
First-principles theoretical study on dry reforming of methane over perfect and boron-vacancy-containing h-BN sheet-supported Ni catalysts†
Yan
Zhang
,
Yi-Fan
Yao
,
Yuan-Yuan
Qiao
and
Gui-Chang
Wang
 *
*
Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education) and the Tianjin Key Lab and Molecule-based Material Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: wangguichang@nankai.edu.cn
First published on 25th November 2020
Abstract
The entire reaction mechanism of the dry reforming of methane (DRM) as well as the competition processes over perfect and boron-vacancy-containing h-BN sheet-supported Ni-catalysts (labeled Ni2/h-BN and Ni2/h-BN-B–D) was studied by density functional theory calculations in the present work. Our calculation results show that B-defected h-BN strongly binds to the Ni2 active sites (i.e., shows a strong metal-support interaction (SMSI) character) due to the better electron transfer between Ni2 sites and the support. It was found that CH4 is easier to activate than molecular CO2. The activation of CO2 occurs on the surface of Ni2/h-BN through a direct route, whereas it is prone to follow a hydrogen-assisted path for Ni2/h-BN-B–D via the COOH* intermediate, and the results show that the oxidant O* is easily formed on the surface of Ni2/h-BN-B–D. It was also found that O* is the main oxidant agent for CHx* intermediates through the CH3–O oxidation mechanism. The reaction kinetic analysis indicated that the reverse water gas shift reaction (RWGS) is much more favorable than DRM (1.30 vs. 1.72 eV) over the Ni2/h-BN system, whereas the RWGS and DRM are comparable on Ni2/h-BN-B–D (1.77 vs. 1.66 eV), suggesting a high DRM activity on Ni2/h-BN-B–D. Moreover, neither methane cracking nor a Boudouard reaction to form C* species is thermodynamically and kinetically unfavorable over Ni2/h-BN-B–D; hence, Ni2/h-BN-B–D has strong resistance to carbon deposition. Compared to Ni(111), both Ni2/h-BN-B–D and Ni2/h-BN show strong resistance to carbon deposition. Our results provide a further mechanistic understanding of the DRM over an Ni-based catalyst through the SMSI characteristic and the SMSI favors strong resistance to carbon deposition.
1. Introduction
The greenhouse effect caused by greenhouse gas emissions has great impact on the ecological environment. Statistics from the Intergovernmental Panel on Climate Change (IPCC) show that carbon dioxide and methane are the main components of greenhouse gases. With respect to reducing emissions, developing methods to efficiently convert greenhouse gases into usable energy is a win-win option. In recent decades, research on the dry reforming (DRM) of methane (CH4(g) + CO2(g) ⇌ 2CO(g) + H2(g)) has received extensive attention,1–6 as this process can simultaneously convert both greenhouse gases CO2 and CH4 into two clean fuel gases: carbon monoxide and hydrogen. Moreover, compared with the syngas produced by other methods such as steam reforming (SRM)7,8 (CH4(g) + H2O(g) ⇌ CO(g) + 3H2(g)), the synthesis gas produced by DRM has a lower H2/CO ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1),9 suitable for further use in the Fischer–Tropsch synthesis of long-chain hydrocarbons.10–14 From the perspectives of economy and the environment, DRM has a broad application space and research value.
:1),9 suitable for further use in the Fischer–Tropsch synthesis of long-chain hydrocarbons.10–14 From the perspectives of economy and the environment, DRM has a broad application space and research value.
However, there are still many problems to be solved to realize the industrialization of the DRM reaction. The occurrence of side reactions is one of the important factors that affect the reactivity of DRM. During the DRM process, the OH* produced by hydrogen-assisted CO2 dissociation and the H* produced by the CH4 dissociation, or the reaction between the O* produced by CO2 dissociation and the H* produced by the CH4 dissociation, produce H2O. As long as H2O is formed during the DRM process, steam reforming may also contribute to the formation of carbon monoxide and H2. Therefore, SRM can affect DRM by changing the ratio of H2 and CO. Due to the high endothermic properties of DRM (ΔH = 247.4 kJ mol−1), the reverse water shift reaction (RWGS) (CH2(g) + H2(g) ⇌ CO(g) + H2O(g)) (ΔH = 41.1 kJ mol−1) is enhanced compared to DRM at lower temperatures.15–17 RWGS can affect DRM by decreasing the ratio of H2 and CO. In addition, the formation of C* is the main cause of DRM catalyst deactivation. During the process, the formation of carbon mainly comes from the cracking of methane (CH4(g) ⇌ C(s) + 2H2(g)) and the Boudouard reaction (2CO(g) ⇌ C(s) + CO2(g)).18,19
The lack of a highly active and stable catalyst is also one of the factors restricting the industrialization of DRM reactions. Although the theoretical and experimental research on DRM catalysts has been very rich, it is still necessary to have a further basic understanding of the reaction in order to rationally design a highly active and coke-resistant catalyst. An important aspect is to understand the preferred reaction path, since it controls the DRM activity as well as the extent of the abovementioned competitive reactions. In general, the mechanism of the methane dry gas reforming reaction is analyzed in the following aspects. The first is the activation mode of carbon dioxide (direct dissociation or hydrogen-assisted CO2 dissociation). A theoretical study by Foppa et al.19 found that direct CO2 dissociation is preferred on Ni; the hydrogen-assisted path presents a similar free energy span for Pd and Pt compared to direct CO2 dissociation. In the DRM reaction, carbon dioxide is generally regarded as an oxidant, because the dissociation of carbon dioxide can generate O*. In addition to O* as oxidant, OH* is also used as an oxidant in some catalytic systems.20 Last but not least, CHx* can couple with O*/OH* to form CHxO*/CHxOH*. When DRM occurs in many catalytic systems, CH4 decomposes to CH* or C* and then oxidizes to CHO* or CO*.19,21–23 It is worth noting that Akri et al. propose that CH3* is oxidized more than it is decomposed on atomically dispersed Ni single atoms.24 Moreover, a recent report on DRM over Ni4 clusters identified the oxidation of CH3* to CH3O* as an alternative route to CO following the initial methane activation,25 as also proposed for Ni1–Ru1/CeO2.26
Nearly three decades of research have found that a nickel-based catalyst is the most competitive catalyst for DRM due to its low cost and its high catalytic activity comparable to noble metal catalysts such as Pd, Rh and Pt.15,27–29 However, the biggest factor restricting the industrialization of Ni-based catalysts for DRM processes is that they suffer from deactivation because of carbon deposition.30–32 To avoid such a problem, many methods have been developed, such as alloying with other less active metals,33–36 making a strong metal-support interaction (SMSI) system by choosing a special reduced metal oxide support to make oxygen defects37 or controlling the size of active sites.38 In fact, a previous study showed that anatase defective TiO2-supported Rh catalyst favors stronger carbon deposition resistance than rutile defective TiO2-supported Rh catalysts for the partial oxidation of methane due to the SMSI properties of the former.39 Moreover, it was also found that a smaller sized active site is much more favorable for carbon deposition resistance, implying a strong size effect.40–42
Very recent experimental results found that Ni nanoparticles embedded on the vacancy defects of hexagonal boron anitride nanosheets (h-BN) show high coke-resistance behavior in the DRM compared to that of perfect h-BN sheets, suggesting the importance of vacancy defects.43 The existence of defects makes the interaction between the metal and the support stronger and the Ni atoms on the surface are more dispersed and not easily aggregated. To shed light on the vacancy defect effect on coke-resistance in the DRM processes, a systemic theoretical study was performed on the detailed reaction mechanism of DRM catalyzed by perfect and defected-h-BN supported Ni catalysts in this work and it was shown that the vacancy defects lead to SMSI and thus high catalytic DRM activity and resistance to carbon deposition.
2. Computational methods and model
2.1. Methods
All the density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP).44–46 Potentials within the projector augmented wave method (PAW)47,48 were used to represent the core–valence electron interaction; the Perdew–Burke–Ernzerhof (PBE)49 functional within the generalized gradient approximation (GGA) was employed to calculate the exchange and correlation energies. The valence electronic states were expanded in plane wave basis sets with a kinetic cutoff energy of 400 eV and the surface Brillouin zone sampling was carried out using a 3 × 3 × 1 Monkhorst–Pack50k-point mesh for better calculation accuracy. During configuration optimization, the self-consistent field calculation was repeated until the absolute force of each field is less than 0.035 eV Å−1. The climbing image nudged elastic band (CI-NEB) method51 was employed to locate the transition states (TSs). A further frequency analysis was carried out to confirm the located TSs; during the frequency calculation, all catalyst atoms remained stationary. Spin-polarized effects were considered for the case of Ni. It is worth mentioning that Tanaka et al.52,53 proposed the scheme about the spin effect on the reaction barrier to improve calculation accuracy.The adsorption energy (Eads), reaction energies (ΔE) and activation energy (Ea) were calculated as follows: Eads= Esystem− Esubstrate− Eabsorbate; ΔE = EFS− EIS; Ea= ETS− EIS. Here, Esystem, Esubstrate, Eadsorbate, EIS, ETS, and EFS represent the energies of the adsorption system, substrate, adsorbate, initial state (IS), transition state (TS), and final state (FS), respectively.
In order to compare the calculation results with the experimental conditions more accurately, the Gibbs adsorption free energy and the Gibbs free energy of the whole reaction were further considered. The Gibbs free energy (G) of a gas-phase species is estimated by considering the contributions of the translation, rotation and vibration of the distribution function and is directly derived from the experimental data. For substances with weaker molecular adsorption, such as methane, we carried out dispersion correction (DFT-D2) and, assuming that they maintain the full rotation and vibration modes of their gas phase materials, the adsorption entropy was calculated using the following formula.54
| Sads(T) = 0.7Sgas(T) − 3.3R | (1) |
Here, R is the ideal gas constant, Sgas is the entropy of the corresponding gas phase material obtained from the thermodynamic database (see Table S1, ESI†) and Sads is the entropy of the adsorbate. For the strongly chemically adsorbed species like CO, we assume that the most important contribution comes from translational entropy and its adsorption entropy change was estimated by using the following formula:55,56
| S(T) = 1.5Rln(2ΠMkT) − 3Rlnh + Rln(kT/P) + 2.5R | (2) |
Here k, T, P, h, R and M represent the Boltzmann constant, temperature, pressure, Planck constant, ideal gas constant, and molecular mass, respectively.
According to the activation energy and reaction energy of each basic reaction, the free energy curve of the reaction can be obtained.19 In this work, we define the total free energy span as the difference between the free energy of the starting reactant and the free energy of the highest transition state. Assuming that the reaction proceeds step by step, the free energy span corresponds to the activation energy of the reaction and can be used to measure the difficulty of the reaction.
2.2. Models
The calculated lattice constant of h-BN is 2.51 Å (the experimental lattice constant is approximately 0.25 nm)57 and the h-BNNS is represented by a single layer slab with a 7 × 7 element of h-BN with a vacuum separation of 15 Å between successive slabs. Periodic boundary conditions were used for all systems. The B-defected h-BN was modeled by removing one B atom from the perfect h-BN (labeled as h-BN-B–D) (see Fig. 1).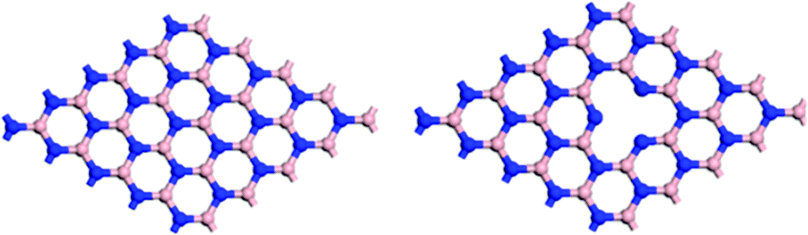 | ||
| Fig. 1 Perfect (left) and B-defected (right) h-BN surfaces. | ||
3. Results and discussion
3.1. Optimization and electronic analysis of perfect and B-defected h-BN supported Ni2 catalysts
The perfect and B-defected h-BN surfaces are shown in Fig. 1. We first examined sites for Ni–Ni adsorption on the perfect h-BN surface. Two top sites for B and N were considered. One bridge site (B–N) and one hollow site were also examined. The most energetically preferred structures are presented in Fig. 2 and the corresponding adsorption energy is −4.28 eV. For B-defected h-BN supported Ni catalysts, we also optimized to find the most stable structure (see Fig. 2) and the corresponding adsorption energy is −8.70 eV. As shown in Fig. 2, when introducing B defects, the distances between Ni atoms and h-BN are shorter than in perfect h-BN, indicating that the introduction of B-defects enhances the interaction between Ni atoms and the carrier. It is worth noting that the defects strongly affect the charge transfer between h-BN and Ni2. The Bader charge analysis58,59 shows that 0.18–0.24 electrons are transferred from the adsorbed Ni2 to pristine h-BN, while the defects in h-BN highly enhance the charge transfer (0.44–1.62 e); it is also apparent that the introduction of B-defects enhances the interaction between Ni atoms and the carrier. The partial densities of states (DOS) of Ni2/h-BN and Ni2/h-BN-BD were also calculated and are shown in supporting Fig. S1 (ESI†). The figure shows that the magnetic moment of Ni2 on the perfect h-BN surface is 1.529 μB and the magnetic moment of Ni2 on the B-defect h-BN surface at the equilibrium lattice constant is 0.75 μB; these results are consistent with the calculated value. On the surface of the B-defect h-BN, the decrease of Ni2 magnetic moment indicates that Ni2 transfers more electrons to the surface, thus showing a strong metal-support interaction character.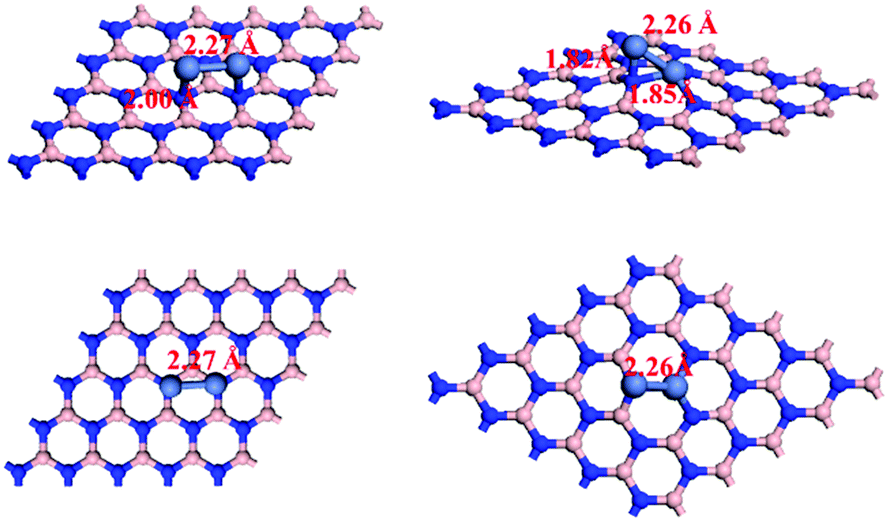 | ||
| Fig. 2 Top and side views of Ni2 adsorption configurations on perfect (left) and B-defected (right) h-BN. | ||
It is worth mentioning that we also considered the adsorption of a single Ni on h-BN (see Fig. S2, ESI†). We found that the activation of methane and carbon dioxide on the Ni1/h-BN and Ni1/h-BN-B–D surfaces becomes more difficult (see Table S2, ESI†). Since the reaction tends to proceed on metallic Ni, there are fewer reactive sites when there is only a single Ni atom on the h-BN surface, which does not match the experimental conditions. Therefore, we did not consider this catalyst configuration. We will conduct a detailed study regarding the effect of Ni cluster size on DRM reactivity in the next work.
3.2. Adsorption configurations and stability of key species involved in DRM
As the adsorption process is the initial step and important for the whole reaction process, it is worth investigating the adsorption strength of reactants like CH4 (CO2) as well as key intermediate species related to DRM.The adsorption energies are shown in Fig. 3 and Table S3 (ESI†) and the corresponding adsorption configurations are displayed in Fig. S4 and S5 (ESI†). First, oxygen and hydroxyl interact strongly with the catalyst; as the proposed hydrocarbon oxidants in the methane reforming reaction, the stabilities of O* and OH* on this Ni2/h-BN-B–D surface are reduced by at least 1 eV compared to those on Ni2/h-BN. Second, the adsorption strengths of methane cracking products CHx* and C* increase in the order Ni2/h-BN-B–D < Ni2/h-BN, so favorable carbon deposition resistance on Ni2/h-BN-B–D can be expected. From Table S3 (ESI†), one can find that most species bind strongly to perfect h-BN supported Ni2 catalyst, generally following the trend Ni2/h-BN > Ni2/h-BN-B–D > Ni(111). The stronger adsorption ability on Ni2-based catalyst may lead to the more facile activation of CH4 and CO2 compared to that on Ni(111) and thus to higher catalytic activity for DRM.
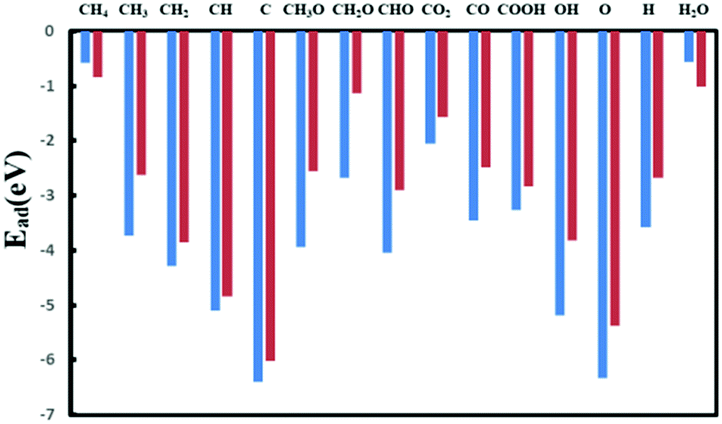 | ||
| Fig. 3 Adsorption energies of key species on Ni2/h-BN-B–D (red) and Ni2/h-BN (blue). | ||
3.3. CH4 and CO2 activation
The activation of methane and carbon dioxide is the starting step of the DRM process, so it is critical to the entire process. We first examine CH4 activation steps on the different catalysts.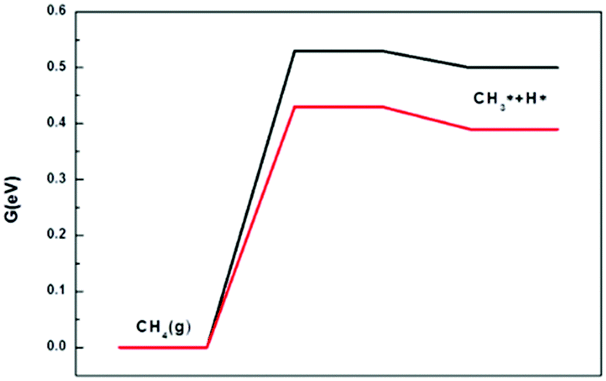 | ||
| Fig. 4 Free energy profiles of CH4 dissociation on the Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K. | ||
| Reaction | Ni2/h-BN | Ni2/h-BN-B–D | |||
|---|---|---|---|---|---|
| E a (eV) | ΔE (eV) | E a (eV) | ΔE (eV) | ||
| 1 | CH4* → CH3* + H* | 0.11 | −0.04 | 0.36 | 0.35 |
| 2 | CH3* → CH2* + H* | 0.95 | 0.79 | 0.95 | 0.94 |
| 3 | CH2* → CH* + H* | 1.00 | 0.87 | 1.20 | 1.15 |
| 4 | CH* → C* + H* | 1.17 | 0.75 | 1.33 | 1.24 |
| 5 | CO2* → CO* + O* | 1.11 | 0.22 | 1.42 | 0.24 |
| 6 | CO2* + H* → COOH* | 1.56 | 0.65 | 0.50 | −0.05 |
| 7 | CO2* + H* → HCOO* | 0.97 | −0.34 | 0.85 | −0.62 |
| 8 | COOH* → CO* + OH* | 0.76 | −1.14 | 0.96 | −1.25 |
| 9 | HCOO* → HCO* + O* | 2.91 | 2.83 | 2.36 | 1.65 |
| 10 | OH* → O* + H* | 1.41 | 1.02 | 1.44 | 1.25 |
| 11 | CH3* + O* → CH3O* | 1.29 | 0.17 | 0.85 | 0.12 |
| 12 | CH3* + OH* → CH3OH* | 1.73 | 1.5 | 1.14 | 0.20 |
| 13 | CH3O* → CH2O* + H* | 0.16 | 0.06 | 0.08 | −0.19 |
| 14 | CH2* + O* → CH2O* | 0.47 | −1.33 | 0.46 | −1.35 |
| 15 | CH2* + OH* → CH2OH* | 1.05 | 0.39 | 1.06 | 0.45 |
| 16 | CH* + O* → CHO* | 0.80 | −2.00 | 0.57 | −2.38 |
| 17 | CH2O* → CHO* + H* | 0.21 | −2.88 | 0.37 | −1.75 |
| 18 | CHO* → CO* + H* | 0.71 | −0.04 | 0.08 | −0.74 |
| 19 | 2H* → H2* | 0.27 | 0.20 | 0.38 | −1.00 |
| 20 | C* + O* → CO* | 1.23 | −3.00 | 0.39 | −4.00 |
| 21 | OH* + H* → H2O* | 1.07 | 0.71 | 0.58 | 0.12 |
| 22 | 2OH* → H2O* + O* | 0.83 | 0.06 | 1.12 | 1.00 |
Different from methane activation, on these two catalysts there is a significant difference between the activation mechanism of CO2 and the energy barrier required by the reaction. Fig. 5 shows two different routes of CO2 activation. In the case of direct decomposition of CO2, the free energy barriers reduce when going from Ni2/h-BN-B–D (1.68 eV) to Ni2/h-BN (1.31 eV). This result is consistent with the adsorption trend where both reactants (CO2) and dissociation products (CO* and O*) are more stable on Ni2/h-BN than Ni2/h-BN-B–D. From the transition state structure in direct CO2 cracking (Fig. S7, ESI†), the C–O fracture bond distances are equal to 1.66 Å on Ni2/h-BN-B–D and 2.01 Å on Ni2/h-BN.
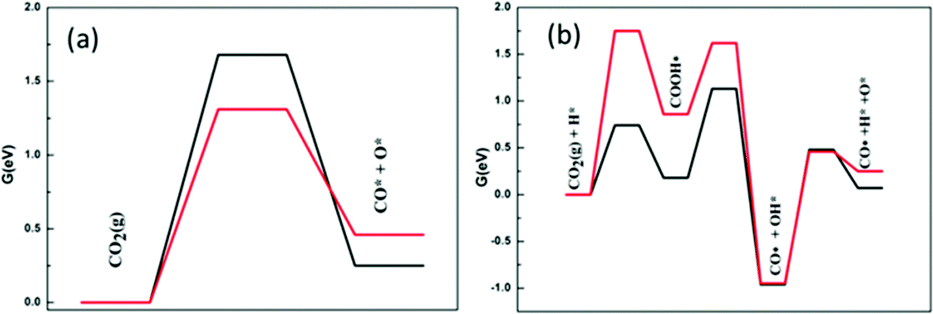 | ||
| Fig. 5 Free energy profiles of CO2 activation on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K: (a) direct route and (b) hydrogen-assisted via COOH* intermediate route. | ||
The hydrogen-assisted path presents different free energy spans for Ni2/h-BN (1.75 eV) and Ni2/h-BN-B–D (1.13 eV) compared to those for direct CO2 dissociation (1.31 eV and 1.68 eV, respectively). These results indicate that the C–O activation pathway is more energetically likely to be a COOH* path on Ni2/h-BN-B–D, but direct CO2 decomposition is preferred on Ni2/h-BN. Due to the introduction of B-defects, the interaction between the support and the metal is enhanced, so the adsorption energy of CO2 on Ni2/h-BN-B–D is less than that on Ni2/h-BN. This reaction of carbon dioxide and hydrogen atoms combining to form COOH* intermediates is easier on Ni2/h-BN-B–D because of the low affinity of this surface for CO2* and H*.
Since the activation of methane is easier than that of carbon dioxide on these two surfaces, we investigated the dissociation of carbon dioxide in the presence of CH3*. IS and TS configurations of CO2 direct activation on Ni2/h-BN in the presence of CH3* are shown in Fig. S3 (ESI†). We found that methane tends to adsorb on the top positions of Ni atoms, while the most stable configuration of carbon dioxide is two-position adsorption between two Ni atoms, so methane and carbon dioxide can be adsorbed simultaneously on the same Ni2 metal cluster. In the presence of CH3*, the reaction energy barrier for direct activation of CO2 is 1.16 eV, which is not significantly different from that in the absence of CH3* (1.11 eV). For the Ni2/h-BN-B–D surface, like the Ni2/h-BN surface, methane and carbon dioxide can be simultaneously adsorbed on the different positions of Ni atoms. The previous calculation results show that, on the Ni2/h-BN-B–D surface, CO2 tends toward H-assisted dissociation, so H atoms are generated after methane activation and the reaction between CO2 and H* can proceed smoothly. Therefore, for these two surfaces, the activation of methane and carbon dioxide can be performed in situ on the Ni dimer. Regarding the influence of the surface coverage of intermediate species on the reaction, we will use kinetic Monte Carlo method in future work to further explain the reaction mechanism and phenomenon.
The comparison between the C–H and C–O activation curves shows that, under the conditions considered, the activation of CO2 is easier on Ni2/h-BN-B–D (1.13 eV) than on Ni2/h-BN (1.31 eV), but CH4 activation is more likely to occur on Ni2/h-BN. This result shows that, compared with the Ni2/h-BN surface, the Ni2/h-BN-B–D surface is more likely to produce O*, but C* is more difficult to generate, showing that the Ni2/h-BN-B–D surface has the potential to resist carbon deposition.
3.4. DRM reaction mechanism
Table 1 shows the activation free energy values of all the steps in the DRM and the competitive reactions network. There are many elementary reactions in DRM. Generally, we consider the reaction process from two major aspects. The first is the activation mode of CO2: (a) direct disintegration of CO2 into CO* and O* or (b) hydrogen-assisted CO2 dissociation (here, the CO2* + H* → COOH* → CO* + OH* → CO* + O* + H* route is considered). The second aspect is the formation of the C–O bond. The continued dissociation of alkanes (CHX*) or the formation of oxides by O* or OH* depends on the competition of the two reactions: CHX* → CHX−1* + H* and CHX* + O*→ CHXO* or CHX* + OH* → CHXOH*.We constructed the free energy curves of the DRM reactions completed through different paths according to the activation energy of each basic reaction. First, we consider the situation of direct dissociation of CO2. There are many paths for the formation of the C–O bond, like CH3, CH2, CH, or C oxidation by O*. As the formations of CH* and C* each have a high energy barrier (see Table 1), the paths of CH* and C* oxidation by O* can be ignored. On these two catalysts, CH3* species are relatively easy to generate, but CH3* → CH2* → CH* is thermodynamically unfavorable, with a high activation barrier. Therefore, we think that the CH3* may be oxidized by O* instead of continuing to decompose. Even though the decomposition of CH3* is a strong endothermic reaction, its reaction activation barrier is lower than the activation energy of CO2 direct decomposition. For the comprehensiveness of the work, we studied the activation pathway of CH2 oxidized by O*. At 973.15 K, the DRM reaction is endothermic and the standard free energy is 1.54 eV. We chose the activation of CH4 instead of CO2 as the starting step of DRM based on which has the lower activation energy.
Fig. 6 shows the different ways of forming C–O bonds on the surfaces of these two catalysts. Fig. 6(a) and (b) respectively show the free energy profiles of the DRM through the CH3–O and CH2–O pathways when CO2 is directly dissociated. On the Ni2/h-BN-B–D surface, the overall free energy spans of DRM through the CH3–O and CH2–O routes are 2.19 eV and 3.13 eV. The results clearly show that DRM on Ni2/h-BN-B–D tends to occur through the CH3–O pathway. On Ni2/h-BN, the overall free energy spans by the CH3–O and CH2–O routes are 1.72 eV and 2.07 eV, respectively; unlike on the Ni2/h-BN-B–D, there is no obvious difference in the overall free energy span of the two paths. Therefore, these two paths have a competitive relationship on the surface of Ni2/h-BN-B–D. This may be because the first step of CH4 activation is faster on Ni2/h-BN than on Ni2/h-BN-B–D (0.11 eV vs. 0.36 eV).
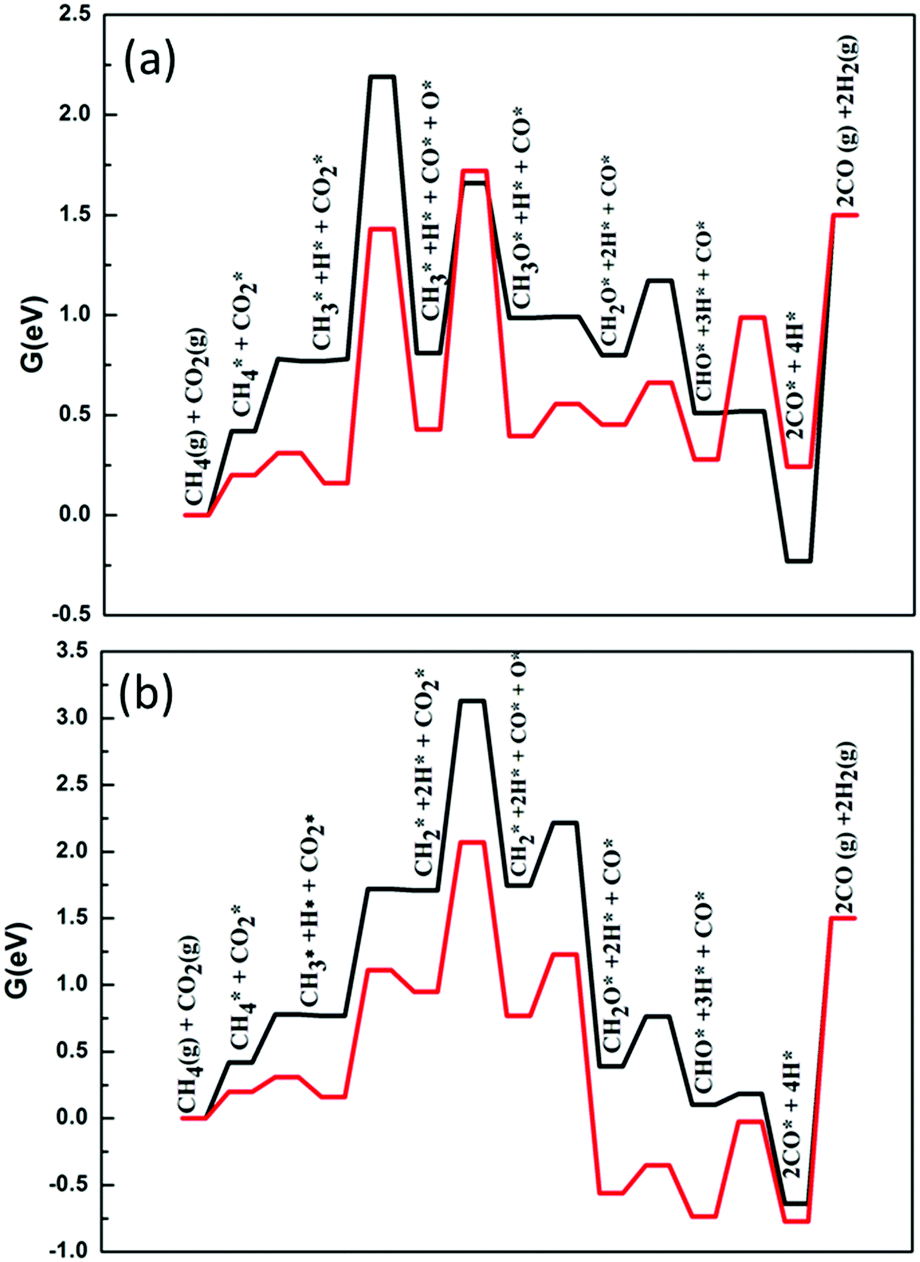 | ||
| Fig. 6 Free energy profiles of DRM on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K. (a) CO2 direct dissociation with CH3–O oxidation and (b) CO2 direct dissociation with CH2–O oxidation. | ||
Secondly, we considered the overall free energy span in the case of the H-assisted CO2 dissociation path and the results are shown in Fig. 7. It should be noted that on the surface of Ni2/h-BN, the activation energy barrier of CO2* + H* → COOH* is much higher than that of the decomposition of CH3*. Therefore, we consider that, on the surface of Ni2/h-BN, CH2–O is the main path of DRM in the case of H-assisted CO2 dissociation via COOH*. Fig. 7(a) shows that the overall free energy span of DRM on Ni2/h-BN is 2.51 eV by H-assisted CO2 dissociation via COOH*, higher than that of CO2 direct dissociation. Consistent with the above analysis, this result shows that the path of CO2 direct dissociation on the surface of Ni2/h-BN is more likely to occur. Fig. 7(b) shows that the overall free energy span of DRM on Ni2/h-BN-B–D is 1.66 eV by H-assisted CO2 dissociation via COOH*, which is significantly lower than the direct dissociation of CO2. This is because, on the surface of Ni2/h-BN-B–D, the activation of CO2 prefers the path of H-assisted dissociation. It is worth mentioning that although OH* was produced in this process (COOH* → CO* + OH*), we did not take OH* as the main oxidant because CHx* + OH* → CHxOH* is a strong endothermic reaction and has a high activation energy barrier (see Table 1).
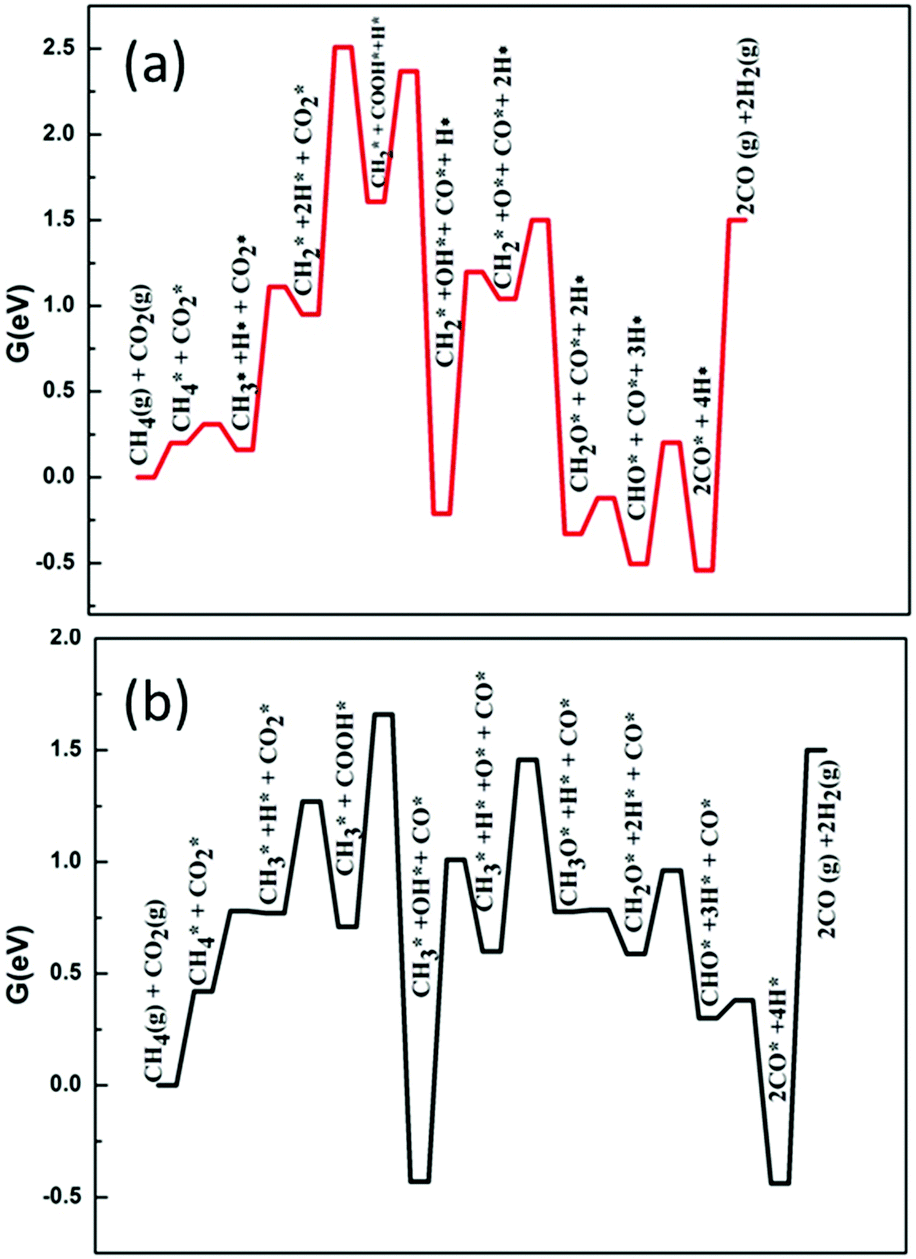 | ||
| Fig. 7 Free energy profiles of DRM by H-assisted CO2 dissociation via COOH* on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K. | ||
On both surfaces, the decomposition of CH4 is the initial step of the reaction. On the surface of Ni2/h-BN, the activation of CO2 tends to be direct decomposition. However, on the surface of Ni2/h-BN-B–D, H-assisted dissociation of CO2 becomes the main method. The oxidation of alkanes mainly occurs through the CH3–O pathway even though there is a competitive relationship on the Ni2/h-BN. On the Ni2/h-BN-B–D, the overall free energy span is lower but there is no significant difference between the two surfaces.
3.5. Reverse water gas shift (RWGS) reaction mechanism
RWGS is a major side reaction during the DRM reaction. When methane cracks to produce H2, it reacts with CO2 in the raw material to produce H2O and CO. Even if the reaction produces CO, by-products (H2O) are also introduced and reduce the ratio of H2 to CO. Therefore, when studying the DRM process, the RWGS reaction should be considered.The standard free energy of the reverse water gas shift reaction at 973.15 K (ΔrG0) is 0.41 eV. The initial step of the RWGS reaction is the activation of carbon dioxide. As in DRM, carbon dioxide may be dissociated directly or through H-assisted dissociation on Ni2/h-BN and Ni2/h-BN-B–D. Fig. 8 shows the free energy profiles of the RWGS calculated for Ni2/h-BN and Ni2/h-BN-B–D. On these two surfaces, there are obvious differences in the pathways of carbon dioxide dissociation. RWGS via direct C–O activation route is preferred on Ni2/h-BN, whereas the reaction proceeds preferentially via the COOH* intermediate on Ni2/h-BN-B–D. The overall free energy span of the RWGS reaction is lower for Ni2/h-BN (1.30 eV) than for Ni2/h-BN-B–D (1.77 eV).
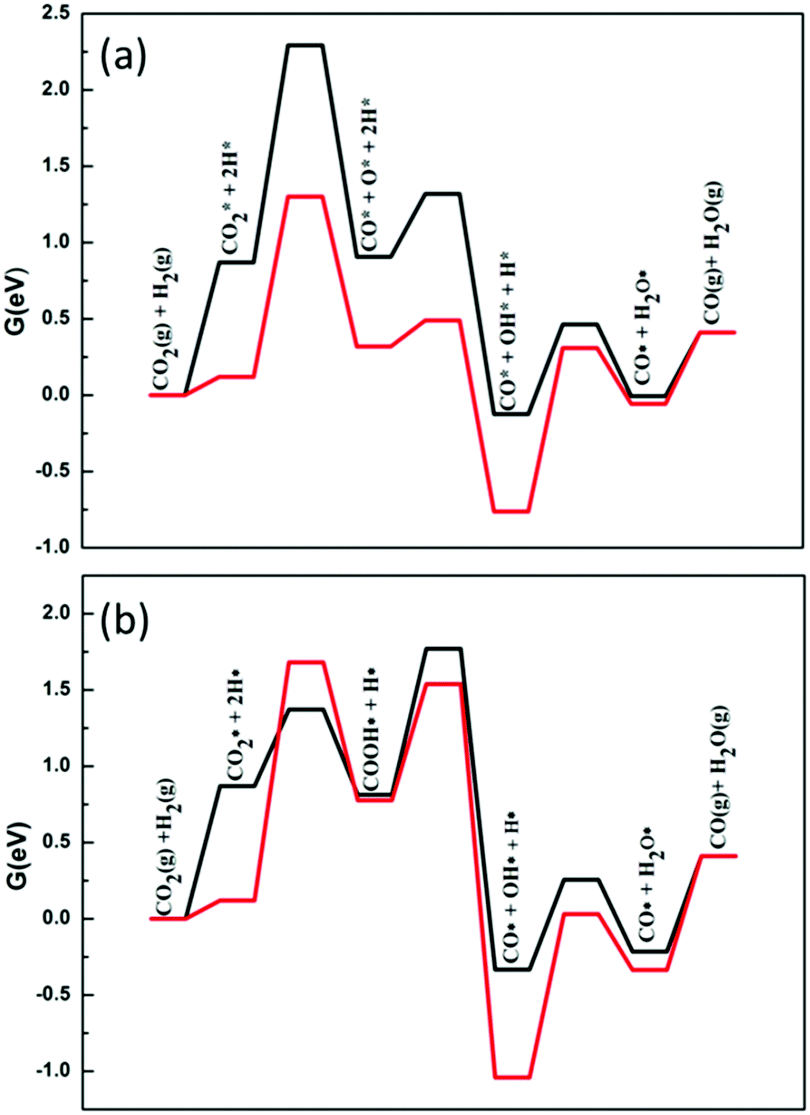 | ||
| Fig. 8 Free energy profiles of RWGS reaction on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K: CO2 direct decomposition path (a) and hydrogen-assisted CO2 dissociation path (b). | ||
As seen in Fig. 8, regardless of the difference in the catalyst and the cracking mechanism of C–O, breaking the C–O bond requires a higher energy barrier than the formation of water; this shows that the cleavage of the C–O bond is the key step that affects the RWGS reaction. Since OH* and O* on Ni2/h-BN-B–D are less stable than on Ni2/h-BN (Fig. 3), it is easier to form water from OH* and H* on the surface of Ni2/h-BN-B–D. Therefore, in the case of Ni2/h-BN-B–D, the H-assisted CO2 activation route, with OH* and H* then forming water, can promote the entire RWGS reaction. It is worth mentioning that the H-assisted C–O cracking pathway on Ni2/h-BN-B–D can significantly reduce the free energy span by 0.52 eV compared to the pathway via direct CO2 activation.
3.6. Steam reforming of methane (SRM) mechanism
The steam reforming reaction must also be considered when evaluating the dry gas reforming reaction. When dry reforming conditions or the reverse water gas shift reaction generates water molecules, methane will react with the water to form CO and H2.Fig. 9 displays the calculated energy profiles for steam reforming of methane on Ni2/h-BN and Ni2/h-BN-B–D. The overall free energy spans of SRM are 3.31 eV and 2.83 eV for the Ni2/h-BN and Ni2/h-BN-B–D surfaces, respectively. Like that of the DRM reaction, the free energy profile for steam reforming is higher in energy on Ni2/h-BN than on Ni2/h-BN-B–D. In steam reforming reactions, lower activation energies are required for the dissociation of H2O on both surfaces (0.37 eV and 0.47 eV for Ni2/h-BN and Ni2/h-BN-B–D). O–H bond breaking reactions occurring in the SRM mechanism display similar free energy barriers on both surfaces. The higher activation energy required on Ni2/h-BN-B–D than Ni2/h-BN is due to the lower affinity of OH* for this surface. From the energy profile (Fig. 9), the combination of CH3* and O* is the rate-limiting step of the steam reforming reaction on Ni2/h-BN; the formation of CH3O* requires higher activation energy on Ni2/h-BN than on Ni2/h-BN-B–D because CH3* and O* are more stable on Ni2/h-BN than on Ni2/h-BN-B–D. It is interesting that the overall energy span of SRM occurring on these two surfaces is higher than that of DRM.
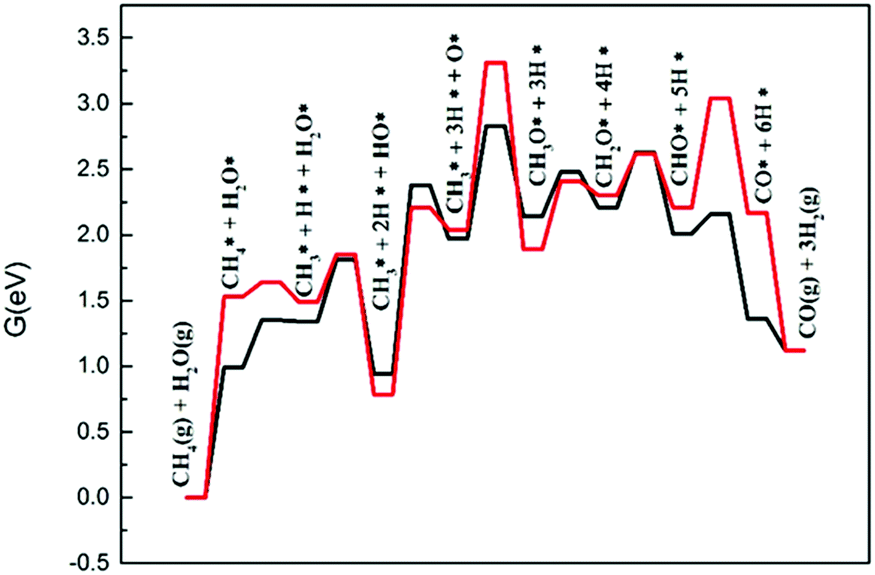 | ||
| Fig. 9 Free energy profiles of steam reforming of methane on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K. | ||
3.7. Coke formation
The ability of the catalyst to resist carbon deposition is an important indicator to measure the stability of the catalyst in the dry and steam reforming reactions. Reforming reactions usually occur at very high temperatures, so the material must resist coke formation and should not sinter at the strict working temperatures. Once coke is formed on the catalyst surface, the activity of the catalyst will be decreased or even deactivated. The ability to resist carbon deposition is an important indicator to measure the Ni-based catalyst in the DRM reaction.There are two main ways to form C* on the catalyst surface: (i) methane cracking (CH4 → C* + 2H2) and (ii) the Boudouard reaction (2CO → C* + CO2). The energy profile of the methane cracking reaction is presented in Fig. 10. Methane cracking requires higher energy on Ni2/h-BN-B–D. In addition to the lower activation energy barrier of methane in the first step, the subsequent decomposition (CH3* → CH2* → CH* → C) energy gradually increases, with an overall free energy span up to 3.85 eV. Less energy is required on the Ni2/h-BN and the overall free energy span is still as high as 3.13 eV.
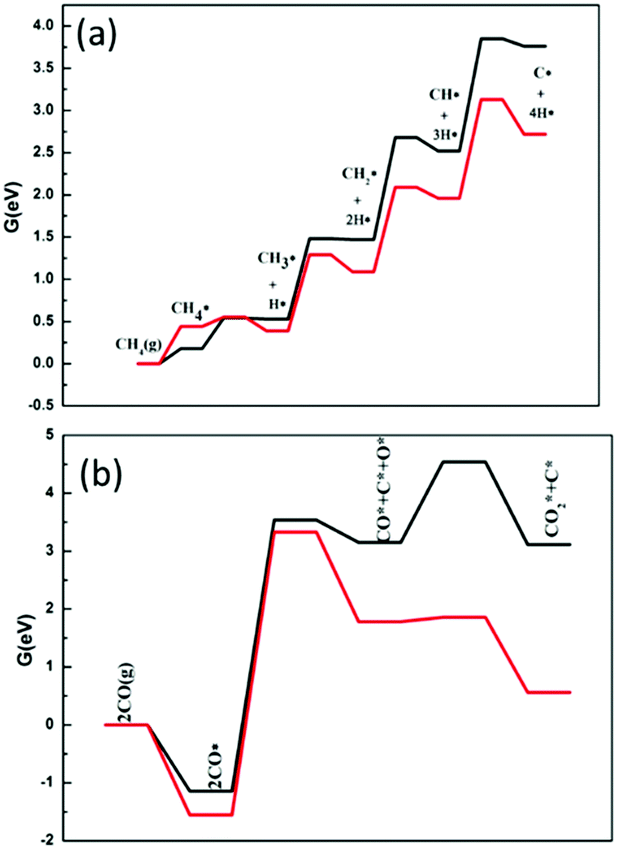 | ||
| Fig. 10 Free energy profiles of the CH4 cracking (a) and Boudouard reaction (b) on Ni2/h-BN (red) and Ni2/h-BN-B–D (black) surfaces at 973.15 K. | ||
C* can also be formed from CO by the Boudouard reaction (as shown in Fig. 10(b)). It is worth noting that the Boudouard reaction (2CO* → CO2* + C*) is not completed in one step, but in two. First, CO* dissociates into C* and O* (CO* → O* + C*), then O* reacts with another CO* to produce CO2* (O* + CO* → CO2*). For Ni2/h-BN and Ni2/h-BN-B–D, the overall free energy spans are 3.10 eV and 4.54 eV, respectively. The free energy span of the Boudouard reaction on Ni2/h-BN is lower than that of methane cracking, which demonstrates that carbon is preferentially formed from CO on this surface. On the Ni2/h-BN-B–D surface, however, cracking of methane is more likely to occur than the Boudouard reaction.
In addition to the reactions that produce carbon deposits, the coupling capacity of C* and the oxidation of C* are also factors that measure the catalyst's ability to resist carbon deposits. The kinetics of carbon deposition on the surface of Ni2/h-BN and Ni2/h-BN-B–D were further studied. We calculated the potential barrier of the surface C-coupling mechanism (C–C coupling) and the potential barrier of forming CO* on the two surfaces.
On Ni2/h-BN, we find that when the catalyst surface is covered with carbon atoms, they tend to form clusters spontaneously. Regardless of the position of the two coupled C atoms in the initial form, a C–C coupling structure is always spontaneously formed after optimization. But for the formation of CO*, the energy barrier of the reaction is 1.26 eV (Table 1), indicating the surface coupling mechanism on this surface is kinetically favored. On Ni2/h-BN-B–D, the barrier for C–C coupling (0.28 eV) is similar to that of CO* formation (0.38 eV), indicating that CO formation and C–C coupling might be kinetically favored on this surface. But we need to pay attention to the fact that the above calculation results show that the formation of C* is more difficult than the formation of O*. Overall, the above results suggest that carbon deposition is preferred on Ni2/h-BN over Ni2/h-BN-B–D.
An experimental study43 found that the Ni content in the defected h-BN catalyst is significantly lower than that of the perfect h-BN catalyst, which further indicates that Ni can be highly dispersed through the introduction of controllable vacancy defects (this has been further confirmed by our recent study on the Ptn/TiO2 system, where it was reported that both the support and defect against thermal sintering of Pt atoms in fact60). Therefore, it is necessary to consider that Ni has a tendency to aggregate on a perfect surface. Here, we constructed the most stable configuration of Ni4/h-BN catalyst to study the effect of Ni accumulation on the resistance to carbon deposition (see Fig. S8, ESI†). The energy span profiles of the methane cracking reaction are presented in Fig. 11, in which the Ni4/h-BN catalyzed cracking reaction presents a much lower energy profile compared to that of Ni2/h-BN (free energy span of 1.43 eV). On the surface of Ni4/h-BN, once methane is activated, C* species are relatively easy to form. As a result, when the amount of nickel at a single site on the h-BN surface increased, the rate of methane decomposition accelerated and C* species were more likely to form on the surface. We studied the kinetics between the deposition and removal of surface carbon on Ni4/h-BN; the results are shown in Fig. 12. On Ni4/h-BN, the barrier for C–C coupling (1.05 eV) is lower than that for CO* formation (1.72 eV), indicating the surface coupling mechanism is kinetically favored on this surface. Therefore, when Ni accumulates on the h-BN surface, the formation of C* becomes easier.
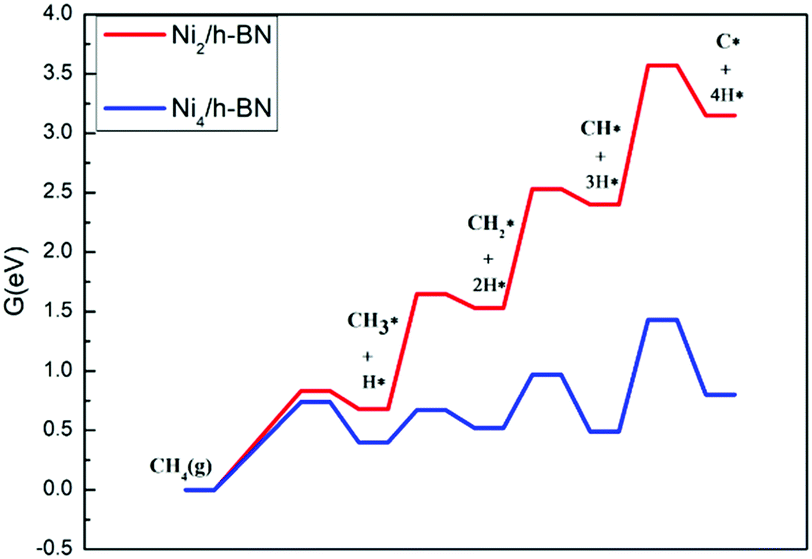 | ||
| Fig. 11 Free energy profiles of CH4 cracking over Ni2/h-BN (red) and Ni4/h-BN (blue) surfaces at 973.15 K. | ||
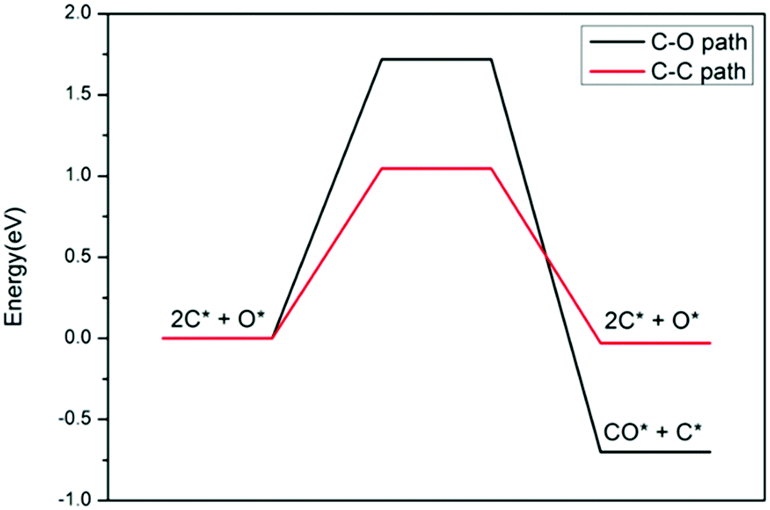 | ||
| Fig. 12 Energy profiles of carbon oxidation to CO (black) and carbon–carbon coupling (red) on Ni4/h-BN. | ||
3.8. Comparisons of DRM activity for Ni2/h-BN, Ni2/h-BN-B–D and Ni(111)
Fig. 13 shows the free energy spans of DRM and the competitive reactions on the Ni2/h-BN and Ni2/h-BN-B–D surfaces at 973.15 K and P = 1 bar. Compared with Ni(111),19 the overall free energy span of the DRM reaction on the Ni2/h-BN and Ni2/h-BN-B–D surfaces is significantly reduced. Also, unlike Ni(111), on Ni2/h-BN and Ni2/h-BN-B–D, methane activation is easier than carbon dioxide activation. On the Ni(111) and Ni2/h-BN surfaces, the RWGS reaction is the reaction with the smallest span of free energy among these reactions, which shows that RWGS easily occurs with the DRM reaction on these two catalysts. Moreover, both Ni2/h-BN and Ni2/h-BN-B–D have significantly improved abilities to resist carbon deposition.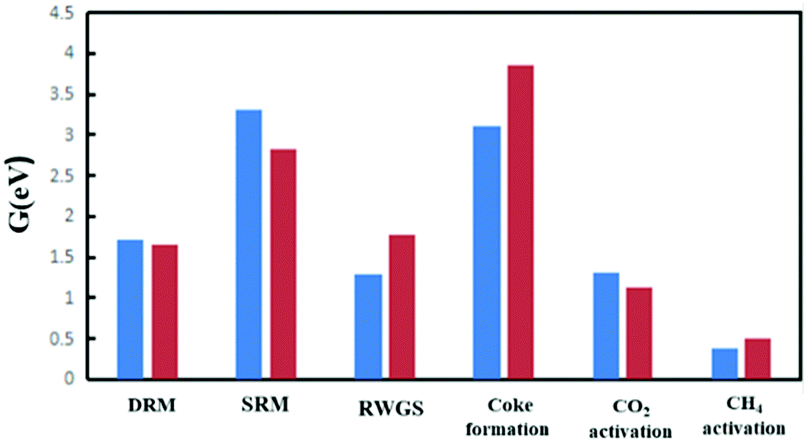 | ||
| Fig. 13 Comparison of free energy spans of DRM and competitive reactions on Ni2/h-BN (red) and Ni2/h-BN-B–D (blue) at 973.15 K and P = 1 bar. | ||
Comparing the reaction paths, on Ni(111) and Ni2/h-BN, CO2 activation tends to be direct dissociation; for the Ni2/h-BN-B–D surface, the overall free energy span required by H-assisted carbon dioxide dissociation is lower. On these three surfaces, O* is the most potent oxide. The difference is that on the Ni(111) surface, the CH-O path has priority, while on the Ni2/h-BN-B–D and Ni2/h-BN surfaces, the free energy span required for CH3–O to occur is lower. Overall, compared with Ni(111), the DRM reaction is more likely to occur on Ni2/h-BN-B–D and Ni2/h-BN and the ability to resist carbon deposition was improved, with the trend being Ni2/h-BN-B–D > Ni2/h-BN > Ni(111). This indicated that a good catalyst for DRM can be rationally designed by performing size and defect engineering through the SMSI properties.
In this work, turnover frequency (TOF) (expressed as the number of cycles performed per unit of time) was used to further measure the efficiency of the Ni-base catalyst for DRM. The TOF can be calculated as follows:61,62
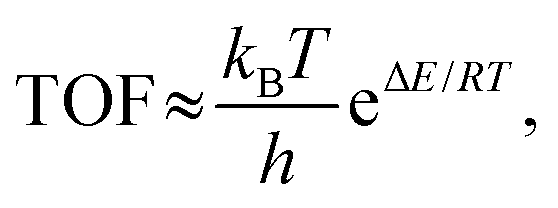
4. Discussion
In this work, we studied the dry reforming of methane (DRM) and a series of competitive reactions over perfect and boron-vacancy-containing h-BN sheet supported Ni-catalysts (Ni2/h-BN and Ni2/h-BN-B–D) using density functional theory calculations and comparison to metallic Ni(111).Compared with the perfect h-BN surface, the introduction of B-defects makes the interaction force between Ni and the surface stronger and thus less binding to adsorbed species like O*, OH*, and CHx (x = 0–3) than Ni2/h-BN. For this reason, DRM reactions involving decomposition reactions, such as the decomposition of carbon dioxide and methane, are more likely to occur on Ni2/h-BN. Recombination reactions (such as oxidation of CHx* species) are more likely to occur on Ni2/h-BN-B–D.
Comparing the activation of DRM reactants on the surface, we find that CH4 is easier to activate than molecular CO2 on both the Ni2/h-BN and Ni2/h-BN-B–D surfaces. The activation of CO2 occurs on the surface of Ni2/h-BN through a direct route, but is prone to following a hydrogen-assisted path on Ni2/h-BN-B–D via the COOH* intermediate. On these two surfaces, O* is the main oxide and the main reaction path is CH3* being oxidized to form CH3–O.
Moreover, comparing the free energy span of DRM on these two catalysts, we found that Ni2/h-BN-B–D is more favorable than Ni2/h-BN (1.66 vs. 1.72 eV). The trend is the same in the SRM reaction. It is worth noting that on the surface of Ni2/h-BN, the trend is the same as that of metal Ni(111) and the RWGS reaction has the smallest free energy span among these reactions. But on the Ni2/h-BN-B–D, DRM has the highest priority.
The calculation results show that on the surface of Ni2/h-BN-B–D, the ability to resist carbon deposition is greatly enhanced. C* formation by Boudouard reaction has a higher energy span than the methane cracking reaction on Ni2/h-BN-B–D, while the latter requires more energy on Ni2/h-BN. From the Ni2/h-BN surface to the Ni2/h-BN-B–D surface, the free energy span formed by C* species increases significantly. And C–C coupling is easier than C–O formation on the Ni2/h-BN surface because the force between Ni and the carrier on the perfect surface is relatively small and Ni tends to aggregate on the surface. When the concentration of Ni increases on the catalytically active sites on the surface, the ability to resist carbon deposition is significantly reduced. Not only is carbon formation easier, but the tendency to form C–C on the catalyst surface is significantly enhanced.
This study analyzed the factors that affect DRM activity from multiple angles. With the introduction of defects on the catalyst, we found that the activity and selectivity of DRM were enhanced and, most importantly, the ability to deposit carbon was greatly improved. It provides theoretical guidance and support for the application of an Ni-based catalyst in the DRM reaction via its SMSI character.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 21973048, 21773123, 91545106).References
- J. Chen, C. Yao, Y. Zhao and P. Jia, Synthesis Gas Production from Dry Reforming of Methane over Ce0.75Zr0.25O2-Supported Ru Catalysts, Int. J. Hydrogen Energy, 2010, 35, 1630–1642 CrossRef CAS.
- A. E. Castro Luna and M. E. Iriarte, Carbon Dioxide Reforming of Methane over a Metal Modified Ni–Al2O3 Catalyst, Appl. Catal., A, 2008, 343, 10–15 CrossRef CAS.
- K. Asai, K. Takane, Y. Nagayasu, S. Iwamoto, E. Yagasaki and M. Inoue, Decomposition of Methane in the Presence of Carbon Dioxide over Ni Catalysts, Chem. Eng. Sci., 2008, 63, 5083–5088 CrossRef CAS.
- Ş. Özkara-Aydınoğlu, E. Özensoy and A. E. Aksoylu, The Effect of Impregnation Strategy on Methane Dry Reforming Activity of Ce Promoted Pt/Zro2, Int. J. Hydrogen Energy, 2009, 34, 9711–9722 CrossRef.
- F. A. J. Al-Doghachi, U. Rashid, Z. Zainal, M. I. Saiman and Y. H. Taufiq Yap, Influence of Ce2O3 and CeO2 Promoters on Pd/MgO Catalysts in the Dry-Reforming of Methane, RSC Adv., 2015, 5, 81739–81752 RSC.
- S. Chen, J. Zaffran and B. Yang, Descriptor Design in the Computational Screening of Ni-Based Catalysts with Balanced Activity and Stability for Dry Reforming of Methane Reaction, ACS Catal., 2020, 10, 3074–3083 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Catalytic Conversion of Biomass to Biofuels, Green Chem., 2010, 12, 1493–1513 RSC.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas, ChemCatChem, 2009, 1, 192–208 CrossRef CAS.
- M. Maestri, D. Vlachos, A. Beretta, G. Groppi and E. Tronconi, Steam and Dry Reforming of Methane on Rh: Microkinetic Analysis and Hierarchy of Kinetic Models, J. Catal., 2008, 259, 211–222 CrossRef CAS.
- J. R. H. Ross, Natural Gas Reforming and CO2 Mitigation, Catal. Today, 2005, 100, 151–158 CrossRef CAS.
- Ş. Özkara-Aydınoğlu, Thermodynamic Equilibrium Analysis of Combined Carbon Dioxide Reforming with Steam Reforming of Methane to Synthesis Gas, Int. J. Hydrogen Energy, 2010, 35, 12821–12828 CrossRef.
- P. Gronchi, P. Centola and R. D. Rosso, Dry reforming of CH4 with Ni and Rh metal catalysts supported on SiO2 and La2O3, Appl. Catal., A, 1997, 152, 83–92 CrossRef CAS.
- S. Golestan, A. A. Mirzaei and H. Atashi, Fischer–Tropsch Synthesis over an Iron–Cobalt–Manganese (Ternary) Nanocatalyst Prepared by Hydrothermal Procedure: Effects of Nanocatalyst Composition and Operational Conditions, Int. J. Hydrogen Energy, 2017, 42, 9816–9830 CrossRef CAS.
- P. Cao, S. Adegbite, H. Zhao, E. Lester and T. Wu, Tuning Dry Reforming of Methane for F–T Syntheses: A Thermodynamic Approach, Appl. Energy, 2018, 227, 190–197 CrossRef CAS.
- D. Pakhare and J. Spivey, A Review of Dry (Co2) Reforming of Methane over Noble Metal Catalysts, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- R. Carrasquillo-Flores, J. M. R. Gallo, K. Hahn, J. A. Dumesic and M. Mavrikakis, Density Functional Theory and Reaction Kinetics Studies of the Water–Gas Shift Reaction on Pt–Re Catalysts, ChemCatChem, 2013, 5, 3690–3699 CrossRef CAS.
- M. Zhang, B. Zijlstra, I. A. W. Filot, F. Li, H. Wang, J. Li and E. J. M. Hensen, A Theoretical Study of the Reverse Water–Gas Shift Reaction on Ni(111) and Ni(311) Surfaces, Can. J. Chem. Eng., 2019, 98, 740–748 CrossRef.
- O. Muraza and A. Galadima, A Review on Coke Management During Dry Reforming of Methane, Int. J. Energy Res., 2015, 39, 1196–1216 CrossRef.
- L. Foppa, M.-C. Silaghi, K. Larmier and A. Comas-Vives, Intrinsic Reactivity of Ni, Pd and Pt Surfaces in Dry Reforming and Competitive Reactions: Insights from First Principles Calculations and Microkinetic Modeling Simulations, J. Catal., 2016, 343, 196–207 CrossRef CAS.
- Y.-A. Zhu, D. Chen, X.-G. Zhou and W.-K. Yuan, Dft Studies of Dry Reforming of Methane on Ni Catalyst, Catal. Today, 2009, 148, 260–267 CrossRef CAS.
- T. Avanesian, G. S. Gusmão and P. Christopher, Mechanism of CO2 Reduction by H2 on Ru(0001) and General Selectivity Descriptors for Late-Transition Metal Catalysts, J. Catal., 2016, 343, 86–96 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Insights into the Mechanisms of CO2 Methanation on Ni(111) Surfaces by Density Functional Theory, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- S.-T. Zhang, H. Yan, M. Wei, D. G. Evans and X. Duan, Hydrogenation Mechanism of Carbon Dioxide and Carbon Monoxide on Ru(0001) Surface: A Density Functional Theory Study, RSC Adv., 2014, 4, 30241–30249 RSC.
- M. Akri, et al., Atomically Dispersed Nickel as Coke-Resistant Active Sites for Methane Dry Reforming, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, Dry Reforming of Methane on Single-Site Ni/MgO Catalysts: Importance of Site Confinement, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- Y. Tang, et al., Synergy of Single-Atom Ni1 and Ru1 Sites on CeO2 for Dry Reforming of CH4, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- L. Foppa, T. Margossian, S. M. Kim, C. Muller, C. Coperet, K. Larmier and A. Comas-Vives, Contrasting the Role of Ni/Al2o3 Interfaces in Water–Gas Shift and Dry Reforming of Methane, J. Am. Chem. Soc., 2017, 139, 17128–17139 CrossRef CAS PubMed.
- C. Fan, Y.-A. Zhu, M.-L. Yang, Z.-J. Sui, X.-G. Zhou and D. Chen, Density Functional Theory-Assisted Microkinetic Analysis of Methane Dry Reforming on Ni Catalyst, Ind. Eng. Chem. Res., 2015, 54, 5901–5913 CrossRef CAS.
- B. Abdullah, N. A. Abd Ghani and D.-V. N. Vo, Recent Advances in Dry Reforming of Methane over Ni-Based Catalysts, J. Cleaner Prod., 2017, 162, 170–185 CrossRef CAS.
- M. Lu, X. Zhang, J. Deng, S. Kuboon, K. Faungnawakij, S. Xiao and D. Zhang, Coking-Resistant Dry Reforming of Methane over Bn–Nanoceria Interface-Confined Ni Catalysts, Catal. Sci. Technol., 2020, 10, 4237–4244 RSC.
- H. Liu, K. Li, R. Zhang, L. Ling and B. Wang, Insight into Carbon Deposition Associated with NiCo/MgO Catalyzed CH4/CO2 Reforming by Using Density Functional Theory, Appl. Surf. Sci., 2017, 423, 1080–1089 CrossRef CAS.
- V. Pawar, D. Ray, C. Subrahmanyam and V. M. Janardhanan, Study of Short-Term Catalyst Deactivation Due to Carbon Deposition During Biogas Dry Reforming on Supported Ni Catalyst, Energy Fuels, 2015, 29, 8047–8052 CrossRef CAS.
- S. Zhang, C. Shi, B. Chen, Y. Zhang and J. Qiu, An Active and Coke-Resistant Dry Reforming Catalyst Comprising Nickel–Tungsten Alloy Nanoparticles, Catal. Commun., 2015, 69, 123–128 CrossRef CAS.
- L. Li, et al., Controlled Surface Segregation Leads to Efficient Coke-Resistant Nickel/Platinum Bimetallic Catalysts for the Dry Reforming of Methane, ChemCatChem, 2015, 7, 819–829 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, Comparative Study of the Kinetics of Methane Steam Reforming on Supported Ni and Sn/Ni Alloy Catalysts: The Impact of the Formation of Ni Alloy on Chemistry, J. Catal., 2009, 263, 220–227 CrossRef CAS.
- S. Zhang, S. Muratsugu, N. Ishiguro and M. Tada, Ceria-Doped Ni/SBA-16 Catalysts for Dry Reforming of Methane, ACS Catal., 2013, 3, 1855–1864 CrossRef CAS.
- J. Yang, C. Q. Lv, Y. Guo and G. C. Wang, A Dft+U Study of Acetylene Selective Hydrogenation on Oxygen Defective Anatase (101) and Rutile (110) TiO2 Supported Pd4 Cluster, J. Chem. Phys., 2012, 136, 104107 CrossRef PubMed.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Uncoupling the Size and Support Effects of Ni Catalysts for Dry Reforming of Methane, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- D. Guo and G.-C. Wang, Partial Oxidation of Methane on Anatase and Rutile Defective TiO2 Supported Rh4 Cluster: A Density Functional Theory Study, J. Phys. Chem. C, 2017, 121, 26308–26320 CrossRef.
- D. Guo, J.-H. Wen and G.-C. Wang, Coordination Dependence of Carbon Deposition Resistance in Partial Oxidation of Methane on Rh Catalysts, Catal. Today, 2020, 355, 422–434 CrossRef CAS.
- J. A. Lercher, J. H. Bitter, W. Hally, W. Niessen and K. Seshan, Design of stable catalysts for methane-carbon dioxide reforming, Stud. Surf. Sci. Catal., 1996, 101, 463–472 CrossRef CAS.
- S. Tang, L. Ji, J. Lin, H. C. Zeng, K. L. Tan and K. Li, CO2 Reforming of Methane to Synthesis Gas over Sol–Gel-Made Ni/Γ-Al2O3 Catalysts from Organometallic Precursors, J. Catal., 2000, 194, 424–430 CrossRef CAS.
- Y. Cao, P. Maitarad, M. Gao, T. Taketsugu, H. Li, T. Yan, L. Shi and D. Zhang, Defect-Induced Efficient Dry Reforming of Methane over Two-Dimensional Ni/H-Boron Nitride Nanosheet Catalysts, Appl. Catal., B, 2018, 238, 51–60 CrossRef CAS.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 0–50 CrossRef CAS.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Tada, H. Koga, Y. Ato, A. Hayashi, M. Okumura and S. Tanaka, Effect of Spin Contamination Error on Surface Catalytic Reaction: No Reduction by Core-Shell Catalysts, Mol. Phys., 2018, 117, 2251–2259 CrossRef.
- K. Tada, T. Maruyama, H. Koga, M. Okumura and S. Tanaka, Extent of Spin Contamination Errors in Dft/Plane-Wave Calculation of Surfaces: A Case of Au Atom Aggregation on a Mgo Surface, Molecules, 2019, 24, 505 CrossRef PubMed.
- C. T. Campbell and J. R. V. Sellers, Correction to “the Entropies of Adsorbed Molecules”, J. Am. Chem. Soc., 2013, 135, 13998 CrossRef CAS.
- S. Kandoi, J. P. Greeley, M. A. Sanchez-Castillo, S. T. Evans, A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, Prediction of Experimental Methanol Decomposition Rates on Platinum from First Principles, Top. Catal., 2006, 37, 17–28 CrossRef CAS.
- M.-R. Li and G.-C. Wang, Differentiation of the C–O and C–C Bond Scission Mechanisms of 1-Hexadecanol on Pt(111) and Ru(0001): A First Principles Analysis, Catal. Sci. Technol., 2017, 7, 743–760 RSC.
- X. Wang, C. Zhi, Q. Weng, Y. Bando and D. Golberg, Boron Nitride Nanosheets: Novel Syntheses and Applications in Polymeric Composites, J. Phys.: Conf. Ser., 2013, 471, 012003 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- H.-Y. Ma and G.-C. Wang, Selective Hydrogenation of Acetylene on Ptn/TiO2 (N = 1, 2, 4, 8) Surfaces: Structure Sensitivity Analysis, ACS Catal., 2020, 10, 4922–4928 CrossRef CAS.
- S. Kozuch and S. Shaik, How to Conceptualize Catalytic Cycles? The Energetic Span Model, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- Y.-X. Wang and G.-C. Wang, A Systematic Theoretical Study of Water Gas Shift Reaction on Cu(111) and Cu(110): Potassium Effect, ACS Catal., 2019, 9, 2261–2274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04732e |
| This journal is © the Owner Societies 2021 |