
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescence and biological stabilization of phosphorus-functionalized mesoporous silica nanospheres modified with a bis(8-hydroxyquinoline) zinc complex†
Norio
Saito
 *a,
Daichi
Noda
b,
Yucheng
Shang
b,
Shota
Yamada
b and
Motohiro
Tagaya
b
*a,
Daichi
Noda
b,
Yucheng
Shang
b,
Shota
Yamada
b and
Motohiro
Tagaya
b
aDepartment of Industrial Chemistry, Faculty of Engineering, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan. E-mail: nsaito@rs.tus.ac.jp
bDepartment of Materials Science and Technology, Nagaoka University of Technology, Kamitomioka 1603-1, Nagaoka, Niigata 940-2188, Japan
First published on 7th September 2021
Abstract
Green-emitting phosphorus-functionalized mesoporous silica (PMPS) nanospheres were fabricated by modifying their surfaces with (8-hydroxyquinoline) zinc (Znq2). A simulated body fluid soaking test and subsequent gas-adsorption measurements revealed that Znq2-modification could dramatically suppress the biodegradation of the nanospheres. This study establishes Znq2 as a novel and potential surface modifier of mesoporous silicas and demonstrates that effective surface design of the hosts will allow the exploitation of potential functionalities of the modifier.
Ordered porous materials such as metal organic frameworks (MOFs), zeolites, and mesoporous silicas (MPSs) have been widely investigated as unique hosts that can incorporate organic molecules and inorganic clusters into well-defined nanospaces.1 In particular, MPSs are promising nanoporous hosts with a large surface area and a uniform and adjustable pore structure, allowing facile modification on their internal and external surfaces with metal ions and functional molecules.2 They have been utilized for biomedical applications,3 catalytic processes,4 and adsorption-based applications.5
Metal 8-hydroxyquinolinate complexes such as tris(8-hydroxyquinoline)aluminum (Alq3) and their related derivatives are known to be typical optoelectronic building blocks.6 This class of molecules effectively interacts with nanoporous materials and is easily immobilized on their surfaces through chemical adsorption.7 It is worth noting that molecular assembly in such small nanospaces restricts the conformation of the guest molecule and induces unique guest–guest and/or host–guest interactions. This, in turn, leads to the emergence of optoelectronic properties that are not exhibited in solutions and bulk crystals. For instance, Fazaeli et al. demonstrated that compared to the case of bulk α-Alq3 crystal, covalent grafting of Alq3 and its substituent molecules on the MPS surface resulted in a significant blue shift of the fluorescence due to changes in the coordination environment of the Al3+ center.8 Similarly, Du et al. successfully synthesized (8-hydroxyquinoline) zinc (Znq2)-modified MPSs and observed a significant blue shift of their greenish fluorescence when the host surface was functionalized with mercapto and sulfonic acid groups.9 These studies suggest that the optical properties of Alq3 and Znq2 can be modulated through assembly in the nanospaces of MPSs and through their interface functionalization.
In this study, we have prepared phosphorus-containing MPS (PMPS) nanospheres using diethyl(2-bromoethyl)phosphonate as a phosphorus source.10 The mesoporous channels of PMPS were modified with Znq2. Adsorption experiments revealed that Znq2 was more favorably adsorbed on the PMPS surface than the case on the MPS surface. Interestingly, Znq2 modification of the PMPS surface efficiently prevented its anionic component dissolution from the surface of the mesopores into the simulated body fluid (SBF). Additionally, it was revealed that Znq2 could be utilized as a surface modifier that could induce fluorescence and biological stability as the host functionalities.
The MPS and PMPS nanospheres were synthesized according to our previously reported method.10 For synthesizing PMPS, the phosphorus to silica (P/Si) molar ratio was set to 6 (i.e., P/Si = 6) by adjusting the amounts of the silicate and phosphorus sources. Adsorption of Znq2 into the prepared nanospheres was induced by admixing the nanospheres and an ethanol solution of Znq2 of several initial concentrations (n) ranging from 0.15 to 0.75 mM. The resulting particles were washed with ultrapure water and dried under reduced pressure for 24 h. The Znq2-modified MPS and PMPS were designated as nZ/MPS and nZ/PMPS, respectively, where n indicates the concentration of Znq2 used for the chemisorption experiments. For the synthesis of PMPS, the phosphorus source diethyl(2-bromoethyl)phosphonate was hydrolyzed together with the hydrolysis reaction of tetraethyl orthosilicate.
The hydrolyzed phosphorus source interacts with the surface of the MPS precursor via the hydrogen bonding of the hydroxyl groups, followed by Si–O–P bond formation via the dehydrated condensation between their hydroxyl groups. Subsequent calcination led to decomposition of the bromoethyl moiety of the phosphorus source,11 and thus, the phosphate group was retained on the surface of the silicate framework. As discussed later, these functional groups strongly enhanced Znq2 adsorption on the PMPS surface. Details of the synthesis procedures are described in the ESI.†
Fourier transform infrared (FT-IR) spectroscopy of MPS and PMPS was performed to ascertain if phosphorus was introduced into the polysiloxane covalent framework. Fig. 1 shows the FT-IR spectra of MPS and PMPS in the range of the siliceous bonding region. Results of the peak deconvolution analysis are also shown in this figure. Total differences between the calculated and observed spectra were 5.0% for MPS and 6.3% for PMPS, suggesting that the sum of the deconvoluted peaks well reproduced the observed data. The spectra of MPS (Fig. 1a) show three shoulder-like peaks at 1204, 1092, and 969 cm−1, and the peak deconvolution analysis revealed that these peaks are assignable to a couple of Si–O–Si asymmetric and symmetric stretching vibrations and Si–OH stretching vibrations.12 On the other hand, in the case of PMPS (Fig. 1b), a new hump was observed at 1174 cm−1, corresponding to the Si–O–P stretching vibration.13 This indicated that a significant amount of phosphorus was successfully introduced into PMPS.
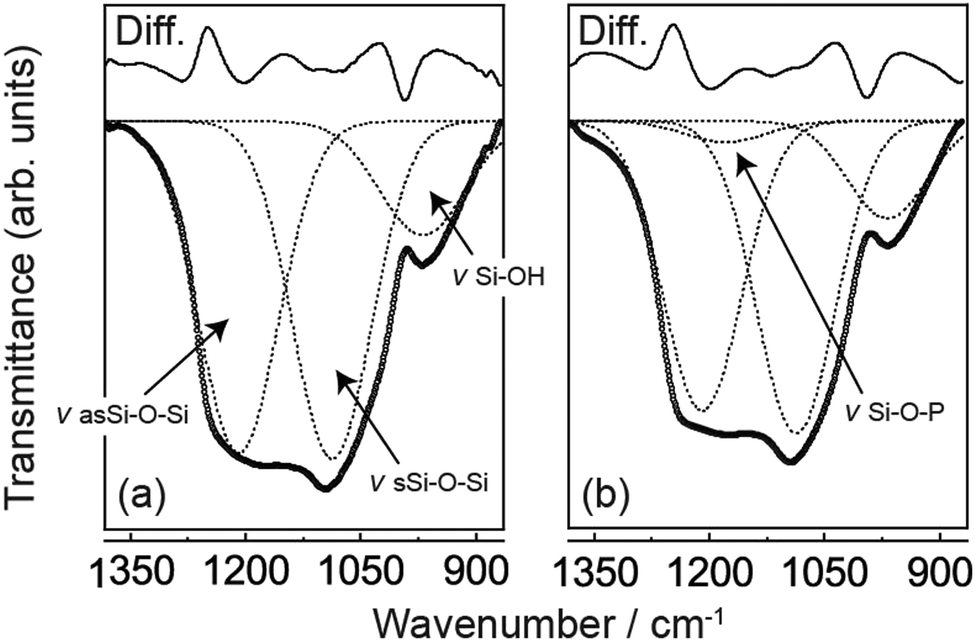 | ||
| Fig. 1 FT-IR spectra of (a) MPS and (b) PMPS in the siliceous bonding region. The open circles show observed spectra, and the dotted lines are simulated ones obtained by the deconvolution calculations. The top solid lines mean differences between the observed and simulated spectra. In spectrum (a), the observed signal was deconvoluted into three characteristic peaks assignable to a couple of Si–OH stretching vibrations and Si–O–Si bonds. In spectrum (b), a new hump was observed at 1174 cm−1, assignable to the Si–O–P stretching vibration. | ||
The prepared MPS and PMPS were modified with Znq2 through chemisorption, and their gas-adsorption properties were examined using N2 adsorption/desorption experiments. The adsorption/desorption isotherms suggested apparently different isotherm characteristics of nZ/MPS and nZ/PMPS (Fig. S1, ESI†). For all n values, nZ/MPS exhibited a type IV isotherm (Fig. S1a–e, ESI†) according to the IUPAC classification,14 and their hysteresis loops exhibited complete reversibility. This suggested that nZ/MPS had cylindrical mesopores that were smaller than ≈4 nm.15 Indeed, low-angle powder X-ray diffraction (PXRD) measurements suggested that the wall thickness was approximately 3 nm for nZ/MPS (Fig. S2a, ESI†). In contrast, the adsorption/desorption isotherms of nZ/PMPS (Fig. S1f–j, ESI†) showed characteristic desorption shoulders with lower closure points at 0.42P0; additionally, a plateau at high P/P0 was absent. These features suggested H3-type hysteresis,15 and possibly, an assembly of slit-shaped mesopores for nZ/PMPS. The PXRD profiles of nZ/PMPS indicated that the wall thickness of the slit-shaped mesopores was ≈4 nm (Fig. S2b, ESI†).
Surface area analyses of nZ/MPS and nZ/PMPS (Fig. 2a) indicate that the latter had a smaller Brunauer–Emmett–Teller (BET) surface area (SBET)16 than the former due to the disordered mesopore distribution induced upon phosphorus incorporation into the polysiloxane framework.10 Additionally, the SBET value decreased slightly with increase in the equilibrium adsorption concentration of Znq2, suggesting that Znq2 adsorption on the host surface closed a portion of their mesopore channels.
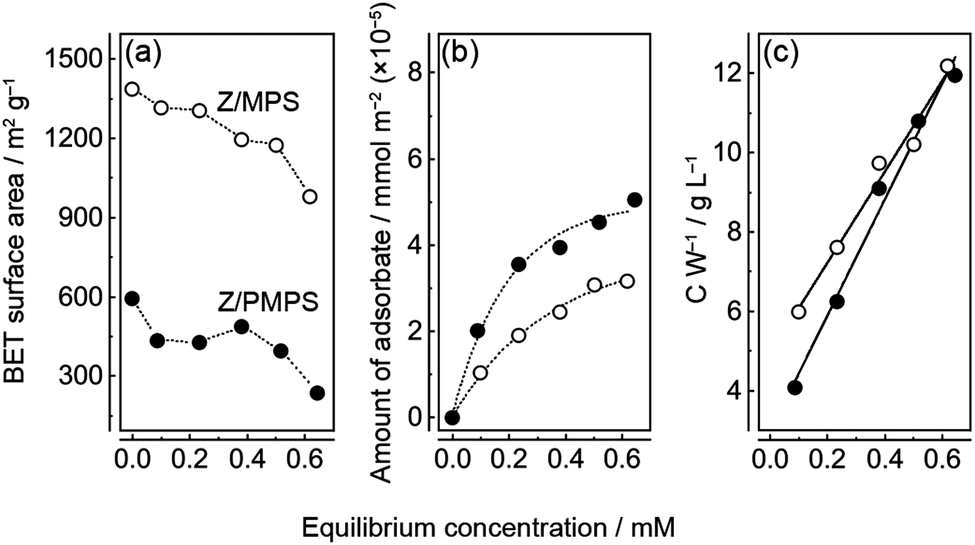 | ||
| Fig. 2 (a) Variation in the SBET values of nZ/MPS and nZ/PMPS and the equilibrium adsorption concentration measured in the chemisorption experiments. (b) Equilibrium adsorption isotherms of Znq2 obtained by the chemisorption experiments of MPS and PMPS. Znq2 was immobilized on the host surfaces by monolayered adsorption. (c) Scatchard plots of MPS and PMPS. | ||
The amount of Znq2 loaded into the hosts was determined from the change in the visible absorbance of the Znq2 solution before and after the chemisorption experiments. The relation between the equilibrium concentration and the amount of adsorbate in the hosts suggested that Znq2 adsorption was consistent with a Langmuir type I isotherm.17 Thus, Znq2 was immobilized in the nanospaces of the hosts via monolayer adsorption. Nevertheless, it must be noted that the surface of the mesoporous channels was not fully covered with Znq2; rather the surface coverage was remarkably low (discussed below) because the molecular size of Znq2 (≈ 1.5 nm)18 is comparable to the size of the nanospaces in the hosts. The Scatchard plots (Fig. 2c) indicate that the maximum amounts of Znq2 (Wmax) loaded into MPS and PMPS were 0.087 and 0.069 mmol g−1, respectively. The difference could be attributed to the higher SBET value of nZ/MPS. The plots also suggest that the adsorption equilibrium constants (Keq) for nZ/MPS and nZ/PMPS were 2.3 and 4.9 M−1, respectively. This implies that Znq2 was more strongly immobilized on the PMPS surface than on the MPS surface. The favorable Znq2 immobilization on the PMPS surface was also confirmed by surface occupation analyses (Fig. S3, ESI†). Here, the surface occupation of Znq2 was estimated from the equilibrium adsorption amounts of the adsorbate and the SBET values of the hosts. Consequently, the maximum surface occupation was calculated to be 2.2% for nZ/MPS and 3.4% for nZ/PMPS, implying a small but significant difference between their molecular immobilization capacities. The above results suggest that Znq2 should be more efficiently immobilized on the PMPS surface than on normal silicate surfaces.
Fig. 3 shows the fluorescence spectra of nZ/MPS and nZ/PMPS upon excitation at 356 nm. A broad, greenish emission centered around 500 nm, which is the characteristic Znq2 fluorescence, was observed.19 Compared to the fluorescence spectrum of Znq2 recorded in the solid state or in ethanol (Fig. S4, ESI†), the spectra of nZ/MPS and nZ/PMPS exhibited apparently broadened emission and a large blue shift of more than 50 nm, clearly suggesting that the adsorption of the guest chromophores into the hosts could modulate their optical properties. Because the degree of the blue shift seemed to be more pronounced in the samples with less loaded Znq2 (Fig. 3a), it can be assumed that the interactions between Znq2 and the host surface were responsible for the optical modulation.
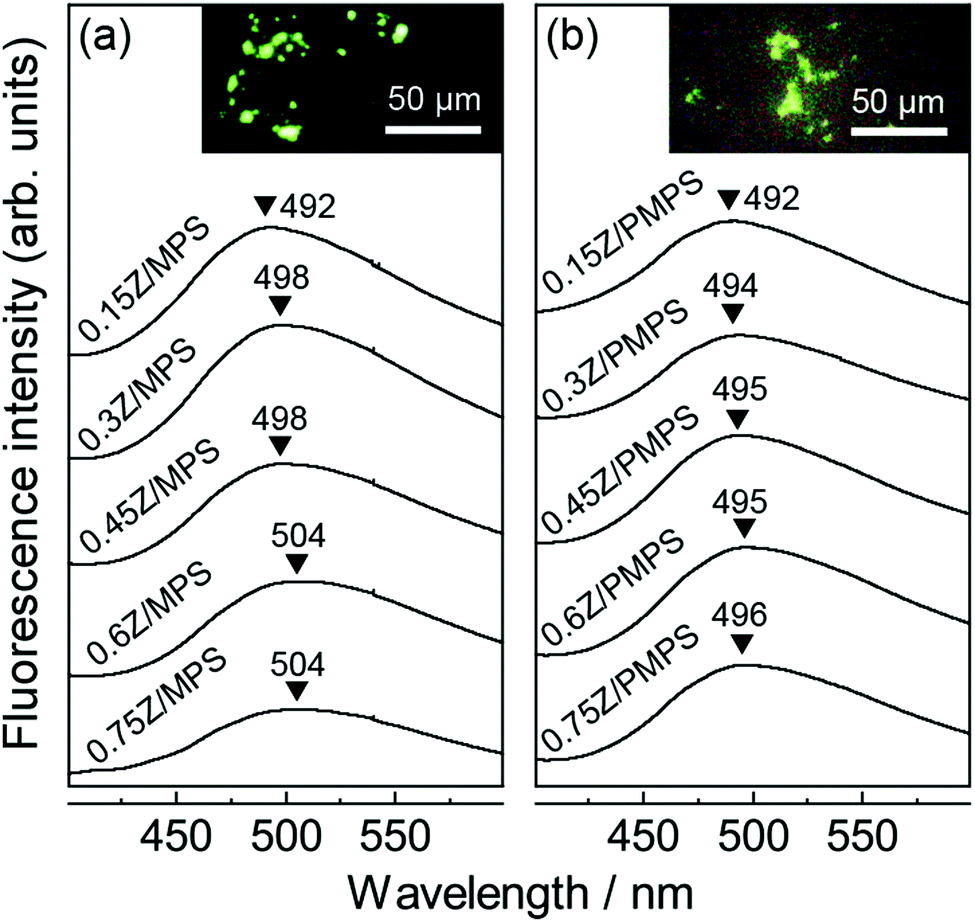 | ||
| Fig. 3 Fluorescence spectra of (a) nZ/MPS and (b) nZ/PMPS. The insets show the fluorescence microscope images for n = 0.75 (λex = 356 nm). | ||
As mentioned before, the phosphate groups on the PMPS surface play an important role during adsorption to ensure the efficient immobilization of Znq2. It should also be noted that the strong host–guest interaction in PMPS is likely to affect the optical properties of Znq2. Indeed, Fig. 3b suggests that the emission of nZ/PMPS was centered at almost a fixed wavelength in the range of 492–496 nm, while nZ/MPS exhibited a red shift from 492 to 504 nm with increase in the amount of loaded Znq2. The fluorescence maxima of metal hydroxyquinoline complexes vary depending on their conformation and aggregation states, and particularly, the red shift in fluorescence can be attributed to the overlapping of the π orbitals of each quinoline ring.20 Generally, the nanospaces in the hosts limit the conformational freedom of the guests and inhibit their aggregation.21 On the other hand, for the case of nZ/MPS, a red shift in fluorescence was observed with increasing concentration of loaded Znq2, indicating local molecular aggregation of Znq2, possibly during the drying process. In contrast, the nearly constant fluorescence of nZ/PMPS suggests the immobilization of the Znq2 monomer. It is speculated that the PMPS surface mainly has two adsorption sites, i.e., the negatively charged silanol (Si–O−) groups and the protonated phosphate (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–OH) groups,22 and that Znq2 interacts with these adsorption sites via electrostatic and hydrogen bonding interactions, respectively. We assume that the multi-point adsorption characteristic of the PMPS surface would inhibit the mobility and re-organization of Znq2, thereby leading to fluorescence stabilization.
P–OH) groups,22 and that Znq2 interacts with these adsorption sites via electrostatic and hydrogen bonding interactions, respectively. We assume that the multi-point adsorption characteristic of the PMPS surface would inhibit the mobility and re-organization of Znq2, thereby leading to fluorescence stabilization.
We examined the biological stability of the Znq2-modified mesoporous hosts. For this experiment, we soaked MPS, PMPS, 0.75Z/MPS, and 0.75Z/PMPS in SBF at 36.5 °C, and then determined the concentrations of the SiO32− anions dissolved in SBF using the molybdenum blue method,10 since the dissolution rate of SiO32− anions is an indicator of the degradation of silicate compounds. The sizes of the MPS and PMPS particles used in this experiment were 207 nm (cv. 9.1%) and 204 nm (cv. 8.6%), respectively.10Fig. 4a shows the time evolution of the SiO32− concentration in the SBF; it is evident that the degradation of the host was significantly dependent on phosphorus incorporation and Znq2 modification. As seen in the figure, MPS shows rapid dissolution until 12 h, following which an equilibrium state is attained. The total amount of the SiO32− anions dissolved in 48 h from MPS was 2.8 wt%. The relatively low degradation rate was attributed to the condensation of the polysiloxane networks upon calcination.23 PMPS also rapidly released the SiO32− anions for the first 12 h; however, a plateau was not observed even after 48 h. The total amount of SiO32− anions dissolved from PMPS was 4.2 wt%. Hence, PMPS is likely to be more prone to degradation in a biological environment. The rapid degradation of PMPS perhaps originates from the strong affinity between the PMPS surface and divalent metal cations. This hypothesis is rationalized by the fact that calcium phosphates were mineralized on the host surface after SBF soaking;10 that is to say, adsorption of the Ca2+ cations on the host surface should be a triggering factor for SiO32− dissolution and subsequent anionic compensation with ambient HPO42−. In this reaction, the oxygen atoms of the hydroxyl and phosphate groups are assumed to predominantly behave as the reaction sites due to their strong ability to coordinate with the metal cations.24 Indeed, we already reported that the PMPS surface forms a hydration layer having strong affinity with the hydrophilic protein molecules.25 In addition, the functional groups on the PMPS surface are negatively charged at a neutral pH.25,26 Hence, PMPS should have more active reaction sites with the metal cations than MPS, leading to the rapid degradation of the former host.
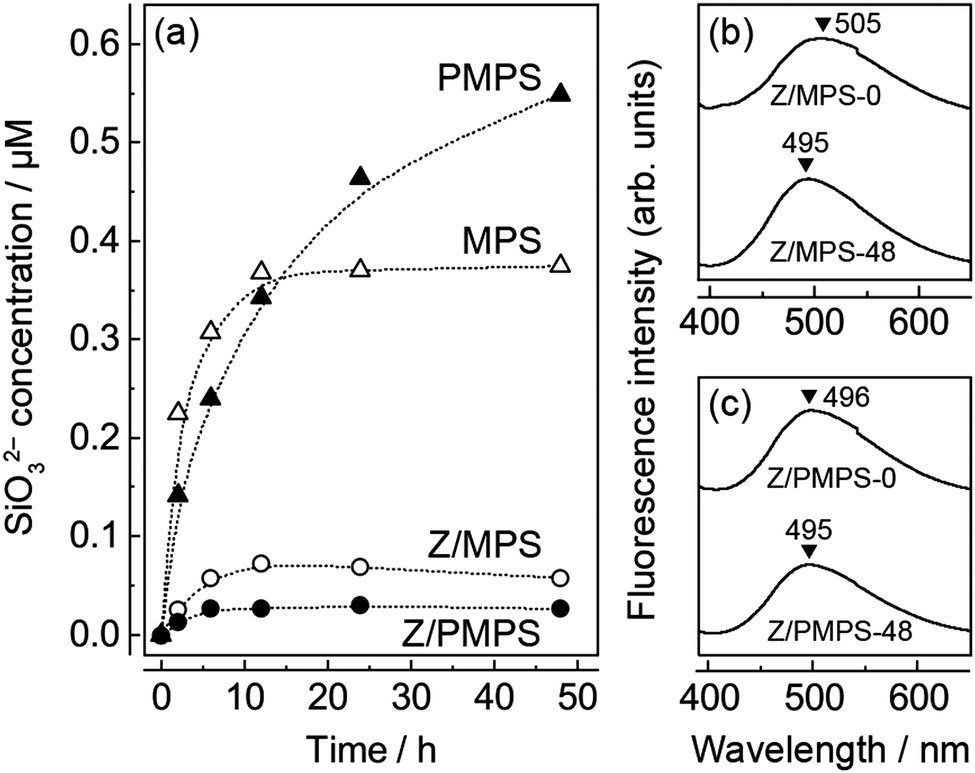 | ||
| Fig. 4 (a) Time evolution of SiO32− concentration after dissolution from MPS, PMPS, 0.75Z/MPS, and 0.75Z/PMPS in SBF. Fluorescence spectra of (b) 0.75Z/MPS and (c) 0.75Z/PMPS before and after SBF soaking. | ||
Fig. 4a also shows the SiO32− dissolution behavior of the Znq2-modified hosts. It is evident that surface modification dramatically reduced the amount of dissolved SiO32− anions, possibly because of the enhanced surface hydrophobicity27 and covering of the adsorption sites. One may be concerned that the Znq2 modification would deteriorate the solubility of MPS and PMPS in aqueous solutions. However, the Znq2 modification minimally affected the surface charge and solubility of the hosts, because Znq2 is preferentially immobilized in the nanospaces of the inner core due to the high density of the adsorptive groups in the nanospaces,28 and the surface state of the outer shell is substantially identical to the unmodified ones. Since the total amounts of the dissolved SiO32− anions were 0.45 and 0.21 wt% for 0.75Z/MPS and 0.75Z/PMPS, respectively, it can be concluded that the Znq2 modification rather stabilized the PMPS surface. The remarkable improvement in the biological stability of 0.75Z/PMPS was also confirmed from the N2 adsorption/desorption isotherms of the samples obtained after SBF soaking. The isotherms and their SBET values (Fig. S5, ESI†) indicate that the adsorption properties of 0.75Z/PMPS did not change remarkably even after SBF soaking, which was in contrast to the significant decline of the adsorption volume observed for 0.75Z/MPS. The mesopore stability was also reflected in their fluorescence spectra (Fig. 4b and c), as the fluorescence emission maximum of 0.75Z/PMPS remained unchanged. Thus, we conclude that the Znq2 modification significantly improved the biological stability of the mesopores in PMPS because of the strong interface affinity between Znq2 and the PMPS surface.
In summary, we demonstrate a new concept of using Znq2 as a surface modifier that can induce greenish fluorescence and biological stability of MPSs. We particularly highlight that the incorporation of phosphorus into the MPS surface allows the immobilization of monomeric Znq2 due to the strong affinity between Znq2 and the phosphate groups, further improving the biological stability of the host. Although there are reports on the modification of Alq3, Znq2, and other complex molecules in the nanospaces of mesoporous hosts,8,9 these studies were mainly focused on the optical and catalytic properties of the hybrids alone. In contrast, the present study focused on how the guest modification altered the properties of the host surface and demonstrates how effective interface design might be utilized to exploit the guest functions as well. We believe that the present concept will be especially useful in the development of biostable fluorescence markers and nanomedicines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 21K14706. M. T. thanks Analysis and Instrumentation Center in Nagaoka University of Technology for providing the facilities.References
- P. Singh, S. Srivastava and S. K. Singh, Sci. Eng., 2009, 5, 4882 Search PubMed; Y. Gao, D. Gao, J. Shen and Q. Wang, Front. Chem., 2020, 8, 598722 CrossRef PubMed.
- X. Feng, G. E. Fryxell, L. Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923 CrossRef CAS; D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448 CrossRef; S. Chen, S. L. Greasley, Z. Y. Ong, P. Naruphontjirakul, S. J. Page, J. V. Hanna, A. N. Redpath, O. Tsigkou, S. Rankin, M. P. Ryan, A. E. Porter and J. R. Jones, Mater. Today Adv., 2020, 6, 100066 CrossRef.
- Q. Lei, J. Guo, A. Noureddine, A. Wang, S. Wuttke, C. J. Brinker and W. Zhu, Adv. Funct. Mater., 2020, 30, 1909539 CrossRef CAS; M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548 CrossRef PubMed.
- M. Opanasenko, P. Stepnicka and J. Cejka, RSC Adv., 2014, 4, 65137 RSC.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef CAS; M. Hartmann, Chem. Mater., 2005, 17, 4577 CrossRef.
- M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812 CrossRef CAS; M. Amati, S. Belviso, P. L. Cristinziano, C. Minichino and F. Lelj, J. Phys. Chem. A, 2007, 111, 13403 CrossRef PubMed.
- L.-N. Sun, H.-J. Zhang, J.-B. Yu, S.-Y. Yu, C.-Y. Peng, S. Dang, X.-M. Guo and J. Feng, Langmuir, 2008, 24, 5500 CrossRef CAS PubMed; A. Badiei, H. Goldooz and G. M. Ziarani, Appl. Surf. Sci., 2011, 257, 4912 CrossRef.
- Y. Fazaeli, M. M. Amini, E. Mohajerani, M. Sharbatdaran and N. Torabi, J. Colloid Interface Sci., 2010, 346, 384 CrossRef CAS PubMed.
- Y. Du, Y. Fu, Y. Shi, X. Lü, C. Lü and Z. Su, J. Solid State Chem., 2009, 182, 1430 CrossRef CAS.
- Y. Shang, Y. Shota, Y. Chai and M. Tagaya, Key Eng. Mater., 2019, 782, 59 Search PubMed; S. Yamada, Y. Shang, I. Yamada and M. Tagaya, Adv. Powder Technol., 2019, 30, 1116 CrossRef CAS.
- Y. Okamoto, T. Kawai and H. Sakurai, Bull. Chem. Soc. Jpn., 1974, 47, 2903 CrossRef CAS.
- E. I. Kamitsos and A. P. Patsis, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 12499 CrossRef CAS PubMed; E. Astorino, J. B. Peri, R. J. Willey and G. Busca, J. Catal., 1995, 157, 482 CrossRef.
- L. Todan, C. Andronescu, D. M. Vuluga, D. C. Culita and M. Zaharescu, J. Therm. Anal. Calorim., 2013, 114, 91 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CAS.
- K. A. Cychosz, R. Guillet-Nicolas, J. Garcia-Martinez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389 RSC.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- I. Abe, J. Oleo Sci., 2002, 2, 275 Search PubMed.
- L. S. Sapochak, F. E. Benincasa, R. S. Schofield, J. L. Baker, K. K. C. Riccio, D. Fogarty, H. Kohlmann, K. F. Ferris and P. E. Burrows, J. Am. Chem. Soc., 2002, 124, 6119 CrossRef CAS PubMed.
- S. Li, J. Lu, H. Ma, D. Yan, Z. Li, S. Qin, D. G. Evans and X. Duan, J. Phys. Chem. C, 2012, 116, 12836 CrossRef CAS; H.-C. Pan, F.-P. Liang, C.-J. Mao, J.-J. Zhu and H.-Y. Chen, J. Phys. Chem. B, 2007, 111, 5767 CrossRef PubMed.
- M. Brinkmann, G. Gadret, M. Muccini, C. Taliani, N. Masciocchi and A. Sironi, J. Am. Chem. Soc., 2000, 122, 5147 CrossRef CAS.
- T. Azaïs, C. Tourné-Péteilh, F. Aussenac, N. Baccile, C. Coelho, J.-M. Devoisselle and F. Babonneau, Chem. Mater., 2006, 18, 6382 CrossRef.
- D. Singappuli-Arachchige and I. I. Slowing, J. Chem. Phys., 2020, 152, 034703 CrossRef CAS PubMed; R. Liu, L. Chi, X. Wang, Y. Sui, Y. Wang and H. Arandiyan, J. Environ. Chem. Eng., 2018, 6, 5269 CrossRef.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef CAS PubMed; Q. He, J. Shi, M. Zhu, Y. Chen and F. Chen, Microporous Mesoporous Mater., 2010, 131, 314 CrossRef.
- O. Francesconi, M. Gentili, F. Bartoli, A. Bencini, L. Conti, C. Giorgi and S. Roelens, Org. Biomol. Chem., 2015, 13, 1860 RSC.
- T. Kobashi, Y. Chai, I. Yamada, S. Yamada and M. Tagaya, Mater. Chem. Phys., 2019, 227, 134 CrossRef CAS; S. Yamada, T. Kobashi and M. Tagaya, J. Mater. Chem. B, 2021, 9, 1896 RSC.
- S. Valetti, A. Feiler and M. Trulsson, Langmuir, 2017, 33, 7343 CrossRef CAS PubMed.
- I. Izquierdo-Barba, M. Colilla, M. Manzano and M. Vallet-Regí, Microporous Mesoporous Mater., 2010, 132, 442 CrossRef CAS.
- C. H. Huang, K.-P. Chang, H.-D. Ou, Y.-C. Chiang and C.-F. Wang, Microporous Mesoporous Mater., 2011, 141, 102 CrossRef CAS; S. Spange, Y. Zimmermann and A. Graeser, Chem. Mater., 1999, 11, 3245 CrossRef; J. Kecht, A. Schlossbauer and T. Bein, Chem. Mater., 2008, 20, 7207 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00636c |
| This journal is © The Royal Society of Chemistry 2021 |