
Zirconium and hafnium polyhedral oligosilsesquioxane complexes – green homogeneous catalysts in the formation of bio-derived ethers via a MPV/etherification reaction cascade†
Shipra
Garg
 ,
Daniel K.
Unruh
and
Clemens
Krempner
*
,
Daniel K.
Unruh
and
Clemens
Krempner
*
Department of Chemistry and Biochemistry, Texas Tech University, Memorial Dr. & Boston, Lubbock, TX 79409, USA. E-mail: clemens.krempner@ttu.edu; Fax: +806 742 1289; Tel: +806 834 3507
First published on 23rd October 2020
Abstract
The polyhedral oligosilsesquioxane complexes, {[(isobutyl)7Si7O12]ZrOPri·(HOPri)}2 (I), {[(cyclohexyl)7Si7O12]ZrOPri·(HOPri)}2 (II), {[(isobutyl)7Si7O12]HfOPri·(HOPri)}2 (III) and {[(cyclohexyl)7Si7O12]HfOPri·(HOPri)}2 (IV), were synthesized in good yields from the reactions of M(OPri)4 (M = Zr, Hf) with R-POSS(OH)3 (R = isobutyl, cyclohexyl), resp. I–IV were characterized by 1H, 13C and 29Si NMR spectroscopy and their dimeric solid-state structures were confirmed by X-ray analysis. I–IV catalyze the reductive etherification of 2-hydroxy- and 4-hydroxy and 2-methoxy and 4-methoxybenzaldehyde and vanillin to their respective isopropyl ethers in isopropanol as a “green” solvent and reagent. I–IV are durable and robust homogeneous catalysts operating at temperatures of 100–160 °C for days without significant loss of catalytic activity. Likewise, I–IV selectively catalyze the conversion of 5-hydroxymethylfurfural (HMF) into 2,5-bis(isopropoxymethyl)furane (BPMF), a potentially high-performance fuel additive. Similar results were achieved by using a combination of M(OPri)4 and ligand R-POSS(OH)3 as a catalyst system demonstrating the potential of this “in situ” approach for applications in biomass transformations. A tentative reaction mechanism for the reductive etherification of aldehydes catalysed by I–IV is proposed.
Introduction
The chemical conversion of cellulosic biomass into liquid fuel, is a subject of current interest from both the industrial as well as the academic perspective.1 Particularly, alkyl ethers have become intensively investigated in the last few years owing to their potential application as high-performance fuel additives.2 One of the most promising synthetic strategies for alkyl ethers involves the reductive etherification of biomass-derived aldehydes via the Meerwein–Ponndorf–Verley (MPV) reaction followed by acid-catalysed etherification (Scheme 1).3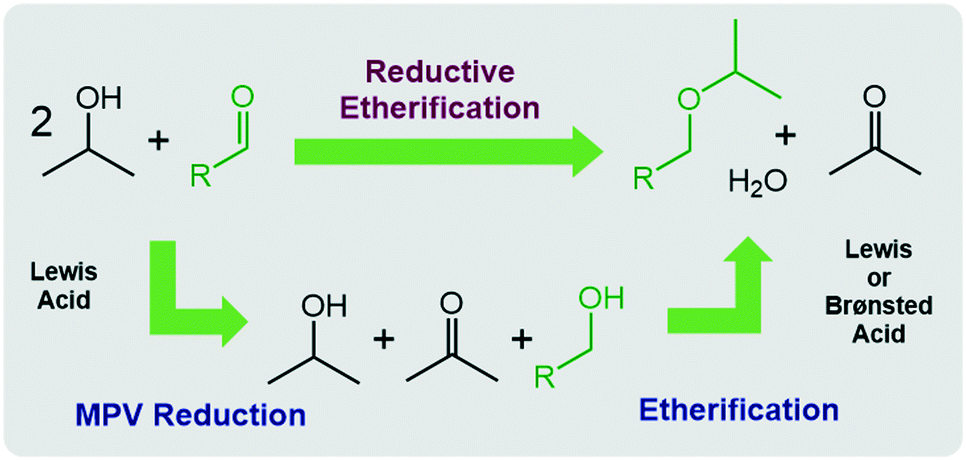 | ||
| Scheme 1 Ether formation from aldehydes via a reductive etherification cascade. | ||
This process is operationally simple and environmentally friendly as it utilizes “green” isopropanol as an inexpensive, safe and non-toxic solvent and hydrogen transfer reagent in combination with cheap and abundant acid catalysts. The MPV reduction,4 the first step in the cascade, proceeds via Lewis acid-catalysed hydrogen transfer usually from a secondary alcohol to the carbonyl substrate with high selectivity, following an outer-sphere mechanism,5 while the etherification proceeds with elimination of water and can be catalysed by both, Brønsted or Lewis acid catalysts. Indeed, some heterogeneous tin, zirconium and hafnium zeolite and oxide based catalysts have been reported to convert 5-hydroxymethylfurfural (HMF), a platform chemical for biofuels, biochemical and biopolymers,6 into 2,5-bis(isopropoxy-methyl)furan (BPMF)7via reductive etherification. In contrast, homogeneous catalysts capable of converting HMF into BPMF with high selectivity and yields have not yet been reported.8
Early transition metal “polyhedral” oligosilsesquioxane (POSS) complexes9 of a tripodal coordination geometry are a largely overlooked class of robust and durable metal complexes that have shown some potential as homogeneous catalysts in alkene epoxidations10 and polymerizations.11 The electron-withdrawing properties of the POSS ligand combined with its steric profile give rise to well-defined metal complexes that possess relatively high Lewis acidity while maintaining good chemical resistance. Herein, we will introduce zirconium and hafnium POSS complexes as a new class of highly efficient and remarkably thermally-stable homogeneous catalysts for selected biomass transformations as demonstrated for the reductive etherification of HMF to biofuel additive, BPMF.
Results and discussion
The synthesis of the targeted zirconium and hafnium POSS complexes I–IV is illustrated in Scheme 2. Reaction of the commercially available trisilanols R-POSS(OH)3 (R = isobutyl or cyclohexyl) with Zr(OPri)4 and Hf(OPri)4, respectively, gave, after removal of solvent and precipitation or recrystallization from isopropanol (IPA), compounds I–IV in 58–66% yield. I–IV are thermally stable crystalline materials, which show good solubility in toluene, hexanes, benzene, dichloromethane, diethyl ether and THF but are sparingly soluble in alcohols and DMSO. In addition to be characterized by 1H, 13C and 29Si NMR spectroscopy and combustion analysis, the solid-state structures of I–IV were determined by X-ray analysis. The results are shown for I and III (Fig. 1); selected bond lengths and angles for I–IV are listed in Table 1.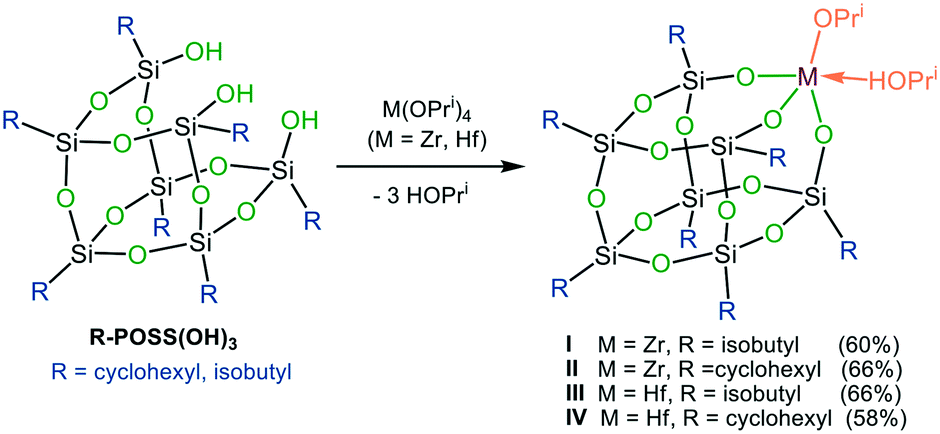 | ||
| Scheme 2 Synthesis of I–IV. | ||
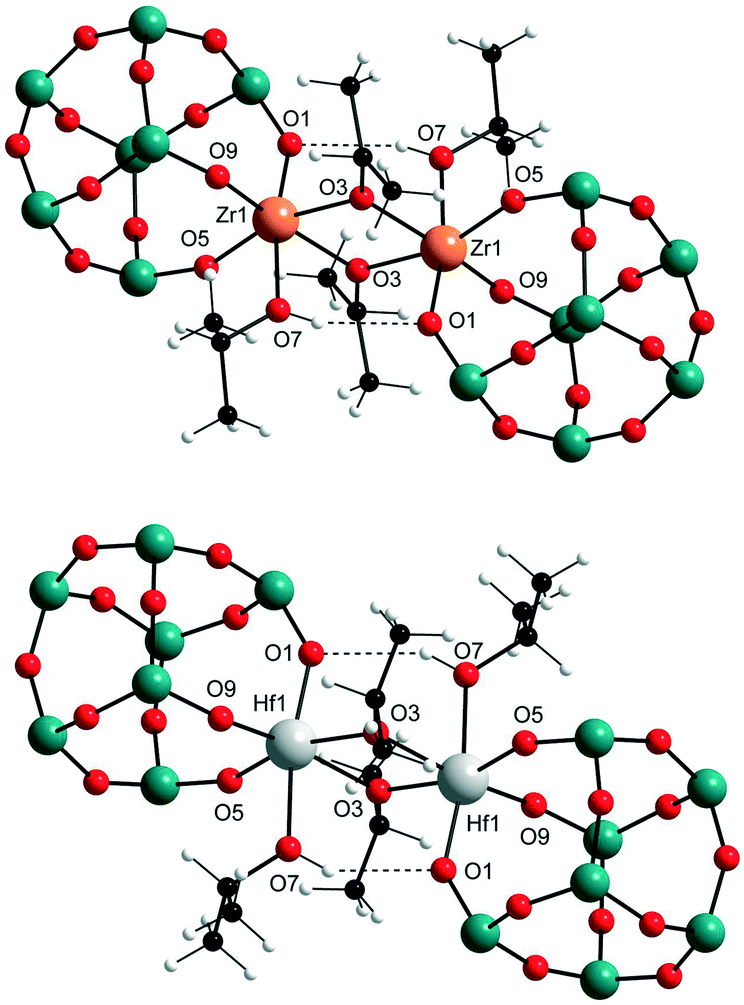 | ||
| Fig. 1 Solid-state structures of I (top) and III (bottom); isobutyl substituents at the silicon atoms are omitted for clarity (blue = silicon, red = oxygen). | ||
| I (M = Zr) | II (M = Zr) | III (M = Hf) | IV (M = Hf) | |
|---|---|---|---|---|
| a Average values. | ||||
| M1–O1 | 2.015(3) | 1.997(1) | 1.993(5) | 1.995(2) |
| M1–O3 | 2.177(3) | 2.156(1) | 2.136(5) | 2.146(2) |
| M1–O5 | 1.982(3) | 1.988(1) | 1.967(5) | 1.984(2) |
| M1–O7 | 2.304(3) | 2.303(2) | 2.257(5) | 2.274(2) |
| M1–O9 | 1.966(3) | 1.987(1) | 1.973(5) | 1.982(2) |
| O1–M1–O7 | 164.8(1) | 170.4(1) | 167.9(2) | 170.3(1) |
| Si–O(M1)a | 1.611(3) | 1.612(2) | 1.606(5) | 1.612(2) |
In the solid state, all compounds are isostructural dimers with distorted octahedral coordination environments for zirconium and hafnium, respectively. The coordination sphere of each metal centre is completed by coordinated isopropanol, similar to what is seen for other titanium and zirconium analogues.12 The dimers are held together via bridging isopropoxide groups and hydrogen bond interaction between the OH group of the coordinated isopropanol and an oxygen from one of the metal binding siloxy groups [O1⋯O7H, 2.99 to 3.12 Å]. There are three types of metal–oxygen bonds that result from this structural arrangement: bridging M–O bonds [M1–O3, 2.14–2.18 Å], M–O bonds of the coordinated isopropanol [M1–O7, 2.26–2.30 Å], and M–O bonds from the supporting POSS ligand [M1–O1/O5/O9, 1.97–2.02 Å]. As expected, the M1–O7 distances are significantly longer than the bridging M1–O3 and POSS related M–O distances, suggesting a relatively weak isopropanol to metal coordination.
With the POSS complexes I–IV in hand, we investigated their catalytic performance in the reductive etherification of various benzaldehydes along with their precursors Zr(OPri)4 and Hf(OPri)4. Initial screening experiments at 25–70 °C did not show any substrate conversion. Therefore, the experiments were carried out in closed reaction vessels at temperatures ranging from 100 °C to 150 °C with a catalyst loading of 1 mol%. Isopropanol (IPA) served as solvent and hydrogen transfer reagent and was used as received (“wet”). Conversions and product yields were determined by 1H NMR spectroscopy using CDCl3 as a solvent and 1,3,5-trimethylbenzene as internal standard. The catalytic experiments were triplicated and conversions and product yields were given as average values.
The moisture sensitive precursors Zr(OPri)4 and Hf(OPri)4 were found to be surprisingly active in selectively reducing benzaldehyde to benzyl alcohol (1) in “wet” isopropanol (IPA) with yields of 57% and 80%, respectively, after 24 hours at 100 °C (Fig. 2). The employment of the new POSS complexes I–IV reproducibly allowed for higher yields with catalysts I and III each producing 91% of 1. At 150 °C, no differences in the performance were noted between I–IV, Zr(OPri)4 and Hf(OPri)4; all quantitatively reduced benzaldehyde to 1 (Table S1†), but did not form benzyl isopropyl ether.
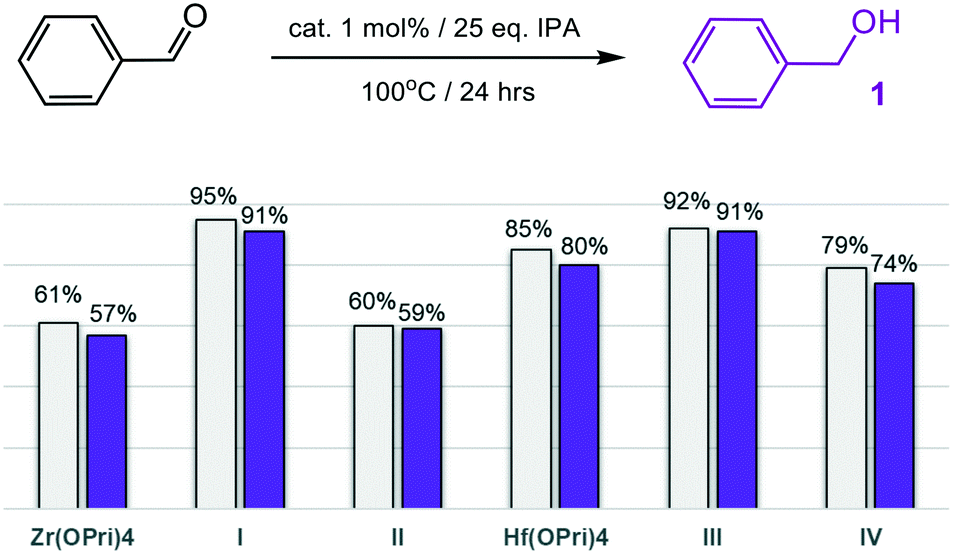 | ||
| Fig. 2 MPV reduction of benzaldehyde to benzyl alcohol (1): conversions (grey bars) and yields of alcohol 1 (purple bars). | ||
Next, the reduction of the more electron-rich 4-methoxy-benzaldehyde was investigated (Fig. 3). Similar to what was seen in the previous case, all zirconium and hafnium based catalysts reduced 4-methoxybenzaldehyde to 4-methoxybenzyl alcohol (2) in good to excellent yields after 24 hours at 100 °C. Hf(OPri)4 and I showed the best catalyst performances generating 94% of alcohol 2, respectively.
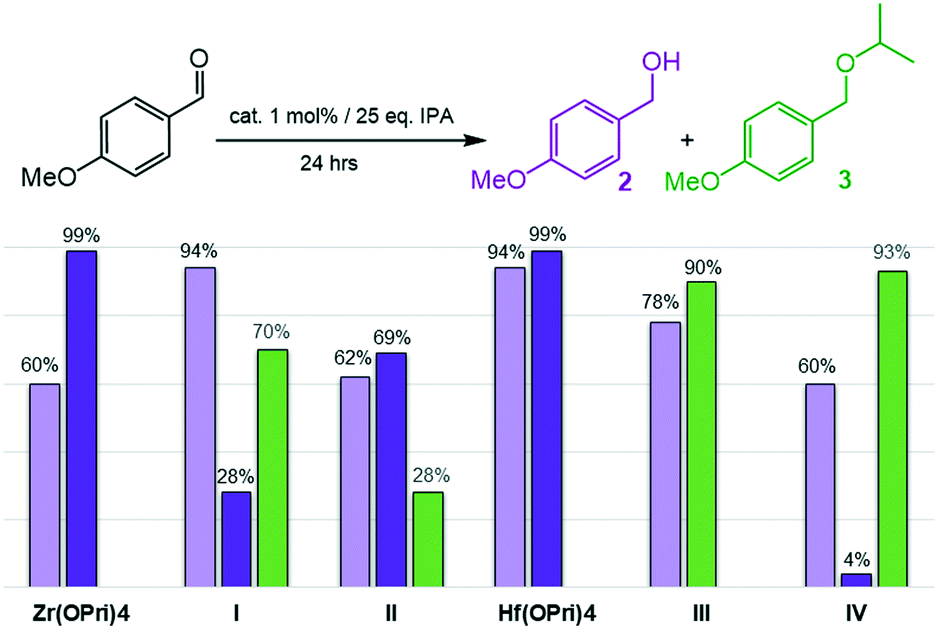 | ||
| Fig. 3 Reductive etherification of 4-methoxybenzaldehyde: yields of alcohol 2 at 100 °C (light purple bars); yields of alcohol 2 (dark purple bars) and ether 3 (green bars) at 150 °C. | ||
Notably, increasing the temperatures from 100 to 150 °C not only improved conversions but also changed the selectivity markedly in favour of the targeted 4-methoxybenzyl isopropyl ether (3). While Hf(OPri)4 and Zr(OPri)4 quantitatively converted 4-methoxybenzaldehyde into alcohol 2 only, the hafnium complexes III and IV selectively produced ether 3 in yields of 90% and 93%, respectively.
2-Methoxybenzaldehyde was converted much faster and with better selectivity into its respective alcohol at 100 °C than 4-methoxybenzaldehyde regardless of the catalyst used (Fig. 4 and Tables S3 and S4†). In fact, isobutyl substituted POSS complex I, Zr(OPri)4 and Hf(OPri)4 quantitatively generated 2-methoxy-benzyl alcohol (4) in only 3 hours at 100 °C. The cyclohexyl substituted complexes II and IV were slightly less active presumably due to their poor solubility in IPA at 100 °C. However, complex III proved to be the most active and selective catalyst. III not only quantitatively produced alcohol 4 in 1 hour, it also converted alcohol 4 into ether 5 upon increasing the temperature to 150 °C. In fact, III generated, after 24 hours, 57% of 5 along with 39% of alcohol 4. After 48 hours, 82% of 5 and 16% of 4 were formed (Fig. 4).
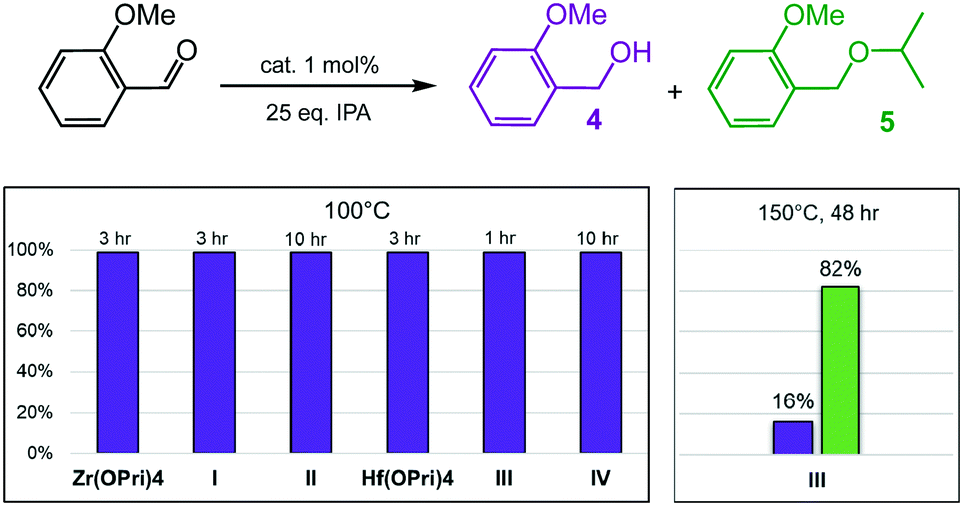 | ||
| Fig. 4 Reductive etherification of 2-methoxybenzaldehyde: (left graph) yields of alcohol 4 (purple bars) at various times; (right graph) yields of alcohol 4 (purple bar) and ether 5 (green bar) after 48 hours. | ||
Given the structural similarities of I–IV (vide supra), it remains unclear why only III generates significant amounts of ether 5. It is known that chelating substrates often cause metal-catalyzed reactions to be sluggish because of the inherent strength of the resulting product–metal bonds, typically the limiting factor in product liberation from the metal catalyst. Therefore, even the subtle changes of the steric profile (isobutyl versus cyclohexyl substituents) and the identity of the metal (zirconium or hafnium) may account for the differences in product selectivity.
Encouraged by the ability to selectively catalyse the formation of ethers, the more challenging substrates 2-hydroxy- and 4-hydroxybenzaldehyde were investigated (Fig. 5 and 6). To our surprise, complexes I–IV smoothly catalysed the formation of the corresponding isopropyl ethers 7 and 9, respectively, which were obtained in good to excellent yields after 24 hours. 2-Hydroxybenzaldehyde was found to convert into 9 faster and at slightly lower temperatures (120 °C) than 4-hydroxybenzaldehyde, which required 125 °C to be converted into ether 7 in acceptable yields. Notably, also the precursors Zr(OPri)4 and Hf(OPri)4 catalysed the formation of ethers 7 and 9, respectively, from their respective aldehydes with similar yields but somewhat lower selectivity.
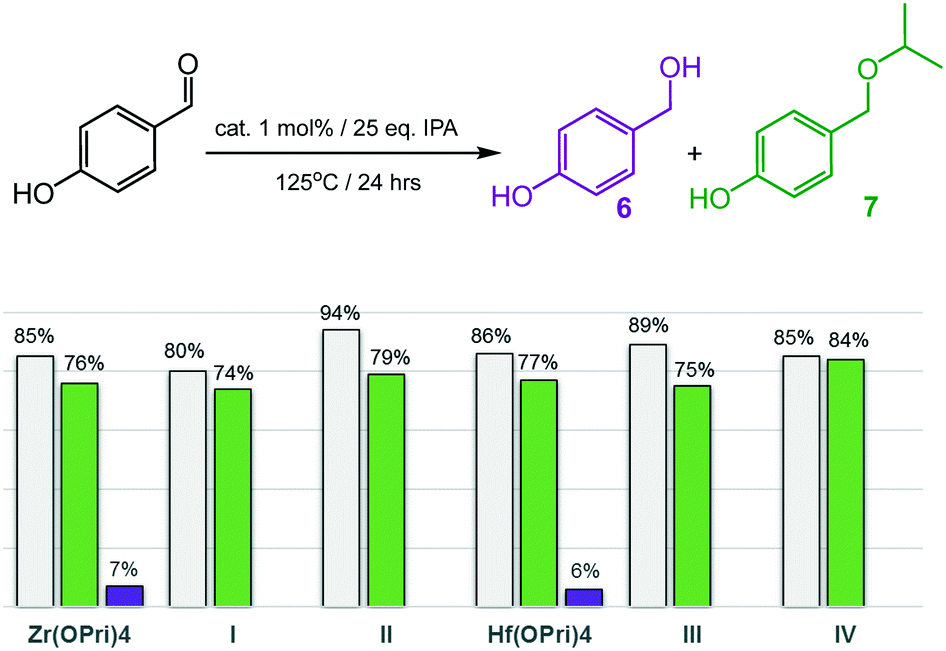 | ||
| Fig. 5 Reductive etherification of 4-hydroxybenzaldehyde. Conversions (grey bars): yields of alcohol 6 (purple bars); yields of ether 7 (green bars). | ||
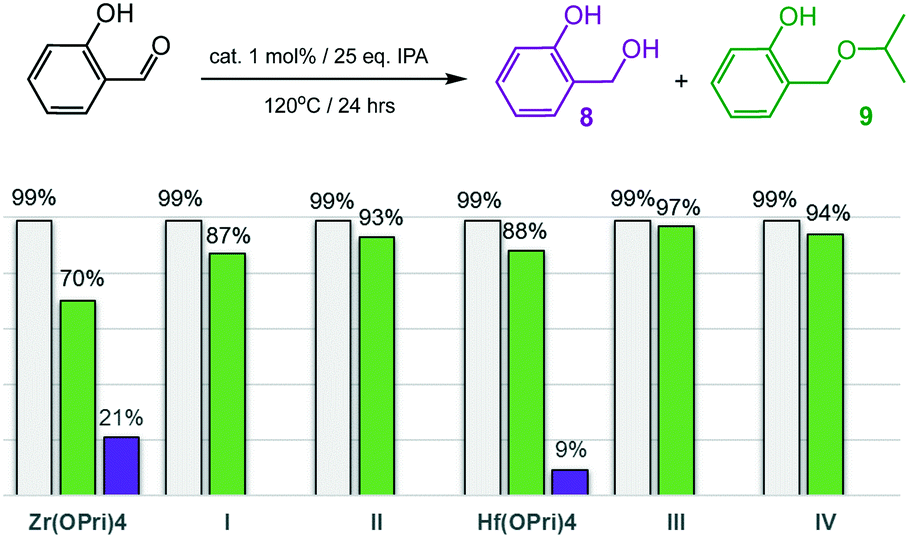 | ||
| Fig. 6 Reductive etherification of 4-hydroxybenzaldehyde. Conversions (grey bars): yields of alcohol 8 (purple bars); yields of ether 9 (green bars). | ||
Having had success in selectively producing hydroxy ethers 7 and 9, vanillin (4-hydroxy-3-methoxybenzaldehyde), one of the most widely used aroma chemicals and fragrances that can be produced from biomass-derived lignin, was examined (Fig. 7). Astonishingly, all zirconium and hafnium catalysts converted vanillin into isopropyl ether 10 with high selectivity and in yields ranging from 68–91%, even though higher temperatures (150 °C) were required relative to the reductive etherification of 2- and 4-hydroxybenzaldehyde. Of all the catalysts used, IV proved to be the most active system as 91% of ether 10 were formed after 24 hours at 150 °C. The catalyst performance of IV is similar to that of Rode's dual heterogeneous catalyst, which is composed of ZrO(OH)2 and Zr-montmorillonite, and gave 10 in 80% yield after 8 hours at 100 °C.7c
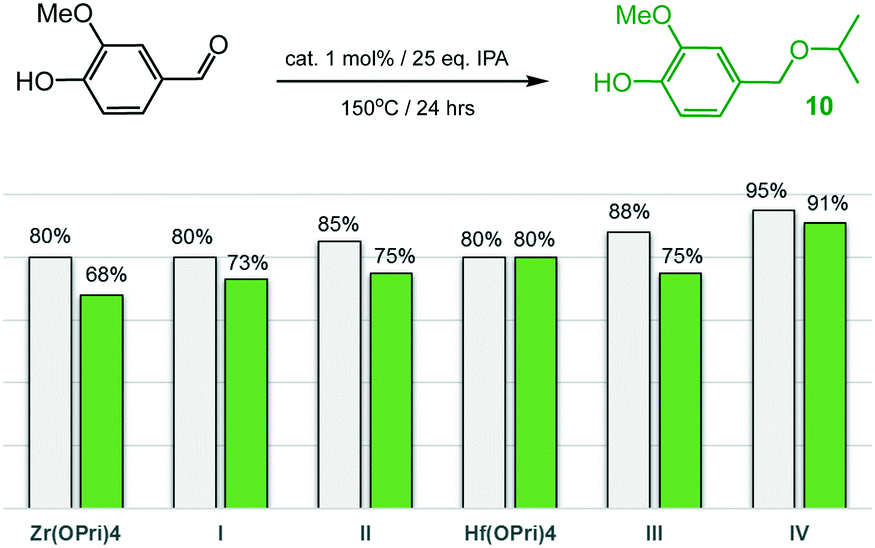 | ||
| Fig. 7 Reductive etherification of vanillin: conversions (grey bars) and yields of ether 10 (green bars). | ||
Unfortunately, efforts to reduce the phenolic aldehydes 3,4-dihydroxybenzaldehyde and 4-hydroxy-3,5-methoxybenzaldehyde (syringe aldehyde) failed. After 2 days at 150 °C, no conversion was noted. We attribute the inability of these multi-functionalized aldehydes to undergo reduction to their metal chelating properties, resulting in deactivation of the catalyst.
In comparing the results described above, we noticed that Zr(OPri)4 and Hf(OPri)4 were fairly active in catalysing the formation of hydroxy ethers 7, 9 and 10, but did not catalyse the formation of methoxy ethers 3 and 5. Moreover, 2- and 4-hydroxybenzaldehyde were converted faster and with better selectivity to the corresponding ethers than 2- and 4-methoxybenzaldehyde regardless of the catalyst used. At first glance, this appeared to be counter intuitive as according to the Hammett sigma constants, OH groups are somewhat stronger electron donors than methoxy groups. This results in lower carbonyl activities for 2- and 4-hydroxybenzaldehyde, and consequently would lead to lower rates of reduction.
To gain more insights, the kinetic profiles of the reductive etherification for all the five benzaldehydes were obtained at 100 °C with 1 mol% of III as the catalyst; the results are shown in Fig. 8 and 9. Consistent with our expectation based on the Hammett constants, the rate of conversion was found to be in the order C6H5CHO (82%; 16 hours) > 4-MeO–C6H4CHO (60%; 16 hours) > 4-HO–C6H4CHO (43%; 16 hours).
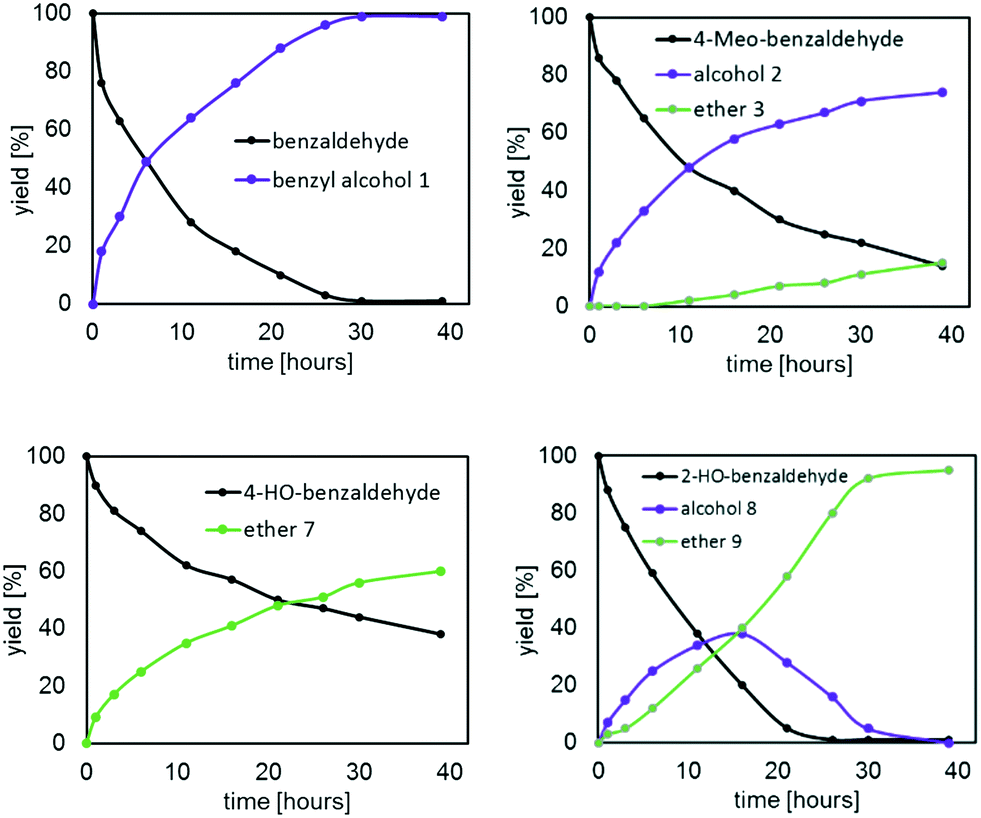 | ||
| Fig. 8 Kinetic profiles of the reductive etherification of benzaldehyde, 4-MeO-benzaldehyde, 4-HO-benzaldehyde and 2-HO-benzaldehyde. Reaction conditions: 100 °C, cat. III (1 mol%), 25 eq. isopropanol (IPA). | ||
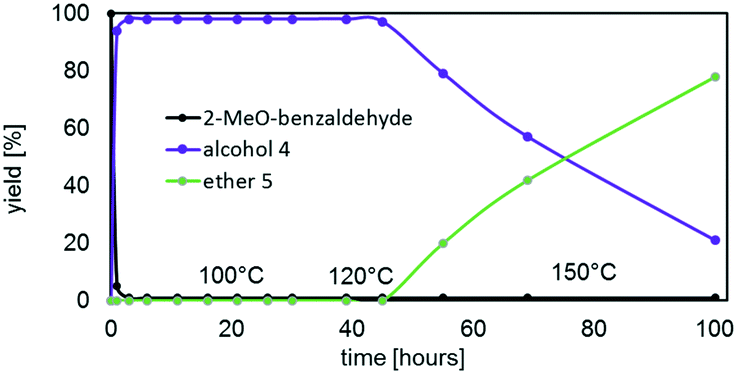 | ||
| Fig. 9 Kinetic profile of the reductive etherification of 2-MeO-benzaldehyde as a function of temperature. Reaction conditions: 100 °C (0–39 hours), 120 °C (39–45 hours), 150 °C (45–99 hours); cat. III (1 mol%), 25 eq. isopropanol (IPA). | ||
The catalytic behaviour of III in the reductive etherification of 2-methoxybenzaldehyde (Fig. 9) warrants an additional comment. After having catalysed the quantitative formation of alcohol 4 in less than an hour, and being further heated at 100 °C for 39 hours and at 120 °C for additional 6 hours, III still preserves its catalytic activity. In fact, upon subsequently increasing the temperature to 150 °C, alcohol 4 slowly converts into ether 5, demonstrating the catalysts' long-term durability and robustness at high temperatures over extensive periods of time and in the presence of water.
We noticed that benzaldehyde and 2-methoxybenzaldehyde exclusively converted into their respective alcohols, and only 4-methoxybenzaldehyde produced small quantities of ether after 30 hours at 100 °C. Therefore, it is reasonable to assume that the slow step of the reductive etherification for these substrates is the metal-catalysed alcohol etherification. Ether formation most likely proceeds via a metal stabilized intermediate of carbo-cationic character generated from proton migration of one of the coordinating IPA molecules (Scheme 3). Additional stabilization arises from the electron-donating 4-MeO group, enabling an intramolecular nucleophilic attack of isopropoxide to form the ether bond.
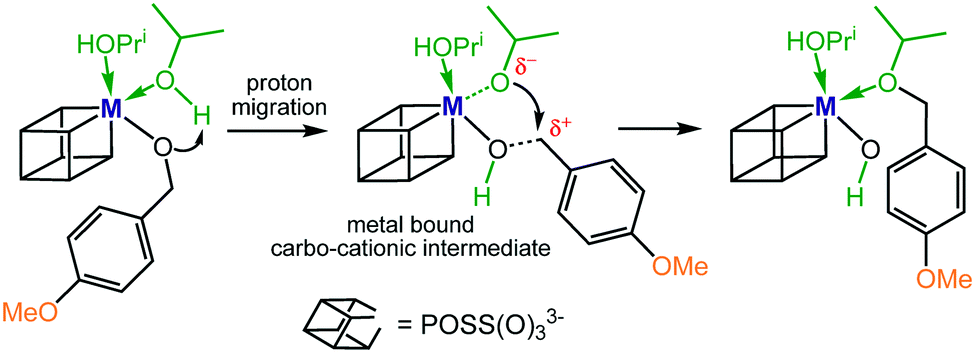 | ||
| Scheme 3 Proposed ether formation via a metal bound carbo-cationic intermediate. | ||
The active involvement of carbo-cationic intermediates is supported by the observation that at 150 °C the methoxy alcohols 2 and 4 convert to the respective ethers but not benzyl alcohol 1, i.e. the cationic benzyl intermediate is markedly less stable than the 2- and 4-methoxy substituted counterparts. One of the key factors that enables rapid and selective ether formation seems to be the Lewis acidity of the catalyst system. This is supported by the observation that I–IV catalyse the etherification of methoxy alcohols 2 and 4 but not Zr(OPri)4 and Hf(OPri)4, i.e.I–IV are stronger Lewis acids than their precursors because of the stronger electron-withdrawing properties of the POSS ligand versus an aliphatic alkoxide ligand.13
By contrast, the precursors Zr(OPri)4 and Hf(OPri)4 not only catalyse the reductive etherification of 2- and 4-hydroxybenzaldehyde and vanillin (Fig. 5–7), their activity is surprisingly similar to those of I–IV. In addition, the kinetic results for catalyst III (Fig. 8 and 9) further confirm that 2- and 4-hydroxybenzaldehyde were converted much faster into their respective ethers than 2- and 4-methoxybenzaldehyde. It seems that these drastic differences in rates are not accounted for by the slightly better ability of the 2- and 4-hydroxy groups to stabilize the carbo-cationic intermediate than the 2- and 4-methoxy groups (Scheme 3). Therefore, we propose an alternative mechanism that involves the formation of ortho- or para-quinone methide intermediates,14 which are formed via proton migration of the phenolic proton followed by cleavage of the benzylic C–O bond (Scheme 4). These reactive Michael acceptors, which have been generated from various organic precursors and used extensively in synthetic organic chemistry,15 can be described as benzylic cations that are strongly stabilized by the resonance electron-donating 4-O− or 2-O− substituents.16 As a result, they are more stable than the cationic methoxybenzyl intermediates involved in the formation of methoxy ethers 3 and 5, nonetheless, can readily be attacked by the external nucleophile isopropanol (present in excess) to form the corresponding hydroxy ethers 7, 9 and 10, respectively.
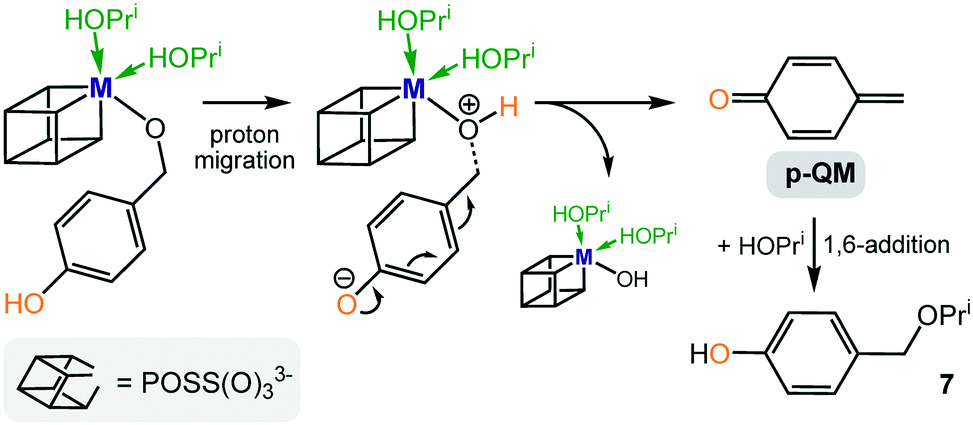 | ||
| Scheme 4 Proposed alternative formation of hydroxy ether 7via a metal stabilized para-quinone methide intermediate (p-QM = para-quinone methide). | ||
We next investigated the reduction of HMF, an important renewable feedstock for various bio-based organic compounds such as 2,5-bis(hydroxymethyl)furan (BHMF) (11), furan-dicarboxylic acid, levulinic acid, ethyl levulinate and derivatives thereof. Employing Sn-Beta as the catalyst, Vlachos and co-worker reported the formation of BPMF (12) from HMF with 87% selectivity and up to 80% overall yield at 180 °C.7b Rode et al. disclosed the conversion of HMF into BPMF (12) in isopropanol at 150 °C with claimed selectivity of up to 95% using the dual catalysts, ZrO(OH)2 and Zr-montmorillonite.7c However, in neither cases have isolated yields been reported.
As the reductive etherification of HMF is a complex chemical transformation that involves the formation of humin polymers17 and various other synthetic intermediates,7c we were pleased to see that the simple precursors Zr(OPri)4 and Hf(OPri)4 selectively generated BHMF (11) in good yields of 58% and 59%, resp., in only 4 hours at 150 °C (Fig. 10).
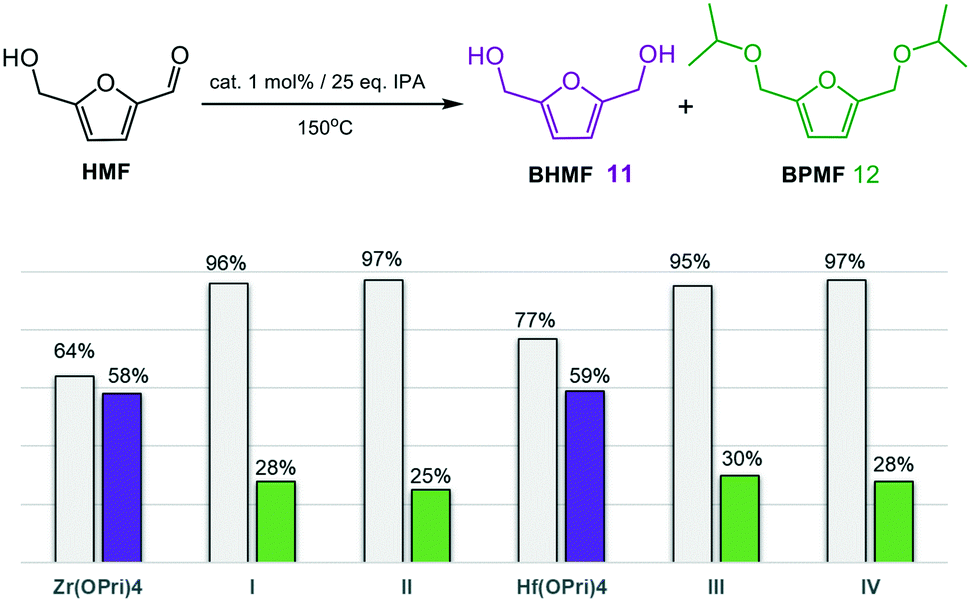 | ||
| Fig. 10 Reductive etherification of 2-hydroxymethylfurfural (HMF). Conversions (grey bars) and product yields (purple bars = 11; green bars = 12) after 4 hours. | ||
In contrast, catalysts I–IV quantitatively converted HMF into primarily humin polymers and the desired BPMF (12), albeit the latter in fairly low yields (25–30%). Increasing the reaction time proved to be counter-productive as the yields of 12 further decreased in favour of humin polymers, a common problem in this chemistry.17 To suppress the formation of humin polymers, a series of experiments was undertaken, where the HMF concentration was gradually decreased by increasing the amounts of isopropanol, from 25 up to 200 eq. using III as the catalyst at 150 °C. The most relevant results are summarized in Fig. 11 (see also Fig. S17–S21†) and revealed the optimum conditions for this process to be ca. 9–10 hours and 100 eq. of isopropanol. Gratifyingly, after 9 hours, both HMF and undesired BHMF (11) were fully consumed, and the reaction mixture only contained ca. 60% BPMF (12) and polymer, which greatly facilitated the purification process. Again, increasing the reaction time led to a slight decrease in the yields of 12 due to polymer formation.
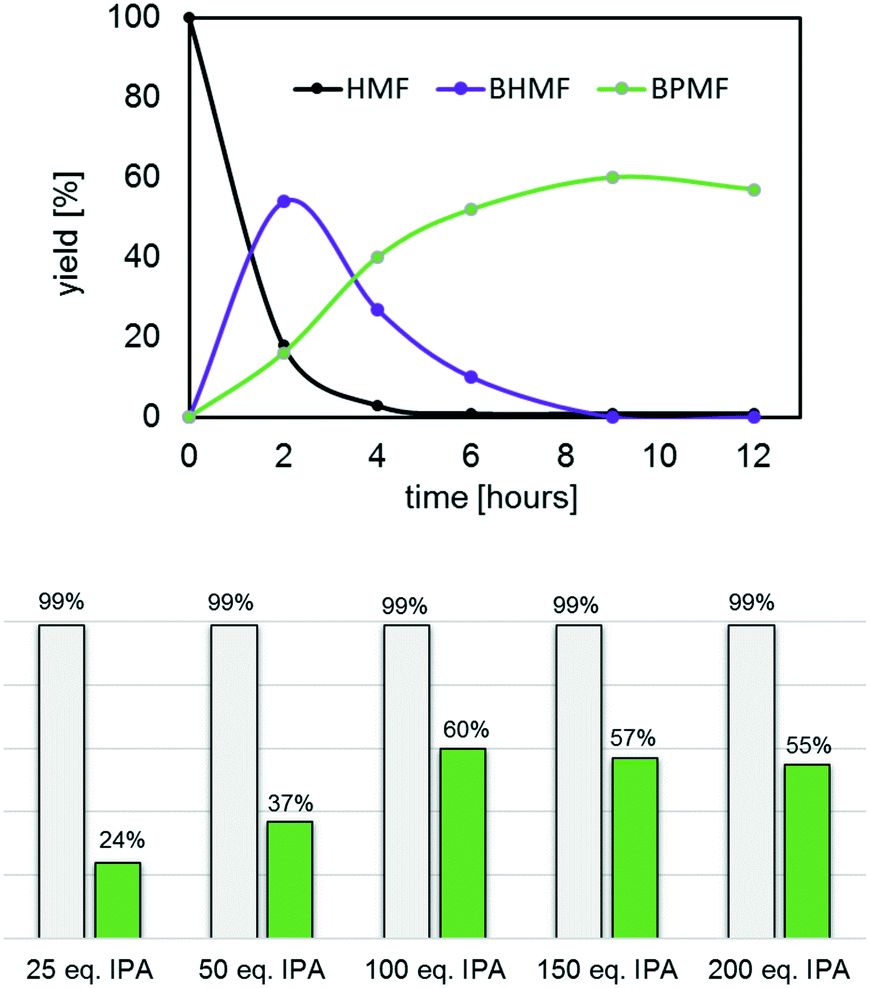 | ||
| Fig. 11 Reductive etherification of HMF as a function of isopropanol (IPA) eq.; cond.: 9 hours, 150 °C, cat. III (1 mol%); top: kinetic profile for 100 eq. of IPA; bottom: conversions (grey bars) and yields of BPMF 12 (green bars) for various eq. of IPA. | ||
To demonstrate the practicality of our homogeneous catalyst approach, scale-up syntheses of ethers 3, 5, 7, 9, 10 and 12 (1 g substrate) were carried out employing the best performing catalysts, resp. (Scheme 5). All products except 7 could easily be purified by vacuum distillation with isolated yields ranging from to 31–94% (ref. 18) without the need of tedious purification by column chromatography.
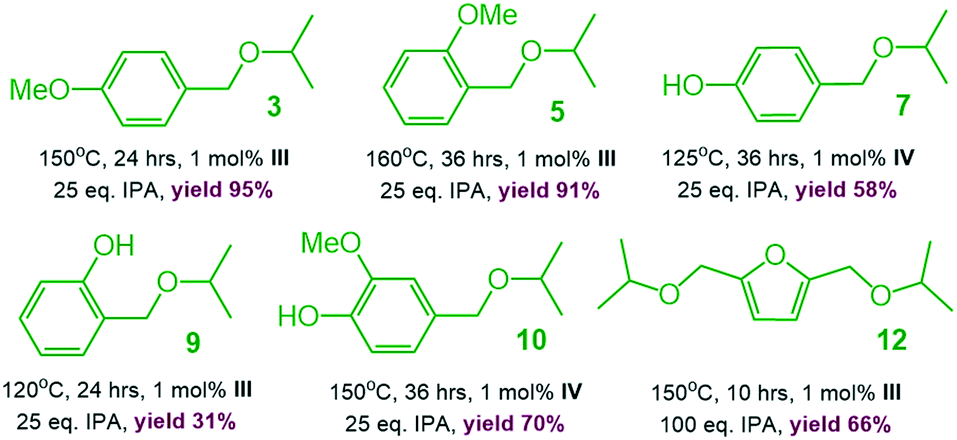 | ||
| Scheme 5 Gram scale synthesis of 3, 5, 7, 9, 10 and 12. | ||
The efficacy and robustness of catalyst III in the conversion of HMF to BPMF (12), prompted us to test the catalytic activity of Hf(OPri)4 (1 mol%) in the presence of ligand Bui-POSS(OH)3 (Scheme 6). Gratifyingly, after ca. 10 hours at 150 °C (100 eq. IPA) and subsequent purification by vacuum distillation, BPMF could be isolated in 69% yield, similar to what was observed with isolated III as the catalyst. Since Hf(OPri)4 itself does not produce BPMF (vide supra), catalyst III appears to have formed during the course of the reaction. This “in situ” catalyst approach could also be applied to the catalytic formation of ethers 3, 5 and 10 with comparable isolated yields but somewhat longer reaction times (see ESI†).
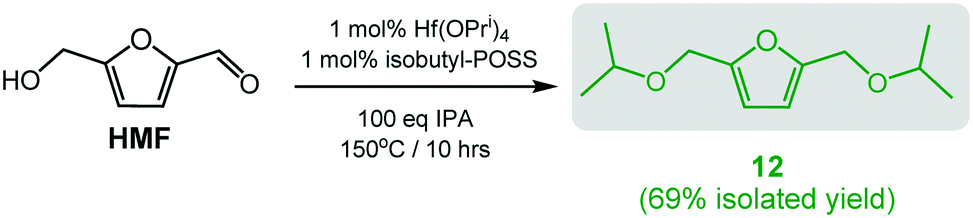 | ||
| Scheme 6 Synthesis of 10 and 12 employing 1 mol% Hf(OPri)4 and 1 mol% Bui-POSS(OH)3. | ||
To study the catalyst structure in the solution, diffusion experiments for hafnium complex III using DOSY-NMR spectroscopy were performed in various solvents at room temperature. The results for THF-D8 and C6D6 suggest III to be dimeric in solution, which is consistent with its solid-state structure. In the more polar solvents CD2Cl2 and acetone-D6, however, III was found to be monomeric (for more details see ESI†).
Based on these observations and those discussed above, we propose a tentative mechanism, where III exists in isopropanol as a hexa-coordinate monomeric complex of type A (Scheme 7). For the MPV reduction to occur, complex A, which bears two coordinated IPA molecules, needs to be in rapid equilibrium with its substrate–metal complex B to allow for an intramolecular transfer of hydride from the isopropoxide to the substrate. Complex C thus generated then converts via replacement of the coordinated acetone product with excess IPA into species D. To enable the second step of the overall process, the etherification, an intramolecular proton transfer in D from the coordinated IPA to the benzyloxide, needs to occur. Complex E thus generated then undergoes an intramolecular nucleophilic substitution to furnish the ether bound metal hydroxide F. Liberation of the coordinated ether product from Fvia replacement with excess IPA produces metal hydroxide G. Subsequent intramolecular proton transfer from the coordinated IPA to the hydroxide generates complex H. The latter finally converts to catalyst A upon replacement of the coordinated water molecule by excess IPA. Note that in the case of 2- and 4-hydroxybenzaldehyde and vanillin as substrates, proton migration in species D will occur involving the phenolic proton. This is followed by C–O bond cleavage and formation of ortho- or para-quinone methide intermediates (see also Scheme 4).
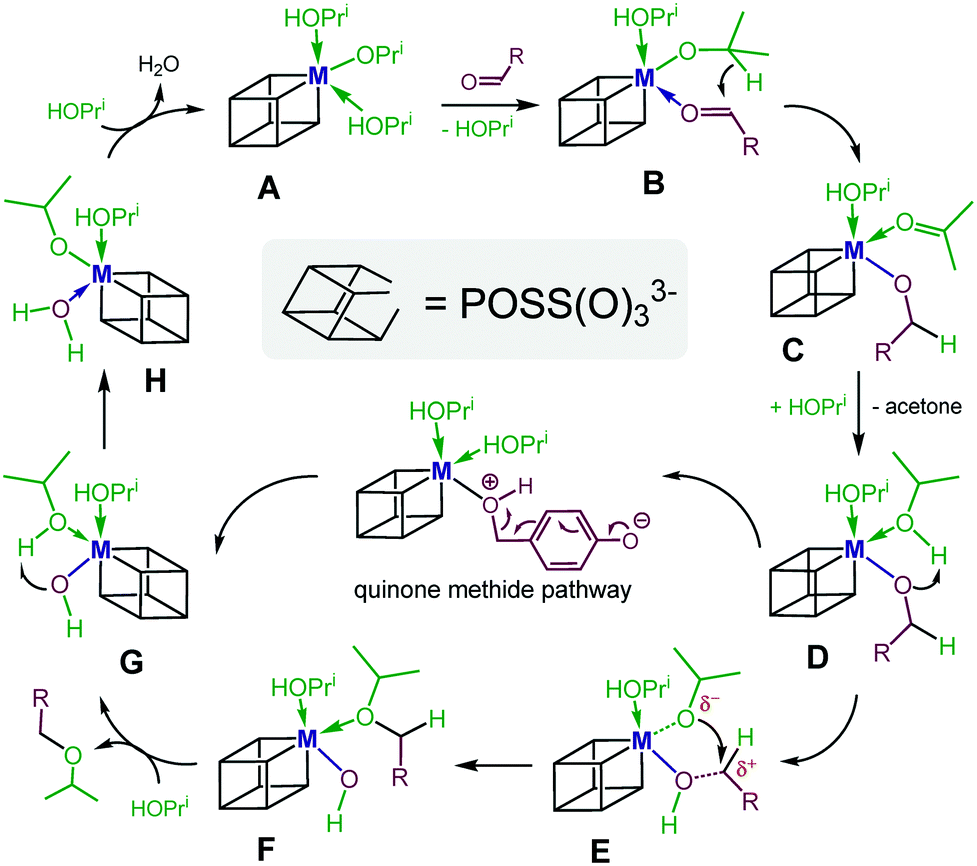 | ||
| Scheme 7 Proposed reaction mechanism of the reductive etherification of aldehydes. | ||
Conclusions
With the goal of developing durable homogeneous catalysts for selective biomass transformations, we have synthesized tripodal zirconium and hafnium isopropoxides I–IV. In these “corner capped” complexes, zirconium and hafnium are rigidly incorporated into the electron-withdrawing POSS(O)33− ligand framework, which provides complexes I–IV with sufficient Lewis acidity, high kinetic stability and thermal robustness. In fact, I–IV are efficient homogeneous catalysts operating at low loadings and high temperatures in the reductive etherification of electron-rich aromatic aldehydes to produce various isopropyl ethers. Zr(OPri)4 and Hf(OPri)4 are active catalysts themselves, showing excellent selectivity toward the formation of alcohols 2, 4 and 11 but are inactive regarding ether formation. These selectivity differences can be interpreted in terms of their different Lewis acidities. In contrast, I–IV and Zr(OPri)4/Hf(OPri)4 generated the hydroxyl ethers 7, 9 and 10 with similar activities and selectivities, which we attribute to the involvement of para- and ortho-quinone methide intermediates. Most importantly, III proved to be the first homogeneous catalyst capable of selectively converting HMF into bio-fuel additive BPMF (12), the latter could be isolated in good yields and purities upon distillation. That similar catalytic performances can be achieved without the need of synthesizing the POSS catalyst, as demonstrated for the selective conversion of HMF into BPMF, highlights the potential of our homogeneous approach for applications in biomass and related transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, under Award DE-SC0019094. We are grateful to Anthony Cozzolino for calculating the van der Waals volume of complex III and Shiva Moaven for assisting in the DOSY-NMR experiments.Notes and references
- (a) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC; (b) J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC; (c) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- (a) A. Liu, B. Liu, Y. Wang, R. Ren and Z. Zhang, Fuel, 2014, 117, 68–73 CrossRef CAS; (b) M. Balakrishnan, E. R. Sacia and A. T. Bell, Green Chem., 2012, 14, 1626–1634 RSC; (c) M. Musolino, M. J. Gines-Molina, R. Moreno-Tost and F. Arico, ACS Sustainable Chem. Eng., 2019, 7, 10221–10226 CrossRef CAS.
- (a) A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS; (b) A. M. Rasero-Almansa, M. Iglesias and F. Sanchez, RSC Adv., 2016, 6, 106790–106797 RSC; (c) G. Li, L. Gao, Z. Sheng, Y. Zhan, C. Zhang, J. Ju, Y. Zhang and Y. Tang, Catal. Sci. Technol., 2019, 9, 4055–4065 RSC.
- (a) C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2013, 15, 2278–2281 CrossRef CAS; (b) B. McNerney, B. Whittlesey, D. B. Cordes and C. Krempner, Chem. – Eur. J., 2014, 20, 14959–14964 CrossRef CAS; (c) X. Wang, J. Hao, L. Deng, H. Zhao, Q. Liu, N. Li, R. He, K. Zhi and H. Zhou, RSC Adv., 2020, 10, 6944–6952 RSC; (d) A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS; (e) K. Flack, K. Kitagawa, P. Pollet, C. A. Eckert, K. Richman, J. Stringer, W. Dubay and C. L. Liotta, Org. Process Res. Dev., 2012, 16, 1301–1306 CrossRef CAS.
- (a) R. Cohen, C. R. Graves, S. T. Nguyen, J. M. L. Martin and M. A. Ratner, J. Am. Chem. Soc., 2004, 126, 14796–14803 CrossRef CAS; (b) O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21–27 RSC; (c) R. S. Assary, L. A. Curtiss and J. A. Dumesic, ACS Catal., 2013, 3, 2694–2704 CrossRef CAS.
- (a) R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS; (b) S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS.
- (a) J. Luo, J. Yu, R. J. Gorte, E. Mahmoud, D. G. Vlachos and M. A. Smith, Catal. Sci. Technol., 2014, 4, 3074–3081 RSC; (b) J. Jae, E. Mahmoud, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 508–513 CrossRef CAS; (c) S. Shinde and C. Rode, ChemSusChem, 2017, 10, 4090–4101 CrossRef CAS.
- H. Nguyen, N. Xiao, S. Daniels, N. Marcella, J. Timoshenko, A. Frenkel and D. G. Vlachos, ACS Catal., 2017, 7, 7363–7370 CrossRef CAS.
- (a) F. J. Feher and T. A. Budzichowski, Polyhedron, 1995, 14, 3239–3253 CrossRef CAS; (b) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS; (c) C. Krempner, Eur. J. Inorg. Chem., 2011, 2011, 1689–1698 CrossRef.
- (a) H. C. L. Abbenhuis, S. Krijnen and R. A. van Santen, Chem. Commun., 1997, 331–332 RSC; (b) T. Maschmeyer, M. C. Klunduk, C. M. Martin, D. S. Shephard, J. M. Thomas and B. F. G. Johnson, Chem. Commun., 1997, 1847–1848 RSC; (c) M. Crocker, R. H. M. Herold and A. G. Orpen, Chem. Commun., 1997, 2411–2412 RSC; (d) F. Carniato, C. Bisio, E. Boccaleri, M. Guidotti, E. Gavrilova and L. Marchese, Chem. – Eur. J., 2008, 14, 8098–8101 CrossRef CAS; (e) E. H. Aish, M. Crocker and F. T. Ladipo, J. Catal., 2010, 273, 66–72 CrossRef CAS; (f) M. D. Skowronska-Ptasinska, M. L.-W. Vorstenbosch, R. A. van Santen and H. C. L. Abbenhuis, Angew. Chem., Int. Ed., 2002, 41, 637–639 CrossRef CAS; (g) L. Zhang, H. C. L. Abbenhuis, G. Gerritsen, N. N. Bhriain, P. C. M. Magusin, B. Mezari, W. Han, R. A. van Santen, Q. Yang and C. Li, Chem. – Eur. J., 2007, 13, 1210–1221 CrossRef CAS; (h) P. Guillo, M. I. Lipschutz, M. E. Fasulo and T. D. Tilley, ACS Catal., 2017, 7, 2303–2312 CrossRef CAS.
- (a) F. J. Feher, J. F. Walzer and R. L. Blanski, J. Am. Chem. Soc., 1991, 113, 3618–3619 CrossRef CAS; (b) F. J. Feher and R. L. Blanski, J. Am. Chem. Soc., 1992, 114, 5886–5887 CrossRef CAS; (c) R. Duchateau, H. C. L. Abbenhuis, R. A. van Santen, A. Meetsma, S. K.-H. Thiele and M. F. H. van Tol, Organometallics, 1998, 17, 5663–5673 CrossRef CAS.
- (a) O. Viotti, A. Fischer, G. A. Seisenbaeva and V. G. Kessler, Inorg. Chem. Commun., 2010, 13, 774–777 CrossRef CAS; (b) O. Viotti, G. A. Seisenbaeva and V. G. Kessler, Inorg. Chem., 2009, 48, 9063–9065 CrossRef CAS.
- (a) F. J. Feher and T. A. Budzichowski, J. Organomet. Chem., 1989, 379, 33–40 CrossRef CAS; (b) R. West and R. H. Baney, J. Am. Chem. Soc., 1959, 81, 6145–6148 CrossRef CAS; (c) R. West, R. H. Baney and D. L. Powell, J. Am. Chem. Soc., 1960, 82, 6269–6272 CrossRef CAS; (d) P. T. Wolczanski, Polyhedron, 1995, 14, 3335–3362 CrossRef CAS; (e) C. Krempner, Eur. J. Inorg. Chem., 2011, 1689–1698 CrossRef CAS.
- (a) Quinone Methides, ed. S. E. Rokita, John Wiley and Sons, 2009 Search PubMed; (b) E. Dorrestijn, M. Kranenburg, M. V. Ciriano and P. Mulder, J. Org. Chem., 1999, 64, 3012–3018 CrossRef CAS; (c) S. J. Gharpure, A. M. Sathiyanarayanan and P. Jonnalagadda, Tetrahedron Lett., 2008, 49, 2974–2978 CrossRef CAS; (d) M. M. Toteva, M. Moran, T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 2003, 125, 8814–8819 CrossRef CAS.
- (a) E. E. Weinert, R. Dondi, S. Colloredo-Melz, K. N. Frankenfield, C. H. Mitchell, M. Freccero and S. E. Rokita, J. Am. Chem. Soc., 2006, 128, 11940–11947 CrossRef CAS; (b) R. W. Van de Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS; (c) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS.
- M. M. Toteva and J. P. Richard, Adv. Phys. Org. Chem., 2011, 45, 39–91 CrossRef CAS.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- The low yield of ether 9 after distillation is the result of a thermal fragmentation to the ortho-quinone methide followed by polymerization to undefined products. See also: (a) S. B. Cavith, H. R. Sarrafizadeh and P. D. Gardner, J. Org. Chem., 1962, 27, 1211–1216 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2009174–2009177. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy01864c |
| This journal is © The Royal Society of Chemistry 2021 |