humidity. A droplet (∼3.5 μL) of the sample was placed on a perforated carbon-coated copper grid, blotted with filter paper, and then the sample was vitrified by plunging the grid into liquid ethane at its freezing point. The vetrified specimens were transferred to a Gatan cryoholder and imaged using a FEI tecnai 20 transmission electron microscope operating at 200 kV.2.5 Fourier transform infrared spectroscopy (FTIR)
FTIR spectra were recorded using a Bruker Tensor 27 FTIR spectrometer with a resolution of 2 cm−1 using a DLATGS detector. The bubble-free samples were sandwiched between two BaF2 windows of an IR cell. Each IR spectrum was an average of 64 scans using a spectral resolution of 4 cm−1. A background spectrum was obtained directly before the sample spectra. The OPUS spectroscopic software was used for data handling.2.6 Molecular dynamics simulation (MDS)
All molecular dynamics simulations were performed using the GROMACS (version 5.1.2) software package, and VMD was used for visualization purposes.24,25 The inter- and intra-molecular interactions in the system were described using the OPLS all-atom force field.26 Details of simulated systems are shown in Table 1. For all the simulations, the temperature was kept constant at 303 K using the Nosé–Hoover thermostat with a coupling constant of 0.1 ps. The pressure was maintained constant at 1 bar using a Berendsen barostat with a coupling constant of 1 ps. The bond lengths were constrained using the LINCS algorithm. An integration time step of 2 fs was used for all the simulations. Electrostatic interactions were evaluated using the particle mesh Ewald method with a real space cutoff of 1.2 nm and a grid spacing of 0.16 nm. Lennard-Jones interactions were truncated at 1.2 nm.| Components (number of molecules in parentheses) | Box size (x × y × z)/nm3 | Number of atoms |
|---|---|---|
(10)P123 + (50)PB + (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000)H2O 000)H2O |
12.38 × 12.38 × 12.38 | 191030 |
| (10)P123 + (50)PP + (60000)H2O |
12.38 × 12.38 × 12.38 | 191080 |
| (10)P123 + (50)PG + (60000)H2O |
12.38 × 12.38 × 12.38 | 191180 |
3. Results and discussion
3.1 Morphological transition of P123 induced by adding small molecules
The self-assembly behaviours of 5.0% (w/w) P123 without or with the tested small molecules are presented in Fig. 2. It is observed that the pure P123 solution is very transparent at 30 °C. Upon addition of small molecules, the solution shows a bluish colour, which is known to have appeared due to the Tyndall effect due to the presence of large scatterers in solution. Upon further increasing the concentration of PP and PG, the scattering intensity of blue light increases, and the solution even becomes opaque, which indicates the formation of larger aggregates such as worm like micelles and vesicles. These observations are similar to the reports by Kim and Davies et al.20,27,28 The size of the micelles is probed by DLS, where a unimodal distribution of Rh in pure P123 solution with an average Rh of 8.3 nm is displayed. This value is in good agreement with the literature value.29 When PB is added into the solution, the values of Rh and polydispersity (PDI) of micelles increase very slightly at all the tested concentration ranges (Fig. 2a), reflecting that PB can gently induce the growth of the micelle, but without any morphological changes. In the case of PP (Fig. 2b), both Rh and PDI of micelles are virtually not altered upon addition of PP below 20 mM. However, a tri-modal distribution of Rh with values ranging from about 10 nm to over 100 nm appears when the concentration of PP reaches 30 mM. This suggests that PP at high concentration would bring about a distinct growth of micelles and consequently a shape transition of micelles, e.g. form spherical micelles to cylindrical or wormlike micelles, as well as vesicles.30,31 Compared with PB and PP, PG with three phenolic hydroxyls starts to induce an obvious turbidity in the P123 solution at a lower concentration, i.e. 20 mM. Moreover, the micellar solution containing PG is much cloudier than those containing PB or PP at each equal concentration. Correspondingly, the size of the micelles increases notably in the presence of 20 mM PG (Fig. 2c). As in the case of PG, the micelle solution is composed of three populations with corresponding hydrodynamic radii of 26.1, 301.1 and 2559 nm when the concentration of PG reaches 40 mM. Such a large increase in micelle dimensions indicates the formation of micelle clusters due to the attractive intermicellar interaction.21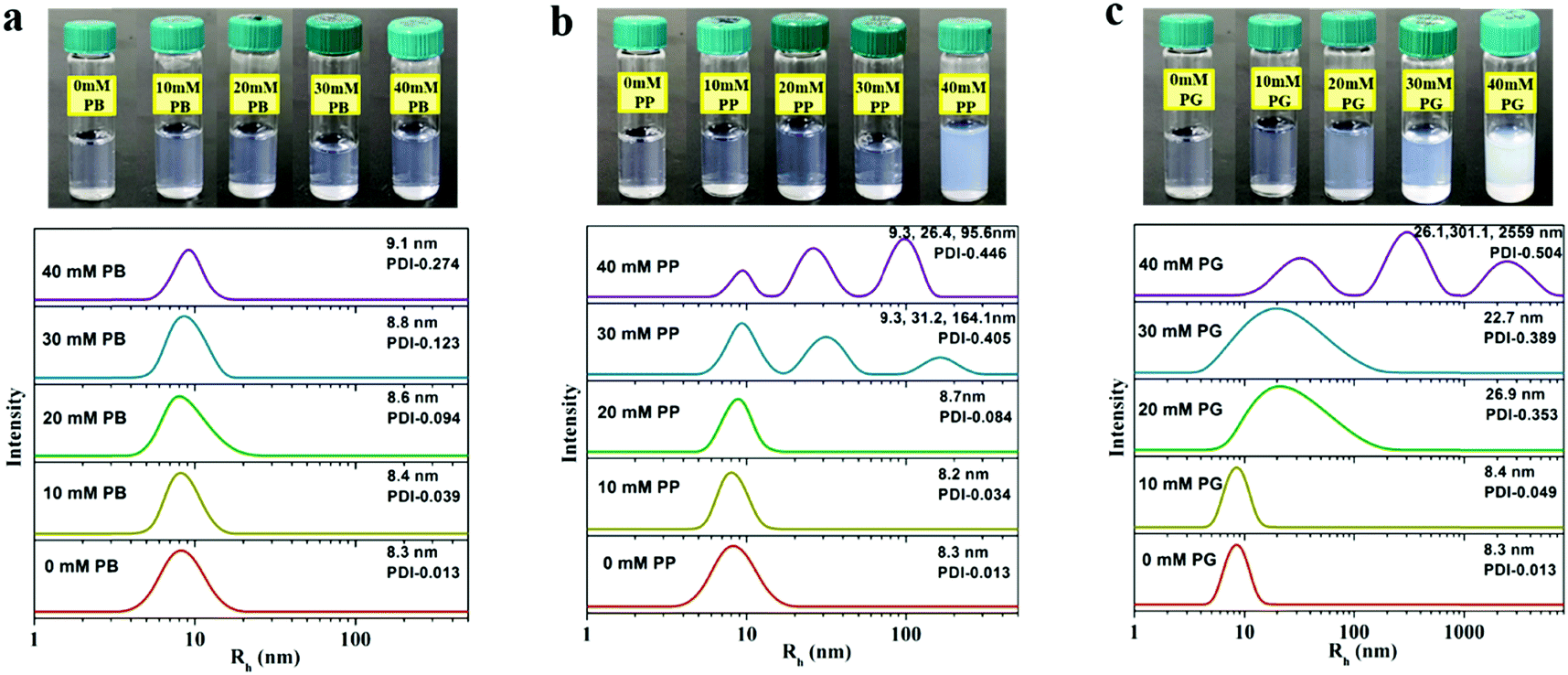 | ||
| Fig. 2 The photos of P123-small molecules mixture and corresponding hydrodynamic radius (Rh) distribution of micelles in solution at 30 °C depending on the concentration of PB (a), PP (b) and PG (c). | ||
It should be noted that the hydrodynamic radius of the aggregate obtained by DLS corresponds to the radius of spheres with the same diffusion coefficient. However, it cannot always be translated directly as the size of aggregates because the attractive potential between aggregates could also decrease the diffusion coefficient, making the aggregates look much larger than they actually are. So it's difficult to clarify whether the increase in the hydrodynamic radius of micelles is caused by the increase of micellar size or the attractions of micelles only by DLS. In order to interpret this, cryo-TEM measurements are employed to investigate visually the morphology of micelles in aqueous solution.
The cryo-TEM images of P123 micelles in the absence and presence of small molecules are shown in Fig. 3. The spherical micelles with a size of about 16 nm are observed in the pure 5.0% P123 solution (Fig. 3a), which is consistent with the DLS measurement. After addition of 40 mM PB, there are mainly spherical micelles with a size of about 18.0 nm in solution (Fig. 3b), which confirms the deduction by DLS data that PB slightly increases the size of the micelles but does not alter the micellar morphology. Considering that the size distribution of P123 micelles starts to change in the presence of 30 mM PP (Fig. 2b), cryo-TEM measurement is performed for this sample. As displayed in Fig. 3c, long wormlike micelles and small spherical micelles coexist in solution. The presentative wormlike micelle could be clearly seen from the inset of Fig. 3c. Meanwhile, some wormlike micelles curl themselves to enclose a few globular micelles (indicated by arrow in Fig. 3c). Upon increasing the concentration of PP to 40 mM, unilamellar vesicles with a wall thickness of about 5 nm are formed (Fig. 3d), and there are also very small amounts of long wormlike micelles surrounding the small spherical micelles (inset of Fig. 3d). These results suggest that the micelles are first transformed from sphere to long wormlike shape, then to the unilamellar vesicles as the concentration of PP increases in P123 solution. Although unilamellar polymer vesicles have many potential applications in nano-, bio-, or medical research, reports on the PEO–PPO–PEO vesicle formation in water remain scanty.32–36 Recently, it has been reported that 5-methylsalicylic acid can also induce the formation of unilamellar vesicles in Pluronic P85 solution.20 However, the mechanism of morphological transition of micelles induced by these small molecules with phenolic hydroxyl groups still remains unknown. In the case of PG, spherical micelles with a size of about 20–40 nm are observed and some of them stick together in the presence of 30 mM PG (Fig. 3e). As the concentration of PG reaches 40 mM, spherical micelles with various sizes are observed. The large spherical micelles are most likely formed by the continuous fusion of small spherical micelles. Besides, micelles with different sizes attract each other to form a large micellar cluster, whose size is above one micrometer (Fig. 3f). The formation of Pluronic micelle clusters induced by inorganic salt has been previously reported, which is ascribed to the specific dehydration of micellar corona.19 In the present work, the small organic molecule PG, not like salt, may have an alternative mode of action on the restructuring of micelles to form micelle clusters.
 | ||
| Fig. 3 Cryo-TEM images of 5.0% P123 (a) and 5.0% P123 with 40 mM PB (b), 30 mM PP (c), 40 mM PP (d), 30 mM PG (e) and 40 mM PG (f) at 30 °C. | ||
It is evident that both the phenolic hydroxyl of the tested small molecules and their concentration exert a significant influence on the morphologies of P123 micelles based on the results of DLS and cryo-TEM. Strikingly, only PP and PG with phenolic hydroxyl can bring about the morphological transition of micelles, while those without phenolic hydroxyl (PB) cannot. It has been reported that phenolic hydroxyl can form hydrogen bonding with an ether oxygen atom of PEO.37,38 One can thus reasonably envisage that a hydrogen bonding interaction between the phenolic hydroxyl of small molecules and PEO blocks would play a significant role in manipulating the micelle morphologies. In order to reveal the underlying mechanism, the intermolecular interaction between small molecules and the P123 block copolymer is investigated by NMR spectroscopy in the following section.
3.2 Interactions between small molecules and P123 copolymers
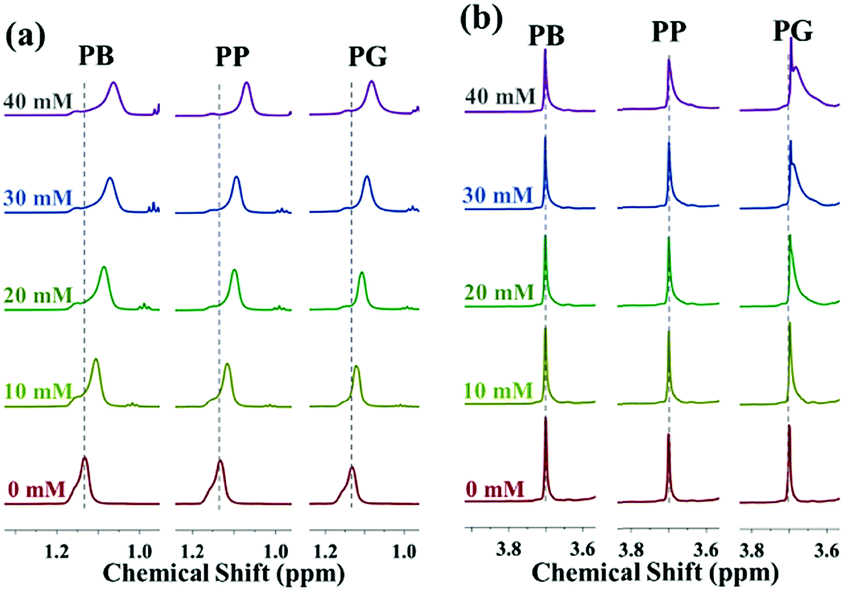 | ||
| Fig. 4 1H NMR spectra of PO methyl protons (a) and EO methylene protons (b) of P123 in the presence of different concentration of additives at 30 °C. The concentration of P123 was constant at 5.0% in solutions. | ||
| Small molecules | Chemical shift/ppm | ||
|---|---|---|---|
| PO-CH3 | EO-CH2 | HDO | |
| 0 mM | 1.133 | 3.701 | 4.722 |
| 10 mM PB | 1.105 | 3.700 | 4.722 |
| 20 mM PB | 1.086 | 3.700 | 4.722 |
| 30 mM PB | 1.071 | 3.700 | 4.722 |
| 40 mM PB | 1.063 | 3.700 | 4.722 |
| 10 mM PP | 1.116 | 3.700 | 4.722 |
| 20 mM PP | 1.099 | 3.700 | 4.722 |
| 30 mM PP | 1.094 | 3.699 | 4.722 |
| 40 mM PP | 1.070 | 3.699 | 4.722 |
| 10 mM PG | 1.122 | 3.699 | 4.722 |
| 20 mM PG | 1.107 | 3.698 | 4.722 |
| 30 mM PG | 1.094 | 3.696 | 4.722 |
| 40 mM PG | 1.083 | 3.695 | 4.722 |
Compared to the variation in the chemical shift of PPO-CH3, the extent of upfield shift of EO-CH2 protons in the presence of small molecules is much less remarkable. There are almost no changes in the chemical shift of EO-CH2 after addition of PB and PP while the proton signals of the EO-CH2 shift upfield slightly in the presence of PG (Fig. 4b). This is because the hydrogen bonding interactions between small molecules and PEO blocks become stronger as the number of phenolic hydroxyl increases, which results in their greater ability to dehydrate the PEO corona. Moreover, it could be deduced that the polar aromatic ring of PG appears to orient towards the PEO corona while the alkyl chain points to the PPO core. Usually, the additives affect the microenvironment of the block copolymer in solution mainly via directly interacting with the copolymer or the indirect interactions (changing the structures of water).42 As shown in Table 2, the chemical shift of HDO in the P123 D2O solution remains unchanged despite the increasing addition of small molecules. This suggests that small molecules interact directly with the block copolymer by either hydrophobic interactions or hydrogen bonding interactions.
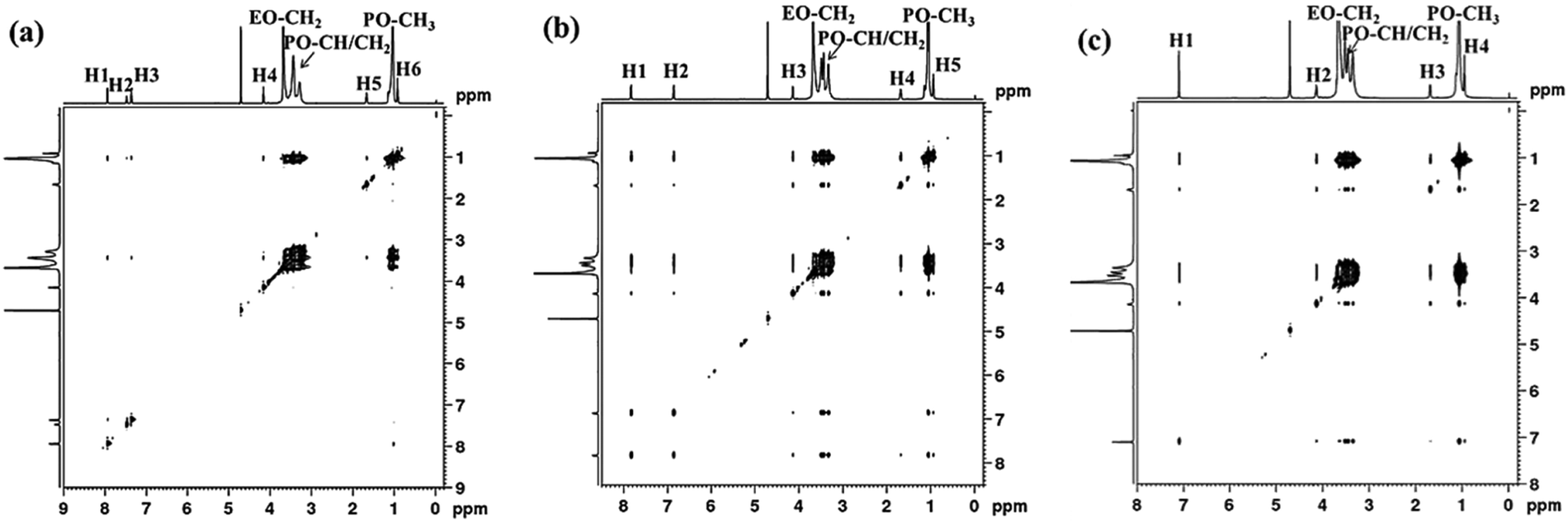 | ||
| Fig. 5 NOESY spectra of 5.0% P123 with 40 mM PB (a), PP (b) and PG (c) at 30 °C. | ||
For the system of P123 with PP, the protons on the aromatic ring (H1, δ ≈ 7.8 ppm, H2, δ ≈ 6.9 ppm) interact with both PEO (–CH2) and PPO (–CH, –CH2, and –CH3) blocks, while the alkyl chain protons (H3, H4, H5) only interact with PPO blocks as shown in Fig. 5b. This result suggests that although PP molecules are mainly located at the PPO core, their aromatic rings are situated at the core/corona interface due to the hydrogen bonding interaction between the phenolic hydroxyl and oxygen atoms of PEO, which is quite different from PB molecules. As for PG, its aromatic protons (H1, δ ≈ 7.1 ppm) and methylene protons (H2, δ ≈ 4.1 ppm) adjacent to the ester interact with both PEO and PPO blocks. Meanwhile, the residual alkyl chain protons H3 (δ ≈ 1.7 ppm) and H4 (δ ≈ 1.7 ppm) interact with only PPO blocks (Fig. 5c). In this case, the hydrogen bonding interaction between phenolic hydroxyl of PG and PEO blocks becomes stronger, dragging PG towards the micelle corona. As a result, the aromatic ring, the ester group and the H2 proton of PG molecules are located at the core/corona interface, while the residual alkyl chains point to the PPO core. In short, the above results demonstrate that the hydrogen bonding interaction between small molecules and PEO blocks pulls the small molecules away from the micelle core to corona, making them locate at different sites within micelles, and consequently result in diverse micelle morphological transitions. The formation of hydrogen bonding between the phenolic hydroxyl and ether groups of PEO has been reported.37,38,46 Our FTIR measurements also confirm the formation of hydrogen bonding between the hydroxyl of PP or PG and P123, while PB could not (Fig. S1, ESI†).
3.3 Molecular dynamics simulations
For quantitatively studying the interaction strength between small molecules and block copolymers and further understanding the underlying mechanism of the morphological transition of Pluronic micelles induced by the tested small molecules, molecular dynamics simulations (MDS), a powerful tool to investigate the interaction between additives and block copolymers, are performed.47,48 The final conformations of the simulation system with ten P123 chains and fifty small molecules in water after 50 ns are shown in Fig. 6. After the simulation system reaches equilibrium, the P123 copolymers self-assemble to form aggregates with the PPO core (green) and the outer PEO corona (red). All PB molecules are incorporated into the internal PPO region (Fig. 6a), which confirms the results of NOESY. The PP and PG molecules are mainly located at the PO/EO interface, while a small amount of them are still dispersed in water (Fig. 6b and c). However, PP molecules are closer to the PPO core as there are no entanglements between PP molecules (black) and PEO segments (red), while the PG molecules (blue) contact with both of PPO segments (green) and PEO segments (red). These results also agree well with the deduction of NMR data. However, due to the relatively smaller system of simulation than the experiments, we can’t obtain the structural change of micelles as acquired by the DLS and cryo-TEM.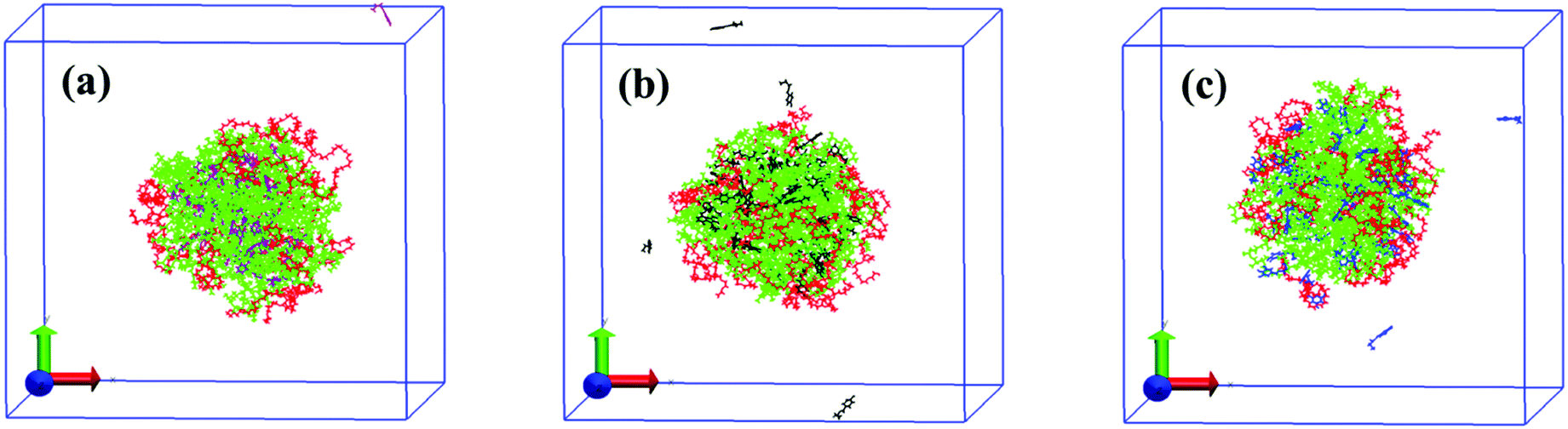 | ||
| Fig. 6 Snapshots of final conformations of the P123 chains and PB (a), PP (b) and PG (c) molecules after a 50 ns simulation. The PEO blocks of P123 is shown in red, PPO blocks in green, PB in purple, PP in black and PG in blue. The water molecules are not shown for clearer views. | ||
In this case, there are two kinds of interactions between the studied three kinds of small molecules and P123 copolymer, i.e. hydrogen bonding and hydrophobic interactions, which determine the equilibrium morphologies of micelles. These interactions are studied by the analysis of the radial distribution functions (RDFs) and interaction energies. RDFs, also known as pair correlation functions g(r), is a powerful statistic mechanic tool to probe the correlation between the positions of two atoms, e.g. α and β, as they are related to the probability to find an atom β at a distance r from an atom α.49 Therefore, RDFs can provide crucial information about the interactions between various atoms. The RDFs between aromatic hydrogen (H2 for PB, p-phenolic hydroxyl for PP and PG), methyl hydrogen of small molecules and oxygen atoms of PPO and PEO are shown in Fig. 7a and b, respectively. The first notable peak at around 0.3 nm and the strong peak at 0.5 nm in Fig. 7a suggest that there are specific interactions between aromatic hydrogen and the oxygen atoms of both PEO and PPO blocks. It also can be seen that the amplitude of the aromatic hydrogen/oxygen atom of PEO enhances as the number of phenolic hydroxyl group increases, suggesting that the more the number of phenolic hydroxyls the molecule possesses, the stronger the hydrogen bonding between small molecules and PEO blocks. In contrast, the magnitude of the aromatic hydrogen/oxygen atom of PPO follows the order PB > PP > PG. In another aspect, it should be noted that the peak of aromatic proton/oxygen of PPO is higher than that of aromatic proton/oxygen of PEO in the case of PB and PP, while they are equivalent in the case of PG. These results suggest that the interactions of the aromatic ring of PB and PP with PPO blocks are stronger than those with PEO blocks, while the aromatic ring of PG exhibits the same interaction strength with both PPO and PEO. Again, this explains why the locations of various molecules vary within the Pluronic micelles.
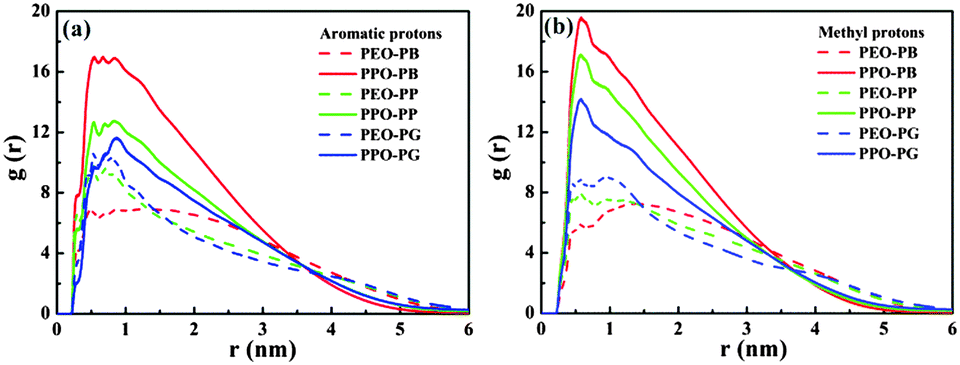 | ||
| Fig. 7 Radial distribution functions of (a) phenolic hydroxyl hydrogen of PB/PP/PG relative to oxygen atoms of PEO and PPO blocks, (b) methyl hydrogen of PB/PP/PG relative to oxygen atom of PEO and PPO blocks. | ||
The peak height of methyl hydrogen/oxygen atoms of PPO is much higher than that of methyl proton/oxygen atoms of PEO blocks for all systems (Fig. 7b), indicating that the methyl of small molecules has stronger attractive interactions with PPO than PEO blocks due to their hydrophobic nature. Unsurprisingly, such an interaction becomes weaker as the number of phenolic hydroxyls of the molecules increases. Similar to the case of aromatic hydrogen, the amplitude of methyl hydrogen/oxygen atoms of PEO increases as the number of phenolic hydroxyls of molecules increases (in the order of PB < PP < PG), which is ascribed to the fact that PG molecules are closer to the PEO corona than PP and PB (Fig. 6). This result is fully consistent with the above NMR measurements.
The interaction energies between small molecules and P123 copolymers are further analyzed and shown in Fig. 8. During the first 20 ns, the total interaction energies between P123 and PB/PP/PG increase with time. From 20 ns to 50 ns, the values of interaction energies virtually remain unchanged, indicative of an equilibrium state. Values of interaction energies of PB/PP/PG with P123 copolymers, as well as with the PPO and PEO blocks estimated from 30 to 50 ns (equilibrium state), are shown in Table 3. The values of interaction energies increase as the number of phenolic hydroxyls of molecules increases, although there are little differences for PP (4608.8 kJ mol−1) and PG (4636.9 kJ mol−1). It is worth noting that the interaction energies between small molecules and PPO blocks are much higher than that with PEO blocks. Moreover, the interaction energies of small molecules with PEO blocks increases notably as the number of phenolic hydroxyls increases while the interaction energies with PPO blocks change oppositely. These results indicate that the hydrogen bonding interactions between small molecules and PEO blocks regulate the formation of various morphologies of micelles, even though the hydrophobic interactions are much stronger (Table 3). Furthermore, the interaction energy of PB/PP/PG with each EO and PO unit indicates that the interaction between PB/PP/PG and PO units is stronger than that of the EO unit, but the difference in interaction strength of small molecules with PO and EO units becomes smaller as the number of phenolic hydroxyls increases, which is due to the increasing strength of hydrogen bonding between phenolic hydroxyl groups and ether oxygen atoms of EO.
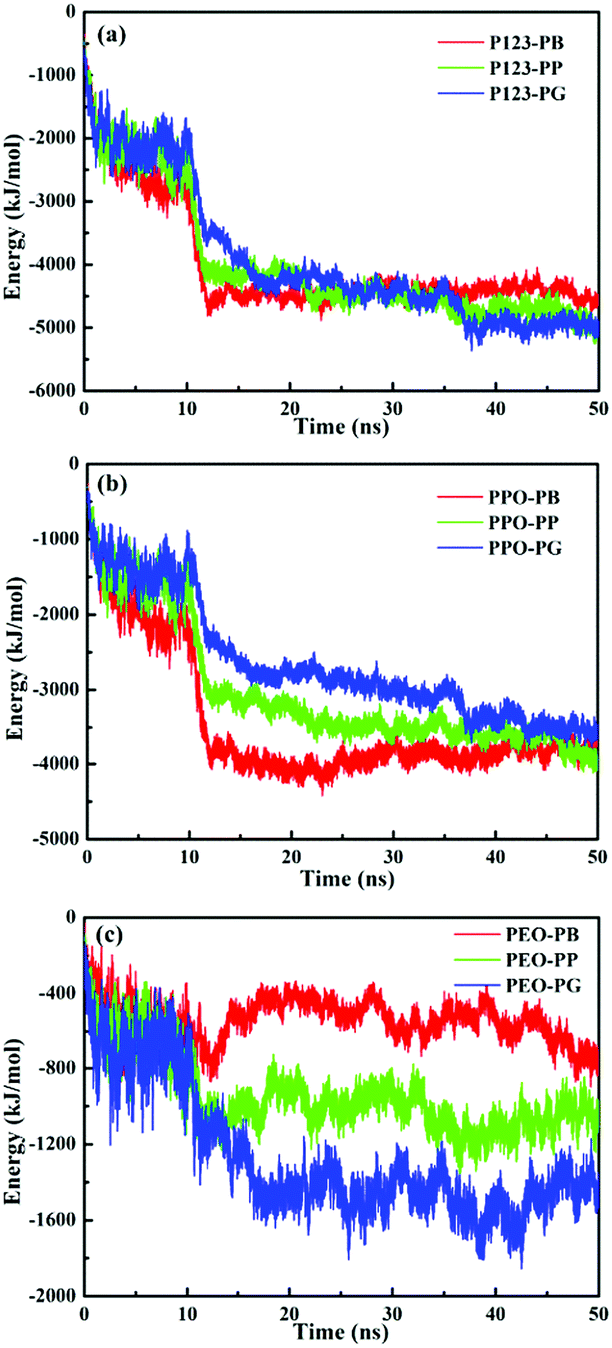 | ||
| Fig. 8 Total interaction energies between PB/PP/PG and P123 (a), PPO blocks (b) and PEO blocks (c) vs. simulation time. | ||
| Molecule | P123 | PPO | PEO | PO | EO |
|---|---|---|---|---|---|
| PB | −4434.4 | −3886.3 | −548.1 | −56.3 | −14.4 |
| PP | −4608.8 | −3568.4 | −1040.4 | −51.7 | −27.4 |
| PG | −4636.9 | −3166.2 | −1470.7 | −45.9 | −38.7 |
3.4 Mechanism of morphological transition of P123 micelles induced by small molecules
Based on the results of experiments and simulations, a mechanism of morphological transition of Pluronic micelles induced by the three tested molecules is proposed in Fig. 9. P123 copolymers form spherical micelles comprising a PPO core (orange) and PEO corona (light-blue) in solution. After introduction of various small molecules, the discrepancy in the intermolecular interaction of small molecules and copolymers led to the different locations of molecules within micelles, resulting in diverse changes in micelle morphologies.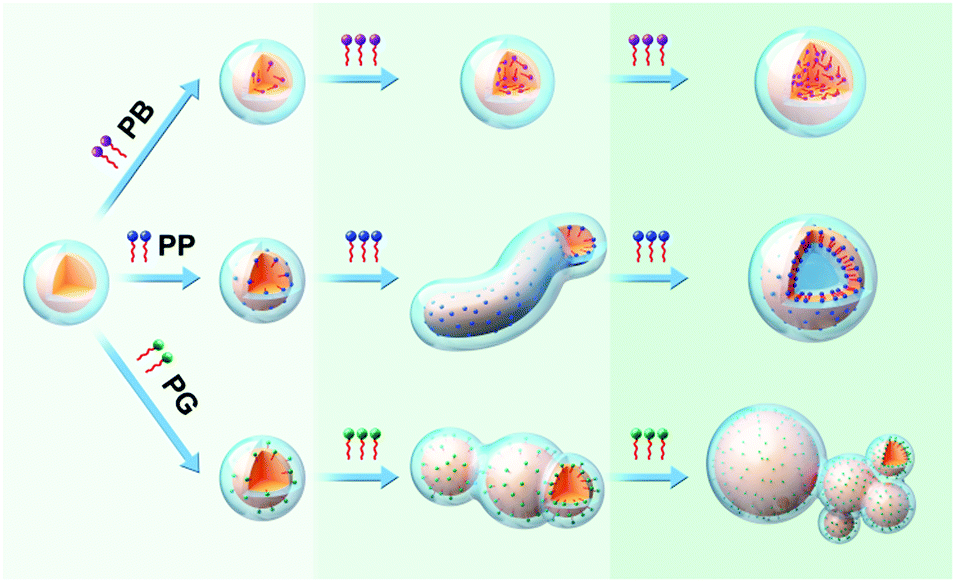 | ||
| Fig. 9 Schematic illustrations of morphological transition of P123 micelles induced by PB, PP and PG, respectively. | ||
As stated in the introduction section, there are mainly three factors (stretching of the core-forming blocks, interfacial energy between core and the outside solvent, and the repulsion interaction among corona) that govern the morphology of block copolymer micelles. Upon addition of the small molecules into the micelle solution, these three factors are affected. PB that only has hydrophobic interactions with PPO blocks of the copolymer is incorporated into the interior of the micelle core. Thus, it may mainly affects the core region. Upon introducing PB into the micelle solution, the H2O molecules around the PPO blocks are replaced by PB molecules, and there is no change in the size of the micelle core and the stretching of PPO blocks. Upon further increasing the PB concentration, more PB molecules are inserted into the micelle core bringing about a slight increase in the core size. Accordingly, the stretching of PPO blocks also enhances but to a very limited extent, which cannot induce the morphological transition of micelles. For PP, apart from hydrophobic interaction, the phenolic hydroxyl of PP can form hydrogen bonding with oxygen atoms of PEO blocks, which makes the PP molecule locate at the outer core with its aromatic ring on the core/corona interface. Therefore, PP can not only influence the micelle core zone but also the core–corona interface. At a low concentration, the effect of PP on the micelle is similar to that of PB. However, as the amount of PP molecules increases, the interfacial energy between the micelle core and the outside solvent is increased besides the increase in the stretching of PPO blocks. Therefore, in order to minimize the total energy of the system, the morphological transformation of sphere-to-wormlike-to-vesicle takes place. In the case of PG, the three phenolic hydroxyl groups endow it much stronger hydrogen bonding with PEO blocks, pulling PG towards the PEO corona, as a result, most parts of the PG molecules are situated at the core/corona interface and only the terminal methyl and its adjacent methylene are located in the PPO core. This specific location of PG within micelles endow them with the ability to influence the micelle core, core/corona interface and micelle corona. The most part of PG in the PPO/PEO interface and the residual alkyl chain in the PPO core could enhance the hydrophobicity of the core/corona interface and the PPO core, respectively. Moreover, a strong hydrogen bonding interaction of the phenolic hydroxyl of PG with PEO decreases the repulsion interactions between PEO chains, therefore the PEO chain around the PPO core cannot shield the hydrophobic PPO core completely since the PEO chains are rather short in P123, resulting in the fusion of micelles through hydrophobic interactions between PPO cores to form large micelles and micelle clusters.
4. Conclusion
In this work, we employed experimental methods and MD simulations to comprehensively investigate the effect of small molecules, i.e. PB, PP and PG on the self-assembly behaviour of Pluronic triblock copolymers. PP possessing one phenolic hydroxyl induces a series of morphological transitions from spheres, to long worm-like aggregates, to unilamellar vesicles by increasing the PP content. The formation of micellar clusters is observed when PG is introduced into the Pluronic aqueous solution. In contrast, PB, with no phenolic hydroxyl, could not alter the morphology of P123 micelles in the studied concentration range. Qualitative and quantitative studies on the interaction between small molecules and the P123 copolymer by NMR and MDS have affirmed that the hydrogen bonding interaction between the phenolic hydroxyl of small molecules and PEO blocks plays a significantly important role in the regulation of micelle morphologies, even though the hydrophobic interaction between them are much stronger. The results obtained in this work disclose new insights into the underlying mechanism of micellar morphology transition induced by hydrogen boding and provide a useful guidance for the regulation of the Pluronic micelle morphology.Conflicts of interest
All authors have no conflict of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21506221 and 21676283) and the National Key R&D Program of China (2016YFB0301702). We would like to thank the Center for Biological Imaging (CBI), Institute of Biophysics, Chinese Academy of Sciences, for our cryo-TEM work.References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- P. Alexandridis and B. Lindman, Amphiphilic block copolymers: self-assembly and applications, Elsevier, Amsterdam, 2000 Search PubMed.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- Y. D. Wang, Q. Gan, C. Y. Shi, X. L. Zheng, S. H. Yang, Z. M. Li and Y. Y. Dai, Chem. Eng. J., 2002, 88, 95–101 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- X. Wan, Y. Min, H. Bludau, A. Keith, S. S. Sheiko, R. Jordan, A. Z. Wang, M. Sokolsky-Papkov and A. V. Kabanov, ACS Nano, 2018, 12, 2426–2439 CrossRef CAS.
- T. Chen, X. Guo, X. Liu, S. Shi, J. Wang, C. Shi, Z. Qian and S. Zhou, Adv. Healthcare Mater., 2012, 1, 214–224 CrossRef CAS.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- Y. Zhu, C. Yi, Q. Hu, W. Wei and X. Liu, Phys. Chem. Chem. Phys., 2016, 18, 26236–26244 RSC.
- L. Fan, M. Degen, N. Grupido, S. Bendle and P. Pennartz, Mater. Sci. Eng., A, 2010, 528, 127–136 CrossRef.
- R. T. Pearson, N. J. Warren, A. L. Lewis and S. P. Armes, Macromolecules, 2013, 46, 1400–1407 CrossRef CAS.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
- L. Zhang, H. Shen and A. Eisenberg, Macromolecules, 1997, 30, 1001–1011 CrossRef CAS.
- P. Bhargava, J. Zheng, P. Li, R. P. Quirk, F. W. Harris and S. Z. D. Cheng, Macromolecules, 2006, 39, 4880–4888 CrossRef CAS.
- L. Yang and P. Alexandridis, Langmuir, 2000, 16, 8555–8561 CrossRef CAS.
- K. Schillén, W. Brown and R. M. Johnsen, Macromolecules, 1994, 27, 4825–4832 CrossRef.
- R. Ganguly, N. Choudhury, V. K. Aawal and P. A. Hassan, J. Phys. Chem. B, 2009, 113, 668–675 CrossRef CAS.
- S. L. Nolan, R. J. Phillips, P. M. Cotts and S. R. Dungan, J. Colloid Interface Sci., 1997, 191, 291–302 CrossRef CAS.
- T.-H. Kim, C. Song, Y.-S. Han, J.-D. Jang and M. C. Choi, Soft Matter, 2014, 10, 484–490 RSC.
- M. Khimani, R. Ganguly, V. K. Aswal, S. Nath and P. Bahadur, J. Phys. Chem. B, 2012, 116, 14943–14950 CrossRef CAS.
- J. Zhu, H. Yu and W. Jiang, Macromolecules, 2005, 38, 7492–7501 CrossRef CAS.
- S. C. Chen, S. W. Guo and F. C. Chang, Langmuir, 2011, 27, 10197–10205 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- T. S. Davies, A. M. Ketner and S. R. Raghavan, J. Am. Chem. Soc., 2006, 128, 6669–6675 CrossRef CAS.
- T.-H. Kim, Y.-S. Han, J.-D. Jang and B.-S. Seong, J. Appl. Crystallogr., 2014, 47, 53–59 CrossRef CAS.
- S. A. Pillai, V. I. Patel, D. Ray, H. Pai, V. K. Aswal and P. Bahadur, Colloids Surf., A, 2018, 559, 314–324 CrossRef CAS.
- P. Parekh, R. Ganguly, V. K. Aswal and P. Bahadur, Soft Matter, 2012, 8, 5864–5872 RSC.
- S. Ghosh, J. Kuchlyan, D. Banik, N. Kundu, A. Roy, C. Banerjee and N. Sarkar, J. Phys. Chem. B, 2014, 118, 11437–11448 CrossRef CAS.
- K. Schillen, K. Bryskhe and Y. S. Mel’Nikova, Macromolecules, 1999, 32, 6885–6888 CrossRef CAS.
- K. Bryskhe, J. Jansson, D. Topgaard, K. Schillén and U. Olsson, J. Phys. Chem. B, 2004, 108, 9710–9719 CrossRef CAS.
- C. Nardin, T. Hirt, J. Leukel and W. Meier, Langmuir, 2000, 16, 1035–1041 CrossRef CAS.
- S. Chen, B. Yang, C. Guo, J. Ma, L. Yang, X. Liang, C. Hua and H. Liu, J. Phys. Chem. B, 2008, 112, 15659–15665 CrossRef CAS.
- G. K. S. Prameela, B. V. N. Phani Kumar, R. Ravikanth Reddy, A. Pan, J. Subramanian, S. Kumar, V. K. Aswal, J. Kohlbrecher, A. B. Mandal and S. P. Moulik, Phys. Chem. Chem. Phys., 2017, 19, 31747–31755 RSC.
- S. Kuo, C. Lin and F. Chang, Macromolecules, 2002, 35, 278–285 CrossRef CAS.
- H. W. Chen, C. H. Jiang, H. D. Wu and F. C. Chang, J. Appl. Polym. Sci., 2004, 91, 1207–1216 CrossRef CAS.
- K. I. Momot, P. W. Kuchel, B. E. Chapman, P. Deo and D. Whittaker, Langmuir, 2003, 19, 2088–2095 CrossRef CAS.
- C. A. Bunton and C. P. Cowell, J. Colloid Interface Sci., 1988, 122, 154–162 CrossRef CAS.
- J. Ma, C. Guo, Y. Tang, H. Zhang and H. Liu, J. Phys. Chem. B, 2007, 111, 13371–13378 CrossRef CAS.
- B. Bharatiya, C. Guo, J. H. Ma, O. Kubota, K. Nakashima and P. Bahadur, Colloid Polym. Sci., 2009, 287, 63–71 CrossRef CAS.
- H. Z. Yuan, S. Zhao, J. Y. Yu, L. F. Shen and Y. R. Du, Colloid Polym. Sci., 1999, 277, 1026–1032 CrossRef CAS.
- V. N. Kim, S. Prévost, K. Seidel, W. Maier, A. K. Marguerre, G. Oetter, T. Tadros and M. Gradzielsi, J. Colloid Interface Sci., 2016, 477, 94–102 CrossRef.
- V. Shah, B. Bharatiya, V. Patel, M. K. Mishra, A. D. Shukla and D. O. Shah, J. Mol. Liq., 2019, 277, 563–570 CrossRef CAS.
- X. Wang, J. Yan, D. Pan, R. Yang, L. Wang, Y. Xu, J. Sheng, Y. Yue, Q. Huang, Y. Wang, R. Wang and M. Yang, Adv. Healthcare Mater., 2018, 7, 1701505 CrossRef.
- S. K. Patel, A. Lavasanifar and P. Choi, Biomaterials, 2010, 31, 345–357 CrossRef CAS.
- S. Samanta and D. Roccatano, J. Phys. Chem. B, 2013, 117, 3250–3257 CrossRef CAS.
- C. Tourné-Péteilh, B. Coasne, M. In, D. Brevet, J. Devoisselle, A. Vioux and L. Viau, Langmuir, 2014, 30, 1229–1238 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01644a |
| This journal is © The Royal Society of Chemistry 2020 |