
An enhanced electrochemical CO2 reduction reaction on the SnOx–PdO surface of SnPd nanoparticles decorated on N-doped carbon fibers†
Sreekanth
Narayanaru
 ab,
Gopinathan M.
Anilkumar
abc,
Masaki
Ito
c,
Takanori
Tamaki
ab and
Takeo
Yamaguchi
*ab
ab,
Gopinathan M.
Anilkumar
abc,
Masaki
Ito
c,
Takanori
Tamaki
ab and
Takeo
Yamaguchi
*ab
aLaboratory for Chemistry and Life Sciences, Tokyo Institute of Technology, R1-17, 4259 Nagatsuta, Midori-Ku, Yokohama, 226-8503 Japan. E-mail: yamag@res.titech.ac.jp
bCore Research for Evolutionary Science and Technology, Japan Science and Technology Agency (JST-CREST), 102-0076 Japan
cR&D Center, Noritake Co., Ltd., 300 Higashiyama, Miyochi-cho, Miyoshi, 470-0293 Japan
First published on 28th October 2020
Abstract
In the electrocatalytic CO2 reduction reaction (CO2RR), tin-based catalysts are known for their high formate faradaic yield. However higher overpotentials are required to attain a high faradaic yield with high partial current density for formate. Here, we describe the increase in the electrocatalytic CO2RR activity of Sn nanoparticles decorated on nitrogen-doped carbon fibers (NCFs) by adding a small amount of Pd. Nitrogen-doped carbon fibers decorated with SnPd nanoparticles (Sn100−yPdy–NCF) of different Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratios were synthesized using the electrospinning method and their electrocatalytic CO2RR activity was studied. The Sn100−yPdy–NCF catalyst with 3 wt% (y = 3) Pd displayed superior activity for the CO2RR and attained a faradaic efficiency of 85%, whereas the NCF with Sn nanoparticles (Sn100–NCF) attained only 57% efficiency at the same potential. The surface electronic configuration, Tafel slope (79 mV dec−1) and bicarbonate reduction activity of the catalyst reveal that the combination of SnOx–PdO on the catalyst surface is responsible for the superior CO2RR activity.
:Pd ratios were synthesized using the electrospinning method and their electrocatalytic CO2RR activity was studied. The Sn100−yPdy–NCF catalyst with 3 wt% (y = 3) Pd displayed superior activity for the CO2RR and attained a faradaic efficiency of 85%, whereas the NCF with Sn nanoparticles (Sn100–NCF) attained only 57% efficiency at the same potential. The surface electronic configuration, Tafel slope (79 mV dec−1) and bicarbonate reduction activity of the catalyst reveal that the combination of SnOx–PdO on the catalyst surface is responsible for the superior CO2RR activity.
1. Introduction
Recent analyses have confirmed that the atmospheric CO2 concentration reached 412 ppm, and its contribution to global warming is alarming.1–4 In this context, reducing the global CO2 emissions and converting CO2 to useful products are the best solutions to control the CO2-related climate change issues.5–8 The electrochemical conversion of CO2 to liquid fuels, such as formic acidt/formate, methanol, and ethanol, or chemical feedstocks such as syngas, CO + H2 (ref. 9–15), is the most promising method for the effective utilization of CO2. Recently, extensive investigations have been conducted across the world for the effective electrochemical reduction of CO2. As a result, the electrocatalytic CO2RR activity of a large number of materials was tested, which include metals,16–18 metal nanoparticles,19–23 bimetallic nanoparticles, metal nanoalloys,24–27 and many types of carbon materials.28–32 These materials have different capabilities and product selectivity for the CO2RR. The product selectivity and efficiency vary with metals,18 the size of the metal particles,33 the roughness of the material surface,34,35 the dopant in the carbon structure28–30 and even the cations present in the electrolytes.36 Therefore, tailor-made materials are important for the selective, efficient, and stable conversion of CO2 to a product.A variety of products have been produced through the electrochemical CO2RR;37 however, techno-economic studies claim that formic acid/formate from CO2 is one of the most economically viable products.38 In the renewable energy industries, formic acid with a volumetric capacity of 53 g L−1 of hydrogen39 and liquid formate-based fuel cells (DFFCs), which have a higher theoretical cell voltage (1.45 V) and low poisoning effect,40 confirm the importance of formic acid/formate as an energy carrier. Recently, researchers have focused on the development of highly efficient electrocatalysts and membranes for the formate oxidation reaction, which may facilitate the commercialization of direct formate fuel cells.41,42 Even though DFFCs are considered as a source of green energy, the industrial production of formic acid/formate by the hydrolysis of methyl formate or formamide is not an eco-friendly method.43,44 In this context, the efficient electrochemical reduction of CO2 to formate using renewable energy sources is a promising method for utilizing CO2 and producing clean and sustainable fuels.
Among the metal catalysts, Sn-based catalysts have an efficient electrochemical CO2 reduction activity. Tin has a high hydrogen overpotential and excellent selectivity for CO2 to formate conversion;19,45–50 tin is also a non-toxic, inexpensive, and eco-friendly element. Furthermore, the native oxide layer of the Sn surface increases the catalytic activity by stabilizing the CO2 radical (CO2˙−).50 Although tin displays high selectivity and efficiency for the CO2 reduction reaction (CO2RR), the requirement of high overpotential is the main drawback. A variety of modifications have been introduced to reduce the overpotential and improve the catalytic activity of Sn,19,45–49 but the ability to achieve high faradaic efficiency and high partial current density remains a substantial challenge. In the present study, we prepared Sn nanoparticles with a low amount of Pd that were supported on N-doped carbon fibers (NCFs) using a simple electrospinning method and investigated the electrocatalytic CO2 reduction activity of these materials. Compared to the usual carbon support for nanoparticles, the N-doped carbon surface is more suitable since pyridinic nitrogen in N-doped CNFs exhibits catalytic CO2 reduction reaction activity.28,29 We chose palladium as an additive to tin to increase the CO2RR activity because the Pd surface is capable of stabilizing and reducing bicarbonate ions to formate at a near thermodynamic potential.51,52 Since the bicarbonate ions are the proton source for the CO2 reduction reaction in bicarbonate solution,19,21 the catalyst surface stabilized bicarbonate ions may enhance the quick protonation of CO2˙−. Based on these aspects, we prepared Sn100−yPdy–NCF catalysts with different amounts of Pd loading. As a result of this rational design of the electrocatalyst, we could achieve a maximum faradaic efficiency of 85% and a partial current density value of 12.75 mA cm−2 during the electrochemical reduction of CO2 on the Sn100−yPdy–NCF catalyst containing 3 wt% (y = 3) Pd. The material characterization and analysis of the electrochemical activity of these catalysts confirm that the combination of SnOx–PdO (where SnOx is the sum of the SnO and SnO2) on the catalyst surface is responsible for this superior activity.
2. Experimental
2.1. Chemicals
N-Doped carbon fibers with Sn and Pd nanoparticles were prepared using SnCl2·2H2O (Wako Chemicals), Pd (CH3COO)2 (Sigma-Aldrich), polyacrylonitrile (PAN, MW = 150000) (Sigma-Aldrich), and DMF (Wako Chemicals) as precursors. Electrolytes were prepared using KHCO3 (Wako Chemicals), H2SO4 (Wako Chemicals), potassium formate (Wako Chemicals), disodium hydrogen phosphate, and monosodium hydrogen formate (Wako Chemicals). DMSO (Sigma-Aldrich) and D2O (Wako Chemicals) were used for NMR analyses. All solutions were prepared in Milli-Q water (18.2 MΩ).
2.2. Synthesis and characterization of metal nanoparticles decorated on N-doped carbon fibers
Tin nanoparticles decorated on N-doped carbon fibers (Sn100–NCF) were prepared by an electrospinning method using a NANON-01A (MECC Co., Ltd.) electrospinning system. A solution containing 1.2 g of polyacrylonitrile (PAN), 1 g of SnCl2·2H2O and 12.8 g of DMF was electrospun to produce Sn100–NCF. The following electrospinning conditions were applied, an applied voltage of 25 kV, a distance of 150 mm that was maintained between the needle and collector, and a precursor solution fed with a feed rate of 0.8 mL per hour. After electrospinning, fibers were dried at 130 °C for 12 hours in a vacuum atmosphere, and the dried fibers were heated at 280 °C for 90 minutes in an air atmosphere at a heating rate of 5 °C per minute to stabilize the PAN fibers. The stabilized composite PAN fibers were then heated at 900 °C for 120 minutes at a ramping rate of 3 °C per minutes under a N2/H2 (100 sccm N2 and 30 sccm H2) atmosphere. Various Sn100−yPdy–NCF catalyst samples with different amounts of Pd were prepared using the method described above. For Sn100−yPdy–NCF the solution for electrospinning was prepared by adding the required amount of metal precursors to PAN (1.2 g) in a DMF (9.8 g) solution. Metal precursor solutions were prepared by separately dissolving Pd (Ac)2 (25, 50, 75, and 100 mg) and SnCl2·2H2O (1 g) in 1.5 mL of DMF, and then the two solutions were mixed and sonicated for 30 minutes.The ICP analysis was performed using a Shimadzu sequential plasma emission spectrometer ICP2-8100 to quantify the total metal loading and the weight percentage distribution of Sn and Pd on the NCF (details are provided in the ESI,† Table S1). Phase identification and crystallinity of the catalyst samples were determined by performing XRD analysis at a scan rate of 1° per minute using an Ultima IV system from Rigaku with a Cu Kα (λ = 1.5406 Å) and X-ray source operating at 40 kV and 40 mA. The surface compositions of the fibers were analyzed using an XPS Quantum 2000, ULVAC-PHI Inc., with a twin-anode X-ray source using Al Kα radiation (hυ = 1486.58 eV). A FESEM (S-4800, Hitachi High Technology) operating at an accelerating voltage of 5 kV and a HR-TEM (TOPCON EM-002BF-J) operating at an accelerating voltage of 200 kV with a twin EDS facility were used to visualize the morphology and to understand the elemental distribution of nanoparticle-decorated fibers.
2.3. Electrochemical characterization
The electrochemical activity of the catalysts was studied using an electrochemical measurement system (Hokuto Denko, HZ5000). Linear sweep voltammetry (LSV), cyclic voltammetry (CV) and constant potential electrolysis were performed in a two-compartment cell separated by a Nafion-117 membrane. An Ag/AgCl (KCl-saturated) reference electrode was used for all measurements, and Pt foil (area: 18 cm2) served as the counter electrode. Catalyst ink was prepared by mixing the required amount of catalyst in a solution containing 5 mL of isopropanol (25 wt%) and 20 μL of the Nafion® solution (5 wt%). The catalyst ink was sonicated for 4 hours in an ice bath to obtain a good dispersion. After achieving a well-dispersed solution, the catalyst was coated on the glassy carbon electrode using the drop-casting method, and the catalyst loading was maintained at 50 μg of metal per cm2 for all the samples. The catalyst-coated glassy carbon electrode (Hokuto Denko) with an electrode area of 0.196 cm2 was used for voltammetric and impedance analyses. The catalyst-coated, 1 cm2 Sigracet 28 BA gas diffusion layer (GDL) electrode was used for electrolysis and stability studies. For the CO2RR, a 0.5 M KHCO3 solution was first saturated with CO2 by bubbling it with CO2 gas for 1 h. During electrolysis, a constant flow of CO2 gas was maintained at the cathode compartment. Similarly, the bicarbonate reduction reaction was performed by passing N2 instead of CO2 in a 0.5 M KHCO3 solution. All applied potential values reported here are iR corrected and with respect to a reversible hydrogen electrode (RHE). The conversion of the applied potential measured using the Ag/AgCl electrode to the RHE was calculated using a method described in the ESI† (section S4).2.4. Product analysis
Liquid product quantification was performed using a Bruker 400 MHz NMR spectrometer with DMSO as the internal standard. The details of the analysis and quantification of the liquid products using NMR are provided in the ESI† (section S6.1).Gas products were quantified using a gas chromatograph (Shimadzu GC-2010 Tracera) equipped with Micropacked ST in series with a Shincarbon ST column and barrier discharge ionization detector (BID). The details of the analysis and quantification of the gas products using gas chromatography are provided in the ESI† (S6.2).
3. Results and discussion
3.1. Synthesis and characterization of the catalyst
In the stabilization process of the as-spun fibers at 280 °C in air, polyacrylonitrile undergoes crosslinking to form a stable fibrous structure.53 Pyrolysis at 900 °C under a N2/H2 atmosphere after stabilizing the fibers with metal precursors produces metal nanoparticle-decorated graphitized N-doped carbon fibers. Based on the amount (wt%) of Sn and Pd present in the NCF, the catalyst samples are designated as Sn100–NCF, Sn97Pd3–NCF, Sn95Pd5–NCF, Sn94Pd6–NCF, and Sn91Pd9–NCF, here the numbers represent the percentage of Sn and Pd in the total metal loaded on the NCF in all other cases.The X-ray diffraction (XRD) patterns of all the samples are presented in Fig. 1a. The XRD pattern of the Sn100–NCF sample indicated that the metallic tin was a major phase, along with a small amount of SnO2. The diffraction patterns correspond to 2θ values of 30.67, 32.06, 43.92, and 44.98° represent the metallic tin (200), (101), (220) and (211) planes, respectively (JCPDS No: 83-7910), and 2θ values of 26.61, 33.89 and 51.79° correspond to the SnO2 (110), (101), and (211) planes, respectively, (JCPDS No: 41-1445). The XRD patterns of Sn97Pd3–NCF and Sn95Pd5–NCF also showed the presence of Sn and SnO2, similar to the Sn100–NCF sample. In addition to Sn and SnO2, the presence of PdO was also observed at 2θ values of 34.8 and 42.4°, representing the PdO (101) and (110) planes, respectively (JCPDS No 43-1024). For the catalysts with comparatively high Pd concentration, Sn94Pd6–NCF and Sn91Pd9–NCF, the XRD peaks correspond to SnO2 and PdO were not observed; however, a slight shift in the diffraction peaks of Sn to higher 2θ values, 0.05 and 0.13 degrees, respectively, was noticed (Fig. 1b) compared to Sn100–NCF, Sn97Pd3–NCF and Sn95Pd5–NCF, indicating the possible formation of an SnPd alloy.54 Although the total amount of Pd present in Sn97Pd3–NCF and Sn95Pd5–NCF is low, the appearance of the PdO peak indicated that most of the Pd is located on the surface and exists as PdO.
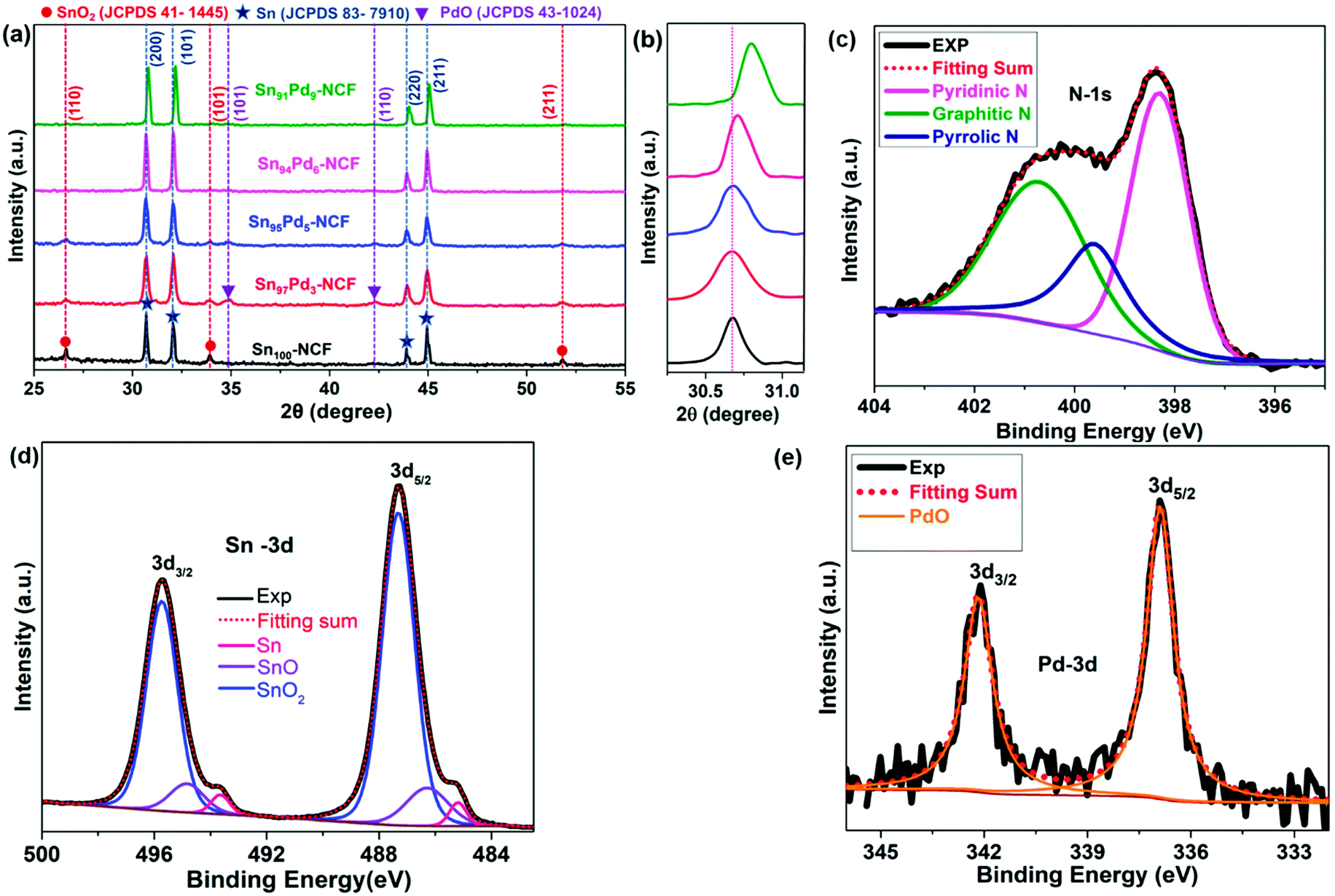 | ||
| Fig. 1 XRD patterns of (a) Sn100–NCF and Sn100−yPdy–NCF catalysts, (b) highlighted, 2θ values between 30 and 31 degrees, and (c–e) the deconvoluted XPS spectra of N, Sn and Pd present in Sn97Pd3–NCF. | ||
XPS analysis of the catalysts was performed to identify the surface composition (Fig. 1c–e, S1, and S2†). The deconvoluted high-resolution XPS N-1s spectra of all catalyst samples indicated the presence of graphitic (BE = 400.76 eV), pyrrolic (BE = 399.60 eV), and pyridinic (BE = 398.30 eV) nitrogen.32 The deconvoluted Sn-3d spectra of all catalysts showed the presence of Sn, SnO, and SnO2. The binding energy values of Sn (3d5/2 – 485.1 eV), SnO (3d5/2 – 486.2 eV), and SnO2 (3d5/2 – 487.2 eV)55 were approximately the same for all catalysts. The atomic percentages (at.%) of Sn, SnO, and SnO2 were calculated from the corresponding peak area of the deconvoluted Sn-3d5/2 spectrum (Table S2a†). For all catalysts, the amount (at.%) of SnOx was greater than 95%, suggesting that the catalyst surface consists of an oxide layer of Sn. The deconvoluted Pd-3d spectrum was different for different Sn100−yPdy–NCF samples (Fig. 1e and S2†); the spectrum of Sn97Pd3–NCF contained only the PdO peak (3d5/2 – 336.9 eV), but peaks for both PdO and Pd (3d5/2 – 335.5 eV) were observed in the spectrum of Sn95Pd5–NCF. For Sn94Pd6–NCF and Sn91Pd9–NCF samples, both Pd and PdO peaks were slightly shifted to higher binding energy.56 For Sn94Pd6–NCF,Pd and PdO peaks were observed at 3d5/2 – 336.2 eV and 3d5/2 – 337.3 eV, respectively, and at 3d5/2 – 336.4 eV and 337.5 eV for Sn91Pd9–NCF, indicating a change in the binding energy values of Pd and PdO probably due to the formation of SnPd alloy.57,58 The ratio of Pd/PdO of the Sn100−yPdy–NCF systems increased with the increase in the Pd concentration (Table S2b†). The XRD and XPS analyses revealed that the core of metal nanoparticles in Sn100–NCF and Sn100−yPdy–NCF consisted of metallic Sn and the surface in the oxidized form. The surface of Sn97Pd3–NCF solely consists of SnOx and PdO, whereas the surface of the Sn95Pd5–NCF system is mainly composed of SnOx, PdO, and a low amount of Pd. For the Sn94Pd6–NCF and Sn91Pd9–NCF samples, the surface concentration of Pd is higher than the PdO concentration. In general, the XPS results corroborate the XRD results.
The morphology of the materials was analyzed using FE-SEM and HR-TEM. The average diameter of N-doped carbon fibers was 650 nm (Fig. 2a), and the nanoparticles were spherical with a size of less than 10 nm uniformly decorating the carbon fibers (Fig. 2b and c). Lattice fringes with a d-spacing of 0.29 nm (inset of Fig. 2d) correspond to the (200) plane of Sn (JCPDS No: 83-7910). The energy-dispersive X-ray spectroscopy (EDS) map revealed the uniform distribution of C, N, O, Sn, and Pd on the Sn100−yPdy–NCF surface (Fig. 2f–j and S3†).
 | ||
| Fig. 2 (a) General morphology of Sn97Pd3–NCF (SEM image). HR-TEM images of (b) a single fiber of Sn97Pd3–NCF and (c) SnPd nanoparticles dispersed on the carbon fiber (d) showing the lattice fringes corresponding to the Sn (200) plane. (e–i) STEM-EDS mapping images of C, N, O, Sn and Pd, respectively. | ||
3.2. Electrochemical reduction of CO2
The electrocatalytic CO2 reduction activity of all materials was primarily evaluated using linear sweep voltammetry (LSV). Fig. 3a and S5† show the voltammograms of Sn100–NCF, Sn100−yPdy–NCF, and NCF, recorded between −0.1 V and −1 V at a scan rate of 10 mV s−1 in CO2 saturated 0.5 M KHCO3. Based on a previous report, the potential at which the catalyst attains a current density value of −0.15 mA cm−2 is considered as the onset potential.29 The onset potential values of the NCF, Sn100–NCF, Sn91Pd9–NCF, Sn94Pd6–NCF, Sn95Pd5–NCF, and Sn97Pd3–NCF were −0.36, −0.21, −0.17, −0.16, −0.14 and −0.12 V, respectively. At −1.0 V the NCF alone attained a current density value of 0.77 mA cm−2, whereas the Sn100–NCF catalyst attained a current density value of 8.23 mA cm−2 at the same potential. Sn97Pd3–NCF, Sn95Pd5–NCF, Sn94Pd6–NCF, and Sn91Pd9–NCF attained a current density value of 19.75, 18.88, 14.12, and 12.07 mA cm−2, respectively, at −1.0 V. However, the current density alone does not ensure the CO2RR activity of the catalysts. So the product analysis was performed after 30 minutes of electrolysis at different potentials and the faradaic efficiency of each product was calculated. The liquid and gaseous products of the CO2RR were qualitatively and quantitatively analyzed using NMR and gas chromatography, respectively. For all the catalysts, formate was the initial CO2RR product obtained. Formate production was observed for Sn100−yPdy–NCF samples at −0.37 V, whereas for Sn100–NCF and the NCF, the potential was −0.57 V, which is 0.2 V higher than the Sn100−yPdy–NCF catalysts. N-Doped carbon can produce CO and formate from CO2,28,29 however in this work the NCF produced only formate. For all other catalysts, the products of the CO2RR were formate and CO, of which formate was the major product. The faradaic efficiency (FECO2RR) and partial current density (jCO2RR) of CO2RR products of all the samples were measured and the results are presented in Fig. 3b and c, S8–S10† and Table 1. The NCF attained a FECO2RR of 56% and jCO2RR of 0.41 mA cm−2 at −0.97 V, whereas at the same potential Sn100–NCF attained a faradaic efficiency of 65% for the CO2RR at −0.97 V with jCO2RR of 4.60 mA cm−2, the formate faradaic efficiency (FEformate) was 57% and the corresponding partial current density of formate (jformate) was 4.03 mA cm−2.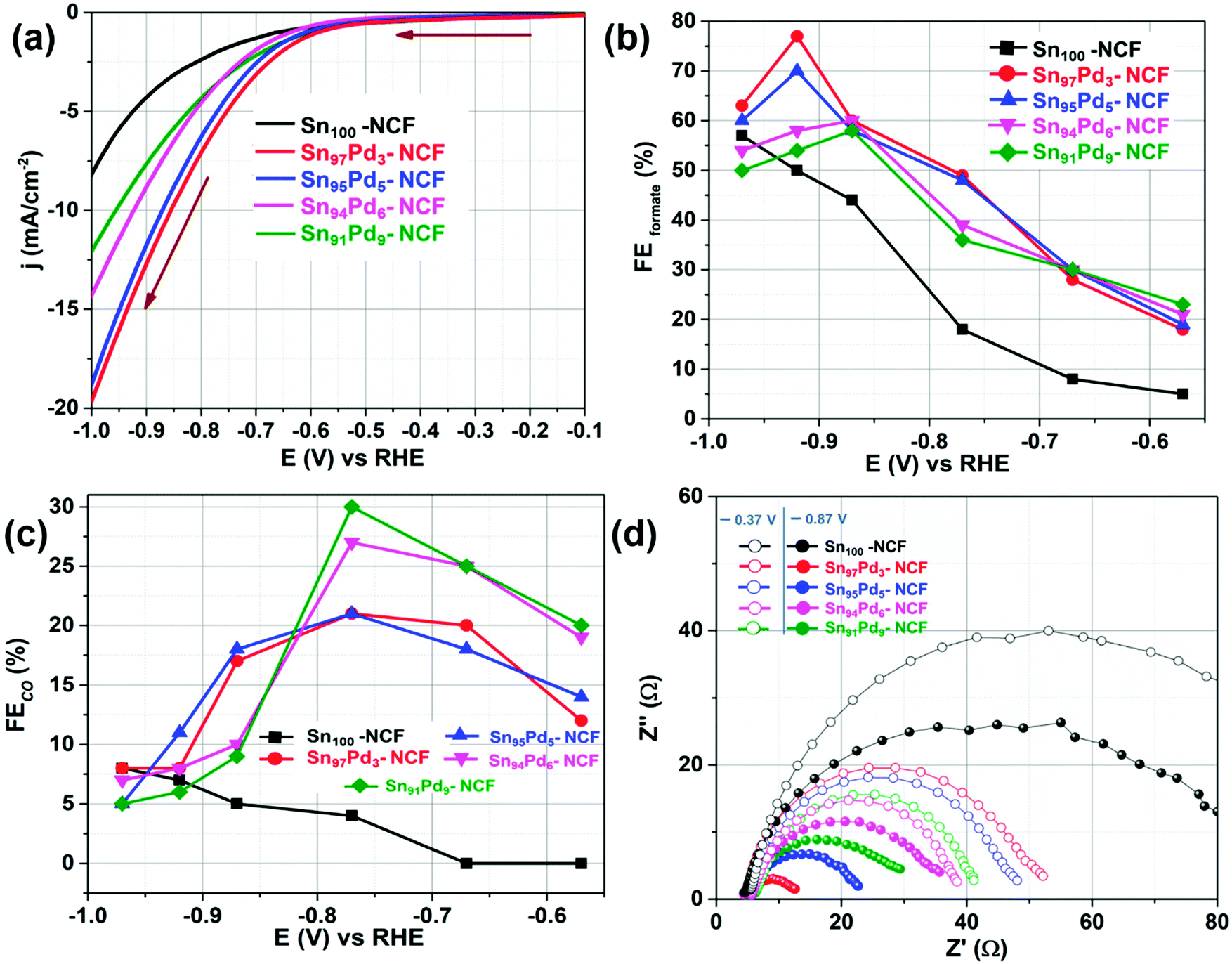 | ||
| Fig. 3 Electrocatalytic CO2 reduction in 0.5 M KHCO3 saturated with CO2 on Sn100–NCF and Sn100−yPdy–NCF catalysts. (a) iR-Corrected linear sweep voltammogram measured at a scan rate of 10 mV s−1, (b) formate faradaic efficiency, (c) CO faradaic efficiency and (d) Nyquist plots measured at −0.37 V and −0.87 V. | ||
| Catalyst | E at maximum FECO2RR (V) | FECO2RR (%) | FEformate (%) | FECO (%) | FEH2 (%) | j CO2RR (mA cm−2) | j formate (mA cm−2) | j CO (mA cm−2) | j H2 (mA cm−2) |
|---|---|---|---|---|---|---|---|---|---|
| Sn91Pd9–NCF | −0.87 | 67 | 58 | 9 | 33 | 3.79 | 3.28 | 0.51 | 1.87 |
| Sn94Pd6–NCF | −0.87 | 70 | 60 | 10 | 30 | 4.20 | 3.60 | 0.60 | 1.80 |
| Sn95Pd5–NCF | −0.92 | 81 | 70 | 11 | 19 | 10.84 | 9.36 | 1.48 | 2.54 |
| Sn97Pd3–NCF | −0.92 | 85 | 77 | 8 | 15 | 12.75 | 11.55 | 1.20 | 2.25 |
| Sn100–NCF | −0.97 | 65 | 57 | 8 | 35 | 4.60 | 4.03 | 0.57 | 2.4 |
| NCF | −0.97 | 56 | 56 | 0 | 44 | 0.41 | 0.41 | — | 0.32 |
The Sn100−yPdy–NCF catalysts have displayed an interesting trend in FECO2RR. Sn94Pd6–NCF and Sn91Pd9–NCF catalysts exhibited a high FECO2RR at the low overpotential region compared to Sn97Pd3–NCF and Sn95Pd5–NCF. However, at slightly higher overpotential, Sn97Pd3–NCF and Sn95Pd5–NCF attained very high FECO2RR and jCO2RR values compared to the other two Sn100−yPdy–NCF catalysts. The faradaic efficiency values of formate and CO of Sn100−yPdy–NCF revealed that formate is the major CO2RR product for all the catalysts. Among the Sn100−yPdy–NCF catalysts Sn97Pd3–NCF has attained high FEformate and jformate values, when evaluating the catalysts in terms of the faradaic efficiency of CO (FECO), the Sn91Pd9–NCF has attained high FECO, 30% at −0.77 V (Fig. 3c). Comparing the activity of efficient Sn100−yPdy–NCF catalysts with Sn100–NCF in terms FEformate, the highly efficient Sn97Pd3–NCF has a 27% higher FEformate and 8 mA cm−2 higher jformate than Sn100–NCF at −0.92 V (Fig. 3b and S9b†). Similarly, comparing the activity in terms of FECO, Sn91Pd9–NCF exhibited a 26% higher FECO and 0.96 mA cm−2jCO than Sn100–NCF at −0.77 V (Fig. 3c, S9c†). These results suggest that a small amount of Pd addition on Sn–NCF, either as Pd or PdO can enhance the CO2RR activity. Based on the material characterization and faradaic efficiency of formate and CO results of the most efficient Sn100−yPdy–NCF catalysts we can say that SnOx–Pd enhances CO production and SnOx–PdO enhances formate production. The CO2RR activity of pure Pd is well studied and it is reported that at low overpotentials Pd produces formate and CO and at higher overpotentials, Pd preferably produces CO,59,60 but the activity of PdO is not well understood. We performed CO2 reduction over the PdO surface (section S8, Fig. S11a†). During the initial stages of electrolysis, the FEformate reached 64% but it quickly reduced and stabilized at 34%, the jformate was <1 mA cm−2. The XRD analysis at different stages of electrolysis confirmed that PdO reduced to Pd during the electrolysis (Fig. S11b†).
Since PdO was quickly reduced to Pd during the electrolysis, we tested the catalytic stability of Sn97Pd3–NCF (Fig. S12†) with periodic analysis of faradaic efficiency, followed by XPS and HR-TEM of the catalyst (Fig. S13†). Interestingly, a stable FECO2RR was retained for Sn97Pd3–NCF for up to 9 hours of electrolysis, and then a decrease in FECO2RR was observed. After 15 hours, the FECO2RR decreased to 70% from 85%. FEformate and FECO were 68% and 2%, respectively. XPS analysis was performed after 9 and 15 hours of electrolysis. After 9 hours of electrolysis the PdO at.% was only reduced to 60 from 100%, the at.% of SnOx remained almost the same, 96.4 from 96.7, (Fig. S13a and b†). The XPS analysis after 15 hours of electrolysis indicated that the at.% of Sn at the surface was increased from 3.26 to 18 and the at.% of SnOx reduced to 82 from 96.74 (Fig. S13c†). A very low-intensity broad Pd-3d peak was obtained. The TEM image of the catalyst after 15 hours of electrolysis shows agglomeration of the particles on the surface (Fig. S13d†), whereas the particles inside the fibers have not changed. The stability test and followed material characterization confirms that the PdO reduction is slower in Sn97Pd3–NCF and the FECO2RR decreases with the reduction in PdO.
Impedance and Tafel slope analyses were performed to understand the charge transfer kinetics and the reaction mechanism. The electrochemical impedance was measured at a lower overpotential, −0.37 V, and a higher overpotential, −0.87 V in a CO2-saturated KHCO3 solution. At −0.37 V, Sn91Pd9–NCF and Sn94Pd6–NCF displayed a lower charge transfer resistance (RCT) than Sn97Pd3–NCF and Sn95Pd5–NCF. The trend changed when the RCT value was measured at −0.87 V, the Sn97Pd3–NCF and Sn95Pd5–NCF samples displayed a lower RCT value than the Sn91Pd9–NCF and Sn94Pd6–NCF catalysts (Fig. 3d and Table S3†). Furthermore, we performed the EIS analysis before and after 30 minutes of electrolysis for the Sn97Pd3–NCF and Sn91Pd9–NCF catalysts (Fig. S14†). The RCT values of Sn91Pd9–NCF increased with the time of electrolysis, whereas the change in RCT values was not significant for Sn97Pd3–NCF. The increase in the RCT values of Sn91Pd9–NCF with the electrolysis time is probably because of the adsorption of intermediates or products on the catalytic surface and hinders the electron transfer. To verify this possibility the Sn91Pd9–NCF electrode was kept at 0.5 V for 30 s after electrolysis and then EIS was performed; the RCT was similar to the initial RCT, suggesting the adsorption of an intermediate or reaction products on the catalyst surface. Since the Pd surface is susceptible to CO poisoning,59–61 the increased RCT values with respect to electrolysis time are possibly due to CO poisoning. The low or insignificant change in the RCT values of Sn97Pd3–NCF after electrolysis revealed that the PdO surface is not susceptible to CO poisoning.62 The increase in RCT values of Sn91Pd9–NCF and Sn94Pd6–NCF at high overpotentials is due to surface poisoning and these results corroborate their low current density and faradaic efficiency at higher overpotentials.
Tafel slopes (Fig. 4 and S15†) were plotted using the log of jformate and overpotentials of each sample to understand the reaction mechanism. Tafel slope values for Sn100–NCF, Sn97Pd3–NCF, Sn95Pd5–NCF, Sn94Pd6–NCF, and Sn91Pd9–NCF were 105, 79, 92, 105 and 117 mv dec−1, respectively. The Tafel slope value of Sn97Pd3–NCF and Sn95Pd5–NCF was less than 120 mv dec−1 and close to 59 mv dec−1. The low Tafel slope value indicates that the first step of the reaction mechanism was the reversible one-electron transfer to CO2 to form CO2˙− then the chemical rate-determining step, i.e. the protonation of C in CO2˙− to form formate.19,48,49 Higher Tafel slope values closer to 120 mv dec−1 suggest that the one-electron transfer to CO2 to form CO2˙− is the rate-limiting step.19,42,45 According to previous reports, the surface electronic structure plays a major role in the selectivity of the CO2 reduction reaction by stabilizing CO2˙− and other intermediates.21,33,34,48 Here, the low Tafel slope, high faradaic efficiency, and partial current density obtained for Sn97Pd3–NCF and Sn95Pd5–NCF confirm that the presence of SnOx–PdO combination at the catalyst surface has an important role in their catalytic activity. A recent study by Zhang et al. on the electrochemical reduction of CO2 using a Pd/SnO2 system explained the role of the Pd–O–Sn combination in promoting the reaction through excellent CO2 adsorption and decreasing CO poisoning on the Pd surface.62
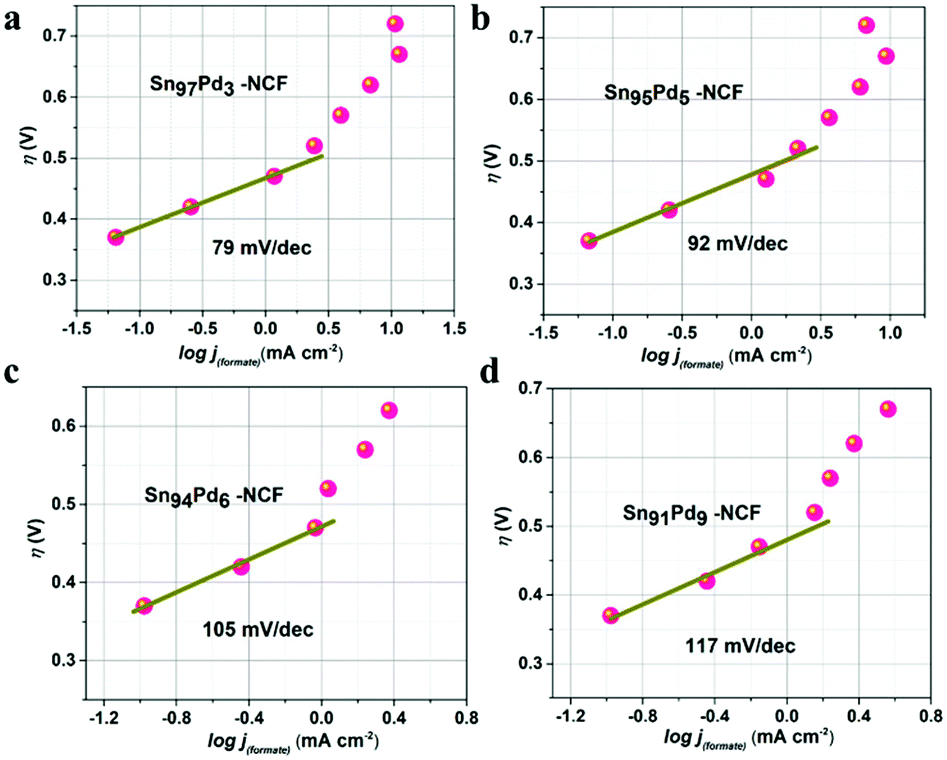 | ||
| Fig. 4 Tafel slopes of (a) Sn97Pd3–NCF (b) Sn95Pd5–NCF (c) Sn94Pd6–NCF and (d) Sn91Pd9–NCF. | ||
In the electrochemical CO2 reduction reaction, bicarbonate ions in the electrolyte provide protons to protonate CO2˙− and produce formate.19,21 A catalyst surface, which can stabilize the bicarbonate ions on its surface, can facilitate the quick protonation of CO2˙− to formate. We believe that the reason for the high current density and faradaic efficiency of the SnPd–NCF catalyst with the SnOx–PdO surface is because the SnOx–PdO surface effectively stabilizes CO2˙− and bicarbonate on its surface and protonates CO2˙− to produce formate. To ensure this possibility, we tested the bicarbonate reduction activity of all catalysts and analyzed the products. The NCF and Sn100–NCF catalysts were unable to reduce bicarbonate to formate, whereas formate was produced when we performed bicarbonate reduction over Sn100−yPdy–NCF. Only a detectable amount of formate (FEformate < 1%) was obtained from Sn91Pd9–NCF and Sn94Pd6–NCF. However, a quantifiable amount of formate (FEformate > 3%) was obtained from the catalysts Sn95Pd5–NCF and Sn97Pd3–NCF (Fig. S16†). A low amount of formate formation suggests that the bicarbonate reduction is not kinetically feasible on the SnOx–PdO surface, but the electrode stabilizes the bicarbonate ions on its surface. Compiling the results obtained from the material characterization, electrochemical CO2RR, and bicarbonate reduction reaction, the catalyst surface with the SnOx–PdO combination appeared to be more active than a catalyst surface with SnOx and SnOx–Pd. Further, we investigated the bicarbonate reduction activity of Pd and PdO by conducting bicarbonate electrolysis on bare Pd and the oxidized Pd electrode. The bicarbonate electrolysis performed on the Pd surface produced no formate, however, formate was obtained when the electrolysis was performed on PdO (section S-14 and Fig. S-17†), revealing that the PdO surface is capable of stabilizing and reducing the bicarbonate ion.
Based on these analyses and previously reported mechanisms,19,62,63 we speculate the probable CO2 reduction reaction mechanism on Sn100–NCF with SnOx (Scheme 1) and Sn100−yPdy–NCF with SnOx–PdO (Scheme 2) as described below, however detailed in situ studies are warranted to confirm this mechanism. The CO2 reduction mechanism on the Sn100–NCF catalyst involves the formation and stabilization of CO2˙− at the catalyst surface. Stabilized CO2˙− further accepts a proton from the bicarbonate ions of the electrolyte to form formate (Scheme 1).19,63 However, on the Sn100−yPdy–NCF surface with SnOx–PdO, the bicarbonate ion adsorbed on the catalyst surface protonates CO2˙− to produce formate (Scheme 2).
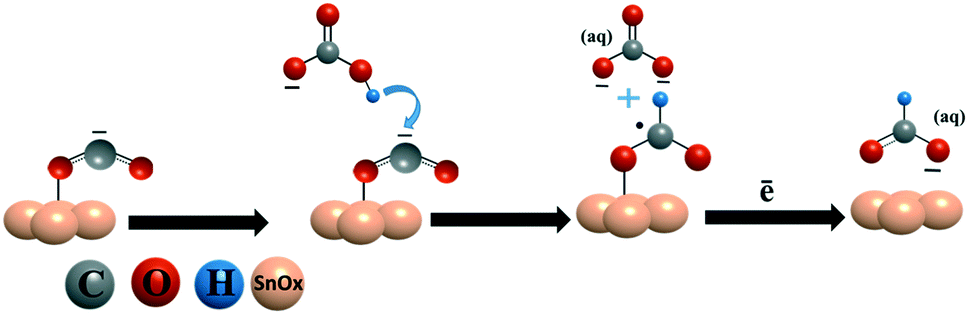 | ||
| Scheme 1 CO2 reduction to formate on the Sn100–NCF surface. | ||
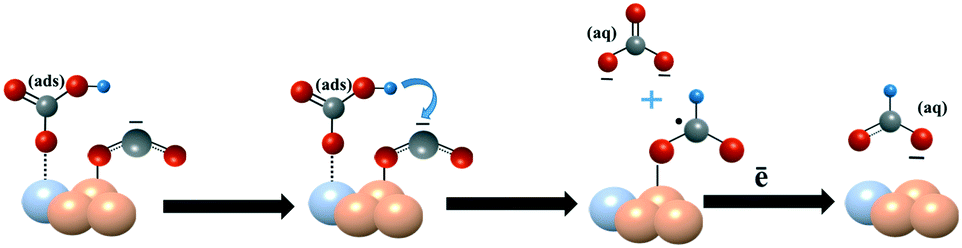 | ||
| Scheme 2 CO2 reduction to formate on the SnOx–PdO surface. | ||
4. Conclusions
In conclusion, the electrocatalytic CO2 reduction reaction activity of SnPd nanoparticles decorated on N-doped carbon fibers with different Pd contents was analyzed. The catalysts were prepared using a simple one-step electrospinning method followed by pyrolysis. The Sn100−yPdy–NCF catalyst with a low Pd content, 3 wt% Pd, showed superior CO2RR activity (total faradaic efficiency of 85% and a partial current density of 12.7 mA cm−2 at −0.92 V compared with RHE) to the Sn100−yPdy–NCF catalysts with a high Pd content. The analysis of the surface composition revealed that Sn100−yPdy–NCF with 3 wt% Pd mainly contained PdO and SnOx on its surface. The Tafel slope analysis and bicarbonate reduction activity of Sn97Pd3–NCF catalysts suggest a possibility that the SnOx–PdO species at the catalyst surface effectively stabilize CO2˙− and bicarbonate on their surface. Simultaneous stabilization of bicarbonate anions and CO2˙− on the catalytic surface can promote the CO2RR by quick protonation of CO2˙− to produce formate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial assistance from the Japan Science and Technology (JST) Core Research for Evolutionary Science and Technology, Japan Science and Technology Agency (JST-CREST, JPMJCR 1543), and this study was also supported in part by the “Five-Star Alliance”. Professor Ken Motokura, Tokyo Institute of Technology, is gratefully acknowledged for providing access to the gas chromatography facility.Notes and references
- Earth System Research Laboratory Global Monitoring Division, Mauna Loa observatory. https://www.esrl.noaa.gov/gmd/webdata/ccgg/trends/co2_data_mlo.png.
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719–1723 CrossRef CAS.
- S. H. Schneider, Science, 1989, 243, 771–781 CrossRef CAS.
- D. Tong, Q. Zhang, Y. Zheng, K. Caldeira, C. Shearer, C. Hong, Y. Qin and S. J. Davis, Nature, 2019, 572, 373–377 CrossRef CAS.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- C. Ampelli, S. Perathoner and G. Centi, Philos. Trans. R. Soc., A, 2015, 373, 1–30 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- S. Back, H. Kim and Y. Jung, ACS Catal., 2015, 5, 965–971 CrossRef CAS.
- G. Centi, C. Ampelli, C. Genovese, B. C. Marepally, G. Papanikolaou and S. Perathoner, Faraday Discuss., 2015, 183, 125–145 RSC.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- H. R. M. Jhong, S. Ma and P. J. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- Z. Chen, P. Kang, M.-T. Zhang and T. J. Meyer, Chem. Commun., 2014, 50, 335–337 RSC.
- P. Kang, Z. Chen, A. Nayak and T. J. Meyer, Energy Environ. Sci., 2014, 07, 4007–4012 RSC.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- Y. Hori, Mod. Aspects Electrochem., 2008, 42, 89–189 CAS.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang, X. Bao, D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang and G. Wang, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS.
- R. Kortlever, I. Peters, S. Koper and M. T. M. Koper, ACS Catal., 2015, 5, 3916–3923 CrossRef CAS.
- X. Zhao, B. Luo, R. Long, C. Wang and Y. Xiong, J. Mater. Chem. A, 2015, 3, 4134–4138 RSC.
- Z. Xu, E. Lai, Y. Shao-Horn and K. Hamad-Schifferli, Chem. Commun., 2012, 48, 5626–5628 RSC.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- S. Pal, S. Narayanaru, B. Kundu, M. Sahoo, S. Bawari, D. K. Rao, S. K. Nayak, A. J. Pal and T. N. Narayanan, J. Phys. Chem. C, 2018, 122, 23385–23392 CrossRef CAS.
- H. R. M. Jhong, C. E. Tornow, B. Smid, A. A. Gewirth, S. M. Lyth and P. J. A. Kenis, ChemSusChem, 2017, 10, 1094–1099 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- S. Narayanaru, J. Chinnaiah, K. L. Phani and F. Scholz, Electrochim. Acta, 2018, 264, 269–274 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- S. Sankar, G. M. Anilkumar, T. Tamaki and T. Yamaguchi, ChemCatChem, 2019, 11, 4731–4737 CrossRef CAS.
- H. Kuroki, S. Miyanishi, A. Sakakibara, Y. Oshiba and T. Yamaguchi, J. Power Sources, 2019, 438, 226997 CrossRef CAS.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, Appl. Energy, 2015, 157, 165–173 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef.
- S. Lee, J. D. Ocon, Y. Il Son and J. Lee, J. Phys. Chem. C, 2015, 119, 4884–4890 CrossRef CAS.
- S. Zhao, S. Li, T. Guo, S. Zhang, J. Wang, Y. Wu and Y. Chen, Nano-Micro Lett., 2019, 11, 62 CrossRef CAS.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS.
- D. H. Won, C. H. Choi, J. Chung, M. W. Chung, E. H. Kim and S. I. Woo, ChemSusChem, 2015, 8, 3092–3098 CrossRef CAS.
- Y. Fu, Y. Li, X. Zhang, Y. Liu, J. Qiao, J. Zhang and D. P. Wilkinson, Appl. Energy, 2016, 175, 536–544 CrossRef CAS.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS.
- M. Spichiger-Ulmann and J. Augustynski, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 713–716 RSC.
- N. Sreekanth and K. L. Phani, Chem. Commun., 2014, 50, 11143–11146 RSC.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS.
- W. Du, K. E. MacKenzie, D. F. Milano, N. A. Deskins, D. Su and X. Teng, ACS Catal., 2012, 2, 287–297 CrossRef CAS.
- Y. Li, J. Qiao, X. Zhang, T. Lei, A. Girma, Y. Liu and J. Zhang, ChemElectroChem, 2016, 3, 1618–1628 CrossRef CAS.
- B. Jiang, X. G. Zhang, K. Jiang, D. Y. Wu and W. Bin Cai, J. Am. Chem. Soc., 2018, 140, 2880–2889 CrossRef CAS.
- S. Sankar, G. M. Anilkumar, T. Tamaki and T. Yamaguchi, ACS Appl. Energy Mater., 2018, 1, 4140–4149 CrossRef CAS.
- Z. Li, Y. Chen, G. Fu, Y. Chen, D. Sun, J. M. Lee and Y. Tang, Nanoscale, 2019, 11, 2974–2980 RSC.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS.
- R. Kortlever, C. Balemans, Y. Kwon and M. T. M. Koper, Catal. Today, 2015, 244, 58–62 CrossRef CAS.
- S. Chatterjee, C. Griego, J. L. Hart, Y. Li, M. L. Taheri, J. Keith and J. D. Snyder, ACS Catal., 2019, 9, 5290–5301 CrossRef CAS.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. Chuong and B. Wu, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01437k |
| This journal is © The Royal Society of Chemistry 2021 |