
Engineering multicomponent metal-oxide units for efficient methane combustion over palladium-based catalysts†
Yelin
Chen
a,
Jia
Lin
*ab,
Xiaohua
Chen
a,
Siqin
Fan
a and
Ying
Zheng
 *a
*a
aCollege of Chemistry and Materials Science, Fujian Provincial Key Laboratory of Advanced Materials Oriented Chemical Engineering, Fujian Normal University, Fuzhou, Fujian 350007, P. R. China. E-mail: linjia1025@163.com; zyingth@sina.com
bCollege of Environmental Science and Engineering, Fujian Normal University, Fuzhou, Fujian 350007, P. R. China
First published on 30th October 2020
Abstract
Multicomponent catalysts have been long known for their potential to improve catalytic performance, whereas rational design proposes profound challenges. Herein, we present a strategy for engineering metal oxide units to realize efficient methane combustion through incorporating Mg into Pd/CexZr1−xO2–Al2O3 catalysts. Catalysts are facilely obtained through a sol–gel method and incipient wetness impregnation process; meanwhile, the surface and structural properties are tuned via Mg modification. The doped Mg component enters the CZ lattice, inducing numerous oxygen vacancies and favourable oxygen migration due to the structural and electronic mismatch between Mg2+ and Ce4+ (or Zr4+). The generated MgAl2O4 spinel and oxygen vacancies conjointly result in weaker Pd–O bonds and improved reducibility. Meanwhile, the efficient oxygen transfer between the metal and support contributes to the reformation of bulk PdO. Consequently, the smooth conversion of Pd ↔ PdO is realized, which is beneficial for methane oxidation. Moreover, the catalytic activity of Pd/5CZA-yM varies with the particle size of palladium species. The as-prepared Pd/5CZA-5M with a moderate Pd particle size demonstrates significantly boosted catalytic activity and long-term stability compared to the unmodified one.
1. Introduction
Natural gas, primarily consisting of methane, has been recognized as an alternative to other automobile fuels (e.g. gasoline and diesel) due to its high thermal efficiency and less pollutant emissions.1–3 However, the unburned methane in the exhaust gases is a more potent greenhouse gas than CO2, which results in a high contribution to global warming.1,4 Currently, catalytic combustion has become a promising technology for the methane abatement; meanwhile, the alumina supported palladium catalyst (Pd/Al2O3) demonstrates excellent catalytic performance for this reaction.5–7 Nevertheless, alumina is susceptible to phase transition under operating conditions, accompanied by coalescence and sintering of active palladium nanoparticles due to the high surface free energy, leading to severe deactivation.6,8,9 Hence, extensive investigations devoted to enhance the thermal stability of alumina and the sinter-resistance of palladium species have been conducted. Rational designing of a multicomponent catalyst modified with promoters is an effective method to realize sustained methane oxidation.Ceria (CeO2) presents excellent oxygen exchange and migration capabilities, making it attractive for applications in the oxidation of methane and other light alkanes,10 but pure CeO2 demonstrates limited thermal stability.11 CeO2–ZrO2 solid solution (CZ) formed via partial replacement of Ce4+ sites by Zr4+ with a smaller ionic radius can effectively inhibit sintering of particles,12 and can also serve as an effective “oxygen buffer” to supply active oxygen for methane oxidation through the facile Ce3+/Ce4+ redox couple.13 Therefore, CZ is regarded as a better additive or support for catalytic combustion compared to CeO2. It was revealed that the CZ support provided abundant active PdO species due to the great oxygen exchange capability,14 which could be further regulated by adjusting the preparation method15 or tuning the phase structure of CZ,16 contributing to a greatly enhanced catalytic performance. In view of the promotion effect brought about by CZ, it has been introduced into the Pd/Al2O3 catalysts. A modified impregnation process has been developed to realize the simultaneous crystallization of CZ and Al2O3.17 The stronger interaction between the CZ component and alumina successfully suppressed the phase transformation of γ-Al2O3 and the phase separation of CZ, resulting in improved thermal stability. However, the catalytic performance of Pd/CexZr1−xO2–Al2O3 (Pd/CZA) catalysts is still insufficient,14,18 thus requiring further advancements in the catalytic activity, in particular, at lower temperatures.
Methane oxidation over supported Pd-based catalysts is supposed to follow the Mars–van Krevelen mechanism,19 where the easy oxygen exchange between active PdO and the support contributes to the Pd/PdO conversion process, leading to the high reactivity of active sites. It is noteworthy that the migration and exchange of oxygen are closely related to the concentration of oxygen vacancies.20,21 Meanwhile, the redox performance and particle size of palladium along with the metal–support interaction also determine the catalytic performance.22–24 Thus, optimizing the distribution and status of active oxygen and palladium species on catalysts through engineering the component becomes a feasible approach to promote the catalytic performance of Pd/CZA.
The introduction of third components with lower valency (e.g. Fe, Bi or Ni) into the CZ solid solution could further promote the oxygen storage/release ability by creating more anion defects.13,25,26 Besides, alkaline earth metals have received widespread attention due to their high alkalinity and excellent electron-donating capacity.27 Efficient Pd-based catalysts have been designed through the tertiary component doping route, where Ba was selectively incorporated into CZA carriers, giving rise to faster oxygen diffusion.28 It has been reported that spinel components in Pd-based catalysts could effectively modulate the dispersion and chemical state of active palladium species.29,30 Since Mg is suitable for creating a spinel phase with Al, the moderate interaction between the MgAl2O4 spinel and PdO supplied a template for the epitaxy of crystalline PdO, which enhanced the reducibility of catalysts.31 Furthermore, the MgAl2O4 spinel phase could stabilize active Pd2+ species over Mg-doped Pd/ZrO2–Al2O3 catalysts; meanwhile, restraining the generation of hydroxyl groups on the surface of the support resulted in improved activity and hydrothermal stability.32 Inspired by the excellent textural property of alumina and great oxygen mobility of the CZ solid solution along with the potential promotion of Mg, it is expected that a newly designed Pd-based catalyst with favourable catalytic performance could be achieved by optimally doping Mg into the CZA carrier. However, the interaction between components could exert complex influences on active palladium species and the catalytic performance, thus the rational design and definite understanding of the structure–activity relationship persist as major challenges.
Herein, a composition modulation strategy of incorporating Mg into Pd/CexZr1−xO2–Al2O3 (Pd/5CZA-yM) was developed for efficient methane abatement. The multicomponent Pd/5CZA-yM catalysts were obtained through a facile sol–gel method and incipient wetness impregnation process; meanwhile, the influence of the Mg content on the structure, properties and catalytic performance of the catalysts was systematically analysed. Encouragingly, the catalyst with a suitable Mg doping content exhibited great low-temperature catalytic activity and stability toward methane combustion under demanding conditions. The enhancement in catalytic performance was well correlated with the generated numerous oxygen vacancies, improved reducibility of palladium, smooth conversion of Pd ↔ PdO and the suitable Pd particle size.
2. Experimental
2.1. Synthesis of supports
Mg-Doped CexZr1−xO2–Al2O3 supports were synthesized via a modified sol–gel method. The obtained xerogels were calcined at 500 °C for 4 h, then calcined at 900 °C for 1 h. Afterwards, the obtained samples were labelled 5CZA-yM, where y represents the mass fraction of magnesium (y = 0, 2, 5, 7 wt%). As for the Al2O3 carrier, the synthesis process was similar to the above, except that no modified elements were added during the preparation process. The detailed information about the synthesis of the supports is given in the ESI† (see Page S2).2.2. Synthesis of catalysts
Taking the as-prepared Al2O3 or 5CZA-yM as carriers, palladium-based catalysts with a theoretical Pd loading amount of 0.5 wt% were synthesized by an incipient wetness impregnation method. After impregnation, the catalyst precursors were dried at 60 °C and 110 °C for 5 h, respectively, and then calcined at 600 °C for 1 h (10 °C min−1). The as-synthesized catalysts were labelled Pd/Al2O3 and Pd/5CZA-yM.2.3. Characterization
The structural performance and physicochemical properties of the Pd/Al2O3 and Pd/5CZA-yM catalysts were characterized by N2 physisorption measurements, powder X-ray diffraction (XRD), Raman spectroscopy, UV-visible (UV-vis) diffuse reflectance spectroscopy, X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), in situ diffuse reflectance FTIR spectroscopy of CO adsorption–desorption experiments (CO-DRIFTS), temperature-programmed reduction of H2 (H2-TPR), temperature-programmed oxidation of O2 (O2-TPO), and temperature-programmed desorption of O2 (O2-TPD), NH3 (NH3-TPD), CO2 (CO2-TPD) and CO (CO-TPD). The detailed information about the characterization techniques is given in the ESI† (see Page S2–S5).2.4. Catalytic performance evaluation
The activity evaluation for methane oxidation was conducted in a fixed-bed quartz reactor and the dosage of the catalysts was 100 mg, which were diluted with 1000 mg inert quartz. The reaction gas consisted of 1 vol% CH4, 5 vol% O2, and N2 (balance gas) with a gas hourly space velocity (GHSV) of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. The inlet and outlet gas concentrations of CH4 were continuously monitored using an online gas chromatograph (Agilent 7820A GC system) equipped with a TCD. The CH4 conversion was recorded and calculated by eqn (1) as follows:
000 mL g−1 h−1. The inlet and outlet gas concentrations of CH4 were continuously monitored using an online gas chromatograph (Agilent 7820A GC system) equipped with a TCD. The CH4 conversion was recorded and calculated by eqn (1) as follows: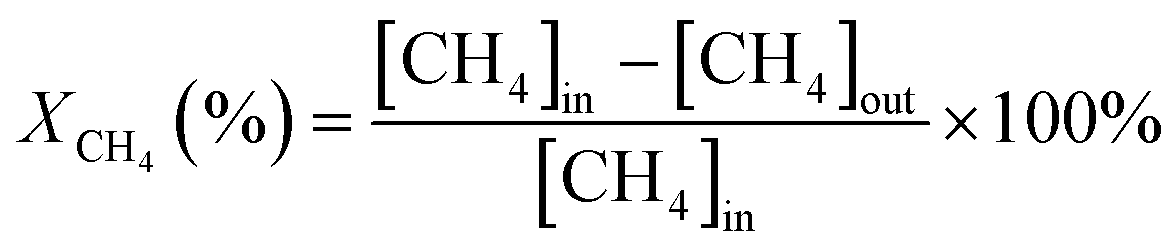 | (1) |
000 mL g−1 h−1.
The kinetic measurements were performed with the methane conversion lower than 15% in order to eliminate the thermal and diffusion effects. The apparent activation energy (Ea) was calculated by the Arrhenius equations, and the reaction rates (r) and turnover frequency (TOF) were calculated by eqn (2) and (3):22
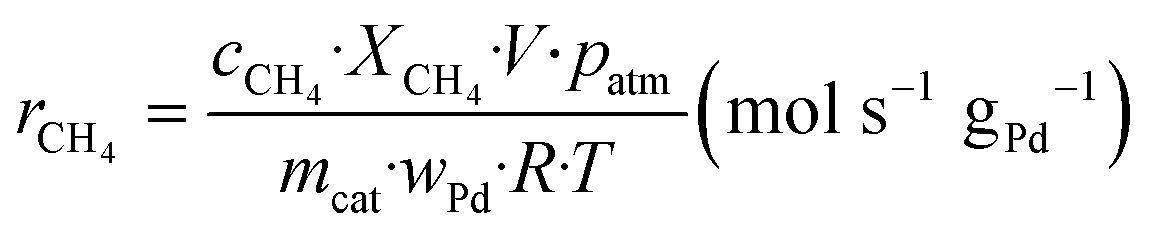 | (2) |
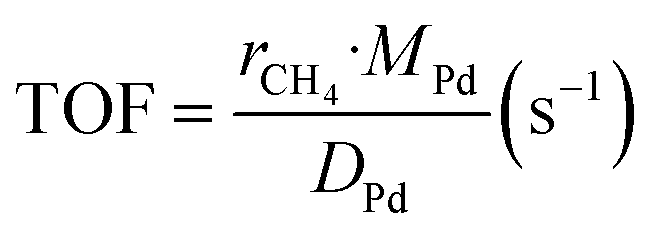 | (3) |
3. Results and discussion
3.1. Evaluation of the catalysts for methane oxidation
The results of the activity evaluation over the Pd/Al2O3 and Pd/5CZA-yM catalysts for methane oxidation are displayed in Fig. 1A and the performance comparison with literature studies is summarized in Table S1.† As compared with Pd/Al2O3 (T99 = 485 °C), the higher initial catalytic activity was observed in Pd/5CZA-0M (T99 = 455 °C), demonstrating the positive effect of the CZ component. The influence of Mg doping on the catalytic performance of Pd/5CZA was further investigated. The results manifested that the methane oxidation activity varied with the Mg amount, and Mg-modified samples presented boosted catalytic activity compared with the unmodified one. Among them, Pd/5CZA-5M exhibited optimal catalytic performance with a T99 of 400 °C, which was decreased by 55 °C relative to Pd/5CZA-0M. However, the excessive doping contents brought about a slightly poorer catalytic activity for Pd/5CZA-7M. The kinetic analysis results of the Pd/Al2O3 and Pd/5CZA-yM catalysts are shown in Fig. 1B and Table 1. Pd/5CZA-2M and Pd/5CZA-5M presented relative lower apparent activation energies (Ea) (114.0 and 92.7 kJ mol−1) and higher reaction rates (r) (14.9 and 16.5 × 10−5 mol gPd−1 s−1) and turnover frequencies (TOFs) (8.4 and 10.9 × 10−2 s−1), which matched well with their better catalytic activity during methane combustion. Additionally, the durability and water resistance of the catalysts were further investigated over representative Pd/5CZA-0M and Pd/5CZA-5M as follows.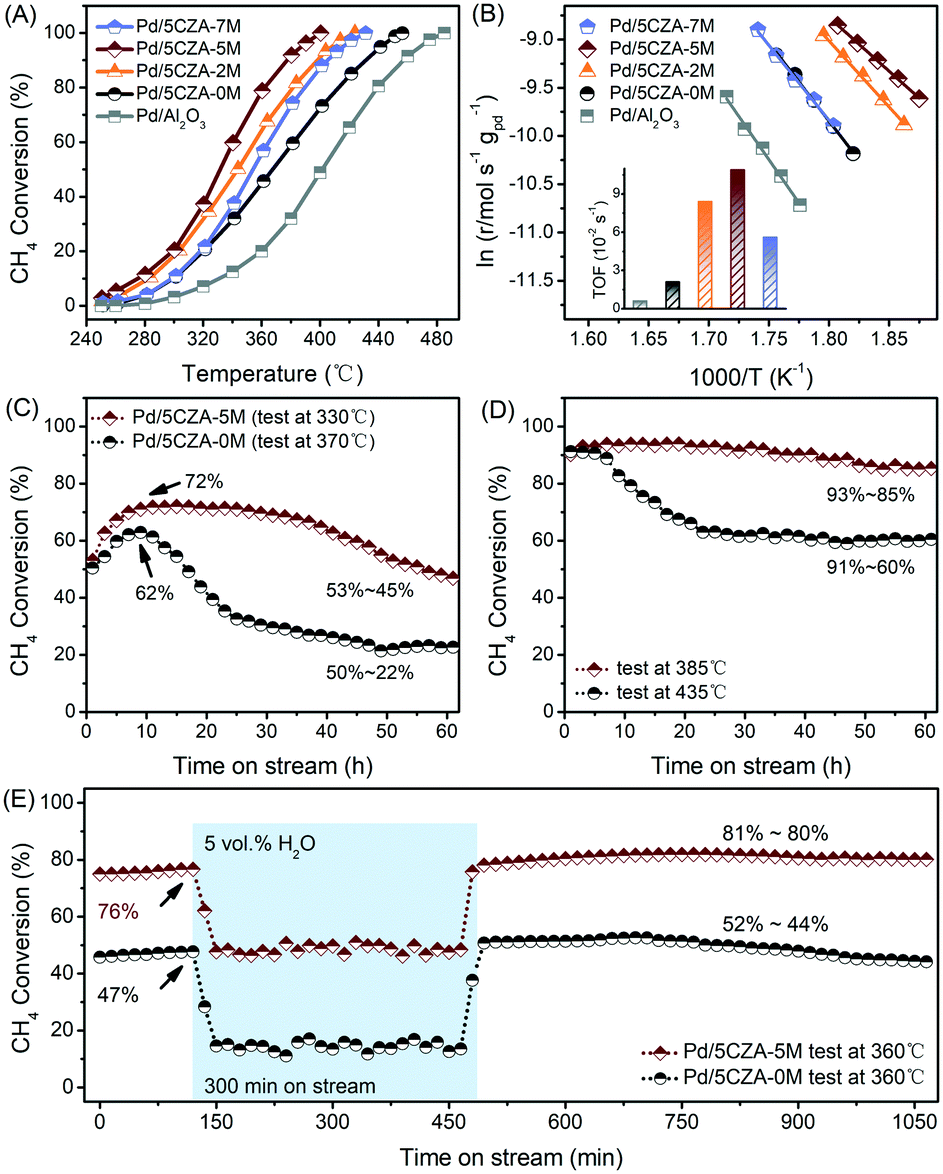 | ||
| Fig. 1 Methane conversion as a function of temperature over the Pd/Al2O3 and Pd/5CZA-yM catalysts (A); Arrhenius plots (inset: TOF at 285 °C) of the catalysts (B); 60 h time on stream test of the Pd/5CZA-0M and Pd/5CZA-5M catalysts at T50 (C) and T90 (D); stability evaluation of the Pd/5CZA-0M and Pd/5CZA-5M catalysts at 360 °C in the absence and presence of 5 vol% water vapor (E). Reaction conditions: 1 vol% CH4, 5.0 vol% O2 in N2 as balance gas, GHSV = 50000 mL h−1 g−1. | ||
| Sample | Pd/Al2O3 | Pd/5CZA-0M | Pd/5CZA-2M | Pd/5CZA-5M | Pd/5CZA-7M |
|---|---|---|---|---|---|
| a The dispersion and particle size of palladium were obtained from CO pulse chemisorption (CO/Pd = 1). | |||||
| T 99 (°C) | 485 | 455 | 420 | 400 | 430 |
| E a (kJ mol−1) | 149.1 | 134.5 | 114.0 | 92.7 | 129.5 |
| r @285 °C (10−5 mol gPd−1 s−1) | 1.7 | 6.6 | 14.9 | 16.5 | 6.6 |
| TOF@285 °C (10−2 s−1) | 0.6 | 2.1 | 8.4 | 10.9 | 5.6 |
| Dispersion of Pda (%) | 32.9 | 33.8 | 18.7 | 16.0 | 12.5 |
| Particle size of Pda (nm) | 3.4 | 3.3 | 5.9 | 6.9 | 8.9 |
As illustrated in Fig. 1C and D, the 60 h on-stream stability tests at T50 and T90 were conducted over Pd/5CZA-0M and Pd/5CZA-5M for further uncovering the role of Mg doping. During the low onset conversion stability experiment, the catalytic activity of Pd/5CZA-0M and Pd/5CZA-5M gradually increased in the first 10 h. Subsequently, the methane conversion over Pd/5CZA-0M dropped significantly from 62% to 22%, while over Pd/5CZA-5M, it could be well maintained at about 70% in the following 25 h, and then gradually restored to the initial activity. Besides, Pd/5CZA-5M also demonstrated favorable stability at 385 °C, with the methane conversion fluctuated by only 8% (93–85%) over the entire period. In contrast, Pd/5CZA-0M presented poor durability with the methane conversion remarkably decreased from 91% to 60% at 435 °C. The obviously enhanced long-term stability at different (high and low) onset conversions over Pd/5CZA-5M could be attributed to the modulation on the surface and the structural properties of the catalyst by Mg modification.
Fig. 1E shows the different steady-state behaviors of CH4 oxidation over Pd/5CZA-0M and Pd/5CZA-5M in the absence and presence of 5 vol% water vapor at a constant temperature of 360 °C, at which the initial methane conversion of Pd/5CZA-0M and Pd/5CZA-5M was 47% and 76%, respectively. When exposed to 5 vol% water vapor, the methane conversion of Pd/5CZA-0M and Pd/5CZA-5M was significantly reduced and kept at about 14% and 48%, respectively, during the 300 minute isothermal process. The deactivation observed in the samples was associated with the formation of inactive Pd(OH)2 due to the fast covering of hydroxyls on the active PdO surface.20,33,34 It could be seen that water vapor had a certain inhibitory effect on the activity of both two samples, whereas the performance of Pd/5CZA-5M decreased to a lesser extent. Notably, the catalytic activity of Pd/5CZA-0M and Pd/5CZA-5M could be completely restored or even improved after removing the water vapor. In the subsequent 10 hour online test, the methane conversion of Pd/5CZA-5M remained almost constant at about 80%, whereas it decreased slightly from 52% to 44% for Pd/5CZA-0M. The incorporation of CZ into Al2O3 endowed the as-prepared catalysts with high oxygen mobility, resulting in a higher rate of surface dehydroxylation, which was responsible for the total recovery of the catalytic activity.20,35,36 Moreover, the further modification of Mg could induce extra oxygen vacancies and allow Pd/5CZA-5M to hold effective methane oxidation after water removal.
3.2. Textural and structural properties
As depicted in Fig. 2A, a weak diffraction peak attributed to the tetragonal CZ phase (PDF-ICDD 38-1436) was observed at 2θ = 33.7° in the Pd/5CZA-yM catalysts. For the Pd/Al2O3 and Pd/5CZA-yM (y = 0, 2) samples, the characteristic peaks indexed to γ-Al2O3 appeared at 2θ = 37.2°, 45.6° and 66.7°, corresponding to the (311), (400) and (440) crystal planes, respectively (JCPDS 10-0425).7 Relative to Pd/Al2O3, peaks attributed to γ-Al2O3 were obviously weaker over Pd/5CZA-0M, demonstrating the decrease in the crystallinity of γ-Al2O3 owing to the introduction of the CZ component. Regarding Pd/5CZA-5M and Pd/5CZA-7M, the peaks at 2θ = 18.9°, 31.5°, 37.2°, 45.3°, 56.5°, 60.1° and 66.2° belonging to the MgAl2O4 spinel phase were clearly visible (PDF-87-0345).37 The intensity of these peaks was gradually increased with the rise of the Mg content, indicating the enhancement of the grain size and crystallinity of the MgAl2O4 spinel. In combination with the activity results, it could be inferred that the improvement of the catalytic performance provided by Mg doping might be associated with the evolution of the MgAl2O4 spinel phase. After the reaction (Fig. 2B), an extra peak attributed to the (220) reflection of γ-Al2O3 appeared over Pd/Al2O3 (JCPDS 10-0425), whereas it was not detected over Pd/5CZA-0M. For the catalysts with Mg doping, it was hard to investigate the above variation due to the overlap of reflections ascribed to the γ-Al2O3 and MgAl2O4 spinel phases. Additionally, no diffraction peaks indexed to palladium species were observed, which might be due to the low palladium loading amount.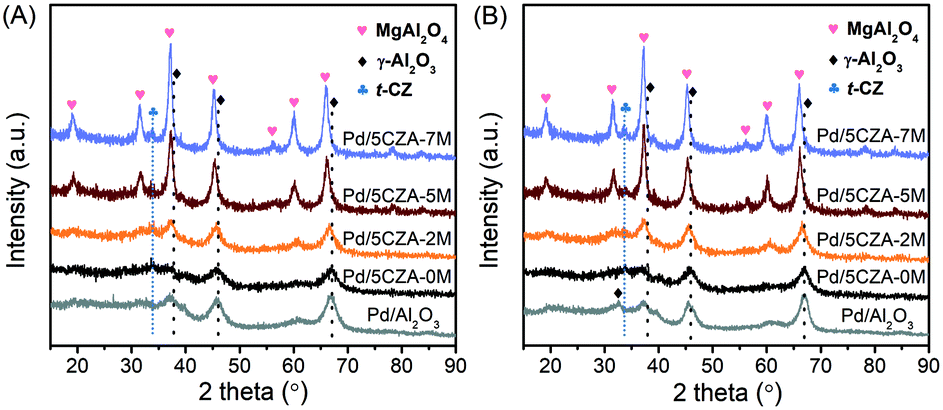 | ||
| Fig. 2 Wide angle XRD patterns of the Pd/Al2O3 and Pd/5CZA-yM catalysts before (A) and after (B) the activity test. | ||
As shown in Fig. S1,† all the catalysts showed type-IV isotherms, manifesting the existence of a mesoporous structure. Pd/Al2O3, Pd/5CZA-0M and Pd/5CZA-yM (y = 2, 5, 7) exhibited H1, H2 and H3-shaped hysteresis loops, which originated from cylindrical pores,3 ink-bottle pores38 and slit-like pores,39 respectively. This supported that both the CZ and Mg doping affected the pore structure. Furthermore, relative to that over Pd/Al2O3 (Table S2†), the surface area of Pd/5CZA-0M was increased, while that of Pd/5CZA-yM (y = 2, 5, 7) was declined. The obvious decrease for Pd/5CZA-7M could be attributed to the larger layered pores caused by the formation of more MgAl2O4 particles.
SEM images (Fig. S2†) showed that the Pd/5CZA-yM catalysts were composed of homogeneous nanoparticles with a uniform size of 40–50 nm, demonstrating that the modification of Mg had a little effect on the morphology of the samples. Meanwhile, Mg incorporation exerted a significant influence on the size and distribution of palladium species. From the HRTEM images of the Pd/Al2O3 and Pd/5CZA-yM catalysts (Fig. 3 and S3†), it was noteworthy that the palladium particle size increased with the rise of the Mg doping content. Specifically, small particles were dispersed homogenously on Pd/Al2O3 and Pd/5CZA-0M (ca. 3–4 nm). The particle size of the palladium species on Pd/5CZA-2M and Pd/5CZA-5M with better catalytic activity was determined as 4.50 ± 0.63 nm and 5.33 ± 0.48 nm, respectively. Meanwhile, obviously larger particles (7.05 ± 0.88 nm) were detected on Pd/5CZA-7M in a more aggregated state. The variation in the particle size and distribution of the palladium species on the catalysts evaluated from CO chemisorption (Table S3†) was well consistent with that from HRTEM observations. A correlation of the TOFs of the catalysts with the average palladium particle size calculated from CO chemisorption is illustrated in Fig. S4.† Notably, the catalytic performance of the Pd/5CZA-yM catalysts was sensitive to the particle size of palladium, showing a volcano-shaped trend with the rise of the particle size from 3.3 to 8.9 nm, among which Pd/5CZA-5M with a moderate palladium particle size of 6.9 nm exhibited a relatively lower apparent activation energy and a higher reaction rate and turnover frequency (Table 1). Although there is no clear consensus regarding the effect of the palladium particle size on the catalytic performance of supported Pd catalysts, many researchers suggest that methane oxidation could be a structure sensitive reaction.40 A similar size-dependent phenomenon toward methane combustion was reported by Murata and co-workers, which was correlated with the variation of active sites and metal–support interactions.5 Besides, as shown in the HRTEM images of Pd/5CZA-5M (Fig. 3), the PdO (101) plane was found to be dominated according to the measured lattice fringes of ca. 0.26 nm. It has been established that the PdO (101) plane was thermodynamically stable and highly active for methane dissociation.41 Therefore, the boosted catalytic performance of Pd/5CZA-5M might be related to the suitable particle size and preferential exposure of the PdO (101) plane.
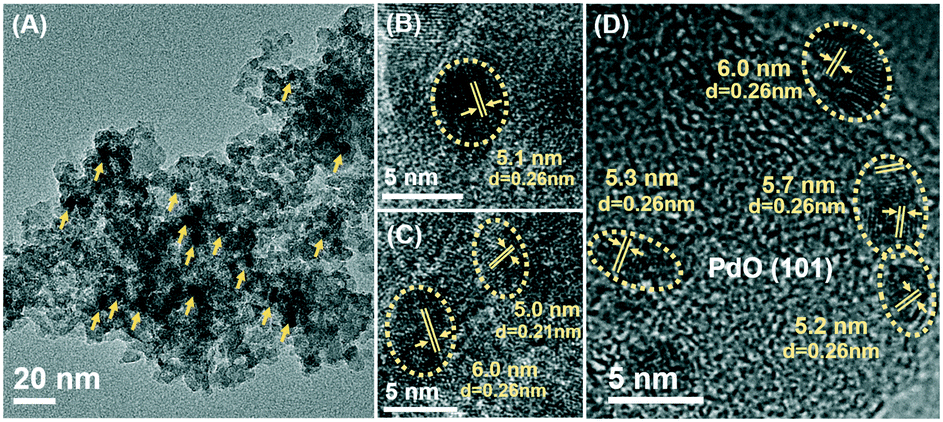 | ||
| Fig. 3 TEM image (A) and HRTEM images (B–D) of the Pd/5CZA-5M catalyst. | ||
3.3. Redox and surface properties of the catalysts
H2-TPR was employed to investigate the reducibility of the samples, and the low temperature section (−10–200 °C) and high temperature section (200–800 °C) of the H2-TPR curves are shown in Fig. 4. All the catalysts exhibited a H2 consumption peak at 0–45 °C, corresponding to the reduction of palladium oxides highly dispersed on the surface of carriers.42 Compared with Pd/Al2O3 and Pd/5CZA-0M, the reduction peaks of the Mg-containing samples were obviously stronger and shifted to lower temperature, among which Pd/5CZA-5M presented the highest amount of H2 consumption and the lowest reduction temperature. Results manifested that the introduction of Mg was beneficial to obtain more palladium oxide species and make them more easily reduced, corresponding to the weaker Pd–O bonds derived from the interaction between the MgAl2O4 spinel and PdO.31,32 Additionally, a negative peak appeared at 60–95 °C for all the samples, which originated from the decomposition of PdHx species formed by adsorbing hydrogen molecules on metal palladium.42,43 The negative peaks gradually enhanced with the rise of the Mg amount owing to the increased size of Pd nanoparticles,32 which was in line with the TEM (Fig. 3 and S3†) and CO chemisorption (Table S3†) results. The reduction peaks of Pd/Al2O3 at the high temperature section (200–800 °C) were assigned to the reduction of surface and subsurface oxygen.8 Meanwhile, for Pd/5CZA-yM, the peak at 225–450 °C was ascribed to the reduction of PdO in close contact with CZ particles accompanied by the reduction of surface or subsurface Ce4+, and the peak at 500–750 °C was attributed to the reduction of bulk Ce4+.35,44 Notably, with increasing Mg content, the reduction peak of bulk Ce4+ moved to lower temperature and overlapped with the reduction peak of surface or subsurface Ce4+, demonstrating the remarkably improved reducibility of Ce4+, which may be attributed to the entry of Mg into the CZ lattice, facilitating the generation of more oxygen vacancies and the increase of bulk oxygen mobility.28 The generated oxygen vacancies may share the oxygen of PdO,20 resulting in the enhanced reducibility of Pd2+ as well.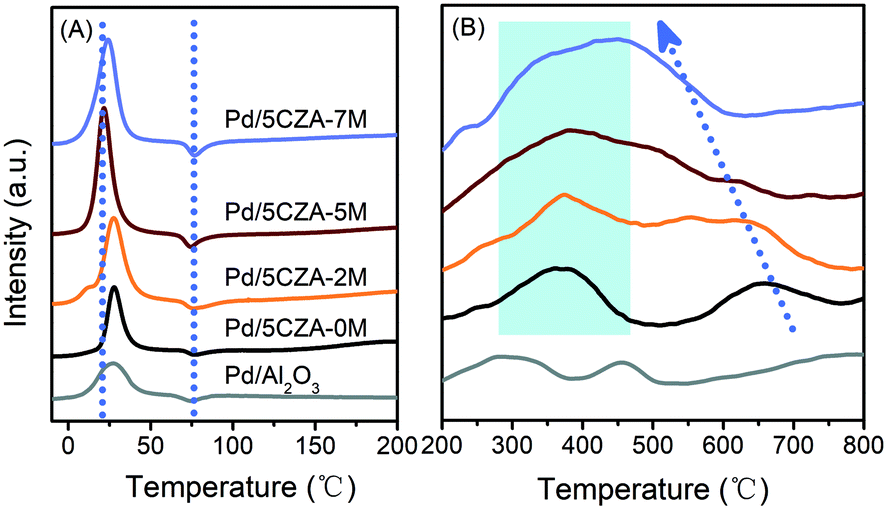 | ||
| Fig. 4 The low temperature section (A) and high temperature section (B) of H2-TPR over the Pd/Al2O3 and Pd/5CZA-yM catalysts. | ||
The thermal reduction and re-oxidation behavior of PdO over the Pd/Al2O3 and Pd/5CZA-yM catalysts were evaluated by O2-TPO. From Fig. 5A, the oxygen-release peaks in the ranges of 230–680 °C and 680–930 °C during the heating ramp corresponded to the desorption of surface oxygen species and the decomposition of palladium oxide, respectively.11,45,46 Notably, the start-up decomposition temperature of PdO on the Mg-containing samples was lower relative to Pd/Al2O3 and Pd/5CZA-0M, reflecting the enhanced thermal reducibility.20 Three peaks could be distinguished after fitting the oxygen-release peaks of PdO with the Gaussian equation, and the quantitative results are listed in Table S4.† Among them, α oxygen-release peaks were associated with the decomposition of PdO in contact with the Pd metal, whereas β and γ oxygen-release peaks belonged to the decomposition of bulk PdO and PdO strongly interacting with supports, respectively.47 It could be observed that bulk PdO species (β peak) were predominant over Pd/Al2O3. Although the incorporation of CZ induced more α peak species, the ratio of β and γ peaks was gradually increased at the expense of α peaks with the rise of the Mg content, manifesting that Mg doping could promote the metal–support interaction of the catalysts. During the cooling ramp, Pd/Al2O3 exhibited only one O2-uptake peak at lower temperature and the hysteresis between PdO decomposition and Pd reoxidation was prominent compared to that of Pd/5CZA-yM, reflecting the poor capacity for the conversion of Pd ↔ PdO. By contrast, two O2-uptake peaks were observed over Pd/5CZA-yM: the one at higher temperature was assigned to the re-oxidation of Pd particles in intimate contact with CZ particles, while the one at lower temperature belonged to the re-oxidation of Pd in contact with alumina.47 The intensity of the uptake peak at the higher temperature gradually enhanced with the rise of the Mg content, resulting in a smaller hysteresis between the decomposition and reformation of PdO. This phenomenon indicated that the introduction of Mg could induce more palladium species to be in contact with CZ, which was beneficial to promote the re-oxidation of Pd and maintain the palladium species in the oxidized form. Thus, the suitable doping content of Mg endowed Pd/5CZA-5M with great stability of the active PdO phase according to the smallest hysteresis, resulting in continuous and effective methane oxidation.
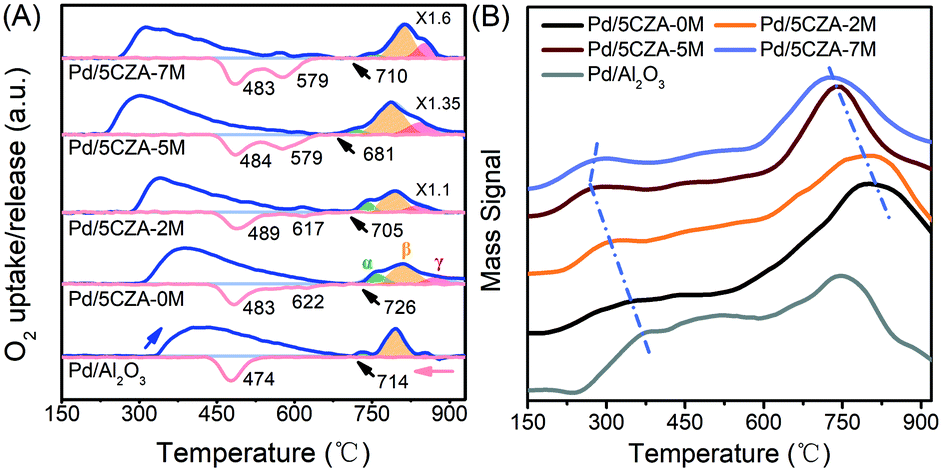 | ||
| Fig. 5 Oxygen uptake and release profiles during the second TPO cycle (A) (blue line: heating; red line: cooling) and O2-TPD profiles (B) of Pd/Al2O3 and Pd/5CZA-yM. | ||
The desorption of surface oxygen species over the catalysts was further identified by O2-TPD. From Fig. 5B, all the catalysts exhibited three desorption peaks in the ranges of 170–400 °C, 400–600 °C and 600–920 °C corresponding to the desorption of chemisorbed oxygen O2−, atomic oxygen O− and lattice oxygen O2− species, respectively.44,48 Among them, the desorption peaks associated with lattice oxygen were prominent over Pd/5CZA-yM. Notably, for the Pd/5CZA-yM catalysts, the desorption peaks of chemisorbed oxygen were located at relatively lower temperature compared with Pd/Al2O3; meanwhile, the desorption peaks of lattice oxygen shifted significantly to lower temperature with the increase of Mg, suggesting high oxygen mobility.48 The enhanced oxygen mobility of the Mg-doped samples could be attributed to the generation of numerous oxygen vacancies, which was derived from the structural and electronic mismatch between the doped Mg2+ and Ce4+ (or Zr4+), facilitating the adsorption and activation of gas-phase oxygen as well as improving the oxygen storage/release capability of the catalysts.44
In addition to the redox performance, the doping of Mg modified the surface acid–base properties of the catalysts. As illustrated in Fig. S5A,† the Pd/5CZA-yM catalysts displayed NH3 desorption peaks at 30–300 °C, 300–550 °C and 550–900 °C, belonging to the desorption of NH3 on weak, moderate and strong acidic sites, respectively.49 The CO2-TPD in Fig. S5B† also presented three desorption peaks of CO2 located at 30–250 °C, 250–550 °C and 550–900 °C, corresponding to the weak, moderate and strong basic sites.49 From Fig. S5 and Table S3,† for Pd/Al2O3, desorption peaks attributed to strong acidic sites and basic sites were absent. Meanwhile, the total acidity and basicity were lower than those for Pd/5CZA-0M, demonstrating the regulating effect of the CZ component on the surface acid–base properties. For Pd/5CZA-yM, both the total acidity and basicity of the samples were declined after Mg doping. Pd/5CZA-5M possessed stronger acidity according to the higher desorption temperature of NH3. The enhanced acidic strength on the catalyst surface was beneficial to break the C–H bonds of methane.50 Meanwhile, the number of basic sites was lower and the strength of basic sites was weaker over Pd/5CZA-5M, benefiting the desorption of reaction product CO2.1 It could be concluded that the surface acid–base properties of the catalysts were further modulated by Mg doping, and the suitable Mg doping content gave rise to an appropriate amount and strength of acid–base sites, contributing to the improved catalytic activity.
3.4. The adsorption property of CO on the catalysts
Fig. 6A and B display the in situ CO-DRIFTS spectra of the catalysts after CO saturation adsorption and Ar purge for 30 min, respectively. As illustrated in Fig. 6A, the band at 1915 cm−1 was assigned to the threefold hollow bonding of CO on metallic Pd0 sites,8 while the band at 1980 cm−1 belonged to the bridge-adsorbed CO on the step sites of Pd particles.5,23 It was noteworthy that these peaks on the Mg-containing samples were shifted toward higher wavenumbers with respect to the Pd/5CZA-0M and Pd/Al2O3 samples, especially for Pd/5CZA-5M. This phenomenon indicated that the C–O bonds detected on the Mg-containing samples were stronger due to the weaker electron back-donation from palladium species into the anti-bonding 2π* orbitals of CO,8 which implied the enhanced metal–support interaction. In addition, the band at 2090 cm−1 was attributed to linear CO adsorbed at the corner or edge sites of Pd particles,5,51 while the band at 2120 cm−1 was ascribed to linearly-bonded CO on ionic Pd+,52 and the band at 2170 cm−1 corresponded to the weak adsorption of gaseous CO.8 It should be noted that the intensity of nonlinear adsorption bands became more intense with the increase of Mg content, which could be correlated to the growth of the palladium particle size, since those bands were typically ascribed to CO adsorbed on the extended metal surface.35,51 From Fig. 6B, after purging with Ar, the peaks of threefold and bridge-bonded CO still existed, while the peaks for the linear adsorption (2090 cm−1, 2120 cm−1, and 2170 cm−1) became weaker or even faded away, inferring that these peaks were associated to CO physical adsorption or weak chemisorption.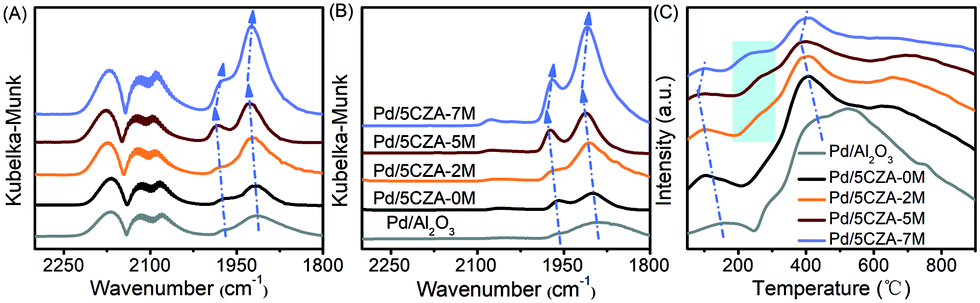 | ||
| Fig. 6 CO-DRIFTS spectra: adsorbed CO until saturation (A) and purged by argon (B). (C) CO-TPD profiles of the Pd/Al2O3 and Pd/5CZA-yM catalysts. | ||
The above results were further verified by CO-TPD. As shown in Fig. 6C, the desorption peak appeared at 110 °C and 410 °C for all the catalysts originated from the desorption of CO from linearly-bonded Pd sites and 3-fold hollow Pd sites, respectively.8,53 When compared with the Pd/5CZA-0M and Pd/Al2O3 samples, the desorption peaks of the Mg-containing samples were located at lower temperature, manifesting the weaker Pd–CO 2π* bonds.8 These variations were in accordance with the CO-DRIFTS results as shown in Fig. 6A. Besides, the desorption peak at around 410 °C gradually broadened with increasing Mg content. Meanwhile, a new CO desorption peak appeared at around 250 °C corresponding to the desorption of bridged-bonded CO on palladium sites,53 which became stronger with the rise of the doping amount. These phenomena further proved that the introduction of Mg strengthened the interaction between the palladium species and carriers, which contributed to the promoted oxygen exchange between PdO and carriers.
3.5. Effect of magnesium on oxygen and palladium species
The distribution and chemical states of oxygen species on the surface of the catalysts were determined by XPS. The O 1s XPS results of fresh and used Pd/Al2O3 and Pd/5CZA-yM are shown in Fig. 7 and Table S5.† The asymmetric O 1s peak could be deconvolved into three components: the binding energy (BE) at 529.9–530.0 eV was ascribed to lattice oxygen species (Olatt), while the BE at 531.1–531.2 eV and 532.5–532.6 eV corresponded to surface adsorbed oxygen (Oads) and hydroxyl or adsorbed water molecule (OOH) species, respectively.8,54 Notably, the surface oxygen vacancies could prompt the adsorption and activation of O2 molecules, which contributed to the generation of more surface electrophilic species (such as O22−, O2− and O−),54 leading to higher surface Oads/Olatt ratios and better catalytic activity.55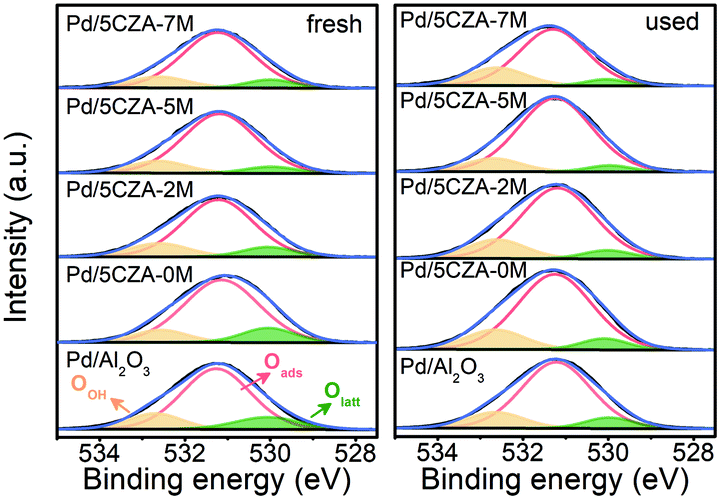 | ||
| Fig. 7 O1s XPS spectra of the fresh and used Pd/Al2O3 and Pd/5CZA-yM catalysts. | ||
As shown in Table S5,† Pd/5CZA-yM (y = 2, 5, 7) presented a higher surface Oads/Olatt ratio with respect to Pd/Al2O3 and Pd/5CZA-0M, reflecting that the incorporation of Mg facilitated the generation of more surface oxygen vacancies along with adsorbed oxygen species. Among them, Pd/5CZA-5M exhibited the highest value of Oads/Olatt whether for the fresh or used sample. Moreover, it was noteworthy that the lattice oxygen of Pd/5CZA-5M declined relatively less after the reaction compared with other samples. In the Mars–van Krevelen mechanism, lattice oxygen participated in the methane oxidation. Therefore, the lower decrease of the lattice oxygen of Pd/5CZA-5M implied that this catalyst possessed abundant oxygen vacancies and efficient lattice oxygen transfer could replenish active oxygen in time,56 corresponding to its favourable catalytic activity. In contrast, the Pd/Al2O3 and Pd/5CZA-0M samples consumed lots of lattice oxygen during the reaction, thus exhibiting poor catalytic performance.
The valence distribution of Ce in Pd/5CZA-yM could offer in-depth information about the formation of oxygen vacancies. From the Ce 3d XPS spectrum (Fig. 8A), the two sets of signals V and U were associated to the spin-orbit splitting of Ce 3d5/2 and Ce 3d3/2, respectively.57 The Ce 3d spectrum could be resolved into 10 peaks: the six peaks labelled V(∼882.6 eV), V2(∼888.2 eV), V3(∼898.3 eV), U(∼901.3 eV), U2(∼906.9 eV) and U3(∼916.9 eV) were characteristic satellite peaks of the tetravalent Ce4+ ion, while the remaining four peaks marked V0(∼881.0 eV), V1(∼885.7 eV), U0(∼899.4 eV) and U1(∼904.2 eV) belonged to the trivalent Ce3+ ion.13,58,59 As shown in Table S5,† the ratio of Ce3+/(Ce3+ + Ce4+) on Pd/5CZA-yM (y = 2, 5, 7) obviously increased compared to that on Pd/5CZA-0M, implying that the generation of Ce3+ was promoted by introducing Mg. It could be inferred that the doping of Mg2+ (0.65 Å) with a smaller radius into the CZ lattice resulted in the lattice contraction of CeO2, inducing Ce4+ (0.97 Å) to convert into Ce3+ (1.10 Å) with a larger radius.57 Notably, the presence of Ce3+ could facilitate the formation of oxygen vacancies.57,59,60 The variation of Ce3+/(Ce3+ + Ce4+) matched well with the Oads/Olatt ratio sequence (Table S5†), further emphasizing that the incorporation of Mg could induce abundant oxygen vacancies. When the Mg doping content increased to 7 wt%, the value of Ce3+/(Ce3+ + Ce4+) slightly declined, which was caused by the aggregation of MgAl2O4 particles on the catalyst surface.
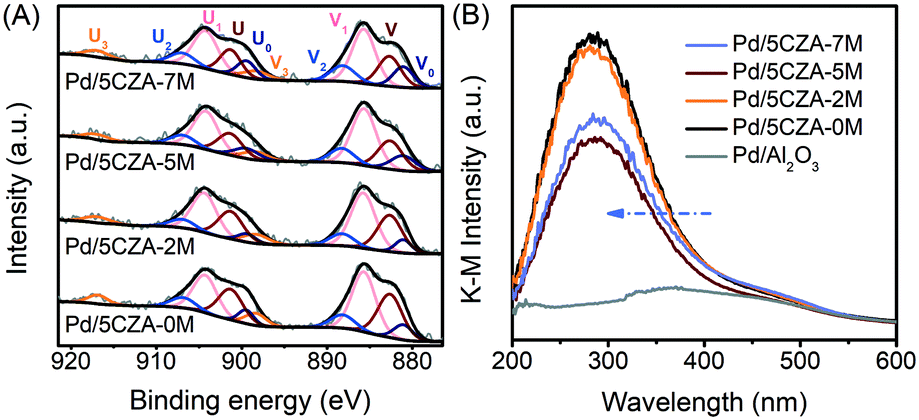 | ||
| Fig. 8 The Ce 3d XPS (A) and UV-vis DRS spectra (B) of the catalysts. | ||
The above conclusions were further confirmed by diffuse reflectance UV-vis spectroscopy, as illustrated in Fig. 8B. The absorption band at about 320–550 nm over Pd/Al2O3 was assigned to the d–d transition of PdO species.3 Meanwhile, for Pd/5CZA-yM, the UV-vis spectra were significantly different from those of Pd/Al2O3. The strong absorption band at around 285 nm was related to the charge-transfer transition of O2− → Ce4+,61 which partly overlapped with the absorption band of PdO. Compared with Pd/5CZA-0M, the edge of the absorption band on the Mg-doped catalysts migrated towards a lower wavelength and the intensity declined. Since the bands ascribed to the charge-transfer transition of O2− → Ce3+ appeared at about 265 nm, the variation implied the conversion of Ce4+ into Ce3+ caused by Mg doping into the CZ lattice,61,62 which could generate abundant oxygen vacancies or lattice defects due to the structural and electronic mismatch between Mg2+ and Ce4+ (or Zr4+).
The modification of Mg also exerted a prominent impact on the distribution and valence states of palladium species. The Pd 3d XPS spectra for the fresh and used Pd/Al2O3 and Pd/5CZA-yM are presented in Fig. 9A. The deconvolution results of Pd 3d5/2 suggested that there were three forms of palladium on the surface of the catalysts: metallic Pd species at BE = 335.8–335.9 eV, PdO species at BE = 336.8–337.0 eV and PdO2 species at BE = 337.8–338.1 eV.63,64 As reported in Table S6,† the addition of CZ would preserve the palladium species in the highly oxidized state corresponding to the large proportion of Pd2+ and Pd4+ over Pd/5CZA-yM, while Pd/Al2O3 possessed more Pd0. For the Pd/5CZA-yM catalysts, with the rising Mg content, the concentration of Pd2+ species increased at the expense of Pd0, while the variation of Pd4+ species was inconspicuous, which demonstrated the regulating effect of Mg doping on the distribution and states of the palladium species. It was generally accepted that Pd2+ species were the catalytically active sites for methane oxidation.34,65 Among the catalysts, the proportion of Pd2+ was the highest for Pd/5CZA-5M, corresponding to its optimal methane activity.
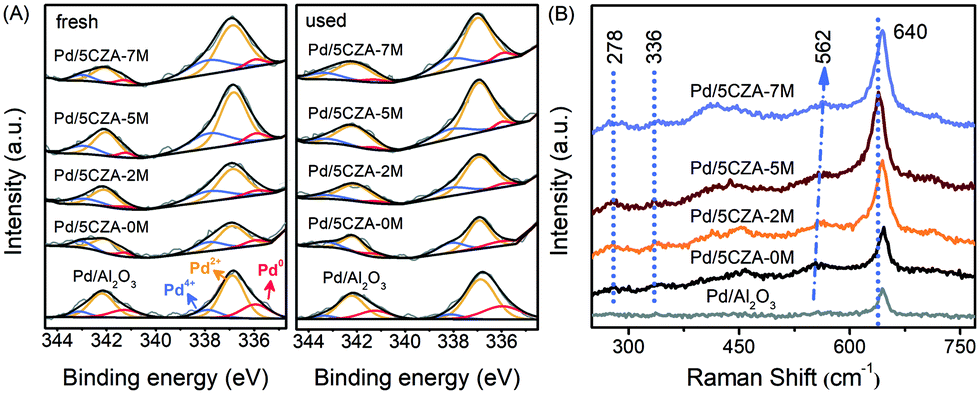 | ||
| Fig. 9 The Pd 3d XPS spectra of the fresh and used catalysts (A) and Raman spectra of the Pd/Al2O3 and Pd/5CZA-yM catalysts (B). | ||
After the reaction, the concentration of the Pd2+ and Pd4+ species for all the catalysts increased and decreased, respectively. Meanwhile, the content of the Pd0 species exhibited a slight increase for the Pd/Al2O3 and Pd/5CZA-0M samples but a dip for the Mg-modified samples. Notably, the palladium species could remain in the active oxidation state over Pd/5CZA-5M during the stability test; meanwhile, the consumed lattice oxygen would be replenished in time (see Tables S5 and S6 and Fig. S6†). Evidence verified that Mg-doping was conducive to further stabilize the active oxidation state of the palladium species owing to the excellent oxygen mobility, while the PdOx phase on the Pd/Al2O3 and Pd/5CZA-0M samples was prone to transforming into the less active Pd0. In consideration of the activity results, Pd/5CZA-5M possessed a steady active PdO phase along with a relatively high Oads/Olatt ratio, cooperatively contributing to its promoted catalytic activity and durability.
Fig. 9B presents the Raman spectra of Pd/Al2O3 and Pd/5CZA-yM. All the samples showed a sharp and symmetrical characteristic peak at around 640 cm−1 corresponding to the B1g vibration mode of tetragonal PdO, and the additional weak peaks appeared at 278 cm−1 and 336 cm−1, which also originated from the crystalline PdO, were observed over Pd/5CZA-yM.66,67 Notably, the strong PdO peaks over the Mg-modified samples arose at a lower wavenumber relative to Pd/Al2O3 and Pd/5CZA-0M, especially for Pd/5CZA-5M, indicating the weaker Pd–O bonds, which might be derived from the synergistic effect of the formed MgAl2O4 phase and induced oxygen vacancies on the active PdO species.20,31,32 This coincided with the results of H2-TPR (Fig. 4). Additionally, the small peak at about 562 cm−1 over Pd/5CZA-yM was associated with the tetragonal CZ phase.68 With the increasing Mg amount, this peak shifted toward a higher wavenumber owing to the generated Mg–O bonds with shorter bond lengths, which was derived from the fact that more Mg2+ with a smaller radius and lower valence entered the CZ phase.28
4. Conclusions
In this work, Mg-promoted multicomponent Pd/5CZA-yM catalysts were rationally designed for the catalytic oxidation of methane. The incorporation of Mg could not only form a MgAl2O4 spinel phase with alumina, but also occupy Ce4+ sites of CZ solid solution and induce Ce4+ to convert into Ce3+. The above structural variations were facilely regulated by the Mg doping content, giving rise to abundant oxygen vacancies and higher oxygen mobility along with better reducibility, which exerted a great influence on the particle size, chemical state and redox properties of active palladium species. Meanwhile, the surface acidity and basicity were tuned by Mg doping. The synergistic effect of the above optimized properties made Pd/5CZA-5M with a moderate doping content (5 wt% Mg) exhibit superior catalytic activity (T99 of 400 °C) and durability for methane combustion. The investigation into Mg doping may shed new light on the synergy of promoters in catalysts, contributing to the well design of high-performance multicomponent catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21872027) and the Natural Science Foundation of Fujian Province (no. 2018J01669, 2020J01200). TEM measurement was performed with the support of the Testing Center of Fuzhou University.References
- K. Murata, D. Kosuge, J. Ohyama, Y. Mahara, Y. Yamamoto, S. Arai and A. Satsuma, ACS Catal., 2020, 10, 1381–1387 CrossRef CAS.
- J. Nilsson, P. Carlsson, N. M. Martin, E. C. Adams, G. Agostini, H. Grönbeck and M. Skoglundh, J. Catal., 2017, 356, 237–245 CrossRef CAS.
- J. Lin, X. Chen, Y. Zheng, F. Huang, Y. Xiao, Y. Zheng and L. Jiang, Catal. Sci. Technol., 2020, 10, 4612–4623 RSC.
- Q. Duan, C. Zhang, S. Sun, Y. Pan, X. Zhou, Y. Liu, K. Chen, C. Li, X. Wang and W. Li, J. Mater. Chem. A, 2020, 8, 7395–7404 RSC.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS.
- X. Yang, Q. Li, E. Lu, Z. Wang, X. Gong, Z. Yu, Y. Guo, L. Wang, Y. Guo, W. Zhan, J. Zhang and S. Dai, Nat. Commun., 2019, 10, 1611 CrossRef.
- O. Mihai, G. Smedler, U. Nylén, M. Olofsson and L. Olsson, Catal. Sci. Technol., 2017, 7, 3084–3096 RSC.
- X. Chen, Y. Zheng, Y. Chen, Y. Xu, F. Zhong, W. Zhang, Y. Xiao and Y. Zheng, Int. J. Hydrogen Energy, 2019, 44, 27772–27783 CrossRef CAS.
- F. Huang, X. Wang, A. Wang, J. Xu and T. Zhang, Catal. Sci. Technol., 2016, 6, 4962–4969 RSC.
- Z. Wang, Z. Huang, J. T. Brosnahan, S. Zhang, Y. Guo, Y. Guo, L. Wang, Y. Wang and W. Zhan, Environ. Sci. Technol., 2019, 53, 5349–5358 CrossRef CAS.
- M. Cargnello, J. J. Delgado Jaén, J. C. Hernández Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS.
- M. Jeong, N. Nunotani and N. Imanaka, Bull. Chem. Soc. Jpn., 2018, 91, 158–164 CrossRef CAS.
- M. Jeong, N. Nunotani, N. Moriyama and N. Imanaka, Catal. Sci. Technol., 2017, 7, 1986–1990 RSC.
- R. Zhou, B. Zhao and B. Yue, Appl. Surf. Sci., 2008, 254, 4701–4707 CrossRef CAS.
- J. Wu, A. Glisenti, J. P. Dacquin, C. Dujardin, C. F. Acevedo, C. S. Castro and P. Granger, Appl. Catal., A, 2020, 598, 117527 CrossRef CAS.
- Y. Ding, Q. Wu, B. Lin, Y. Guo, Y. Guo, Y. Wang, L. Wang and W. Zhan, Appl. Catal., B, 2020, 266, 118631 CrossRef.
- L. Lan, S. Chen, H. Li, J. Wang, D. Li and Y. Chen, Mater. Des., 2018, 147, 191–199 CrossRef CAS.
- B. Yue, R. Zhou, X. Zheng and W. Lu, Mater. Chem. Phys., 2009, 114, 722–727 CrossRef CAS.
- J. Xu, L. Ouyang, W. Mao, X. J. Yang, X. C. Xu, J. J. Su, T. Z. Zhuang, H. Li and Y. F. Han, ACS Catal., 2012, 2, 261–269 CrossRef CAS.
- Y. Wu, J. Chen, W. Hu, K. Zhao, P. Qu, P. Shen, M. Zhao, L. Zhong and Y. Chen, J. Catal., 2019, 377, 565–576 CrossRef CAS.
- Y. Xiao, W. Zhu, G. Cai, M. Chen, Y. Zheng, F. Zhong and L. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 30418–30428 RSC.
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X. M. Cao, Y. Guo, P. Hu and G. Lu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS.
- K. Murata, E. Eleeda, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Phys. Chem. Chem. Phys., 2019, 21, 18128–18137 RSC.
- C. Xiang, Y. Xiao, H. Bai, X. Yin, M. Peng, X. Yang, Y. Ding and Z. Du, Nanoscale, 2020, 12, 3663–3667 RSC.
- K. Yasuda, T. Masui, T. Miyamoto and N. Imanaka, J. Mater. Sci., 2011, 46, 4046–4052 CrossRef CAS.
- M. Jeong, N. Nunotani, N. Moriyama and N. Imanaka, J. Asian Ceram. Soc., 2016, 4, 259–262 CrossRef.
- J. Lin, L. Yang, T. Wang and R. Zhou, Phys. Chem. Chem. Phys., 2017, 19, 7844–7852 RSC.
- L. Lan, S. Chen, Y. Cao, S. Wang, Q. Wu, Y. Zhou, M. Huang, M. Gong and Y. Chen, J. Mol. Catal. A: Chem., 2015, 410, 100–109 CrossRef CAS.
- X. Zou, J. Chen, Z. Rui and H. Ji, Appl. Catal., B, 2020, 273, 119071 CrossRef CAS.
- Z. Zhang, X. Hu, Y. Zhang, L. Sun, H. Tian and X. Yang, Catal. Sci. Technol., 2019, 9, 6404–6414 RSC.
- L. Yang, C. Shi, X. He and J. Cai, Appl. Catal., B, 2002, 38, 117–125 CrossRef CAS.
- W. Hu, G. Li, J. Chen, F. Huang, M. Gong, L. Zhong and Y. Chen, Fuel, 2017, 194, 368–374 CrossRef CAS.
- X. Chen, Y. Zheng, F. Huang, Y. Xiao, G. Cai, Y. Zhang, Y. Zheng and L. Jiang, ACS Catal., 2018, 8, 11016–11028 CrossRef CAS.
- P. Velin, C.-R. Florén, M. Skoglundh, A. Raj, D. Thompsett, G. Smedler and P.-A. Carlsson, Catal. Sci. Technol., 2020, 10, 5460–5469 RSC.
- A. Toso, S. Colussi, S. Padigapaty, C. de Leitenburg and A. Trovarelli, Appl. Catal., B, 2018, 230, 237–245 CrossRef CAS.
- D. Ciuparu, E. Perkins and L. Pfefferle, Appl. Catal., A, 2004, 263, 145–153 CAS.
- X. Duan, Z. Wen, Y. Zhao, J. Zhou, H. Fang, Y. Cao, L. Jiang, L. Ye and Y. Yuan, Nanoscale, 2018, 10, 3331–3341 CAS.
- J. Wang, Y. Wang, J. Wen, M. Shen and W. Wang, Microporous Mesoporous Mater., 2009, 121, 208–218 CAS.
- W. Zhao, X. Zheng, S. Liang, X. Zheng, L. Shen, F. Liu, Y. Cao, Z. Wei and L. Jiang, Green Chem., 2018, 20, 4645–4654 RSC.
- T. V. Choudhary, S. Banerjee and V. R. Choudhary, Appl. Catal., A, 2002, 234, 1–23 CrossRef CAS.
- J. J. Willis, A. Gallo, D. Sokaras, H. Aljama, S. H. Nowak, E. D. Goodman, L. Wu, C. J. Tassone, T. F. Jaramillo, F. Abild-Pedersen and M. Cargnello, ACS Catal., 2017, 7, 7810–7821 CrossRef CAS.
- S. Lin, L. Yang, X. Yang and R. Zhou, Appl. Surf. Sci., 2014, 305, 642–649 CrossRef CAS.
- X. Yang, Q. Gao, Z. Zhao, Y. Guo, Y. Guo, L. Wang, Y. Wang and W. Zhan, Appl. Catal., B, 2018, 239, 373–382 CrossRef CAS.
- J. Liu, B. Liu, Y. Fang, Z. Zhao, Y. Wei, X. Q. Gong, C. Xu, A. Duan and G. Jiang, Environ. Sci. Technol., 2014, 48, 12403–12410 CrossRef CAS.
- W. Lin, Y. X. Zhu, N. Z. Wu, Y. C. Xie, I. Murwani and E. Kemnitz, Appl. Catal., B, 2004, 50, 59–66 CrossRef CAS.
- X. Zou, Z. Rui and H. Ji, ACS Catal., 2017, 7, 1615–1625 CrossRef CAS.
- S. Colussi, A. Trovarelli, E. Vesselli, A. Baraldi, G. Comelli, G. Groppi and J. Llorca, Appl. Catal., A, 2010, 390, 1–10 CrossRef CAS.
- Y. Zheng, K. Li, H. Wang, Y. Wang, D. Tian, Y. Wei, X. Zhu, C. Zeng and Y. Luo, J. Catal., 2016, 344, 365–377 CrossRef CAS.
- L. Zhang, X. Wang, C. Chen, X. Zou, W. Ding and X. Lu, Int. J. Hydrogen Energy, 2017, 42, 11333–11345 CAS.
- W. Kumsung, M. Chareonpanich, P. Kongkachuichay, S. Senkan and A. Seubsai, Catal. Commun., 2018, 110, 83–87 CAS.
- J. H. Carter, S. Althahban, E. Nowicka, S. J. Freakley, D. J. Morgan, P. M. Shah, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2016, 6, 6623–6633 CrossRef CAS.
- X. Wang, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2012, 115-116, 7–15 CrossRef CAS.
- S. Komeili, M. T. Ravanchi and A. Taeb, Appl. Catal., A, 2015, 502, 287–296 CrossRef CAS.
- S. Xie, J. Deng, S. Zang, H. Yang, G. Guo, H. Arandiyan and H. Dai, J. Catal., 2015, 322, 38–48 CrossRef CAS.
- J. Chen, W. Shi and J. Li, Catal. Today, 2011, 175, 216–222 CrossRef CAS.
- J. Lin, Y. Chen, X. Liu, X. Chen, Y. Zheng, F. Huang, Y. Xiao, Y. Zheng and L. Jiang, Appl. Catal., B, 2020, 263, 118269 CrossRef CAS.
- B. Liu, C. Li, G. Zhang, X. Yao, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- X. Zhang, S. D. House, Y. Tang, L. Nguyen, Y. Li, A. A. Opalade, J. C. Yang, Z. Sun and F. F. Tao, ACS Sustainable Chem. Eng., 2018, 6, 6467–6477 CrossRef CAS.
- J. Ma, Y. Lou, Y. Cai, Z. Zhao, L. Wang, W. Zhan, Y. Guo and Y. Guo, Catal. Sci. Technol., 2018, 8, 2567–2577 RSC.
- C. Wen, Y. Zhu, Y. Ye, S. Zhang, F. Cheng, Y. Liu, P. Wang and F. Feng Tao, ACS Nano, 2012, 6, 9305–9313 CrossRef CAS.
- L. Liu, Z. Yao, B. Liu and L. Dong, J. Catal., 2010, 275, 45–60 CrossRef CAS.
- V. B. Mortola, S. Damyanova, D. Zanchet and J. M. C. Bueno, Appl. Catal., B, 2011, 107, 221–236 CrossRef CAS.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57–71 CrossRef CAS.
- J. Xiong, Q. Wu, X. Mei, J. Liu, Y. Wei, Z. Zhao, D. Wu and J. Li, ACS Catal., 2018, 8, 7915–7930 CrossRef CAS.
- W. Huang, E. D. Goodman, P. Losch and M. Cargnello, Ind. Eng. Chem. Res., 2018, 57, 10261–10268 CrossRef CAS.
- D. K. Chlebda, R. J. Jędrzejczyk, P. J. Jodłowski and J. Łojewska, J. Raman Spectrosc., 2017, 48, 1871–1880 CAS.
- A. Baylet, P. Marécot, D. Duprez, P. Castellazzi, G. Groppi and P. Forzatti, Phys. Chem. Chem. Phys., 2011, 13, 4607–4613 RSC.
- Q. Yuan, Q. Liu, W. G. Song, W. Feng, W. L. Pu, L. D. Sun, Y. W. Zhang and C. H. Yan, J. Am. Chem. Soc., 2007, 129, 6698–6699 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: More details about synthetic methods and characterization techniques, comparison with literature studies about the catalytic performance, and additional characterization and evaluation results. See DOI: 10.1039/d0cy01742f |
| This journal is © The Royal Society of Chemistry 2021 |