
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh toughness fully physical cross-linked double network organohydrogels for strain sensors with anti-freezing and anti-fatigue properties†
Li
Tang‡
 a,
Shaoji
Wu‡
a,
Yue
Xu
a,
Ting
Cui
a,
Yuhua
Li
b,
Wu
Wang
a,
Liang
Gong
*a and
Jianxin
Tang
*a
a,
Shaoji
Wu‡
a,
Yue
Xu
a,
Ting
Cui
a,
Yuhua
Li
b,
Wu
Wang
a,
Liang
Gong
*a and
Jianxin
Tang
*a
aHunan Key Laboratory of Biomedical Nanomaterials and Devices, College of Life Sciences and Chemistry, Hunan University of Technology, Zhuzhou 412007, China. E-mail: jxtang0733@163.com
bCollege of Packaging and Materials Engineering, Hunan University of Technology, Zhuzhou 412007, China
First published on 20th August 2021
Abstract
Flexible sensors based on conductive hydrogels have been of wide interest in the field of smart wearable electronics due to the excellent stretchability and strain-responsive ability. However, lacking harsh environment tolerance and self-recovery properties seriously limit their practical applications. Therefore, the development of anti-fatigue hydrogels with anti-freezing and water-retaining abilities is urgently required. In this study, we constructed a fully physically cross-linked gelatin/poly(N-hydroxyethyl acrylamide)/glycerin/lithium chloride double network (gelatin/pHEAA/Gly/LiCl DN) organohydrogel based on a hydrogen bond crosslinking strategy using a facial one-pot method. The dynamic hydrogen bond in the DN organohydrogels provided an effective energy dissipation pathway, which produced gels with high tensile strength/strain (2.14 MPa/1637.49%), fast self-recovery properties and strong interfacial toughness. The introduction of binary solvents of water and glycerin endowed the DN organohydrogels with excellent anti-freezing and water-retaining properties. Furthermore, a simple flexible sensor was fabricated based on the organohydrogel for detecting human motions. The sensor not only showed remarkable sensitivity (GF = 14.54), broad strain range (0–1600%) and high response speed (0.2 s), but also presented accurate and reliable signals under different mechanical deformations and low temperature (−20 °C). This work provides a feasible way to build high mechanical and sensing performance organohydrogel-based sensors with anti-freezing and water-retaining abilities, which greatly promotes the application of flexible sensors in the field of smart wearable electronic devices, electronic skin and human/machine interface.
Introduction
With the rapid development of smart wearable electronics, flexible sensors have been widely applied in human motion monitoring,1–4 healthcare diagnosis5–7 and post-operative observation.8,9 The key points in making flexible sensors are how to design and synthesize conductive and stretchable materials, which can convert applied stress or strain into electrical signals. Conductive elastomers are typical flexible sensors which can efficiently convert tensile strain into electrical signals.10–14 However, the poor tensile ability (2–300%) of elastomers doesn’t meet the demand of flexible wearable sensors.15 On the contrary, conductive hydrogels show obvious advantages in the application of flexible wearable sensors due to their large stretchability (>1000%), ionic conductivity, high water-retaining property and high biocompatibility.The flexible wearable sensors constructed using conductive hydrogels need to be able to sense repeatedly and stably under various deformations (stretching, compression and bending) in practical applications. Generally, conventional chemically cross-linked hydrogels have low resilience and poor shape recovery capacity due to the irreversibility of covalent bonds. To meet the challenge, introduction of reversible dynamic bonds, including hydrogen bonds,16,17 electrostatic interactions,18,19 hydrophobic associations20,21 or other physically cross-linked methods22,23 into networks to construct physically–chemically hybrid hydrogels or fully physically cross-linked hydrogels is a good choice. Unfortunately, only a few physically cross-linked hydrogels have been reported that have high mechanical properties.24–33
In addition, under extreme conditions, traditional conductive hydrogels will be dehydrated or frozen, leading to a significant reduction in stretchability and conductivity. To solve this problem, enhancing anti-freezing and water-retention properties of conductive hydrogels is a highly effective strategy. The most common strategies are the introduction of inorganic salts34,35 or organic solvents36,37 into hydrogels to inhibit evaporation of water or formation of ice crystals. For example, Zhang and co-workers prepared zwitterionic hydrogels by soaking hydrogels in zwitterionic solution to replace the water in the hydrogel. The resultant hydrogel showed excellent ionic conductivity (∼2.7 S m−1) at −40 °C, and maintained good flexibility in a wide temperature range (−40 °C to 25 °C).38 Nevertheless, too much salt in a hydrogel can lead to undesirable mechanical properties, which is not conducive to practical application of flexible hydrogel sensors. Wu and co-workers synthesized organohydrogels with anti-freezing and water-retention capabilities by using a solvent replacement strategy to introduce ethylene glycol and glycerol into κ-carrageenan/PAAm hydrogels.39 The organohydrogel exhibited good stability and repeatability for strain signals in the range of 0.5–50% at −18 to 25 °C. However, new problems arose, as the introduction of organic solvent reduced the ionic conductivity of the gel, which led to the obtained organohydrogel sensor exhibiting low sensitivity (GF = 4.5). Hydrogels with anti-freezing and water-retention capabilities always possess disadvantages, including low toughness, poor sensitivity and time-consuming solvent replacement steps. Therefore, it is particularly necessary to design anti-freezing and water-retaining hydrogels with simple preparation steps, and high mechanical and sensing properties.
Herein, we successfully prepared an anti-freezing, water-retaining, anti-fatigue and self-adhesive ultra-stretchable flexible sensor based on gelatin/poly(N-hydroxyethyl acrylamide)/glycerin/lithium chloride double network (gelatin/pHEAA/Gly/LiCl DN) organohydrogels. Gelatin is a soluble protein derived from skin, cartilage or connective tissue which has excellent biocompatibility.40–42 Moreover, gelatin has thermally reversible gel–sol transformation properties between 25 °C and 60 °C, which allows it to change from liquid to solid macroscopically. Therefore, gelatin was chosen to construct the first rigid network of DN hydrogels. HEAA is a monomer containing hydrophilic functional groups –OH, –NH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which is able to form a soft network spontaneously through hydrogen bond interactions without a chemical crosslinker under photopolymerization. Besides, glycerin and LiCl were doped to provide abundant hydrogen bonds and electrostatic interactions to reconstruct the original pure hydrogen bond network, and they also endowed the gels with anti-freezing and water-retaining capacities. The resultant organohydrogel exhibited outstanding mechanical properties (tensile stress of 2.14 MPa, failure strain of 1637.49%), fast self-recovery (recovery efficiency with 78.59% after 5 min resting at room temperature without any external stimuli), high interface toughness on hogskin (∼256 J m−2), repeatable adhesion on Ti (reversible interface toughness of ∼200 J m−2), remarkable sensitivity (GF = 14.54 at strain of 1500%) and excellent anti-freezing and water-retaining abilities. Hence, this work provides a feasible strategy for constructing high performance flexible sensors with anti-freezing and water-retaining abilities, which greatly promotes the application of flexible sensors in the field of wearable electronic devices.
O, which is able to form a soft network spontaneously through hydrogen bond interactions without a chemical crosslinker under photopolymerization. Besides, glycerin and LiCl were doped to provide abundant hydrogen bonds and electrostatic interactions to reconstruct the original pure hydrogen bond network, and they also endowed the gels with anti-freezing and water-retaining capacities. The resultant organohydrogel exhibited outstanding mechanical properties (tensile stress of 2.14 MPa, failure strain of 1637.49%), fast self-recovery (recovery efficiency with 78.59% after 5 min resting at room temperature without any external stimuli), high interface toughness on hogskin (∼256 J m−2), repeatable adhesion on Ti (reversible interface toughness of ∼200 J m−2), remarkable sensitivity (GF = 14.54 at strain of 1500%) and excellent anti-freezing and water-retaining abilities. Hence, this work provides a feasible strategy for constructing high performance flexible sensors with anti-freezing and water-retaining abilities, which greatly promotes the application of flexible sensors in the field of wearable electronic devices.
Results and discussion
Fabrication of the gelatin/pHEAA/Gly/LiCl DN organohydrogels
The fabrication process of gelatin/pHEAA/Gly/LiCl DN organohydrogels was schematically illustrated in Fig. 1. In short, all reactants (including gelatin, HEAA, glycerin, LiCl and I2959) were mixed together in deionized water/glycerin solution. The mixed solution was stirred at 60 °C until gelatin was completely dissolved. After that, it was gradually cooled to 25 °C so that gelatin converted from an irregular curled state to a triple helix state to form the first physically crosslinked gelatin network. Then, the second physically crosslinked pHEAA network was formed by hydrogen bonds between the inter- and intropolymer chain after photopolymerization under UV light. The fully physically crosslinked structure provided an efficient energy dissipation pathway and provided the hydrogel with high mechanical and fast self-recovery properties. During synthesis, LiCl was dissolved in the mixed solution and confined in the gel network to provide excellent electrical conductivity. Glycerin contained abundant hydroxyl groups, which not only inhibited the formation of ice crystals, but also formed hydrogen bonds with the hydrogel network to improve the mechanical properties of the hydrogel. | ||
| Fig. 1 Schematic of the synthesis of gelatin/pHEAA/Gly/LiCl DN organohydrogels. | ||
The organohydrogels also exhibited excellent plasticity and toughness. They could be injected into various molds to form desired 3D shapes, such as a HUT (abbreviation of Hunan University of Technology) logo (Fig. 2a), could also bear crossover stretching without breaking (Fig. 2b) and easily lift up to 500 g of weight (Fig. 2c). More importantly, it could withstand knotting and stretching to 12 times the original length at −10 °C without breaking (Fig. 2d).
 | ||
| Fig. 2 The DN organohydrogel can (a) adapt different shapes by injecting into a mold, (b) bear crossover stretching, (c) lift 500 g of weight and (d) withstand knotting and stretching up to 12 times its original length at −10 °C. | ||
High mechanical performance of gelatin/pHEAA/Gly/LiCl DN organohydrogels
In our previous studies, the gelatin/HEAA hydrogel formed by 10 wt% gelatin and 50 wt% HEAA exhibited the best mechanical performance,43 so the ratio of gelatin to HEAA was fixed (the ratio was used in subsequent experiments, and abbreviated as g10H50, where g means gelatin and H means HEAA) to explore the influences of LiCl concentration and water/glycerin ratios on the mechanical performance of gelatin/pHEAA/Gly/LiCl DN organohydrogels. A series of gelatin/pHEAA/Gly/LiCl DN organohydrogels (abbreviated as g10H50Gx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y-Lz, where G means glycerin, L means LiCl, and x and y respectively represent the proportion of water and glycerin in the solvent, and z represents the concentration of LiCl) have been prepared and tested by varying the concentrations of LiCl from 1 wt% to 7 wt% and ratios of water/glycerin from 3:1 to 1:1 (Table 1).
:y-Lz, where G means glycerin, L means LiCl, and x and y respectively represent the proportion of water and glycerin in the solvent, and z represents the concentration of LiCl) have been prepared and tested by varying the concentrations of LiCl from 1 wt% to 7 wt% and ratios of water/glycerin from 3:1 to 1:1 (Table 1).
| Sample | Water (wt%) | Glycerin (wt%) | LiCl (wt%) | Tensile stress (MPa) | Tensile strain (%) |
|---|---|---|---|---|---|
| g10H50G1:1-L1 |
19.5 | 19.5 | 1 | 1.95 | 1007.05 |
| g10H50G1:1-L3 |
18.5 | 18.5 | 3 | 2.00 | 1236.72 |
| g10H50G1:1-L5 |
17.5 | 17.5 | 5 | 2.14 | 1637.49 |
| g10H50G1:1-L7 |
16.5 | 16.5 | 7 | 1.06 | 1442.32 |
| g10H50G2:1-L5 |
23.33 | 11.67 | 5 | 1.62 | 1331.16 |
| g10H50G3:1-L5 |
26.25 | 8.75 | 5 | 1.61 | 1141.93 |
The influence of LiCl on the mechanical properties of gelatin/pHEAA/Gly/LiCl DN organohydrogels was determined using a tensile test, as shown in Fig. 3a. It can be seen that with the increase of LiCl concentration from 1 wt% to 5 wt%, the tensile stress of the gelatin/pHEAA/Gly/LiCl DN organohydrogels increased from 1.95 MPa to 2.14 MPa (the corresponding tensile strain increased from 1007.05% to 1637.49%), and toughness increased from 9.85 MJ m−3 to 13.7 MJ m−3 (Fig. 3b). The results indicated that the introduction of a small amount of LiCl would reduce the crosslinking density of hydrogen bonds by enhancing electrostatic interactions, and improved the stretching ability and toughness of the organohydrogels. It's worth noting that the introduction of a large amount of LiCl will severely destroy the original hydrogen bond network. When the concentration of LiCl was 7 wt%, the mechanical properties of the organohydrogels were significantly reduced.4
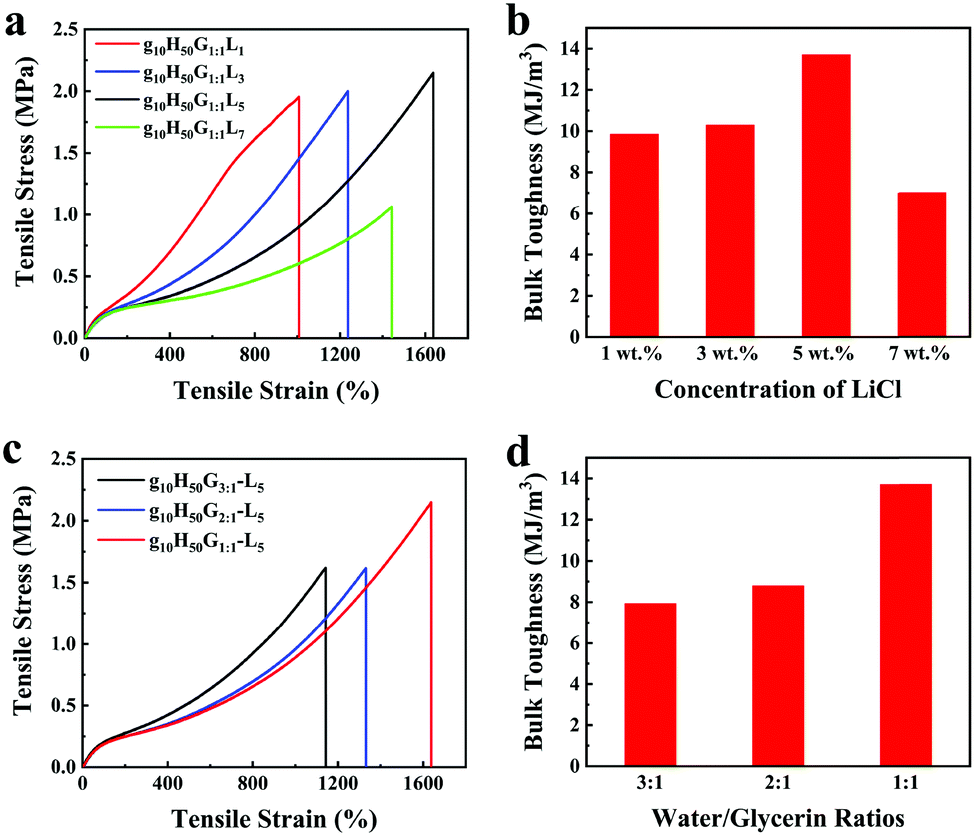 | ||
| Fig. 3 The influence of (a and b) LiCl concentration and (c and d) water/glycerin ratio on the mechanical properties and bulk toughness of gelatin/pHEAA/Gly/LiCl DN organohydrogels, respectively. | ||
Furthermore, the influence of water/glycerin ratios on the mechanical properties of organohydrogels was investigated. The tensile stress, tensile strain and toughness of the gelatin/pHEAA/Gly/LiCl DN organohydrogels increased from 1.61 MPa to 2.14 MPa, 1141.93% to 1637.49%, and 7.93 MJ m−3 to 13.7 MJ m−3 with an increasing proportion of glycerin (Fig. 3c and d), respectively. This could be attributed to the plasticizing effect of glycerol, which was conducive to improving the amount of stretching and toughness of the organohydrogels.44 In summary, after testing the influences of LiCl concentration and water/glycerin ratios, it is found that g10H50G1:1-L5 could attain a high tensile strength of 2.14 MPa, a tensile strain of 1637.49% and bulk toughness of 13.7 J m−3. Unless otherwise stated, g10H50G1:1-L5 was used for the following tests.
Hysteresis, energy dissipation, and self-recovery performance of gelatin/pHEAA/Gly/LiCl DN organohydrogels
Herein, the energy dissipation mode of the organohydrogel was explored by using loading–unloading tests. The same organohydrogel was subjected to successive loading–unloading tests (Fig. 4a), and there was no rest time between any two successive tests. The organohydrogel underwent fifteen loading–unloading cycles as the strain increased from 100% to 1500%, and it broke at the sixteenth cycle. Obviously, the dissipated energy of the organohydrogel increased regularly from 0.07 MJ m−3 to 3.22 MJ m−3 with a continuous increase in the number of cycles (Fig. 4b). At the same time, it can be seen that even if the strain interval between successive loading–unloading curves was 200%, the adjacent hysteresis loops still partially overlapped, indicating that the organohydrogels possess good self-recovery performance.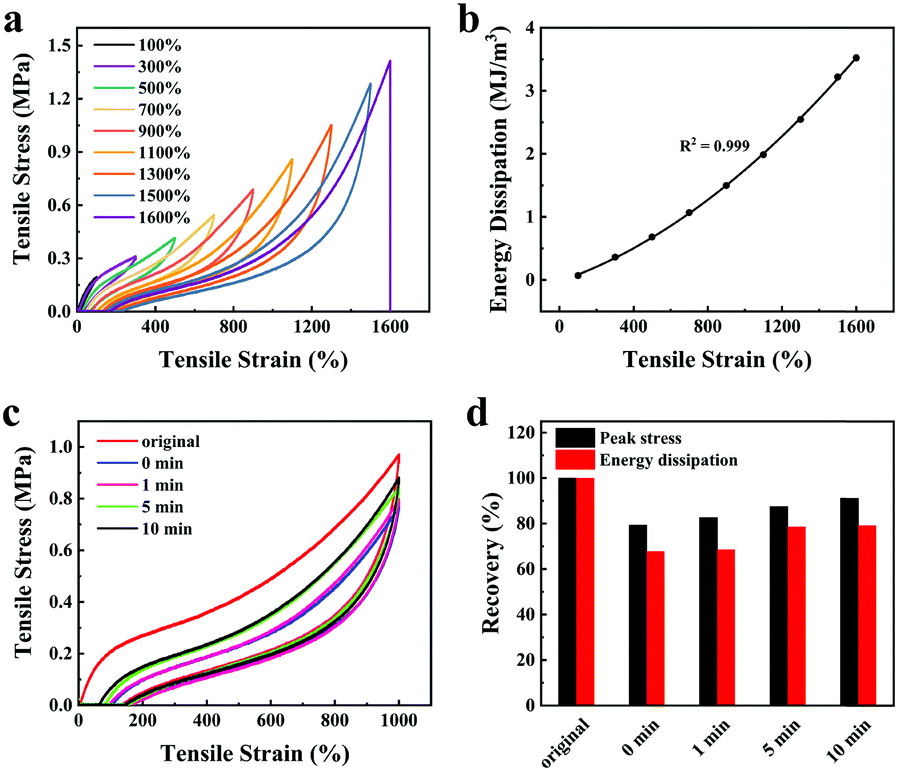 | ||
| Fig. 4 (a) Loading–unloading curves and (b) the corresponding energy dissipation of the same organohydrogel at 100–1600% strains. (c) The self-recovery ability of the organohydrogel estimated by four successive loading–unloading cycles with a strain of 1000%, and the rest time was fixed at 0 min, 1 min, 5 min and 10 min, respectively. (d) Quantitative analysis of peak stress and energy dissipation recovery ratios, which were calculated from the loading–unloading curves in (c). | ||
Therefore, we explored the influence of rest time (i.e. 0, 1, 5, and 10 min) on the self-recovery efficiency of the organohydrogel. Fig. 4c showed the loading–unloading curves of the organohydrogel at different rest times. Under different rest times, the self-recovery efficiency of the organohydrogel peak stress and energy dissipation were quantitatively analyzed to obtain Fig. 4d. As the rest time increased, the peak stress recovery efficiency of the organohydrogel increased from 79.28% to 91.17%, and the energy dissipation recovery increased from 67.75% to 79.21%. Moreover, the self-recovery efficiency of energy dissipation from 0 to 5 min was much higher than that from 5 to 10 min, which meant that the dynamic hydrogen bonds in the network have a much higher reconstructed speed within 5 min. The high self-recovery efficiency endowed the organohydrogel with a better anti-fatigue ability than other hydrogels under the same conditions.
Interfacial toughness of gelatin/pHEAA/Gly/LiCl DN organohydrogels
Adhesiveness on various surfaces is very significant for flexible hydrogel sensors to achieve interface bonding, which is beneficial in improving their portability and comfort. Repeatable adhesion is useful as it enables sensors to be peeled and bound at different parts many times and also prolongs the service life of sensors. Therefore, the interfacial toughness of gelatin/pHEAA/Gly/LiCl DN organohydrogels was investigated.First, the interfacial toughness of gelatin/pHEAA/Gly/LiCl DN organohydrogels was simply investigated by visual inspection through direct contact with different solid surfaces like glass, foam, ceramic, steel, paper, rubber and human skin (Fig. 5a).
 | ||
| Fig. 5 (a) The gelatin/pHEAA/Gly/LiCl DN organohydrogel can directly adhere to various solid surfaces without any surface modification. The interfacial toughness of the gelatin/pHEAA/Gly/LiCl DN organohydrogel on a glass substrate as a function of (b) LiCl concentration and (c) water/glycerin ratio. (d) Peeling force/width curves of g10H50G1:1-L7 organohydrogels on Al, Ti, steel, ceramic, hogskin, PET, glass and PTFE substrates at a peeling rate of 100 mm min−1. (e) Reversible interfacial toughness of the g10H50G1:1-L7 organohydrogel by 10 adhesion/peeling cycles on a Ti substrate at a peeling rate of 100 mm min−1. | ||
Second, quantitative analysis of interfacial toughness was further carried out by using a 90° peeling test at a peeling rate of 100 mm min−1 on a negatively charged glass substrate. The results of the influence of LiCl concentration on the interfacial toughness of organohydrogels were shown in Fig. 5b. Gradually increasing the concentration of LiCl in a low concentration range (from 1 wt% to 5 wt%) caused the interfacial toughness of organohydrogels to reduce slightly. This could be attributed to the fact that the electrostatic interactions of LiCl reduced the number of hydrogen bonds in the system, resulting in the decrease of interface toughness. At higher LiCl concentration (7 wt%), the interfacial toughness of organohydrogels has rebounded. This was because the high concentration of LiCl provided a stronger electrostatic effect, which allowed organohydrogels to adhere more firmly to the negatively charged glass substrate.45Fig. 5c showed that increasing the concentration of polyhydroxyl glycerin could improve the interface toughness of the organohydrogel. The results indicated that glycerin is helpful in the formation of hydrogen bonds between organohydrogel and the surface of substrates.
Third, the interfacial toughness of organohydrogels on various substrates was further examined using the same peeling test method. As shown in Fig. 5d, the interface toughness of the organohydrogel on hydrophilic surfaces (aluminum (Al), titanium (Ti), steel, ceramic, glass, polyethylene terephthalate (PET) and hogskin) was about 256–470 J m−2, which meant that the organohydrogel strongly adhered to various hydrophilic substrates. In addition, it can be seen that the surface of the hogskin sample was slightly pulled up when the organohydrogel was peeled from it, and no hydrogel fragments were left on its surface after the peeling test (Movie S1, ESI†). However, the interfacial toughness of organohydrogels on hydrophobic polytetrafluoroethylene (PTFE) was only 77 J m−2. This phenomenon was due to the fact that the formation of hydrogen bonds was related to the hydrophilicity of substrates.46 On the surface of hydrophilic substrates, a large number of oxide-rich groups exist, which was beneficial to the formation of strong hydrogen bonds and results in high interface toughness. Conversely, was difficult for the hydrophobic substrate to form hydrogen bonds and it presented weaker interface toughness.
Finally, as the gel–substrate interface was mainly adhered by hydrogen bonds, it can be predicted that the gelatin/pHEAA/Gly/LiCl DN organohydrogel may be able to repeatedly adhere to the hydrophilic surface of substrates. Therefore, 10 adhesion/peeling cycles of the organohydrogel were performed on the Ti substrate to estimate the reproducible adhesion. As shown in Fig. 5e, the interface toughness of the gel–substrate interface was about 470 J m−2 at the first peeling cycle. In subsequent cycles, the interface toughness was stable at about 200 J m−2, and there was no major fluctuation with the increase in number of peelings. These results indicated that the hydroxyl, amino and amide groups of the organohydrogel were easily combined with the oxide-rich groups of the Ti substrate to form hydrogen bonds. The dynamic reversibility of hydrogen bonds allowed the interface toughness to be quickly restored.
Anti-freezing and water-retaining performance of the gelatin/pHEAA/Gly/LiCl organohydrogels
As we all know, traditional hydrogels will freeze at sub-zero temperatures and lose water in dry environments, resulting in a decrease in the conductivity and flexibility of the hydrogel, which severely limit the practical application of flexible hydrogel sensors. In this work, glycerin was incorporated into a hydrogel to enhance its anti-freezing and water-retaining capabilities. Both the gelatin/pHEAA hydrogel (g10H50) and the gelatin/pHEAA/Gly/LiCl organohydrogel (g10H50G1:1-L5) were twisted and stretched after being kept at −40 °C for 30 min. As shown in Fig. 6a, the g10H50 hydrogel broke when it was twisted, but the g10H50G1:1-L5 organohydrogel could not only be twisted but also stretched, and still maintained good flexibility. Fig. 6b showed the tensile stress–strain curves of organohydrogels at −10 °C, −20 °C, −30 °C and −40 °C, respectively. It can be seen that with the decrease of temperature, the tensile stress of organohydrogels sharply increased from 2.82 MPa to 13.19 MPa, and the tensile strain decreased from 1174.46% to 130.01%. This was due to the formation of ice crystals in organohydrogels at low temperatures, which had a nano-toughening effect on the organohydrogels and toughened them.47 It was evident that the organohydrogels maintained good flexibility even at extremely harsh low temperatures. At the same time, the DSC curve of the organohydrogel from −100 °C to 10 °C further proved that the organohydrogels had an excellent anti-freezing ability (Fig. S1, ESI†).
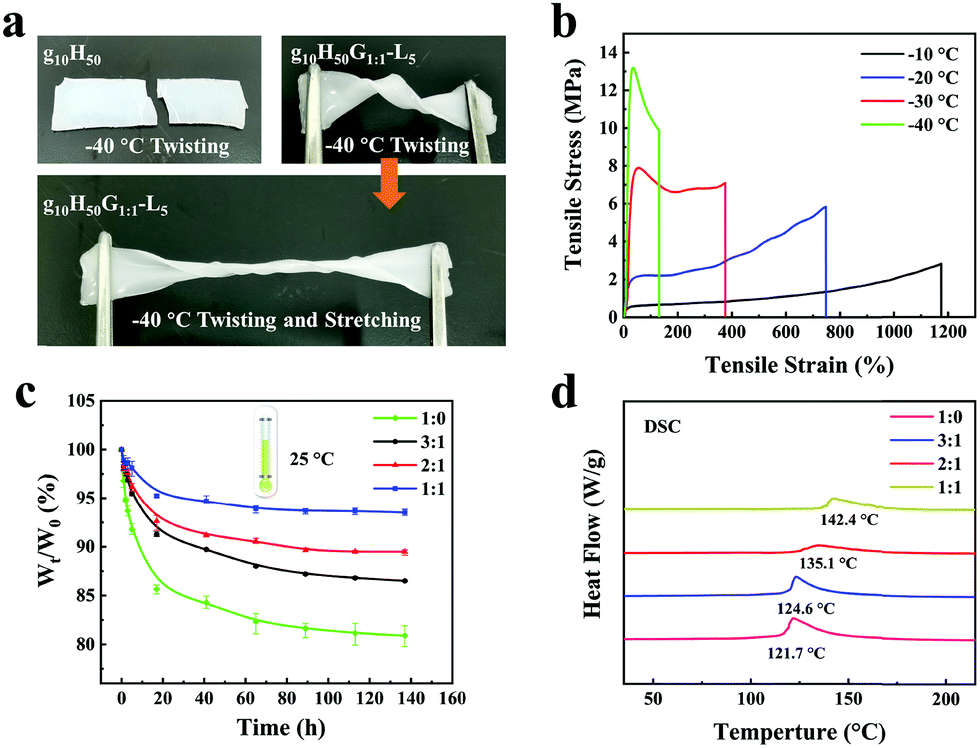 | ||
| Fig. 6 (a) Twisting and stretching of the g10H50 hydrogel and the g10H50G1:1-L5 organohydrogel at −40 °C. (b) Tensile stress–strain curves of organohydrogels at −10 °C, −20 °C, −30 °C and −40 °C, respectively. (c) The weight changes of gels with different water/glycerin ratios under long-term storage at 25 °C and 40%RH. (d) DSC curves of gelatin/pHEAA/Gly/LiCl gels with different water/glycerin ratios from 30 °C to 220 °C. | ||
Water-retaining performances of the organohydrogels with different water/glycerin ratios were monitored and the results were illustrated in Fig. 6c. The obtained organohydrogels were stored in a dry environment (25 °C and 40%RH) for different amounts of time. When the water/glycerin ratio changed from 1:0 to 1:1, the mass retention ratio of the gels increased from 81.45% to 93.53% after storing for 135 h. On the one hand, glycerin had a lot of hydroxyl groups, which could convert part of the free water in gels to bound water and improved the water-retaining ability. On the other hand, glycerin had a strong water absorption capacity, which was conducive to inhibit the loss of water.
Fig. 6d showed the DSC curves of the gelatin/pHEAA/Gly/LiCl gels with different water/glycerin ratios from 30 °C to 220 °C. It can be seen that with the change in water/glycerin ratio from 1:0 to 1:1, the endothermic peak of the gels shifted from 121.7 °C to 142.4 °C. This indicated that an increase in the proportion of glycerin is helpful to enhance the boiling point of the solvent in the gels and to improve the water-retaining ability of gels.
Conductivity and sensing performance of gelatin/pHEAA/Gly/LiCl organohydrogels for pressure/strain sensors
In order to study the conductivity of gelatin/pHEAA/Gly/LiCl DN organohydrogels, the conductivity of organohydrogels with different water/glycerin ratios was measured using linear sweep voltammetry at a voltage from −0.5 V to 0.5 V at 10 °C. The current and voltage of all organohydrogels showed a linear correlation (Fig. S2, ESI†), indicating that the organohydrogel was an ohmic conductor, which conformed to Ohm's law like metal. Meanwhile, the effect of temperature on the conductivity of the organohydrogel was not negligible. With the increase of temperature, the conductivity of the organohydrogel also increased and exhibited an extremely strong linear relationship (Fig. S3, ESI†). This result also meant that the organohydrogel could be used in the field of temperature monitoring. In addition, the water/glycerin ratio also had a remarkable influence on the conductivity. As the water/glycerin ratio changed from 1:0 to 1:1, the conductivity of organohydrogels decreased from 0.13 S m−1 to 0.01 S m−1 (Fig. S4, ESI†). This could be attributed to the fact that the high viscosity of glycerin reduced the migration rate of ions in the organohydrogel. The organohydrogels were then used as a conductor to light up a light-emitting diode (LED) with a constant voltage of 9 V (Fig. 7a). The conductivity of organohydrogels in different states (bending, origin and stretching) was also measured (Fig. 7b–d). The LED could be lit in all the above mechanical states. When the organohydrogel was stretched, the LED became dark, indicating that the strain of the organohydrogel has a positive correlation with the resistance. Macroscopically, the cross-sectional area of organohydrogels became narrow and the length became longer, which restricted the movement of ions and resulted in an increase of resistance. In addition, the organohydrogel also demonstrated a repairable electrical conductivity. As the organohydrogel recovered through self-healing, the conductivity of the organohydrogel could be restored to the original level in 2.2 s as shown in Fig. 7e.
 | ||
| Fig. 7 (a) Schematic diagram of a circuit using the gelatin/pHEAA/Gly/LiCl DN organohydrogel as a conductor with a constant voltage of 9 V and an LED. The LED lighting conditions of organohydrogels in a state of (b) bending, (c) original length, and (d) stretching. (e) The disconnected organohydrogel can re-light an LED through self-healing without external stimulation. | ||
Taking advantage of the good ionic conductive, anti-freezing and water-retaining abilities of the organohydrogel, organohydrogel-based flexible sensors were prepared to resist extreme weather conditions. Fig. 8a showed the changes of relative resistance of the organohydrogel-based pressure sensor under different compression strain. It can be seen that the gauge factor (GF) values were 3.6 and 0.71 under the compression strain range of 0–5% and 5–50%, respectively. The sensing performance of the pressure sensors was also tested by adding different weights (Fig. 8b). With an increase in weight from 0 to 100 g, the changes in the relative resistance of the sensors decreased from 100% to 78%. Furthermore, the organohydrogel-based pressure sensor was used as a handwriting touchpad. Fig. 8c showed a continuous signal that was obtained by handwriting “OK”. It was proved that the organohydrogel-based pressure sensor had an excellent pressure response ability.
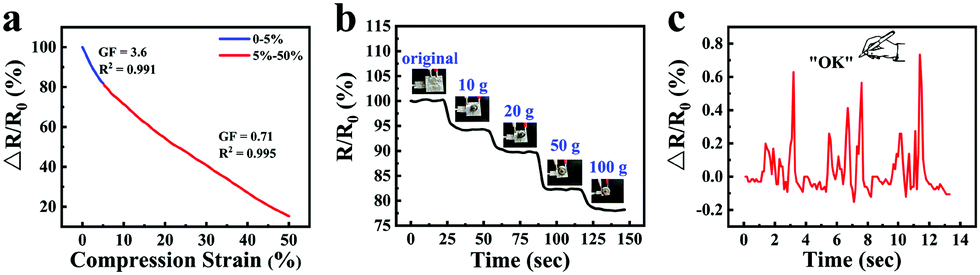 | ||
| Fig. 8 The changes in the relative resistance of organohydrogel-based pressure sensors as (a) the compression strain increases from 0 to 50% and (b) the weight increases from 0 to 100 g. (c) The organohydrogel-based pressure sensor was used for handwriting “OK”. | ||
The sensitivity of the strain sensor based on organohydrogels was also assessed. The GF values were 1.33, 2.94, 4.18, 6.66 and 14.54 in the tensile strain range of 0–100%, 100–300%, 300–1100%, 1100–1500% and 1500–1600% (Fig. 9a), respectively, which were much higher than mostly previously reported hydrogel-based strain sensors (Table 2). Particularly, the rapid and obvious stability signals were successfully obtained in the process of stretching and unloading under an extremely small strain of 1% (Fig. 9b). To demonstrate the stability of organohydrogel-based strain sensors, the signals under small strain (10–50%) and large strain (100–500%) were also collected, as shown in Fig. 9c and d, respectively. Additionally, the response time of the electrical signal and the exact time of applied 10% tensile strain was simultaneously recorded at the frequency of 0.5 Hz (Fig. 9e). The results showed that the response of the electrical signal almost synchronously changed with the applied strain, and the changes in relative resistance time was only a 0.2 s delay compared with the stretching time. It was proven that organohydrogel-based strain sensors could immediately respond to changes in external strain.
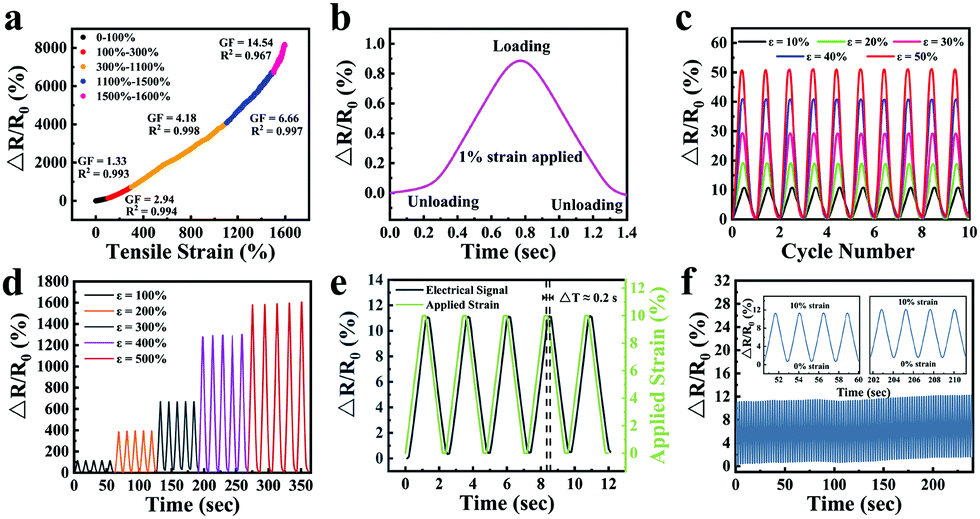 | ||
| Fig. 9 (a) Dynamic stretching sensitivity of the gelatin/pHEAA/Gly/LiCl DN organohydrogel-based strain sensor. (b) The change in the relative resistance of the organohydrogel-based strain sensor during the loading–unloading cycles with a strain of 1%. The changes in the relative resistance of repeated stretching tests of the organohydrogel-based strain sensor at (c) small strain (10%, 20%, 30%, 40% and 50%) and (d) large strains (100%, 200%, 300% and 400%). (e) Time-resolved responses of the change in the relative resistance of the organohydrogel-based strain sensor upon 0–10% strains. (f) The change in the relative resistance of the organohydrogel-based strain sensor during 100 loading–unloading cycles of 10% strain. | ||
| Composition | Maximal strain (%) | Maximal strength (kPa) | Gauge factor | Sensing ranges (%) | Anti-freezing | Ref. |
|---|---|---|---|---|---|---|
| Note: “—” indicates “not available” in the references. | ||||||
| Gelatin/pHEAA/Gly/LiCl | 1637 | 2140 | 14.54 | 0–1600 | Yes | This work |
| PAA/CNC/AlCl3 | 1600 | 350 | 4.9 | 0–80 | No | 48 |
| PAAm/PDA/KCl | 1000 | 25 | 0.7 | 0–1000 | No | 49 |
| Cellulose/NaCl | 236 | 50 | 0.29 | 0–230 | Yes | 50 |
| PAAm/Casein/LiCl | 1465 | 170 | 0.4 | 0–100 | Yes | 51 |
| PSBMA/PVA | 400 | 600 | 1.5 | 0–300 | No | 52 |
| PHEA/SA/KCl/CaCl2 | 410 | 200 | 1.87 | 0–200 | No | 53 |
| PAAm/PDA/Casein | 2100 | 160 | <4 | 0–300 | No | 54 |
| PAAm/PAA/CS/NaCl/FeCl3 | 1400 | 2750 | 3.62 | 0–500 | No | 55 |
| PAAm/PAA/NaCl/FeCl3 | 1360 | 469 | 3.18 | 0–500 | Yes | 56 |
| PAA/GO/FeCl3 | 630 | 400 | 1.32 | 0–500 | No | 57 |
| PAAm/PSBMA/NaCl | 1150 | 600 | <1 | 0–700 | No | 58 |
| PAAm/Alginate/CaCl2 | 1700 | 375 | 0.3 | 20–800 | No | 59 |
| Agar/Alginate/PAAm/CaCl2 | 250 | 500 | 3.83 | 0–200 | No | 60 |
| PVA/Alginate/PAAm/CaCl2 | 959 | 512 | <3 | 0–300 | No | 61 |
| PEG/PAMAA/FeCl3 | 1800 | 460 | — | 0–800 | No | 62 |
| Eg/Gl/PAAm/KCl | 950 | — | 6 | 0.5–400 | Yes | 39 |
| Poly(LysMA-co-AAm)/LiCl | 2422 | 60 | — | — | Yes | 63 |
| PVP/PVA/FeCl3 | 1160 | 2100 | 0.478 | — | No | 64 |
| PAA/GO/CaCl2 | — | — | <1 | 0–500 | No | 65 |
| PAAm/PDMS/LiCl | — | — | 0.84 | 0–40 | Yes | 66 |
In order to study the stability and repeatability of the strain sensor, the change of relative resistance was recorded using 100 successive loading–unloading cycles with 10% strain. As shown in Fig. 9f, the maximum relative resistance was maintained almost unchanged during the cyclic deformations. Four cycles of early and late periods were compared (Fig. 9f inset). The deviation of the maximum relative resistance was less than 1%, which meant the strain sensor showed high repeatability. The reason for this phenomenon was that the all-physical cross-linking strategy endowed the organohydrogel-based strain sensors with excellent self-recovery and anti-fatigue properties. Furthermore, the organohydrogel-based strain sensors had a good water-retaining ability, which could reduce the evaporation of water and further prevent the unstable performance of organohydrogel-based strain sensors caused by drying.
Motion monitoring performance of the gelatin/pHEAA/Gly/LiCl organohydrogel strain sensors at room temperature and −20 °C
In order to visually show the potential application of gelatin/pHEAA/Gly/LiCl DN organohydrogels as strain sensors for monitoring human movement and physiological activities, the organohydrogel-based strain sensor was pasted on the elbow, wrist, knee and finger, which were regularly bent, respectively. Fig. 10a–c showed the changes of the relative resistance of the elbow, wrist and knee, respectively. It can be seen that the signal curves of different bending parts were obviously different and highly identifiable. In addition, signals were recorded with finger bending angles of 30°, 60° and 90°, which were held for a certain period of time (Fig. 10d). The detected constant relative resistances were 10%, 20% and 33% respectively, and the relative resistance could return to the original state after the angle of the finger returned to 0°. Moreover, the organohydrogel-based strain sensor could also capture the vibrations of human vocal cords and facial movements (Fig. 10e and f). These results showed that the organohydrogel-based strain sensor was fully qualified for monitoring human sports and physiological activities at room temperature.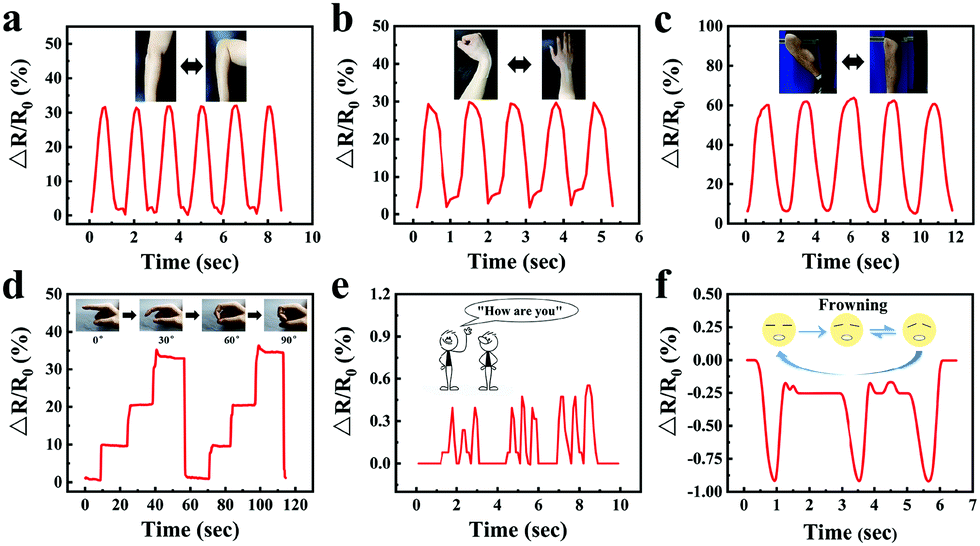 | ||
| Fig. 10 The gelatin/pHEAA/Gly/LiCl DN organohydrogel-based strain sensor was used to monitor the movement of an (a) elbow, (b) wrist, (c) knee and (d) finger. The changes in the relative resistance when (e) saying “How are you” and (f) frowning. | ||
Compared with a traditional conductive hydrogel, the gelatin/pHEAA/Gly/LiCl DN organohydrogel had better conductivity and flexibility in an extremely wide temperature range. As shown in Fig. 11a, the organohydrogel could still light an LED even after being treated at room temperature, −20 °C and −80 °C for 30 min, respectively, indicating that the organohydrogel still had conductivity at extremely low temperature. Furthermore, the sensing performance of human motion at −20 °C was conducted (Fig. 11b–e). Obviously, the changes of relative resistance at low temperature were higher than that at room temperature. This showed that the organohydrogel-based strain sensor had greater sensitivity at low temperature, because the stiffness of the organohydrogel increased at low temperature, leading to greater changes in the conducting path of the organohydrogel under the same strain. In any case, the results showed that the gelatin/pHEAA/Gly/LiCl organohydrogel can detect human motion in an extremely wide range, and it has great application potential in smart wearable electronic devices.
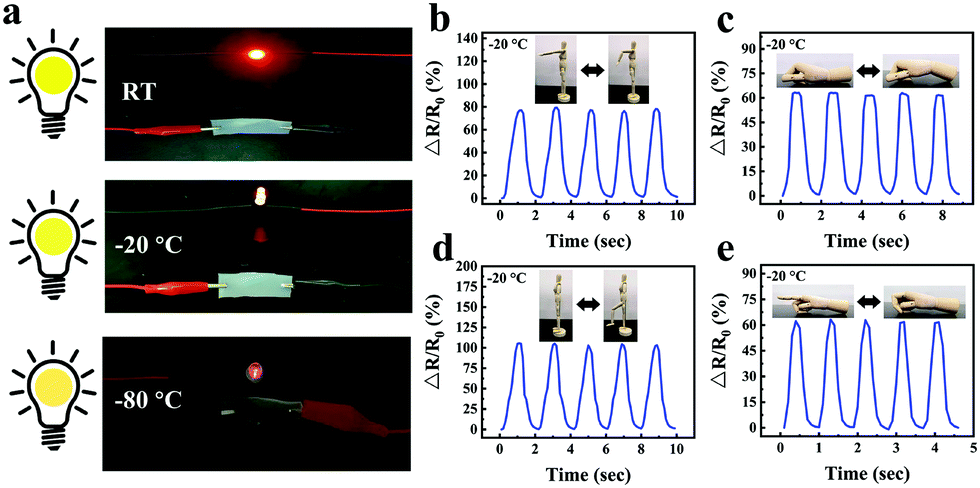 | ||
| Fig. 11 (a) The LED lighting conditions of the gelatin/pHEAA/Gly/LiCl organohydrogels at room temperature, −20 °C and −80 °C. (b) The changes in the relative resistance of the gelatin/pHEAA/Gly/LiCl DN organohydrogel strain sensor were used to monitor the movement of an (b) elbow, (c) wrist, (d) knee and (e) finger at −20 °C. | ||
Experimental
Materials
Gelatin from hogskin (type A and gel strength ∼300 g bloom), glycerin (≥99%), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I2959, ≥98%), and LiCl·H2O (reagent grade, >95%) were purchased from Aladdin. N-Hydroxyethyl acrylamide (HEAA, >98%) was purchased from TCI China. Deionized water used in this study was purified via a water purification system.Gelatin/pHEAA/Gly/LiCl DN organohydrogel preparation
A gelatin/pHEAA/Gly/LiCl DN organohydrogel was prepared via a simple one-pot heating–cooling-photopolymerization method as previously reported.43 Firstly, 10 wt% gelatin, 50 wt% HEAA, I2959 (1 mol% of HEAA), 5 wt% LiCl and 35 wt% water/glycerin (1:1, g/g) mixed solution were added into a glass container. The glass container was heated at 60 °C until all the reactants completely dissolved to form a clear and transparent solution. Then, the solution was poured into a glass mold and left to cool at 25 °C. In the cooling process, gelatin molecules changed from an irregular curled state to a triple helix state, and formed the first physically cross-linked gelatin network. After that, the mold was exposed to 365 nm UV light with a power of 8 W for 1 hour, and HEAA molecules were polymerized to form pHEAA chains. The second physically cross-linked pHEAA network was formed under hydrogen bond interactions between pHEAA chains. Finally, the organohydrogel was taken out from the mold for performance tests.
Tensile test
Tensile tests were carried out on a universal tester (AGS-X) equipped with 1000 N load cell at a speed of 100 mm min−1. The gel samples were cut into dumbbell shapes with a gauge length of 30 mm, width of 4 mm and thickness of 1 mm. Tensile strain (ε) was the length of stretching (Δl) of the sample divided by the initial length (l0), multiplied by 100% (ε = Δl/l0 × 100%). Tensile stress (σ) was the applied force (F) divided by the initial cross-sectional area (A0) of the sample (σ = F/A0). For tensile cycle tests, samples were stretched to a preset strain, and then unloaded to 0 strain at the same rate of 100 mm min−1. Dissipated energy was estimated according to the area between the loading and unloading cycle.Interfacial toughness measurement
In order to form stable interfacial toughness between the organohydrogel and non-porous substrates, the surface of solid substrates were washed with acetone, ethanol and deionized water in turn for 30 minutes in an ultrasonic instrument, and then dried in an oven. Furthermore, organohydrogels were synthesized on a clean substrate with a silica gel gasket (80 mm × 15 mm × 3 mm). To allow it to be easily removed from the mould, transparent PET film was used to cover the mold. The subsequent method of preparing organohydrogels was the same as mentioned above. Then, according to the test requirements, the samples were tested with a standard 90° peeling test at a peeling rate of 100 mm min−1. The interfacial toughness (g) was calculated by g = Fmax/w, where Fmax is the maximum force during the peeling process and w is the width of the tested organohydrogel.Tensile test at subzero temperature
The tensile stress and tensile strain of organohydrogels below zero temperature were tested by a universal tester (Zwick Roell Z010) with an environmental chamber. Before stretching, each sample was kept at the test temperature for 20 minutes to ensure that the temperature of the sample was the same as that of the external environment. The stretching rate was also controlled at 100 mm min−1.Differential scanning calorimetry analysis
Differential scanning calorimetry (DSC) tests were performed using NETZSCH DSC 200F3 at a heating rate of 10 °C min−1 in a set temperature range under nitrogen protection.Determination of water retention capacity
The water-retaining capacity of the organohydrogels was evaluated by measuring their weight loss in an environment with a relative humidity of 40% at 25 °C for a long time.Measurement of conductivity
The conductivity of organohydrogels was measured by using the linear sweep voltammetry of an electrochemical workstation (CHI660D, China), and the conductivity (σ) can be calculated as σ = Δl/(R × S), where Δl represents the length of the organohydrogel, R is the bulk resistance, and S is the cross-sectional area of the organohydrogel.Strain- and pressure-sensing tests
Both the relative resistance and applied strain of the organohydrogel sensor were monitored simultaneously using a universal tester (AGX-S) with an electrochemical workstation (CHI660D, China). The sensitivity defined as the gauge factor (GF) can be estimated as GF = (ΔR/R0)/ε, where ΔR is the change in resistance with strain, R0 is the resistance of the organohydrogel at the original length, and ε is the applied strain.Conclusions
In summary, we synthesized a full physically cross-linked gelatin/pHEAA/Gly/LiCl double network organohydrogel via a simple one-pot heating–cooling-photopolymerization method. The resultant gelatin/pHEAA/Gly/LiCl double network organohydrogels showed high mechanical properties (tensile stress of 2.14 MPa, tensile strain of 1637.49%), fast self-recovery (peak stress/energy dissipated recovery of 91.17%/79.21% after 10 min resting at room temperature) and strong interfacial toughness (256–470 J m−2). The organohydrogel also presented high flexibility even in cold (−40 °C) and drying conditions (25 °C, stored for 135 h at 40%RH). Furthermore, the extraordinary sensitivity of the gelatin/pHEAA/Gly/LiCl organohydrogel-based flexible sensor was expanded to an extremely broad strain window (0–1600%) with a high GF of 14.54 at 1500%, which allowed accurate and reliable detection of different mechanical deformations. The gelatin/pHEAA/Gly/LiCl organohydrogel-based strain/pressure sensor is expected to meet the complex application needs in practice and adapt to a variety of harsh environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 51774128), and the Scientific Research Project of Hunan Provincial Department of Education (No. 20C0585). The authors would like to thank Jialing Wu from Shiyanjia Lab (http://www.shiyanjia.com) for the DSC analysis.Notes and references
- Q. Yu, Z. Qin, F. Ji, S. Chen, S. Luo, M. Yao, X. Wu, W. Liu, X. Sun, H. Zhang, Y. Zhao, F. Yao and J. Li, Chem. Eng. J., 2021, 404, 126559 CrossRef CAS.
- Q. Zhang, X. Liu, X. Ren, F. Jia, L. Duan and G. Gao, Chem. Mater., 2019, 31, 5881–5889 CrossRef CAS.
- J. Huang, S. Peng, J. Gu, G. Chen, J. Gao, J. Zhang, L. Hou, X. Yang, X. Jiang and L. Guan, Mater. Horiz., 2020, 7, 2085–2096 RSC.
- B. Yang and W. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 16765–16775 CrossRef CAS PubMed.
- A. D. Valentine, T. A. Busbee, J. W. Boley, J. R. Raney, A. Chortos, A. Kotikian, J. D. Berrigan, M. F. Durstock and J. A. Lewis, Adv. Mater., 2017, 29, 1703817 CrossRef PubMed.
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC.
- Z. Wang, J. Chen, Y. Cong, H. Zhang, T. Xu, L. Nie and J. Fu, Chem. Mater., 2018, 30, 8062–8069 CrossRef CAS.
- G. Y. Bae, S. W. Pak, D. Kim, G. Lee, D. H. Kim, Y. Chung and K. Cho, Adv. Mater., 2016, 28, 5300–5306 CrossRef CAS PubMed.
- Y. Khan, A. E. Ostfeld, C. M. Lochner, A. Pierre and A. C. Arias, Adv. Mater., 2016, 28, 4373–4395 CrossRef CAS PubMed.
- C. Pang, G. Y. Lee, T. i. Kim, S. M. Kim, H. N. Kim, S.-H. Ahn and K.-Y. Suh, Nat. Mater., 2012, 11, 795–801 CrossRef CAS PubMed.
- S. Peng, Y. Li, L. Wu, J. Zhong, Z. Weng, L. Zheng, Z. Yang and J.-T. Miao, ACS Appl. Mater. Interfaces, 2020, 12, 6479–6488 CrossRef CAS PubMed.
- S. C. B. Mannsfeld, B. C. K. Tee, R. M. Stoltenberg, C. V. H. H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed.
- H. Liu, Y. Li, K. Dai, G. Zheng, C. Liu, C. Shen, X. Yan, J. Guo and Z. Guo, J. Mater. Chem. C, 2016, 4, 157–166 RSC.
- Z. Yang, H. Liu, S. Wu, Z. Tang, B. Guo and L. Zhang, Chem. Eng. J., 2018, 342, 112–119 CrossRef CAS.
- M. Amjadi, K.-U. Kyung, I. Park and M. Sitti, Adv. Funct. Mater., 2016, 26, 1678–1698 CrossRef CAS.
- H. Chen, Y. Liu, B. Ren, Y. Zhang, J. Ma, L. Xu, Q. Chen and J. Zheng, Adv. Funct. Mater., 2017, 27, 1703086 CrossRef.
- H. Fan, J. Wang and Z. Jin, Macromolecules, 2018, 51, 1696–1705 CrossRef CAS.
- S. C. Huang, X. X. Xia, R. X. Fan and Z. G. Qian, Chem. Mater., 2020, 32, 1937–1945 CrossRef CAS.
- H. Fan, J. Wang, Z. Tao, J. Huang, P. Rao, T. Kurokawa and J. P. Gong, Nat. Commun., 2019, 10, 5127 CrossRef PubMed.
- X. Liu, X. He, B. Yang, L. Lai, N. Chen, J. Hu and Q. Lu, Adv. Funct. Mater., 2021, 31, 2008187 CrossRef CAS.
- X. Xu, V. V. Jerca and R. Hoogenboom, Mater. Horiz., 2021, 8, 1173–1188 RSC.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- X. Du, H. Cui, Q. Zhao, J. Wang, H. Chen and Y. Wang, Research, 2019, 2019, 6398296 CAS.
- Z. Tang, Q. Chen, F. Chen, L. Zhu, S. Lu, B. Ren, Y. Zhang, J. Yang and J. Zheng, Chem. Mater., 2019, 31, 179–189 CrossRef CAS.
- H. Chen, F. Yang, R. Hu, M. Zhang, B. Ren, X. Gong, J. Ma, B. Jiang, Q. Chen and J. Zheng, J. Mater. Chem. B, 2016, 4, 5814–5824 RSC.
- J. Li, Z. Suo and J. J. Vlassak, J. Mater. Chem. B, 2014, 2, 6708–6713 CAS.
- S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 29749–29758 CrossRef CAS PubMed.
- F. Chen, Z. Tang, S. Lu, L. Zhu, Q. Wang, Q. Gang, J. Yang and Q. Chen, Polymer, 2019, 168, 159–167 CrossRef CAS.
- M. X. Wang, Y. M. Chen, Y. Gao, C. Hu, J. Hu, L. Tan and Z. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 26610–26617 CrossRef CAS PubMed.
- Y. Yang, X. Wang, F. Yang, L. Wang and D. Wu, Adv. Mater., 2018, 30, 1707071 CrossRef PubMed.
- S. Xia, S. Song and G. Gao, Chem. Eng. J., 2018, 354, 817–824 CrossRef CAS.
- X. H. Wang, F. Song, D. Qian, Y. D. He, W. C. Nie, X. L. Wang and Y. Z. Wang, Chem. Eng. J., 2018, 349, 588–594 CrossRef CAS.
- X. Li, H. Wang, D. Li, S. Long, G. Zhang and Z. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 31198–31207 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Y. Ren, G. Jin, C. Zhang, W. Chen and F. Yan, Mater. Horiz., 2020, 7, 919–927 RSC.
- X. F. Zhang, X. Ma, T. Hou, K. Guo, J. Yin, Z. Wang, L. Shu, M. He and J. Yao, Angew. Chem., Int. Ed., 2019, 58, 7366–7370 CrossRef CAS PubMed.
- D. Lou, C. Wang, Z. He, X. Sun, J. Luo and J. Li, Chem. Commun., 2019, 55, 8422–8425 RSC.
- Y. Wei, L. Xiang, H. Ou, F. Li, Y. Zhang, Y. Qian, L. Hao, J. Diao, M. Zhang, P. Zhu, Y. Liu, Y. Kuang and G. Chen, Adv. Funct. Mater., 2020, 30, 2005135 CrossRef CAS.
- X. Sui, H. Guo, P. Chen, Y. Zhu, C. Wen, Y. Gao, J. Yang, X. Zhang and L. Zhang, Adv. Funct. Mater., 2020, 30, 1907986 CrossRef CAS.
- J. Wu, Z. Wu, X. Lu, S. Han, B. R. Yang, X. Gui, K. Tao, J. Miao and C. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 9405–9414 CrossRef CAS PubMed.
- Z. L. Yu, W. C. Zeng, W. H. Zhang, X. P. Liao and B. Shi, Ultrason. Sonochem., 2016, 29, 495–501 CrossRef CAS PubMed.
- Z. He and W. Yuan, ACS Appl. Mater. Interfaces, 2021, 13, 1474–1485 CrossRef CAS PubMed.
- Z. Qin, D. Dong, M. Yao, Q. Yu, X. Sun, Q. Guo, H. Zhang, F. Yao and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 21184–21193 CrossRef CAS PubMed.
- L. Tang, D. Zhang, L. Gong, Y. Zhang, S. Xie, B. Ren, Y. Liu, F. Yang, G. Zhou and Y. Chang, Macromolecules, 2019, 52, 9512–9525 CrossRef CAS.
- A. Railanmaa, S. Lehtimäki and D. Lupo, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 459 CrossRef.
- X. Su, Y. Luo, Z. Tian, Z. Yuan, Y. Han, R. Dong, L. Xu, Y. Feng, X. Liu and J. Huang, Mater. Horiz., 2020, 7, 2651–2661 RSC.
- Y. Zhang, B. Ren, S. Xie, Y. Cai, T. Wang, Z. Feng, J. Tang, Q. Chen, J. Xu, L. Xu and J. Zheng, ACS Appl. Polym. Mater., 2019, 1, 701–713 CrossRef CAS.
- Y. Yang, Y. Yang, Y. Cao, X. Wang, Y. Chen, H. Liu, Y. Gao, J. Wang, C. Liu, W. Wang, J. K. Yu and D. Wu, Chem. Eng. J., 2021, 403, 126431 CrossRef CAS.
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- X. Jing, H. Y. Mi, Y. J. Lin, E. Enriquez, X. F. Peng and L. S. Turng, ACS Appl. Mater. Interfaces, 2018, 10, 20897–20909 CrossRef CAS PubMed.
- R. Tong, G. Chen, D. Pan, J. Tian, H. Qi, R. a. Li, F. Lu and M. He, ACS Sustainable Chem. Eng., 2019, 7, 14256–14265 CrossRef CAS.
- L. Guan, S. Yan, X. Liu, X. Li and G. Gao, J. Mater. Chem. B, 2019, 7, 5230–5236 RSC.
- Z. Wang, J. Chen, L. Wang, G. Gao, Y. Zhou, R. Wang, T. Xu, J. Yin and J. Fu, J. Mater. Chem. B, 2019, 7, 24–29 RSC.
- J. Song, S. Chen, L. Sun, Y. Guo, L. Zhang, S. Wang, H. Xuan, Q. Guan and Z. You, Adv. Mater., 2020, 32, 1906994 CrossRef CAS PubMed.
- J. Xu, G. Wang, Y. Wu, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 25613–25623 CrossRef CAS PubMed.
- J. Xu, R. Jin, X. Ren and G. Gao, J. Mater. Chem. A, 2019, 7, 25441–25448 RSC.
- S. Li, H. Pan, Y. Wang and J. Sun, J. Mater. Chem. A, 2020, 8, 3667–3675 RSC.
- X. Jing, H.-Y. Mi, X. F. Peng and L. S. Turng, Carbon, 2018, 136, 63–72 CrossRef CAS.
- B. Yang and W. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 40620–40628 CrossRef CAS PubMed.
- H. Sun, K. Zhou, Y. Yu, X. Yue, K. Dai, G. Zheng, C. Liu and C. Shen, Macromol. Mater. Eng., 2019, 304, 1900227 CrossRef CAS.
- J. Wang, Y. Liu, S. Su, J. Wei, S. E. Rahman, F. Ning, G. Christopher, W. Cong and J. Qiu, Polymers, 2019, 11, 1873 CrossRef CAS PubMed.
- S. Li, X. Bu, L. Wu, X. Ma, W. Diao, Z. Zhuang and Y. Zhou, Polym. Eng. Sci., 2019, 59, 1657–1666 CrossRef CAS.
- S. Liu, O. Oderinde, I. Hussain, F. Yao and G. Fu, Polymer, 2018, 144, 111–120 CrossRef CAS.
- Y. Gao, F. Jia and G. Gao, Chem. Eng. J., 2019, 375, 121915 CrossRef CAS.
- Y. J. Liu, W. T. Cao, M. G. Ma and P. Wan, ACS Appl. Mater. Interfaces, 2017, 9, 25559–25570 CrossRef CAS PubMed.
- S. Lin, X. Zhao, X. Jiang, A. Wu, H. Ding, Y. Zhong, J. Li, J. Pan, B. Liu and H. Zhu, Small, 2019, 15, 1900848 CrossRef PubMed.
- K. Tian, J. Bae, S. E. Bakarich, C. Yang, R. D. Gately, G. M. Spinks, M. in het Panhuis, Z. Suo and J. J. Vlassak, Adv. Mater., 2017, 29, 1604827 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00618e |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |