
Insight into the chemoselective aromatic vs. side-chain hydroxylation of alkylaromatics with H2O2 catalyzed by a non-heme imine-based iron complex†
Barbara
Ticconi
a,
Giorgio
Capocasa
a,
Andrea
Cerrato
a,
Stefano
Di Stefano
 a,
Andrea
Lapi
a,
Beatrice
Marincioni
a,
Giorgio
Olivo‡
b and
Osvaldo
Lanzalunga
*a
a,
Andrea
Lapi
a,
Beatrice
Marincioni
a,
Giorgio
Olivo‡
b and
Osvaldo
Lanzalunga
*a
aDipartimento di Chimica, Università degli Studi di Roma “La Sapienza” and, Istituto CNR per i Sistemi Biologici (ISB-CNR), Sezione Meccanismi di Reazione, c/o Dipartimento di Chimica, Università degli Studi di Roma “La Sapienza”, P.le A. Moro 5, I-00185 Rome, Italy. E-mail: osvaldo.lanzalunga@uniroma1.it
bInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, C/ Maria Aurèlia Capmany 69, 17003 Girona, Spain
First published on 3rd November 2020
Abstract
The oxidation of a series of alkylaromatic compounds with H2O2 catalyzed by an imine-based non-heme iron complex prepared in situ by reaction of 2-picolylaldehyde, 2-picolylamine, and Fe(OTf)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio leads to a marked chemoselectivity for aromatic ring hydroxylation over side-chain oxidation. This selectivity is herein investigated in detail. Side-chain/ring oxygenated product ratio was found to increase upon decreasing the bond dissociation energy (BDE) of the benzylic C–H bond in line with expectation. Evidence for competitive reactions leading either to aromatic hydroxylation via electrophilic aromatic substitution or side-chain oxidation via benzylic hydrogen atom abstraction, promoted by a metal-based oxidant, has been provided by kinetic isotope effect analysis.
:2:1 ratio leads to a marked chemoselectivity for aromatic ring hydroxylation over side-chain oxidation. This selectivity is herein investigated in detail. Side-chain/ring oxygenated product ratio was found to increase upon decreasing the bond dissociation energy (BDE) of the benzylic C–H bond in line with expectation. Evidence for competitive reactions leading either to aromatic hydroxylation via electrophilic aromatic substitution or side-chain oxidation via benzylic hydrogen atom abstraction, promoted by a metal-based oxidant, has been provided by kinetic isotope effect analysis.
Introduction
The conversion of aromatic compounds to phenolic products by hydroxylation of aryl rings represents one of the most challenging transformation in synthetic organic chemistry. The relevance of phenolic moieties in a variety of natural products, pharmaceutical compounds and fine chemicals obtained in industrial processes1,2 calls for the development of simple synthetic protocols for the direct insertion of hydroxyl functions in aromatic rings. Notwithstanding, only a few oxidizing systems are available for the catalytic conversion of aromatic substrates to phenols.3–10Among these catalytic systems, a special attention has been devoted to those involving the environmentally friendly and sustainable oxidant H2O2 and catalysts based on abundant and low toxic metals, such as iron and manganese. Along this line, good results have been obtained in the aromatic oxidation catalyzed by heme and non-heme metal complexes which are biomimetic models of natural oxygenases, although most of them are stoichiometric transformations.11–31
We have recently reported that iminopyridine complex 1, easily obtained by an in situ self-assembly of 2-picolylamine, 2-picolylaldehyde and iron(II) triflate in a 2:2:1 ratio in acetonitrile (Scheme 1),32–36 efficiently catalyzes the hydroxylation of aromatic rings with H2O2 under mild conditions.37
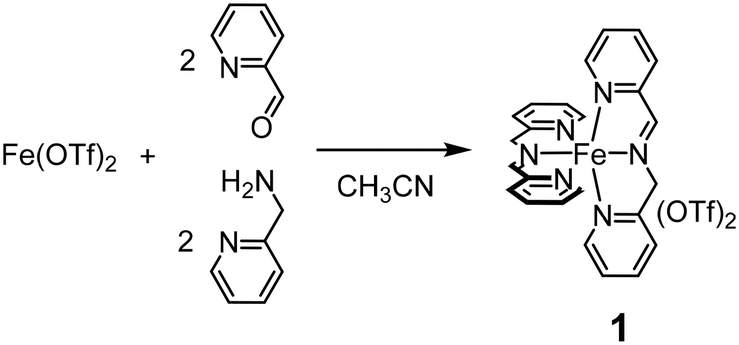 | ||
| Scheme 1 Self-assembled imine-based non-heme iron(II) complex 1. | ||
Kinetic isotope effect (KIE) studies, the use of radical scavengers, substituent effects on inter- and intramolecular selectivity and rearrangement experiments converged to the operation of a metal-based electrophilic aromatic substitution (SEAr) pathway with no involvement of free radical species (Scheme 2).35,37 Concerning the nature of the active species (metal-based oxidant), we were not able to isolate the intermediate, formed upon addition of H2O2 to 1, which is competent for the oxidation.35 Generally, activation of H2O2 by non-heme iron complexes is due to the formation of FeIII–OOH intermediates which undergo either heterolytic O–O bond cleavage to produce a highly reactive FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complex or homolytic cleavage of the same bond leading to a FeIVO complex and HO˙.35,38–45 Both intermediates mediate aromatic hydroxylation, albeit with different rates, selectivities and KIEs.11–24,45,46 With 1 and H2O2, we observed a KIE and a selectivity consistent with those reported in (stoichiometric) aromatic hydroxylation mediated by putative FeVO species.37 On this basis, we proposed the formation of a similar high-valent iron–oxo complex after the initial oxidation of Fe(II) complex 1 to Fe(III) followed by detachment of a pyridine arm, and coordination and activation by H2O2.34,37
O complex or homolytic cleavage of the same bond leading to a FeIVO complex and HO˙.35,38–45 Both intermediates mediate aromatic hydroxylation, albeit with different rates, selectivities and KIEs.11–24,45,46 With 1 and H2O2, we observed a KIE and a selectivity consistent with those reported in (stoichiometric) aromatic hydroxylation mediated by putative FeVO species.37 On this basis, we proposed the formation of a similar high-valent iron–oxo complex after the initial oxidation of Fe(II) complex 1 to Fe(III) followed by detachment of a pyridine arm, and coordination and activation by H2O2.34,37
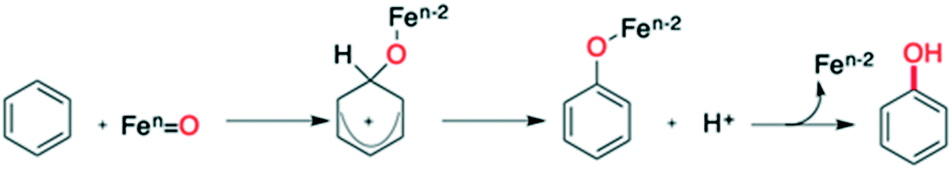 | ||
| Scheme 2 Proposed mechanism for aromatic hydroxylation of C–H bonds promoted by imine-based non-heme iron(II) complex/H2O2. | ||
One of the major challenges in aromatic hydroxylation of substrates bearing benzylic C–H bonds is the competition of side-chain oxidation. Most of the oxidation processes and in particular those involving free radical species such as HO˙ and HOO˙ are characterized by poor selectivity9,13,14,16,47–51 with a certain preference for the aromatic side-chain in view of the relatively low bond dissociation energies (BDEs) of the benzylic C–H bonds especially when secondary or tertiary benzylic C–H bonds are present.13,14,47–49,52,53
When applied to the oxidation of alkylaromatics (toluene, ethylbenzene, and cumene), the 1/H2O2 system showed a marked preference (chemoselectivity) for aromatic ring hydroxylation over aliphatic side-chain oxidation. Phenolic compounds were obtained in satisfactory yields accompanied by small amounts of side-chain oxygenated products.37,54 Remarkably, the chemoselectivity for aromatic hydroxylation was significantly higher than that displayed by other iron catalysts,20,47,55–57 allowing the inclusion of 1/H2O2 system in the restricted family of the most selective catalytic systems.4,8–10,24
In this context, it is notable that the 1/H2O2 system allowed the direct conversion of phenylalanine to tyrosine isomers without overoxidation or side-chain functionalization.36
In order to gain more insight into this high chemoselectivity and define its potential, in this work, we focused our attention on the oxidation chemoselectivity of polycyclic alkylaromatic substrates containing more reactive benzylic C–H bonds characterized by relatively low BDE values (75–82 kcal mol−1, Chart 1).59
 | ||
| Chart 1 Benzylic C–H BDE values for some alkylaromatic substrates. | ||
In particular, the variation of ring/side-chain oxidation product ratio has been analyzed in terms of substrate benzylic C–H BDE values. Deuteration of the benzylic hydrogen atoms provided additional insights into the mechanism of aliphatic side-chain oxidation.
Results and discussion
Oxidation reactions were carried out by mixing Fe(OTf)2(CH3CN)2 (2.5 μmol, TfO− = CF3SO3−), 2-picolylaldehyde and 2-picolylamine (5.0 μmol each) followed by the substrate (250 μmol) in CH3CN at 25 °C. A solution of H2O2 (50 μmol, diluted from a 35% w/w H2O2 solution) was then added using a syringe pump in 30 minutes and the reaction was left under vigorous stirring for 90 minutes. After addition of the internal standard, the reaction mixture was filtered over a short pad of SiO2 eluting with AcOEt, and analyzed by GC and 1H NMR (see ESI†). Reactions have been carried out under oxidant-deficient conditions (0.2 eq. with respect to the substrate) in order to minimize the formation of overoxidation products which may alter the aromatic vs. side-chain selectivity values.60 Control experiments were performed in the presence of H2O2 and Fe(OTf)2(CH3CN)2 only, with no ligand added, to compare the differences in terms of reactivity and selectivity between the metal-based mechanism catalyzed by complex 1 and the Fenton-type reactions promoted by Fe(OTf)2/H2O2.61,62 Products and yields, referring to the amount of oxidant, are reported in Table 1. With all the substrates, the mass balance was satisfactory (>90%).| Entry | Substrate | Recovered substrateb | Productsc | TNd | ||||
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 20 mol% H2O2, 1 mol% catalyst 1, CH3CN at 25 °C, reaction time: 90 minutes, oxidant added using a syringe pump (30 min). The reported results are the average of at least two runs. Recovered substrates and product yields (%), determined by GC and/or 1H NMR analysis. Error ±5%. b Refers to the initial amount of substrate. c Yields refer to the amount of oxidant. d TN defined as the moles of oxidation products/mole of Fe. Ketones and aldehydes were considered as the double oxidation products and quinones as the triple oxidation products. | ||||||||
| 1 |
|
95 |
 o-6%
o-6% |
 m-, p-5%
m-, p-5% |
 <1% <1% |
2.6 | ||
| 2 |
|
80 |
 o-37%
o-37% |
 m-, p-45%
m-, p-45% |
 4% 4% |
 2% 2% |
 3% 3% |
20 |
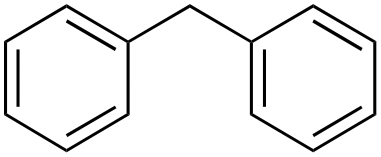
| 3 |
|
82 |
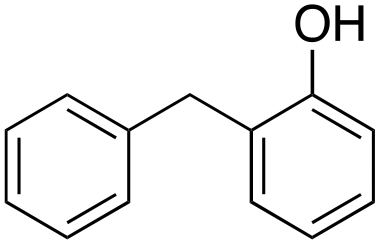 o-27%
o-27% |
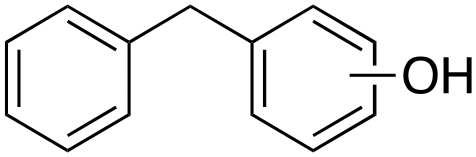 m-, p-42%
m-, p-42% |
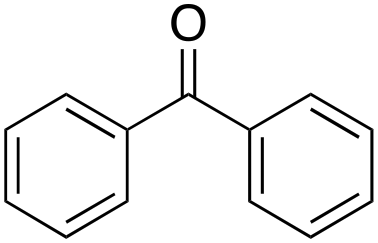 1% 1% |
14 | ||
| 4 |
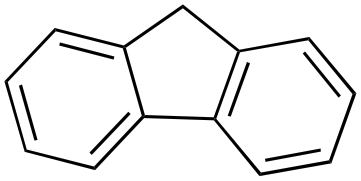
|
89 |
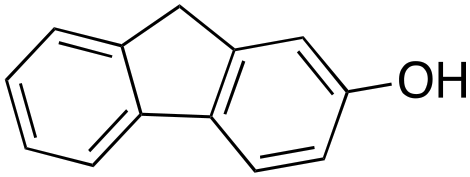 13% 13% |
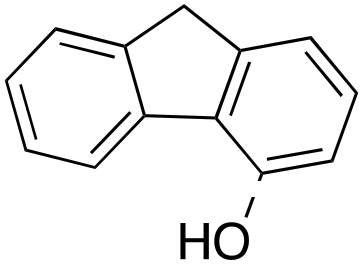 14% 14% |
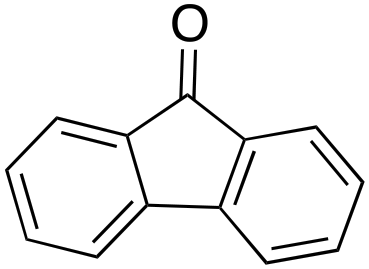 14% 14% |
11 | ||
| 5 |
|
90 |
 16% 16% |
 8% 8% |
 13% 13% |
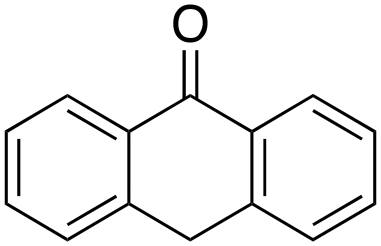 1% 1% 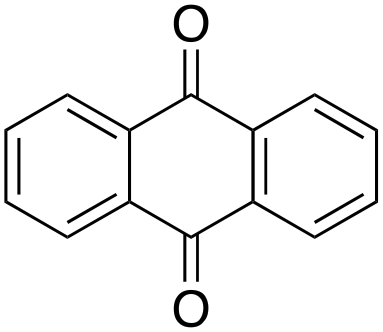 2% 2% |
9 | |
| 6 |
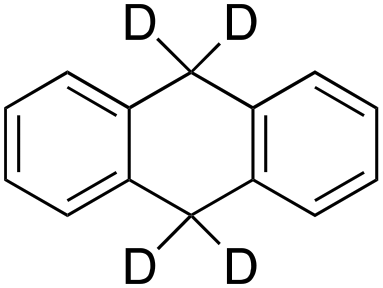
|
85 |
 34% 34% |
 26% 26% |
 8% 8% |
 1% 1% 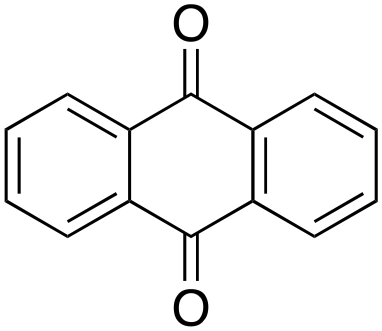 2% 2% |
13 | |
| 7 |
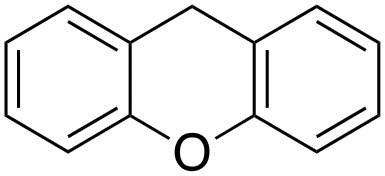
|
88 |
 4% 4% |
 5% 5% |
 29% 29% |
13 | ||
| 8 |
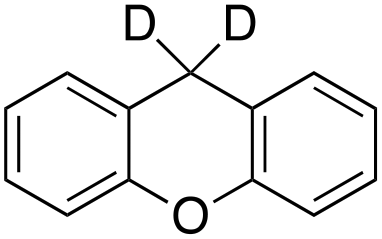
|
95 |
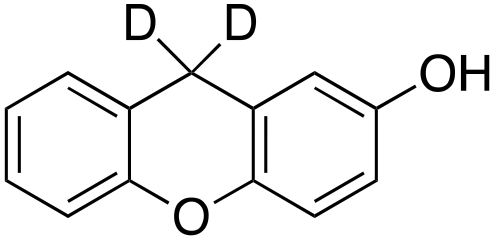 5% 5% |
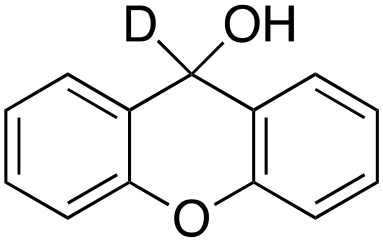 4% 4% |
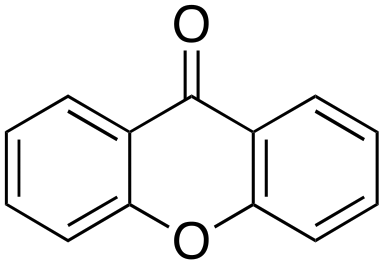 9% 9% |
5.4 | ||
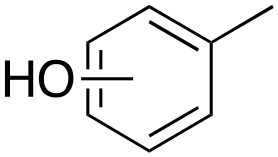
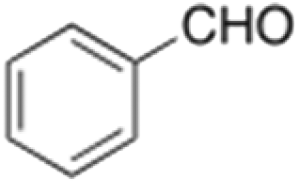
Oxidation of toluene led to the formation of o-cresol (6%), a mixture of m- and p-cresols (5%), as the main reaction products. Remarkably, only a small amount (<1%) of benzaldehyde (Table 1, entry 1) deriving from benzylic hydrogen atom abstraction was detected, indicating a high selectivity of the 1/H2O2 system for the aromatic hydroxylation. Control experiments in the absence of the imine ligand showed that no phenolic products are formed with the substrate quantitatively recovered.
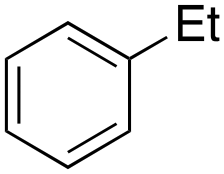
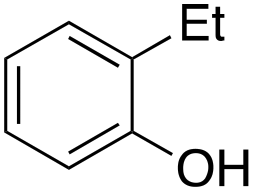
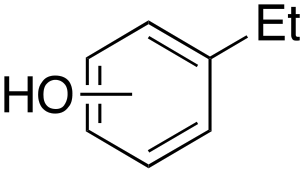
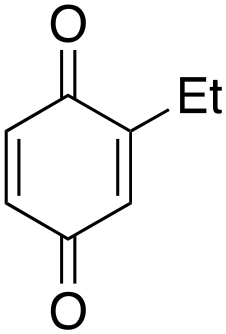
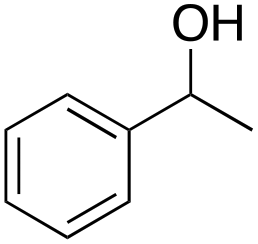
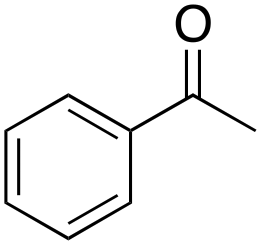
Ethylbenzene was found to be more reactive than toluene and the oxidation reaction gave a mixture of o-, m- and p-ethylphenol (37% of ortho and 45% of meta + para), accompanied by the overoxidation product 2-ethyl-p-benzoquinone (4%) (entry 2). Small amounts of the side-chain oxidation products, 1-phenylethanol (2%) and acetophenone (3%), were also observed. Thus, even in the oxidation of ethylbenzene, which bears weaker secondary benzylic C–H bonds, the 1/H2O2 system is highly selective for aromatic over aliphatic oxidation with a 94:6 ring/side-chain chemoselectivity (ring-oxygenated products/side-chain-oxygenated products). Control experiments, carried out in the absence of imine ligand (Fenton conditions), showed the exclusive formation of side-chain oxidation products, with 1-phenylethanol and acetophenone forming in 8% and 4% yields, respectively. This result supports the operation of a metal-based oxidant in the reaction of ethylbenzene with the 1/H2O2 system and excludes any involvement of HO˙ radicals.49,58,62
In the oxidation of diphenylmethane (entry 3), almost exclusive formation of aromatic oxidation products, o-, m- and p-benzylphenol (27% of ortho and 42% of meta + para), was again observed accompanied by only a small amount (1%) of the benzylic oxidation product benzophenone. Also with this substrate, a high ring/side-chain chemoselectivity is observed (>98). The higher chemoselectivity for aromatic hydroxylation compared to that observed for ethylbenzene is likely a consequence of the higher steric hindrance of the benzylic C–H bonds of diphenylmethane. As a matter of fact, the oxidizing species formed upon treatment of 1 with H2O2 has been found to be rather sensitive to steric effects.33,34 Blank experiments with the Fe(OTf)2/H2O2 system showed the exclusive formation of benzophenone (8%).
In a control experiment, the reaction product p-benzylphenol was used as a substrate in the oxidation with 1/H2O2 system. No products have been detected and the substrate was recovered quantitatively. This result is somewhat surprising in view of the activation of phenolic compounds towards electrophilic aromatic substitutions by the electron-releasing hydroxyl group and may be due to the binding of the phenolic products on the imine-based iron catalyst which prevents further oxidation as previously reported for related systems.11,12,17 The oxidation of fluorene led to the formation of 2-hydroxyfluorene (13%) and 4-hydroxyfluorene (14%) due to aromatic oxidation and 9-fluorenone (14%) due to benzylic oxidation (entry 4). Again, aromatic hydroxylation prevails over benzylic oxidation, but the ring/side-chain chemoselectivity drops to 66:34.
The decrease of selectivity for the aromatic oxidation observed with this substrate can be rationalized on the basis of a higher HAT reactivity of the benzylic C–H bonds of fluorene (benzylic BDEC–H = 80 kcal mol−1). As a matter of fact, 9-fluorenone is the exclusive oxidation product observed when the reaction is carried out in the absence of the imine ligand in the control experiments as well as in previous reports with iron catalysts14,58,63 or well-defined FeIV–oxo complexes.38,39,41,64
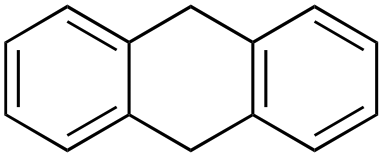
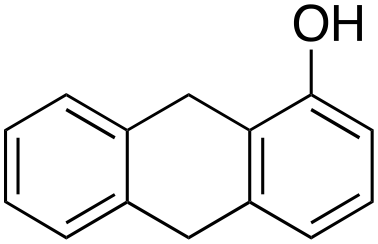
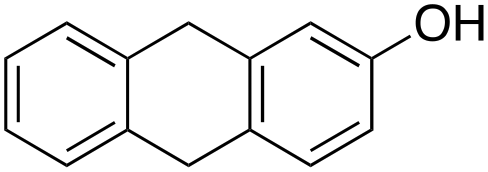
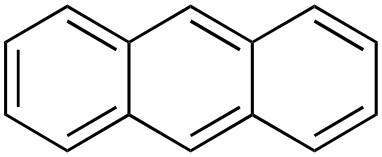
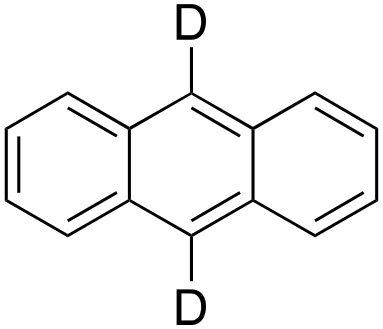
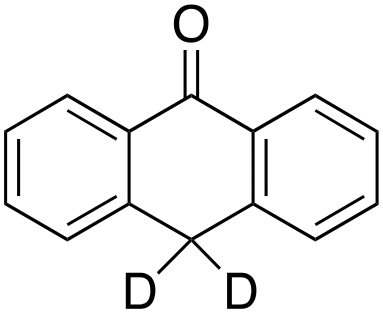
Product analysis of the oxidation of 9,10-dihydroanthracene (DHA) showed formation of the aromatic oxidation products, 1-hydroxydihydroanthracene (16%) and 2-hydroxydihydroanthracene (8%) (entry 5). As observed with fluorene, benzylic oxidation competes with the aromatic one leading to the formation of anthracene (13%), anthrone (1%) and anthraquinone (2%). Remarkably, the product yields and selectivity for hydroxylation under argon were almost the same as those under air, showing that the possibility of participation by molecular oxygen in air can be excluded. Again, aromatic hydroxylation prevails over the benzylic oxidation, but a lower ring/side-chain chemoselectivity is observed (60:40). Control experiments showed that no oxidation of 9,10-dihydroanthracene occurs when the reaction is performed either in the absence of the oxidant or of the catalyst, while using Fe(OTf)2 and H2O2 in the absence of the imine ligand, side-chain oxidation products are exclusively formed in relatively high yields: anthracene (43%), anthrone (30%) and anthraquinone (9%). The increase of HAT reactivity of the benzylic positions in the Fenton type reaction can be again rationalized on the basis of the relatively low benzylic BDEC–H value of DHA (78 kcal mol−1). Indeed, HAT from 9,10-dihydroanthracene to FeIVO complexes is usually facile and fast to eventually provide anthracene or anthrone.38,39,41,64
In the case of 9,10-dihydroanthracene, the competition between aromatic and side-chain oxidation and the mechanism of the hydrogen atom transfer from the benzylic C–H bonds have been investigated in more detail. Formation of the oxidation products of DHA with 1/H2O2 system may be described according to the mechanism reported in Scheme 3. In the metal-based electrophilic aromatic substitution (path a), the first step involves the electrophilic attack of the putative iron(V)–oxo active species on the aromatic ring to give the Wheland complex. In the next step, the aromaticity is restored by deprotonation, leading to the formation of 1- and 2-hydroxydihydroanthracene and the iron(III) catalyst.
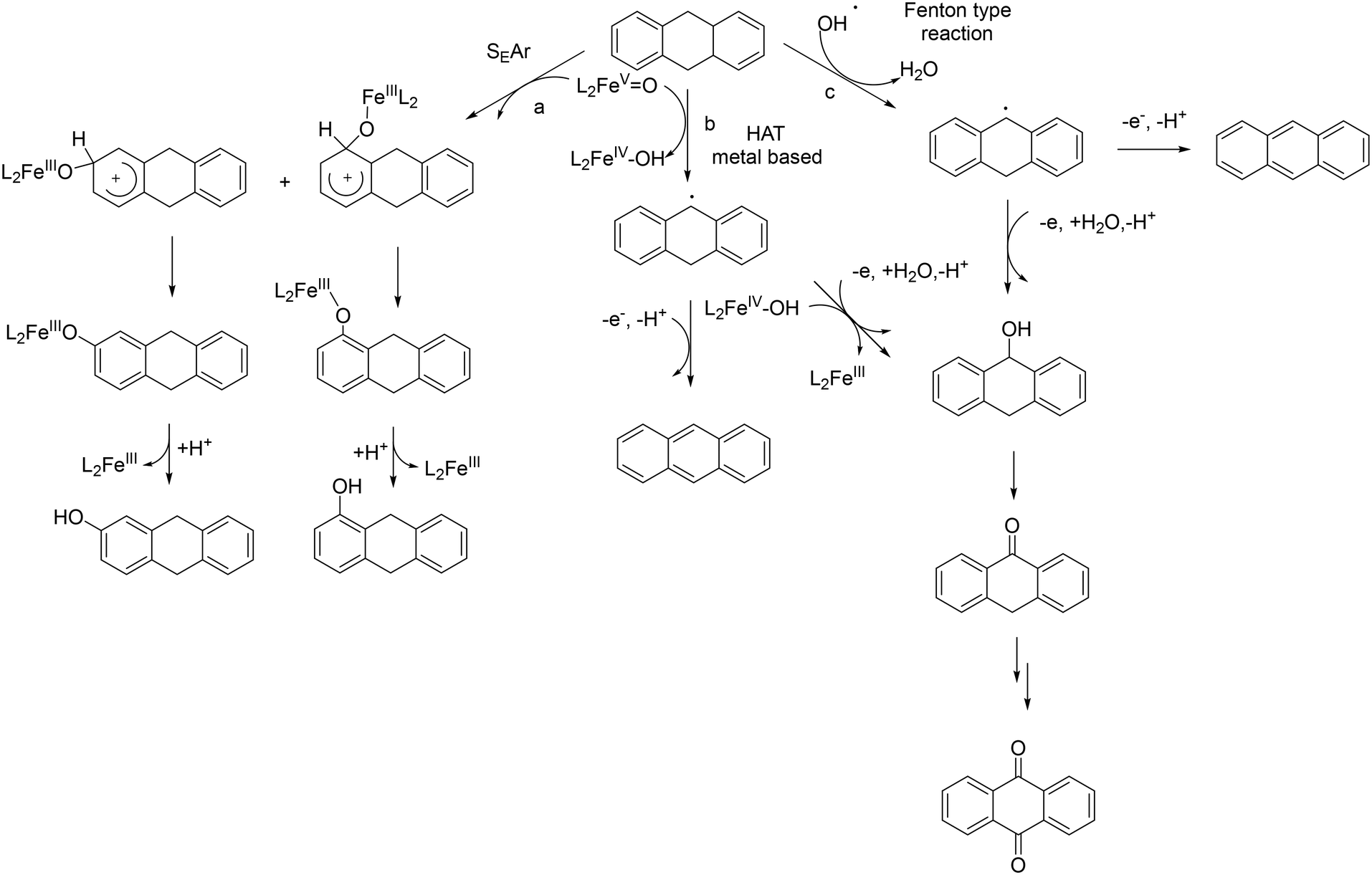 | ||
| Scheme 3 Proposed mechanism for DHA oxidation by the 1/H2O2 system. | ||
Concerning the side-chain oxidation process, in principle, two possible mechanistic pathways are feasible: a metal-based HAT process promoted by a high-valent iron–oxo species (path b) or a HAT process promoted by oxygen-centered radicals (HO˙) formed through Fenton-type reactions of the iron complex with H2O2 (path c).65
In the HAT metal-based process, initial hydrogen atom abstraction from the substrate to the iron(V)–oxo complex leads to the formation of an Fe(IV)–OH complex and a benzylic radical which, in turn, can produce i) anthracene by a desaturation process or ii) 9-hydroxy-9,10-dihydroanthracene by reaction with the Fe(IV)–OH complex (oxygen rebound) or by oxidation and reaction with a water molecule in a non-rebound process.66,67 9-Hydroxy-9,10-dihydroanthracene can be further oxidized to anthrone and anthraquinone. Alternatively, a Fenton-type reaction promoted by the highly reactive and rather unselective HO˙ (or HOO˙) can occur.51,62,68–70 This species can abstract a hydrogen atom from the benzylic C–H bond of the substrate leading to the formation of a benzylic radical, which can be easily oxidized to a cation. The latter can react with H2O leading to 9-hydroxy-9,10-dihydroanthracene which then follows the pathways described above.
In order to gain more insight into this reaction mechanism, a product isotope effect study has been carried out. Product analysis of the oxidation of DHA and its deuterated counterpart DHA-d4 by 1/H2O2 system has been compared. While the aromatic oxidation occurs with the same rate constants kAr, benzylic oxidation is characterized by a rate constant, kHBenz or kDBenz, for DHA or DHA-d4, respectively, as shown in Scheme 4.
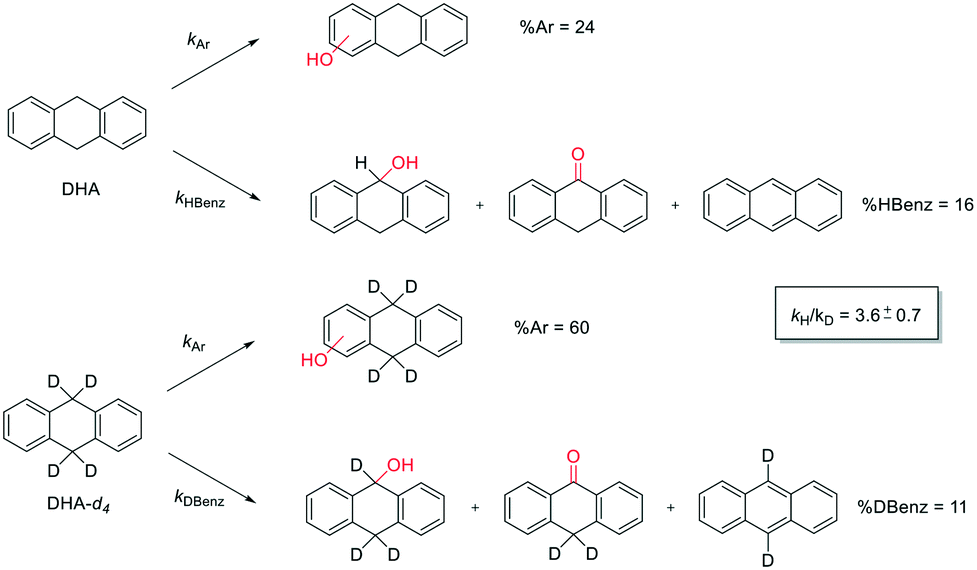 | ||
| Scheme 4 Aromatic and benzylic product distribution in the oxidation of DHA and DHA-d4 by the 1/H2O2 system. | ||
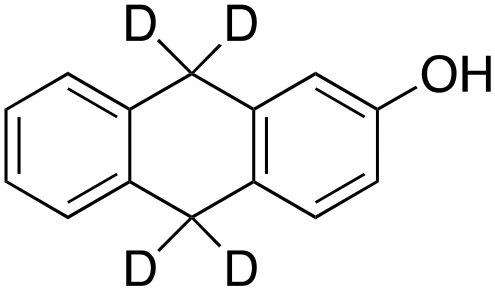
According to Scheme 4, the ratio of the overall yields of benzylic (%Benz) and aromatic (%Ar) oxidation products, (%Benz)/(%Ar), can be considered equal to the ratio of the corresponding rate constants, kHBenz/kAr and kDBenz/kAr, for DHA and DHA-d4, respectively. Under the reasonable assumption that the kAr values are the same for both DHA and DHA-d4, from the ratios (%Hbenz)/(%HAr) and (%Dbenz)/(%DAr), it is possible to determine the kH/kD value for the benzylic HAT process. From the product yields of the oxidation of DHA and DHA-d4 reported in Table 1, entries 5 and 6, respectively, a KIE (kH/kD) value of 3.6 ± 0.7 is obtained.
This result rules out a Fenton type HAT process but supports the involvement of a selective metal-based oxidant that is able to discriminate between the C−H and C−D bonds. This KIE value is also in accordance with that previously observed in cyclohexane hydroxylation catalyzed by complex 1 (KIE = 3.3).33 In fact, reactions initiated by highly reactive oxygen-centered radicals, such as hydroxyl radicals, are generally characterized by kH/kD values between 1 and 2.35,61,71–73 A value of 3.6 for kH/kD is instead in line with those obtained with other non-heme amine-based iron complexes reported in the literature (KIE values in the range of 3–4) for which the involvement of a FeVO species has been proposed,40,44,45 but lower than that reported for characterized FeIVO species.38,39,41,64 Furthermore, when the oxidation of DHA was performed under argon, no significant variation of the reaction selectivity was observed. The poor sensitivity to the presence/absence of O2 also does not support the operation of a free radical-based oxidation since the presence of O2 is known to affect both the efficiency and the selectivity of oxidative processes with Fenton-type systems.35,58,72,74 Thus, the results obtained with the non-heme imine-based iron complex clearly indicate that both aromatic and aliphatic side-chain oxidations are performed by the same high valent iron–oxo species, similar to that observed in non-heme PDP iron complexes.30,45
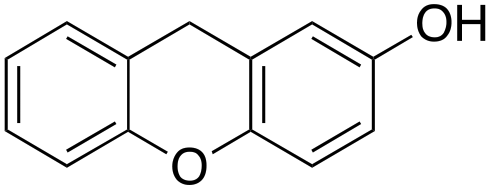
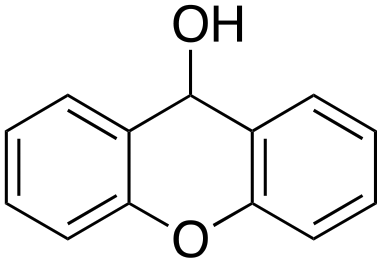
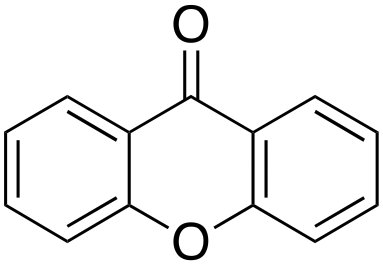
Eventually, xanthene oxidation was considered. This substrate has the lowest benzylic BDEC–H value (75 kcal mol−1) in the series of Table 1. The oxidation of xanthene led to the formation of 2-hydroxyxanthene (4%) as the only phenolic product, accompanied by larger amounts of 9-hydroxyxanthene (5%) and xanthone (29%), which are the products of benzylic oxidation (entry 7). The ring/side-chain chemoselectivity observed for this substrate (33:64) is in accordance with the most reactive benzylic C–H bond. Blank experiments in the absence of the imine ligand show the exclusive formation of benzylic oxidation products, 9-hydroxyxanthene (1%) and 9-xanthone (65%).
In order to confirm that the HAT from the benzylic C–H bonds occurs via the metal-based process, also with this substrate, a KIE study has been carried out (see Scheme 5). Products and yields of the oxidation of xanthene and xanthene-d2 are reported in Table 1, entries 7 and 8, respectively. From the product yields, a KIE value of 3.3 ± 0.7 is obtained confirming that the benzylic HAT process from xanthene is promoted by a metal-based oxidant.
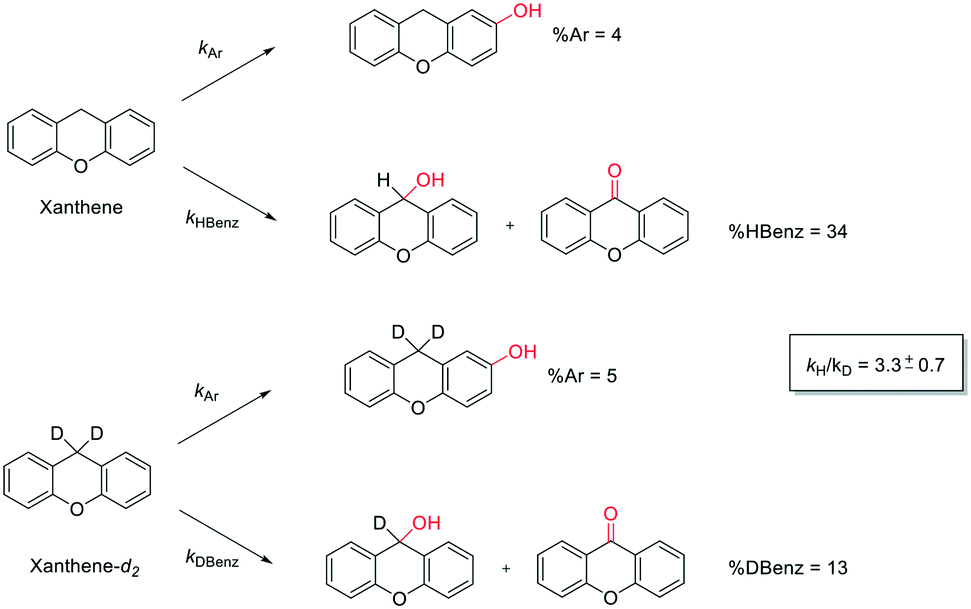 | ||
| Scheme 5 Aromatic and benzylic product distribution in the oxidation of xanthene and xanthene-d2 by the 1/H2O2 system. | ||
Table 2 summarizes the aromatic vs. side-chain chemoselectivity together with the benzylic BDEC–H values of the alkylaromatic substrates. The data reported clearly indicate that a high selectivity for aromatic hydroxylation is observed for most alkylaromatic substrates with benzylic BDEC–H higher than 82 kcal mol−1. This implies that the oxidation will selectively occur on the aromatic ring for most benzylic substrates. A fair correlation between the selectivity data and the strength of the benzylic C–H bonds is outlined by the ring/side-chain chemoselectivity, which regularly decreases on decreasing benzylic BDEC–H values (Fig. 1). With xanthene, the low benzylic BDEC–H value (75 kcal mol−1) determines an inversion of selectivity with benzylic oxidation prevailing over the aromatic one.
| Substrate | BDE (kcal mol−1) | Ring/side-chain chemoselectivity |
|---|---|---|
| Toluene | 88 | >98:2 |
| Ethylbenzene | 84 | 94:6 |
| Diphenylmethane | 82 | 98:2 |
| Fluorene | 80 | 66:34 |
| 9,10-Dihydroanthracene | 78 | 60:40 |
| Xanthene | 75 | 33:67 |
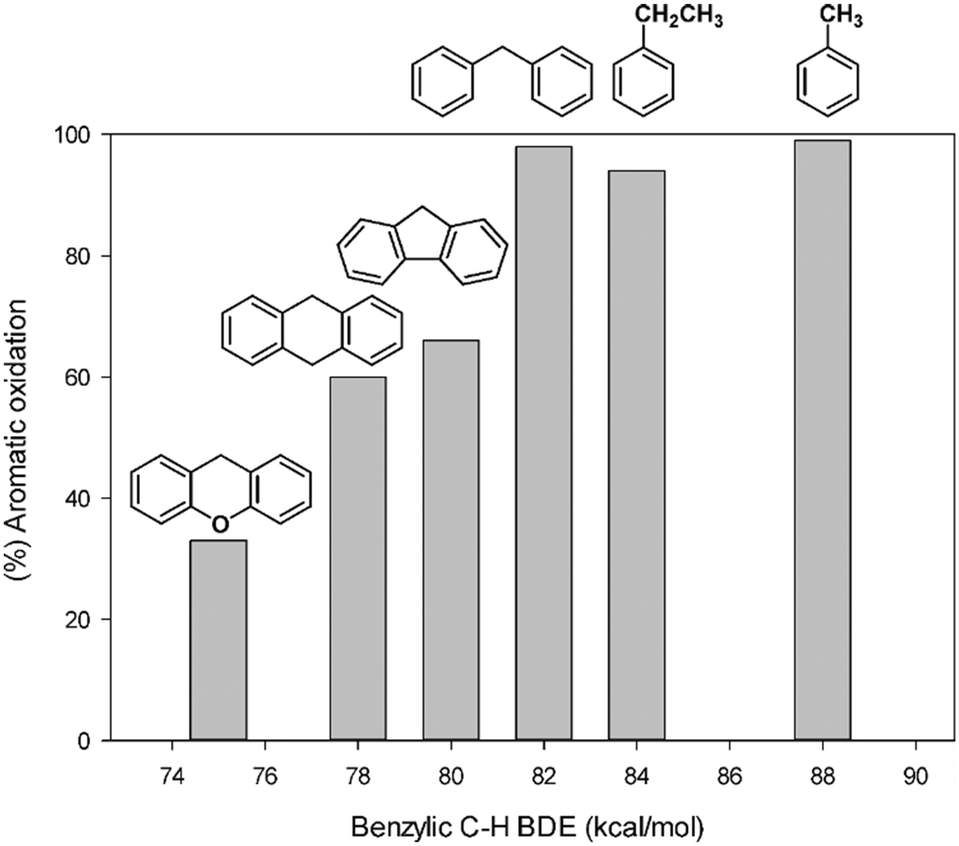 | ||
| Fig. 1 Percentage of aromatic oxidation as a function of benzylic BDEC–H of alkylaromatics in the oxidation promoted by the 1/H2O2 system. | ||
This result is in qualitative accordance with the general correlation between the amount of side-chain oxygenated products and the strength of aliphatic C–H bonds reported in several iron-catalyzed alkylbenzene oxidations.11–24,39,64 However, the 1/H2O2 system shows a remarkably high chemoselectivity for aromatic hydroxylation, higher than that found with most of the catalytic systems reported in related studies.20,47,55–57
Conclusions
Imine-based iron complex 1 prepared in situ by reaction of 2-picolylaldehyde, 2-picolylamine, and Fe(OTf)2 efficiently catalyzes the oxidation of alkylaromatic compounds with H2O2 as oxidant under mild conditions. A remarkable chemoselectivity for the aromatic oxidation is observed with monocyclic aromatic systems. With more activated polycyclic substrates, the side-chain/ring oxygenated product ratio regularly increases on decreasing bond dissociation energy (BDE) of the benzylic C–H bond in line with expectation. An inversion of chemoselectivity is found with xanthene where benzylic oxidation products are more abundant than phenolic ones. Clear evidence for metal-based competitive reactions leading either to aromatic hydroxylation (electrophilic aromatic substitution) or side-chain oxidation (benzylic hydrogen atom abstraction) has been provided by the analysis of deuterium kinetic isotope effects and the poor sensitivity of the product yields to the presence/absence of O2.Experimental
Oxidation procedure
Oxidations were carried out by mixing Fe(CF3SO3)2(CH3CN)2 (1.09 mg, 2.50 μmol), 2-picolylamine (50 μL of a 0.1 M solution in CH3CN, 5.0 μmol) and 2-picolylaldehyde (50 μL of a 0.1 M solution in CH3CN, 5.0 μmol) in a vial at 25 °C. Substrate (250 μmol) and CH3CN were then added up to a total volume of 1 mL. A solution of H2O2 in CH3CN (1.74 M diluted from a 35% w/w H2O2 solution) was added over 30 minutes using a syringe pump under vigorous stirring and left reacting for additional 1 hour. Finally, an internal standard was added and the reaction mixture was filtered over a short pad of SiO2 with 10 mL of AcOEt and analyzed by GC, GC–MS and 1H NMR. Oxidation products were identified by comparing with authentic specimens (o-cresol, m-cresol, p-cresol, benzaldehyde, 2-ethylphenol, 3-ethylphenol, 4-ethylphenol, 1-phenylethanol, acetophenone, 2-benzylphenol, 3-benzylphenol, 4-benzylphenol, benzophenone, 2-hydroxyfluorene, 9-fluorenone, anthracene, anthrone, anthraquinone, and 9-xanthone) or by comparing of their spectral data with those reported in the literature (2-ethyl-p-benzoquinone,75 4-hydroxyfluorene,76 2-xanthenol,9 9-hydroxyxanthene77).1-Hydroxydihydroanthracene and 2-hydroxydihydroanthracene, were isolated from the crude reaction mixture of an oxidation reaction carried out on a semipreparative scale by column chromatography (see ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministero dell'Università e della Ricerca for financial support. This work was also partially supported by the Università di Roma “La Sapienza” (Progetti di Ricerca 2018). We also thank the CIRCC, Interuniversity Consortium of Chemical Catalysis and Reactivity.Notes and references
- S. Fukuzumi and K. Ohkubo, Asian J. Org. Chem., 2015, 4, 836–845 CrossRef CAS.
- J. Genovino, D. Sames, L. G. Hamann and B. B. Tour, Angew. Chem., Int. Ed., 2016, 55, 14218–14238 CrossRef CAS.
- S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105–107 CrossRef CAS.
- J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026–16031 CrossRef.
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321–323 CrossRef CAS.
- B. Lücke, K. V. Narayana, A. Martin and K. Jähnisch, Adv. Synth. Catal., 2004, 346, 1407–1424 CrossRef.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS.
- K. Kamata, T. Yamaura and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7275–7278 CrossRef CAS.
- T. Tsuji, A. A. Zaoputra, Y. Hitomi, K. Mieda, T. Ogura, Y. Shiota, K. Yoshizawa, H. Sato and M. Kodera, Angew. Chem., Int. Ed., 2017, 56, 7779–7782 CrossRef CAS.
- M. P. Jensen, M. P. Mehn and L. Que Jr., Angew. Chem., Int. Ed., 2003, 42, 4357–4360 CrossRef CAS.
- O. V. Makhlynets, P. Das, S. Taktak, M. Flook, R. Mas-Ballesté, E. V. Rybak-Akimova and L. Que Jr., Chem. – Eur. J., 2009, 15, 13171–13180 CrossRef CAS.
- A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem. – Eur. J., 2012, 18, 2715–2724 CrossRef CAS.
- L. Carneiro and A. R. Silva, Catal. Sci. Technol., 2016, 6, 8166–8176 RSC.
- E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2008, 14, 7123–7126 CAS.
- J.-N. Rebilly, W. Zhang, C. Herrero, H. Dridi, K. Sénéchal-David, R. Guillot and F. Banse, Chem. – Eur. J., 2020, 26, 659–668 CrossRef CAS.
- O. V. Makhlynets and E. V. Rybak-Akimova, Chem. – Eur. J., 2010, 16, 13995–14006 CrossRef CAS.
- S. Taktak, M. Flook, B. M. Foxman, B. L. Que Jr. and E. V. Rybak-Akimova, Chem. Commun., 2005, 5301–5303 RSC.
- S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS.
- A. Raba, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2014, 50, 11454–11457 RSC.
- A. C. Lindhorst, S. Haslinger and F. E. Kühn, Chem. Commun., 2015, 51, 17193–17212 RSC.
- P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC.
- N. V. Tkachenko, R. V. Ottenbacher, O. Y. Lyakin, A. M. Zima, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, ChemCatChem, 2018, 10, 4052–4057 CrossRef CAS.
- O. Y. Lyakin, A. M. Zima, N. V. Tkachenko, E. P. Talsi and K. P. Bryliakov, ACS Catal., 2018, 8, 5255–5260 CrossRef CAS.
- R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Appl. Organomet. Chem., 2020, 34, e5900 CrossRef CAS.
- J.-F. Bartoli, V. Mouries-Mansuy, K. Le Barch-Ozette, M. Palacio, P. Battioni and D. Mansuy, Chem. Commun., 2000, 827–828 RSC.
- M.-J. Kang, W. J. Song, A.-R. Han, Y. S. Choi, H. G. Jang and W. Nam, J. Org. Chem., 2007, 72, 6301–6304 CrossRef CAS.
- Y. Kang, H. Chen, Y. J. Jeong, W. Lai, E. H. Bae, S. Shaik and W. Nam, Chem. – Eur. J., 2009, 15, 10039–10046 CrossRef CAS.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS.
- M. Asaka and H. Fujii, J. Am. Chem. Soc., 2016, 138, 8048–8051 CrossRef CAS.
- D. Kumar, G. N. Sastry and S. P. de Visser, J. Phys. Chem. B, 2012, 116, 718–730 CrossRef CAS.
- G. Olivo, G. Arancio, L. Mandolini, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2014, 4, 2900–2904 RSC.
- G. Olivo, M. Nardi, D. Vìdal, A. Barbieri, A. Lapi, L. Gómez, O. Lanzalunga, M. Costas and S. Di Stefano, Inorg. Chem., 2015, 54, 10141–10152 CrossRef CAS.
- G. Olivo, S. Giosia, A. Barbieri, O. Lanzalunga and S. Di Stefano, Org. Biomol. Chem., 2016, 14, 10630–10635 RSC.
- G. Olivo, O. Lanzalunga and S. Di Stefano, Adv. Synth. Catal., 2016, 358, 843–863 CrossRef CAS.
- B. Ticconi, A. Colcerasa, S. Di Stefano, O. Lanzalunga, A. Lapi, M. Mazzonna and G. Olivo, RSC Adv., 2018, 8, 19144–19151 RSC.
- G. Capocasa, G. Olivo, A. Barbieri, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2017, 7, 5677–5686 RSC.
- L. Que Jr., Acc. Chem. Res., 2007, 40, 493–500 CrossRef.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS.
- W. N. Oloo and L. Que Jr., Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS.
- K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS.
- G. Olivo, O. Cussó, M. Borrell and M. Costas, J. Biol. Inorg. Chem., 2017, 22, 425–452 CrossRef CAS.
- S. Kal, S. Xu and L. Que, Angew. Chem., Int. Ed., 2019, 59, 7332–7349 CrossRef.
- S. Kal, A. Draksharapu and L. Que, J. Am. Chem. Soc., 2018, 140, 5798–5804 CrossRef CAS.
- G. C. Silva, N. M. F. Carvalho, A. Horn Jr., E. R. Lachter and O. A. C. Antunes, J. Mol. Catal. A: Chem., 2017, 426, 564–571 CrossRef CAS.
- M. Bianchi, M. Bertoli, R. Tassinari, M. Ricci and R. Vignola, J. Mol. Catal. A: Chem., 2003, 200, 111–116 CrossRef.
- M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856–2863 RSC.
- K. Hirose, K. Ohkubo and S. Fukuzumi, Chem. – Eur. J., 2016, 22, 12904–12909 CrossRef CAS.
- A. Kunai, S. Hata, S. Ito and K. Sasaki, J. Am. Chem. Soc., 1986, 108, 6012–6016 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS.
- G. Capocasa, M. Di Berto Mancini, F. Frateloreto, O. Lanzalunga, G. Olivo and S. Di Stefano, Eur. J. Org. Chem., 2020, 23, 3390–3397 CrossRef.
- A. Kejriwal, P. Bandyopadhyay and A. N. Biswas, Dalton Trans., 2015, 44, 17261–17267 RSC.
- V. Balland, D. Mathieu, N. Pons-Y-Moll, J. F. Bartoli, F. Banse, P. Battioni, J.-J. Girerd and D. Mansuy, J. Mol. Catal. A: Chem., 2004, 215, 81–87 CrossRef CAS.
- A. Thibon, J. F. Bartoli, R. Guillot, J. Sainton, M. Martinho, D. Mansuy and F. Banse, J. Mol. Catal. A: Chem., 2008, 287, 115–120 CrossRef CAS.
- R. R. Fernandes, M. V. Kirillova, J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2009, 353, 107–112 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, Florida, USA, 2007 Search PubMed.
- Oxidation of toluene and ethylbenzene carried out using stoichiometric amounts or an excess of oxidant (ref. 37) showed a significant increase of turnover number (TN) accompanied by formation of overoxidation products. For example, in the oxidation of toluene, TN observed are 2.6 and 33 using 0.2 equiv. and 2.5 equiv. of H2O2, respectively. 2-Methyl-1,4-benzoquinone is only observed using an excess of oxidant (3% with respect to the initial amount of substrate, see ref. 37).
- C. Walling, Acc. Chem. Res., 1998, 31, 155–157 CrossRef CAS.
- T. Kurata, Y. Watanabe, M. Katoh and Y. Sawaki, J. Am. Chem. Soc., 1988, 110, 7472–7478 CrossRef CAS.
- P. Shejwalkar, N. P. Rath and E. B. Bauer, Dalton Trans., 2011, 40, 7617–7631 RSC.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS.
- Formation of OH˙ may derive from the homolytic cleavage of the O–O bond in the intermediate iron(III)–OOH bis-imine complex. See ref. 28 and 30.
- K. Bin Cho, X. Wu, Y. M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef.
- A. Barbieri, O. Lanzalunga, A. Lapi and S. Di Stefano, J. Org. Chem., 2019, 84, 13549–13556 CrossRef CAS.
- C. Walling and R. A. Johnson, J. Am. Chem. Soc., 1975, 97, 363–367 CrossRef CAS.
- D. I. Metelitsa, Russ. Chem. Rev., 1971, 40, 563–580 CrossRef.
- A. Mitarai, K. Hikino, M. Hirama and K. Sasaki, J. Org. Chem., 1992, 57, 6937–6941 CrossRef.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- M. Costas, K. Chen and L. Que Jr, Coord. Chem. Rev., 2000, 200-202, 517–544 CrossRef CAS.
- A. F. Trotman-Dickenson, Adv. Free Radical Chem., 1965, 1, 1–38 CAS.
- K. H. Mitchell, C. E. Rogge, T. Gierahn and B. G. Fox, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3784–3789 CrossRef CAS.
- B. E. Love, B. C. Duffy and A. L. Simmons, Tetrahedron Lett., 2014, 55, 1994–1997 CrossRef CAS.
- M. Lukeman and P. Wan, J. Am. Chem. Soc., 2002, 124, 9458–9464 CrossRef CAS.
- K. Okuma, A. Nojima, N. Matsunaga and K. Shioji, Org. Lett., 2009, 11, 169–171 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Instruments and general methods, selected GC chromatograms and 1HNMR spectra of some oxidation reactions. See DOI: 10.1039/d0cy01868f |
| ‡ Current address: Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Spain. |
| This journal is © The Royal Society of Chemistry 2021 |