
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the defects of sub-100 nm multipodal titanium nitride/oxynitride nanotubes for efficient water splitting performance†
Kamel
Eid‡
 *a,
Mostafa H.
Sliem‡
b and
Aboubakr M.
Abdullah
*b
*a,
Mostafa H.
Sliem‡
b and
Aboubakr M.
Abdullah
*b
aGas Processing Center, College of Engineering, Qatar University, P. O. Box 2713, Doha, Qatar. E-mail: kamel.eid@qu.edu
bCenter for Advanced Materials, Qatar University, P. O. Box 2713, Doha, Qatar. E-mail: bakr@qu.edu.qa
First published on 12th July 2021
Abstract
Deciphering the photocatalytic-defect relationship of photoanodes can pave the way towards the rational design for high-performance solar energy conversion. Herein, we rationally designed uniform and aligned ultrathin sub-100 nm multipodal titanium nitride/oxynitride nanotubes (TiONxNTs) (x = 2, 4, and 6 h) via the anodic oxidation of Ti-foil in a formamide-based electrolyte followed by annealing under ammonia gas for different durations. XPS, XPS imaging, Auger electron spectra, and positron annihilation spectroscopy disclosed that the high nitridation rate induced the generation of a mixture of Ti-nitride and oxynitride with various vacancy-type defects, including monovacancies, vacancy clusters, and a few voids inside TiOxNTs. These defects decreased the bandgap energy to 2.4 eV, increased visible-light response, and enhanced the incident photon-to-current collection efficiency (IPCE) and the photocurrent density of TiONxNTs by nearly 8 times compared with TiO2NTs, besides a quick carrier diffusion at the nanotube/electrolyte interface. The water-splitting performance of sub-100 nm TiON6NT multipodal nanotubes was superior to the long compacted TiONxNTs with different lengths and TiO2 nanoparticles. Thus, the optimization of the nitridation rate tailors the defect concentration, thereby achieving the highest solar conversion efficiency.
Introduction
Biofuel,1,2 gas conversion reactions,3–7 anodic and cathodic reactions (i.e., water splitting,8,9 methanol10/ethanol oxidation,1 and oxygen reduction11,12) driven by porous catalysts are highly promising green and sustainable energy sources. Porous titanium nanostructures, especially nanotubes, have grabbed appreciable attention in photocatalytic water oxidation, owing to their excellent activity, high surface area, low-density, and abundant active sites.13,14 However, the wide bandgap and lower visible light-harvesting characteristics are critical, thus making TiO2 impractical for solar-driven energy conversion devices.15,16 As such, various efforts have been devoted to defeating such barriers, which are based on reducing the bandgap and delaying the recombination of the photogenerated electrons and holes.17–19 This can be achieved throughout the creation of various defects inside the lattice structure of TiO2via its doping with non-metal dopants such as H2, N, S, Cu, and F.20–22 These vacancy-type defects act as electron scavengers that retard the electron–hole pair recombination and enhance the number of charge carriers without diminishing the crystallinity and/or photocurrent density of TiO2.23,24 This is in addition to significantly decreasing the bandgap energy of TiO2, as well as enhancing the UV-visible light responses. To this end, N-doped porous TiO2, in the form of titanium oxynitride (TiON), is of great importance in solar-driven water splitting, owing to the excellent visible-light absorption and suitable bandgap.25–28 For example, mesoporous TiON films formed through combining sol–gel chemistry, evaporation-induced self-assembly, flash crystallization, and ammonia treatment displayed superior photocatalytic activity as compared to TiO2 for the degradation of methylene blue and lauric acid.29 Unlike other porous morphologies, nanotubes have higher surface areas, more active sites, quick charge collection efficiencies, greater UV-visible light responses, and enhanced mass transfer. Also, nanotubes provide various active sites that enhance the adsorption of reactants and the desorption of products.30,31 However, the fabrication of porous TiON nanotubes has rarely been reported, and they are not sufficiently emphasized for water-splitting as compared with other nanostructures. For instance, TiON-NTs prepared by the annealing of TiO2NTs under ammonia exhibited a significant enhancement in the photocurrent density and UV-visible light responses of TiO2NTs.32Likewise, TiON-NTs obtained using the anodization of TiN alloy enhanced the incident photon-to-current collection efficiency (IPCE) and visible-light responses substantially as compared to TiO2.33 Despite the significant advances in the fabrication of TiON-NTs, their photocatalytic-defect relationship was not emphasized and is still unknown. Most of the previous reports focused only on the hydrogenated and/or oxygenated TiO2 nanocrystals to induce the formation of oxygen vacancies and defects inside the TiO2 nanostructures.34–38 Some studies reported that the photocatalytic efficiency of TiO2 nanocrystals could be enhanced as the concentration of bulk defects to surface defects decreased due to the improved isolation efficiency of the photogenerated electrons–holes.37,39,40 The defects can generate trapping centers that accelerate the electron–hole combination besides deteriorating the crystallinity and/or photocurrents of TiO2.37,39,41 Thus, the photocatalytic-defect relationship of TiO2 is hitherto ravelled and ambiguous. Consequently, defect engineering in TiON-NTs and deciphering the defects-photocatalytic relationship can boost their charge carrier collection efficiency while preserving the crystallinity.42 This can also pave the way towards integrating TiON-NTs into cost-effective and efficient, practical solar-driven water-splitting devices.
Herein ultrathin sub-100 nm multipodal TiONxNTs nanotubes were fabricated using anodic oxidation followed by annealing under ammonia. The nitridation rate was optimized to tailor the concentration and types of the vacancy-type defects inside TiO2NTs. The nature and concentration of the vacancy-type defects were confirmed using various characterization techniques like transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), and positron annihilation spectroscopy (PALS). In addition, the photoelectrochemical water splitting performance of the ammonia-annealed TiONxNTs samples was benchmarked relative to the air-annealed TiO2NTs to elucidate the relationship between the photocatalytic performance of the ammonia-annealed TiONxNTs and air-annealed TiO2NTs. Unlike previous reports, our newly designed TiONxNTs possess various inimitable structure and composition merits, including a porous one-dimensional ultrashort sub-100 nm multipodal nanotube structure that provides a short electron pathway and maximizes the charge collection efficiency. The N-doping in TiO2NTs forms a mixture of Ti-nitride and oxynitride with various defects such as monovacancies, vacancy clusters, and a few voids that can reduce the valence band edge and enhance the visible light response of TiONxNTs. Furthermore, our obtained materials were formed as solid nanosheets ornamented with nanotubes that could be directly used as anodes without additional steps for the activation and/or conversion into working electrodes, which are highly essential features for practical applications.
Experimental
Chemicals and materials
Polyvinylpyrrolidone (PVP, 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), titanium foil (0.25 mm thick, purity 99.8%), ammonium fluoride (NH4F), and formamide were purchased from Sigma-Aldrich Chemie GmbH (Munich, Germany).
000), titanium foil (0.25 mm thick, purity 99.8%), ammonium fluoride (NH4F), and formamide were purchased from Sigma-Aldrich Chemie GmbH (Munich, Germany).
Synthesis of TiONxNTs
A Pt sheet was used as the counter electrode during the anodization of Ti foil in an aqueous formamide solution containing 1.2% of NH4F, 1.3 wt% PVP, and 3 mL H2O.43 The anodization process was carried out for 2.5 h in an ice bath at 0 °C under 36 V (0.1 V s−1). First, the current was maintained at 25–27 mA under acidic conditions using 0.1 M acetic acid.43 Then, the as-anodized foils were rinsed thoroughly with distilled water and annealed at 450 °C (2 °C min−1) for 1.5 h under air to obtain TiO2NTs. Next, the resultant TiO2NTs were annealed under ammonia flow (200 sccm) at 600 °C for 2, 4, and 6 h to form TiON2NTs, TiON4NTs, and TiON6NTs, respectively. The heating and cooling rates were 1 °C min−1.Material characterization
The morphology and composition of typically synthesized nanotubes were characterized by SEM (Hitachi S-4800, Hitachi, Tokyo, Japan) equipped with energy-dispersive X-ray spectroscopy (EDX) and TEM (TecnaiG220, FEI, Hillsboro, OR, USA). The XPS was measured on an Ultra DLD XPS Kratos, Manchester, UK spectrometer equipped with a monochromatic Al Kα radiation source (1486.6 eV) under a UHV environment (ca. 5 × 10−9 torr). The Auger electron spectra (AES) were measured on an Ultra DLD XPS spectrometer (Kratos, Manchester, UK) using a high-energy Ag Lα monochromatic X-ray source. The XRD patterns were obtained using an X'Pert-Pro MPD diffractometer (PANalytical Co., Netherlands) with a Cu Kα X-ray source (λ = 1.540598 Å). The ultraviolet-visible (UV-Vis) diffuse reflectance spectra were obtained using a diffuse reflectance accessory of a UV-vis spectrophotometer (UV2550, Shimadzu, Japan). The positron annihilation lifetime spectroscopy (PALS) was recorded using a 22Na (∼10 μCi) radioactive positron source.Water-splitting reaction
The photoelectrochemical measurements were carried out on a Gamry electrochemical analyzer (reference 3000, Gamry Co., USA) using a conventional three-electrode photo-glass cell involving a platinum wire as the counter electrode, reversible hydrogen electrode (RHE) (reference electrode), and our prepared samples were used as working electrodes. The sunlight was simulated with a xenon ozone-free lamp (100 W) (Abet Technologies, USA) and AM 1.5G filter at 100 mW cm−2. All the measured currents were normalized to the geometric area of the working-electrodes, and all potentials were recorded after the IR-drop correction.Results and discussion
TiO2NTs were initially synthesized via the electrochemical anodization of titanium foils in the formamide-based electrolyte and then were annealed under ammonia gas for different durations (Scheme 1).42,43 The formamide, with its remarkable ability to control the electrolyte solution's viscosity and conductivity, prevents the current oscillation due to the “in-pore” local pH variation providing favorable conditions for tailoring the fabrication of ordered, aligned, and oriented thin-walled multipodal nanotubes. The solution pH was adjusted by adding 0.1 M acetic acid, and 1 wt% of PVP was used for structural stability.43 The electrolyte solution should be mixed well under heating at 100 °C before the anodization process to ensure the mixing of the precursors and avoid the precipitation of NH4F. Fig. 1a reveals the SEM image of the typically synthesized TiO2NTs. The nanotube arrays were formed in well-ordered, homogeneous, and vertically aligned nanotubes without noticeable debris or distortion. The side view shows that the nanotubes are multipodal with two or more legs, as indicated by the dashed circles (Fig. 1b). The average nanotube length was about 80 ± 2 nm, the pore diameter was 12 ± 1 nm, and the wall thickness was 3–5 nm (Fig. 1b). The as-formed TiO2NTs were annealed under ammonia gas for 2, 4, and 6 h to form TiON2NTs, TiON4NTs, and TiON6NTs, respectively (Fig. 1c–e). Interestingly, the ammonia-annealed samples retained their porous nanotube structures without any alteration in the morphology and/or dimensions but with only a slight tight packing at a high nitridation rate (6 h). This confirmed the structural stability of the as-synthesized NTs.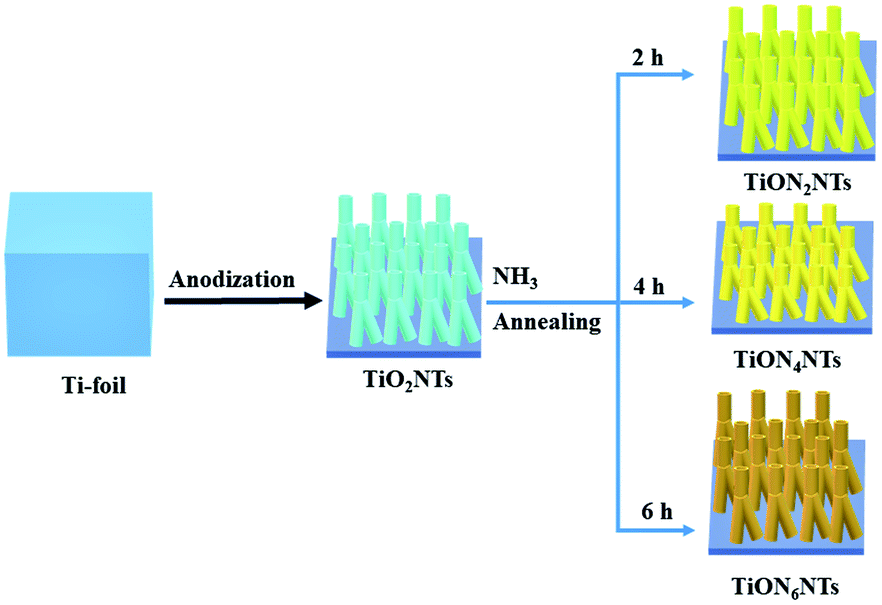 | ||
| Scheme 1 The formation process of TiO2NTs and TiON2NTs, TiON4NTs, and TiON6NTs. | ||
 | ||
| Fig. 1 SEM image of the typically prepared (a and b) TiO2-NTs, (c) TiON2NTs, (d) TiON4NTs, and (e) TiON6NTs. (f) EDX of the typically synthesized materials. | ||
The composition of the as-synthesized materials was investigated by the EDX analysis, which demonstrated the presence of Ti and O in TiO2NTs with atomic content of 33.9 and 66.1%. The ammonia-annealed samples revealed Ti, O, and an additional N peak. The intensity of the N peak increased with increasing the nitridation time, as indicated by the dashed square (Fig. 1f). From EDX analysis, the estimated N-content in TiON2NTs, TiON4NTs, and TiON6NTs was determined to be 1.7, and 3.2, and 4.9%, respectively. The as-made NTs were immersed in hydrochloric acid to peel off the NTs from the sheets. Fig. 2a shows the TEM image of TiO2NTs, which demonstrates the porous nanotube morphology. The high-resolution TEM (HR-TEM) image of TiO2NTs showed the crystalline anatase phase structure. The interplanar distance (d-spacing) was determined to be 3.57 Å, assigned to the 101 facets of TiO2 anatase (Fig. 2b). The selected area electron diffraction (SAED) pattern disclosed the set of the typical rings attributed to the (011), (004), (020), (015), (024), (220), and (125) facets of anatase TiO2, agreeing with previous reports.44 After nitridation, the TiON2NTs, TiON4NTs, and TiON6NTs, preserved their porous nanotube morphology (Fig. 2c, e and g). The HRTEM images of ammonia-annealed samples showed their amorphous crystalline structure. Intriguingly, the determined d-spacings were about 0.354, 0.352, 0.350, and 0.346 nm in TiO2NTs, TiON2NTs, TiON4NTs, and TiON6NTs, respectively, assigned to the facets of anatase (Fig. 2d, f and h).25 The slight decrease in the d-spacing of ammonia-annealed NTs relative to air-annealed NTs plausibly originated from the Ti–N and/or TiON legend effect, which compresses the lattice spacing of TiO2. Notably, although the surface-disordered domain was boosted clearly at a high nitridation rate, there was no phase transformation in the lattice structure, suggesting their stability. Simultaneously, the SAED exhibited the typical rings associated with the (110), (111), (020), (015), (022), (220), and (113) facets of a mixture of anatase, TiN, and TiON phases (Fig. 2d, f and h).
 | ||
| Fig. 2 (a, c, e and g) TEM images and (b, d, f and h) HRTEM images of TiO2NTs, TiON2NTs, TiON4NTs, and TiON6NTs, respectively, and their SAED patterns are shown as insets. The indicated scale bars in the insets of (b, d, f and h) are 5 nm. | ||
The crystalline structures of the as-synthesized TiONxNTs as compared to TiO2NTs were investigated via XRD analysis (Fig. 3). The air-annealed TiO2NTs showed the main characteristic diffraction peaks attributed to the tetragonal phase matched with (ICSD: 172914) along with some peaks of tetragonal rutile (ICSD: 33838) (Fig. 3a). After the nitridation, there was a notable change in the XRD patterns in the formed TiOxNNTs. In the TiOxNNTs the diffraction patterns of TiO2(011), (020), and (220) were disappeared, whereas the (131) and (222) of TiON (ICSD: 426340) were observed, along with the enhancement in the intensity of (111), (020), (022), and (113) of TiN (ICSD: 236801) (Fig. 3a).
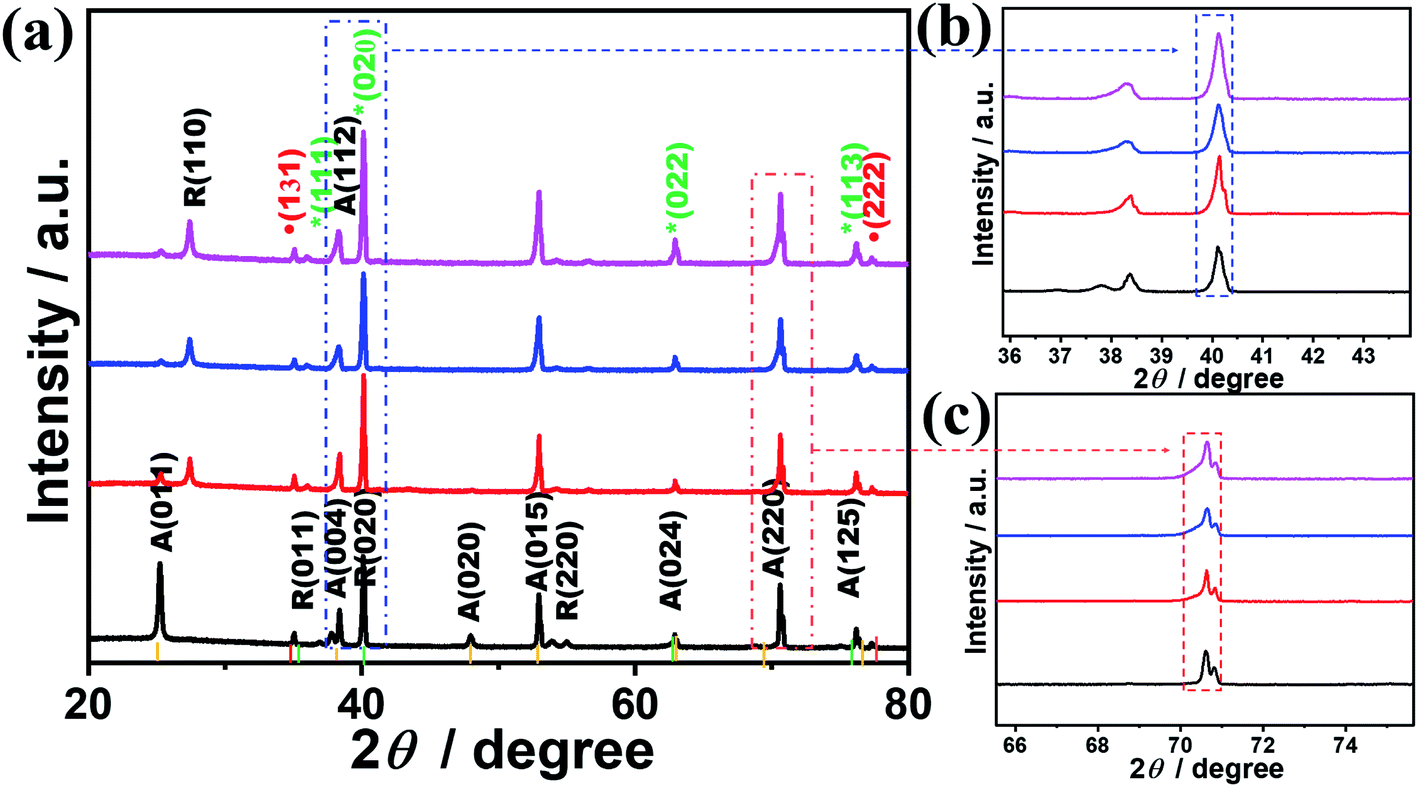 | ||
| Fig. 3 (a) XRD analysis of TiO2NTs and TiONxNTs, (b) magnification of the (020) peak and (c) magnification of A (220). The yellow line represents TiO2 anatase (ICSD: 172914), green line represents TiN (ICSD: 236801), and the red line represents TiON (ICSD: 426340). | ||
The TiN peaks overlapped with the TiO2 peaks, which is in line with other reports.33,42,45 However, the intensity of the (020) (Fig. 3b) and (220) peaks of TiN (Fig. 3c) and their full width at the half maximum (FWHM) increased significantly with the increasing nitridation time as indicated by the dashed lines, due to the incorporation of N into the TiO2 lattice structure resulting in the formation of crystal defects. The estimated FWHM of TiON6NTs (0.142°) is larger than that of TiON4NTs (0.139°), TiON2NTs (0.135°) and TiO2NTs (0.13°) from (022) peak. That is also obvious in the increment of the lattice parameters of TiON6NTs (4.245 Å), TiON4NTs (4.223 Å), and TiON2NTs (4.173 Å) relative to TiON2NTs (4.112 Å), implying the expansion of the crystal lattice with the increased N content, in agreement with previous reports.45
The average crystallite size of TiON6NTs (12.1 nm) is larger than that of TiON4NTs (10.5 nm), TiON2NTs (9.1 nm), and TiO2NTs (7.2 nm) as determined from the half-peak width of the XRD peak by applying the Scherrer equation (L = 0.89λ/βcosθ). The increment of the crystallite size is due to the increase of the annealing time as reported elsewhere.43,46
The valence states, surface composition, and defect-states of the typically fabricated samples were investigated by XPS analysis (Fig. 4). The binding energies of Ti, O, and N and the N-content of the as-synthesized materials are given in (Table S1†). The XPS of TiO2NTs revealed the Ti 2p and O 1s core-level, while the ammonia-annealed TiONxNTs showed the core-level of Ti 2p, O 1s, and N 1s (Fig. 4a). The intensity of the N 1s peak increased with increasing the nitridation time as indicated by the dashed square and the inset in Fig. 4a. The high-resolution Ti 2p spectra of TiO2NTs revealed only two peaks assigned to Ti 2p3/2 (458.4 eV) and Ti 2p1/2 (464.3 eV) representing a Ti4+ oxidation state (Fig. 4b). The high-resolution XPS spectra of Ti 2P in the ammonia-annealed NTs exhibited additional peaks of TiN (455.5 and 461.3 eV) assigned to the Ti3+ defect sites, and TiON peaks (457.2 and 462.7 eV) attributed to the Ti2+ defect sites, and a small shoulder for TiO2 (Ti2p3/2 at 458.4 and Ti2p1/2 at 464.3 eV) (Fig. 4b and Table S1†), indicating that Ti3+ and Ti2+ are the main phases in TiONxNTs samples. The intensities of Ti3+ and Ti2+ defect sites peaks of TiON samples were significantly higher as compared with Ti4+, lower than those of TiN and TiON, and most of the oxide was transformed into oxynitride after ammonia-annealing. Also, the defects of Ti3+ were greater as compared with both Ti2+ and Ti4+, as indicated by its FWHM. Interestingly, the FWHM of TiN and TiON peaks in TiONxNTs increased significantly with increasing the nitridation times, implying the increase in Ti3+ and Ti2+ defect sites as indicated by the dashed lines in (Fig. 4b). This was attributed to the insertion of N atoms into the lattice structure of TiO2, resulting in the replacement of oxygen atoms or the formation of the nitride and oxynitride phase, as was shown by the red-shift in the Ti 2p spectra of ammonia-annealed TiONxNTs at longer nitridation times. This was due to the N-dopant with less electronegativity being introduced into the crystal lattice of TiO2, resulting in the increase in the electron cloud density around Ti and converting Ti4+ into Ti3+ and Ti2+ (Fig. 4b). The O 1s spectra in air-annealed TiO2NTs showed three peaks ascribed to the lattice oxygen (Ti–O) at 530.2 eV, the oxygen in the adsorbed hydroxyl (Ti–OH) at 532.1 eV, and Oads at 532.4 eV. The ammonia-annealed TiONxNTs revealed TiO, O–N, and Oads (Fig. 4c). The disappearance of the Ti–OH peak in TiONxNTs is due to the neutralization of the oxygen vacancies in TiO2, which decreased the concentration of Ti4+ and increased the Ti3+ and Ti2+ defects at a high nitridation rate. This is seen in the lower stoichiometry of ammonia-annealed samples TiON2NTs (1.5), TiON4NTs (1.42), and TiON6NTs (1.4) as compared to the air-annealed TiO2NTs (1.8).
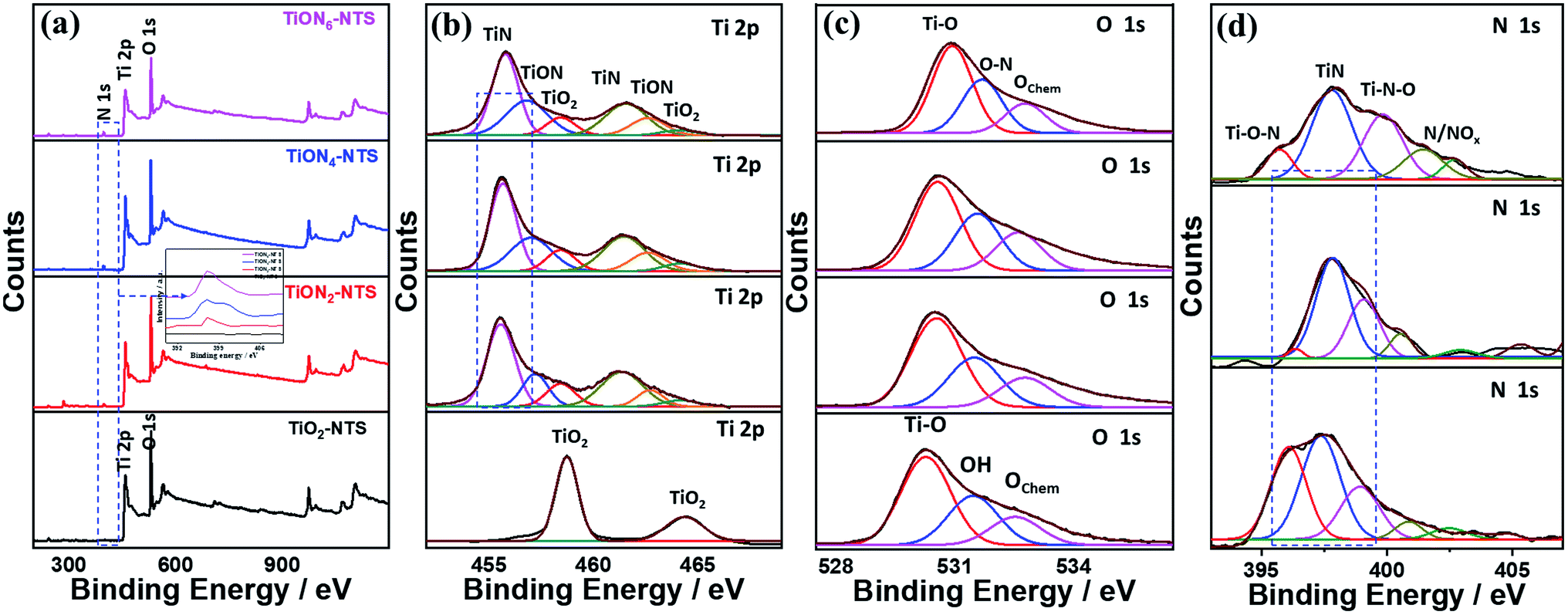 | ||
| Fig. 4 (a) XPS surveys of the as-synthesized materials and their high-resolution XPS spectra of (b) Ti 2p, (c) O 1s, (d) N 1s of TiO2NTs and TiONxNTs before etching. | ||
The N 1s spectra of TiONxNTs showed mainly doublet peaks attributed to TiN at 397.3 eV and TiON at 399.0 eV, along with small shoulders around 399 eV, attributed to γ-N, indicating the chemisorption of N2, and at 402.0 eV corresponding to β-N states state, indicating the atomic N-doping in the form of the oxynitride TiON shoulder (Fig. 4d and Table S1†). Notably, increasing the annealing time resulted in a significant enhancement in the intensity and FWHM of TiON and TiN peaks, indicating the higher N content. Thus, annealing under ammonia led to the substitution of some O-sites by N, in line with previous reports on N-doped TiO2,28,33,42 as shown by the increase in the N content with increasing the nitridation rate. The estimated amounts of N were about 1.9, 3.1, and 4.5% in TiON2NTs, TiON4NTs, and TiON6NTs, respectively, in line with the EDX. To gain further insight into the electronic structure and valence states of the formed materials, XPS was conducted after etching 4 times (Fig. S1 and Table S1†). The XPS surveys showed the same peaks as before etching, including the Ti 2p and O 1s core-level in TiO2NTs, and Ti 2p, O 1s, and N 1s in TiONxNTs (Fig. S1a†). The high-resolution Ti 2p spectra of TiO2NTs revealed two peaks assigned to Ti 2p3/2 and Ti 2p1/2, representing a Ti4+ oxidation state (Fig. S1b†). Interestingly, the high-resolution XPS spectra of Ti 2P in TiONxNTs displayed TiN assigned to Ti3+ defect sites and TiON attributed to Ti2+ defect sites, and a small shoulder for TiO2 (Fig. S1b and Table S1†). The TiN and TiON still existed after etching, contributing to the presence of Ti3+ and Ti2+ defect sites in the bulk, which is important for proving the various active sites, trapping sites, and delaying the electron–hole recombination during water oxidation. The spectra after etching became wider with lower intensities than before etching, which is plausible due to the lower N-content in the bulk. However, the intensity of Ti3+ and Ti2+ defect sites of TiONxNTs samples remained significantly higher than that of Ti4+. This is seen in the red-shifted Ti 2p spectra in the ammonia-annealed TiONxNTs at higher nitridation times. The O 1s spectra in air-annealed TiO2NTs showed Ti–O and Oads, while ammonia-annealed TiONxNTs revealed TiO, O–N, and Oads (Fig. S1c†). The N 1s spectra of TiONxNTs displayed TiN and TiON and adsorbed γ-N (Fig. S1d†). The estimated amount of N was about 1.1, and 1.9, and 2.6% in TiON2NTs, TiON4NTs, and TiON6NTs, respectively, which indicated the homogenous distribution of N in the bulk and surface.
The XPS mapping was used to further investigate the composition of the obtained materials (Fig. 5a–d). The results indicate the good distribution of Ti and O in the air-annealed TiONTs (Fig. 5a), while Ti, O, and N were homogeneously distributed in ammonia-annealed TiONxNTs (Fig. 5b–d). The N content in TiONxNTs increased with increasing the annealing time, as shown by the increasing number of red dots in (Fig. 5b–d). These imply the significant effect of ammonia-annealing on the creation of various Ti3+ and Ti2+ defects. That is also seen in the Auger electron spectroscopy recorded using a 3 kV electron beam with a current of 300 nA (Fig. 5e–g). Fig. 5e shows the AES spectra of TiLMM for air annealed TiO2NTs and TiONxNTs, which all displayed the presence of doublet peaks assigned to TiL3M2,3M4,5 at (413 and 418 eV).47,48 The intensity of TiL3M2,3M4,5 in the TiONxNTs increased with increasing the annealing time under ammonia, which was plausibly attributed to the increasing Ti3+ and Ti2+ defect sites resulting from the integration of N atoms. This is seen in the appearance of a small shoulder at 397 eV, assigned to TiN in the TiONxNTs.49,50 The intensity of the TiN peak increased slightly with increasing the nitridation time as shown in the inset in Fig. 5e. The difference in the intensities of the TiN peak is not high because the N contents in the ammonia-annealed TiONxNTs are too close. The OKLL spectra revealed OKL1L2,3(1p) and OKL2,3L2,3 with a kinetic value of 492 eV and 511 eV, respectively (Fig. 5f). Additionally, two small humps were seen at 479 and 501 eV, assigned to OKL1L1 and OKL1L2,3(3p), respectively, due to the replacement of O with N to form the Ti–ON bond and Ti–Nbond.51–53 This is seen in the resolution of the N KVV spectra, including N KL1L1, KL1L23, and KL23L23 at 352, 362, and 384 eV, respectively in the TiONxNTs, and the intensity of N KVV increased with increasing the nitridation time (Fig. 5g) resulting from the incorporation of N into the TiO2 lattice to form Ti3+ TiN, and Ti2+ TiON defect sites.28,54,55 Notably, the nitridation for 2, 4, and 6 h are the optimum periods to allow N-doping without affecting the nanotube shape. This is because the nanotubes showed significant debris and distortion when the nitridation time increased to 8 h.
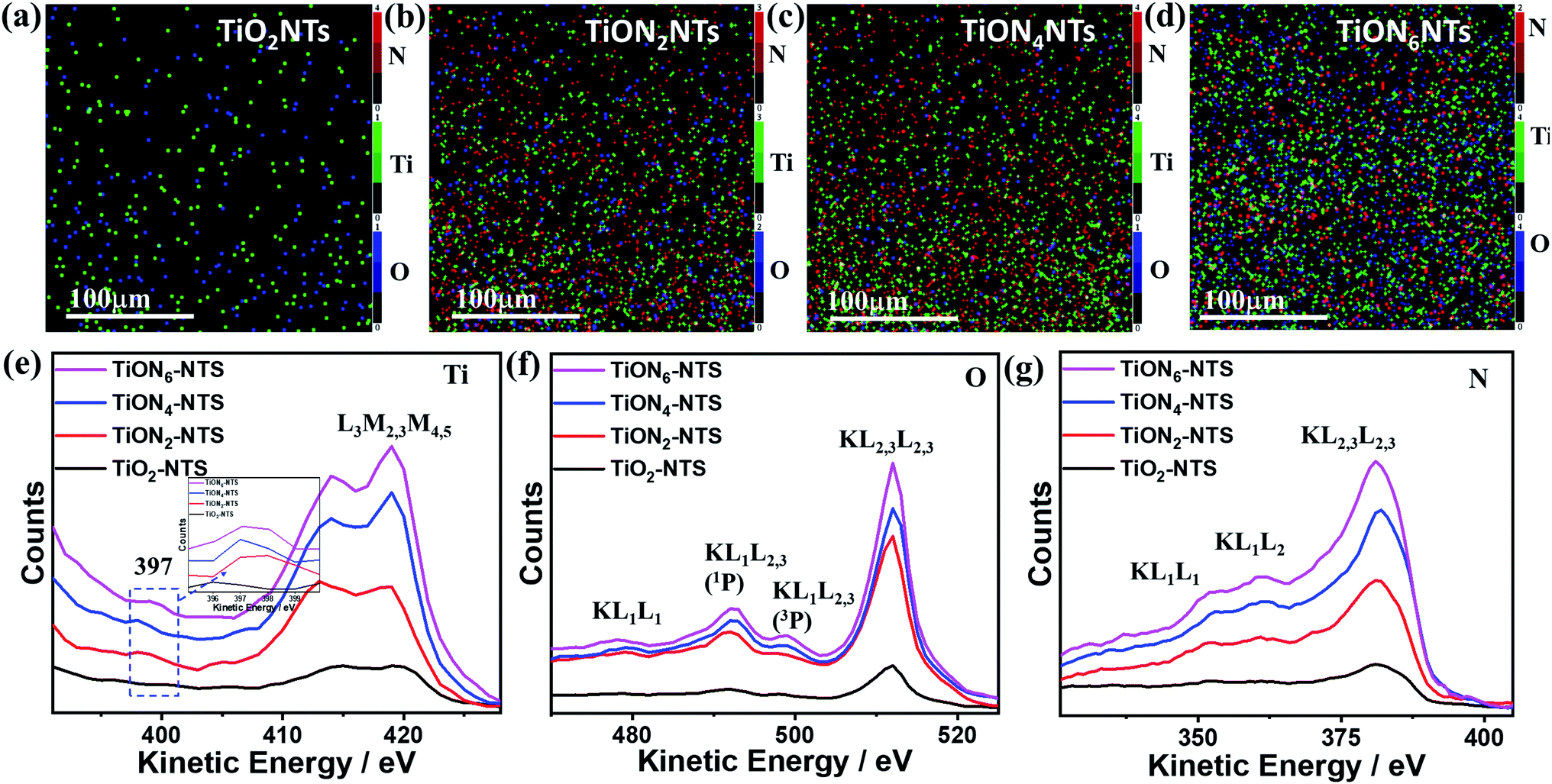 | ||
| Fig. 5 XPS elemental mapping analysis for (a) TiO2NTs, (b) TiON2NTs, (c) TiON4NTs, and (d) TiON6NTs. The green, blue, and red dots represent Ti, O, and N, respectively. Auger electron spectra (AES) of (e) Ti LMM, (f) O KLL, (g) N kLL. | ||
Fig. 6a exhibits the UV-Vis absorption spectra of typically synthesized air-annealed and ammonia-annealed TiONxNTs. The air-annealed TiO2NTs showed an absorption wavelength of around 410 nm. The ammonia-annealed NTs revealed a substantial blue shift in the absorption wavelength from 590 to 520 nm in the visible region. Intriguingly enough, the positive shift in the absorption wavelengths increased remarkably at a high nitridation rate. This is due to the substitution of some oxygen sites by nitrogen, enhancing the visible light absorption efficiency of ammonia-annealed NTs as compared to TiO2NTs. This was further observed in the lower bandgap energy of ammonia-annealed NTs as compared with TiO2NTs. Regarding this, the bandgap energy of TiO2NTs (3.1 eV) was substantially greater than that of TiON2NTs (2.8 eV), TiON4NTs (2.6 eV), and TiON6NTs (2.4 eV) as calculated using Tauc plots (Fig. S2†).
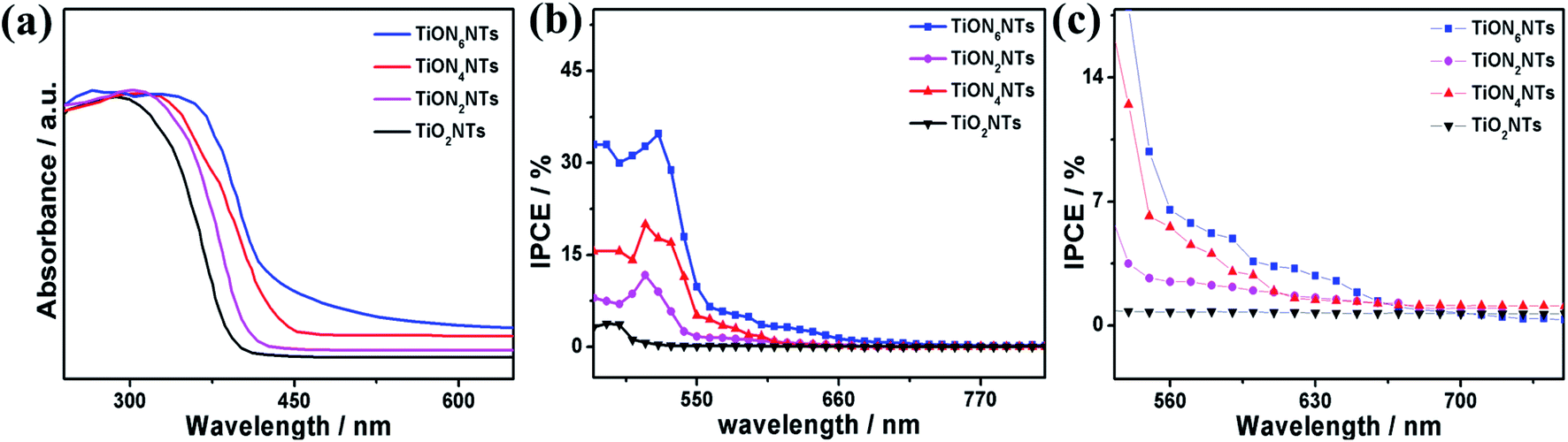 | ||
| Fig. 6 (a) UV-vis transmittance, (b) IPCE spectra of the as-formed material measured in 0.1 M KOH under no applied bias and (c) zooming in on the area between 550 and 750 nm. | ||
The incident photon to current conversion (IPCE) was measured in a two-electrode cell using air-annealed and ammonia-annealed NTs as working electrodes and a platinum wire as a counter electrode in 0.1 M KOH solution. The IPCE was calculated using the following equation:
| IPCE = [1240 × jph/λ × I] × 100 |
The ammonia-annealed NTs were significantly superior to air-annealed TiO2NTs. In particular, at 1.99 VRHE, TiO2NTs produced a photocurrent of 0.57 mA cm−2, whereas the ammonia-annealed TiONxNTs revealed photocurrents ranging from 1.75 to 4.5 mA cm−2, implying the significant effect of N-doping on the enhancement of the photocatalytic activity (Fig. 7a). It is noteworthy that ammonia-annealed NTs showed a superior activity at earlier potentials than that of TiO2NT. Moreover, the ammonia-annealed NTs produced greater photocurrents than TiO2NTs under applied potential (Fig. 7b). The measured onset potential (Eonset) on TiON2NTs (1.45 V), TiON4NTs (1.4 V), and TiON6NTs (1.2 V) were significantly more negative than that of TiO2NTs (1.52 V) (Fig. 7c). This plausibly emanated from the substitution of some O sites by N, which led to introducing a high concentration of vacancy-type defects inside the lattice structure of TiO2NTs. These defects provide various electron scavenger sites facilitating the water oxidation kinetics at lower potentials, as further confirmed in the lower Tafel slope of ammonia-annealed NTs (0.44–0.55 V dec−1) relative to TiO2NTs (0.8 V dec−1) (Fig. 7d).
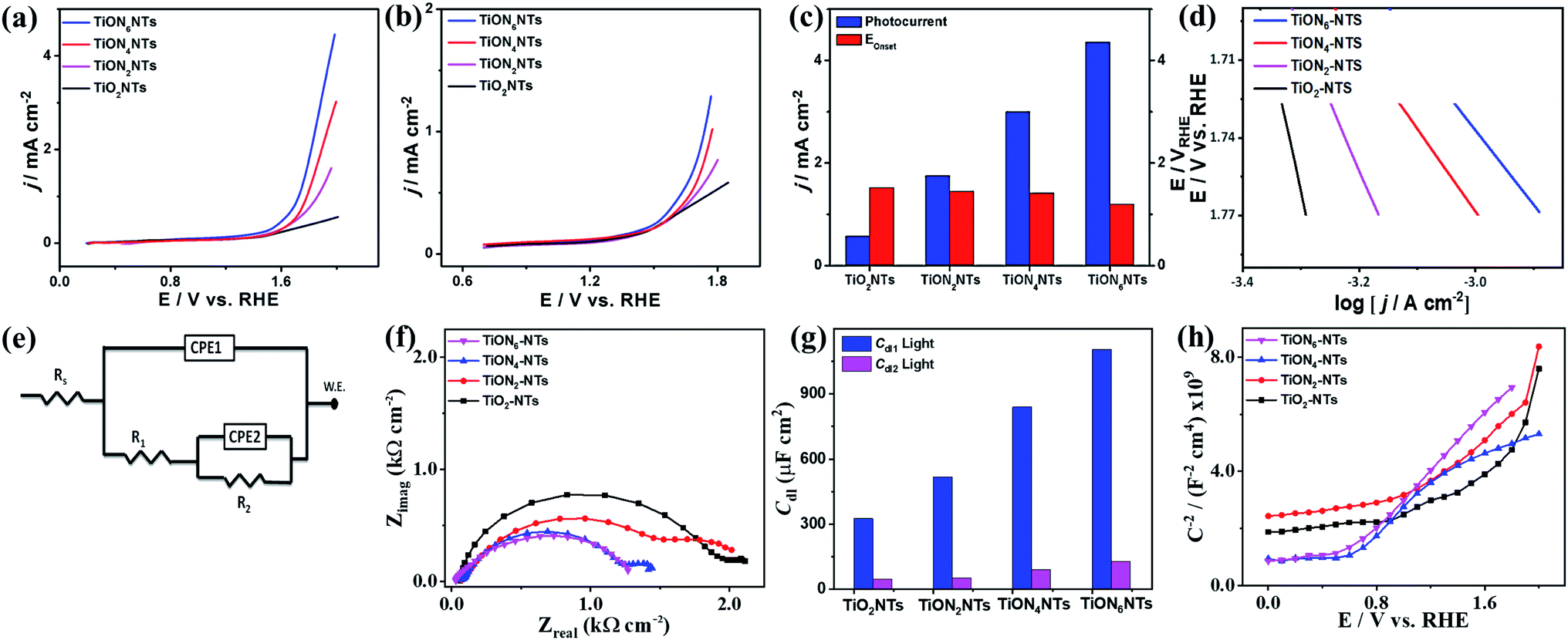 | ||
| Fig. 7 (a and b) LSV of water oxidation on different catalysts measured in an aqueous solution of 0.1 M KOH at a scan rate of 10 mV s−1 at room temperature. (c) Comparisons of photocurrent densities and Eonset. (d) Tafel plot of TiO2NTs and TiONxNTs. (e) The utilized equivalent circuit (f) electrochemical impedance spectroscopy (EIS) Nyquist plots measured in 0.1 M KOH in a frequency range of 10 mHz to 100 kHz and a perturbation amplitude of 5 mV; (g) double layer, and (h) Mott–Schottky plots of TiO2NTs and TiONxNTs. All the tests were performed under light illumination. | ||
Compared with other TiO2 or TiON nanostructures, the photocurrent obtained on our developed ammonia-annealed multipodal (80 nm) TiON6NTs (4.5 mA cm−2) was superior to that reported for Co–TiON nanowires (80–100 nm) (0.61 mA cm−2),57 TiON nanowires (4.4 μm) (0.23 mA cm−2),58 and compacted TiO2NTs (70 ± 10 nm) (0.45 mA cm−2),43 titanium nitride/titanium oxide (TiN/TiO2) nanocomposite (0.12 mA cm−2),59 and long TiON nanotubes (1.3 μm) (2.25 mA cm−2)42 measured under similar conditions in KOH electrolyte under light illumination (a xenon ozone-free lamp 100 or 300 W).
Moreover, we measured the photocatalytic water splitting performances of ammonia-annealed NTs relative to ammonia-annealed TiO2 nanotubes with different lengths of 1.2 μm (denoted TiONNTs 1.2) and 500 nm (denoted TiONNTs 500), as well as commercial TiO2 nanoparticles (Fig. S3†). The current densities of ammonia-annealed TiON6NTs (4.5 mA cm−2) were 3.19, 2.14, 11.25, and 20.4 times higher than that of TiONNTs-1.2 (1.41 mA cm−2), TiONNTs-500 (2.1 mA cm−2), N-free TiO2NTs-1.2 (0.4 mA cm−2), and TiO2 nanoparticles (0.225 mA cm−2) (Fig. S3†). This is due to the uniformity, multipodal nanotube shape, and high defect concentration of TiON6NTs, which plausibly enhanced the conductivity and accelerated charge mobility. It is universally accepted that sub-100 nm nanotubes have various unique merits relative to traditional compacted long nanotubes, including but not limited to greater surface area, shorter aspect ratio, and directional/quick charge transfer, as well as a short electron pathway.43 This is in addition to the effective electron–hole separation and better light-harvesting ability that drive the water-splitting at a lower potential relative to traditional long nanotubes. Meanwhile, the multipodal shape provides a graded refractive index, large surface, and easier charge separation at the core-leg interface enabling greater volume ratio and higher scattering.60 In contrast, the long diameter of the compact nanotube (over 170 nm) can act as a sub-wavelength structure that scatters light in Rayleigh fashion as reported before.60
That was furthered proved by measuring the electrochemical impedance spectroscopy (EIS) under light for TiO2NTs and TiONxNTs. The equivalent circuit (EC) was utilized to fit and analyze the measured data (Fig. 7e). A two-time constant equivalent circuit was employed, which is primarily used for electrodes with coatings or porous oxide layers on top. The EIS parameters, such as the electrolyte resistance (Rs), the charge transfer resistance at the oxide or the oxynitride/solution interface (R1), the charge transfer resistance at the metal solution interface (R2), the two constant phase elements (CPE1 and CPE2) associated with the resistances mentioned above, and the deviation parameters (n1 and n2), are listed in Table S2.† A constant phase element is used instead of a pure capacitor to express the non-ideal behavior (indicated by the depressed semicircles) of the double-layer capacitance because of the non-uniform (i) thickness of the oxide/nitrided layer, (ii) reaction kinetics on the surface, (iii) current distribution, in addition to (iv) surface roughness.61 The diameters of the depressed semicircles of TiOxNTs relative to TiO2NTs are shown in Fig. 7f. Increasing the N-content leads to decreasing the diameter of the thus-obtained TiOxNTs, which indicates their quick charge mobility and electrolyte–electrode conductivity relative to air-annealed TiO2NTs. Fig. 7g depicts the double-layer capacitance (Cdl) derived from the variables in Table S2† using the equation below:
| Cdl = [(Yo × Rct)1/n]/Rct | (1) |
It can be noted that the double layer capacitance is inversely proportional to the impedance values (Fig. 7g); the Cdl values in TiONxNTs were higher than that in TiO2NTs, indicate the dielectric nature of the TiO2NTs substrate. This is more obvious in the decreasing R1,R2, Rs, and n1, and n2 in TiONxNTs as compared to TiO2NTs (Table S2†).
Mott–Schottky plots were utilized to elucidate the charge distribution on the as-synthesized interface versus the electrolyte by measuring the electrode capacitance C as a function of electrode potential E (Fig. 7h). It is worth mentioning that all samples provided a positive slope, which is characteristic of the n-type TiO2 semiconductor. Hence, the donor density (Nd) was calculated using the equation below:
| C−2 = (2/eεε0ANd) × (E − EFb − (KbT/e)) | (2) |
 | ||
| Fig. 8 Energy level diagrams for (a) TiO2NTs and (b) TiONxNTs, as well the proposed water splitting mechanism. | ||
Upon light irradiation, the electrons and holes are generated in the conduction band and the valence band, respectively. The electrons and holes are scavenged by the O2 molecules to produce superoxide radical anions (O2˙) and hydroxyl radicals (OH˙), which are highly active and act as oxidative species facilitating water oxidation under lower potential (Fig. 8b).
The type and concentration of the defects in the air-annealed TiO2NTs and ammonia-annealed TiONxNTs were benchmarked using positron annihilation spectroscopy (PALS). The injected positrons through TiO2 are thermalized and annihilated by electrons, which lead to the emission of γ rays and allows the measurement of the lifetime of positrons. The positrons are distributed in the area with low electron density (i.e., vacancy-type defects, vacancy clusters, and microvoids). Thus, the electron-positron annihilation photon allows the measurement of the lifetimes (t1, t2, and t3) of positrons and their intensities (I1, I2, and I3) as indicators of the defects. The results showed the resolved positron lifetimes, including the shorter lifetime (τ1), longer positron lifetime (τ2), and longest lifetime components (τ3) along with their related intensities I1, I2, and I3 (Table 1). The respective τ1 values attributed to the free annihilation of positrons in the defect-free crystals of TiON2NTs (178 ps), TiON4NTs (181 ps), and TiON6NTs (188 ps) were significantly higher than that of TiO2NTs (174 ps). The increment in τ1 is attributed to the higher content of monovacancies and/or shallow positron traps in the ammonia-annealed samples, which decreased the surrounding electron density and subsequently increased the lifetime of τ1 in TiOxNNTs as compared to in air-annealed TiO2NTs. Therefore, the prolonged τ1 of TiONxNTs validated that a huge number of small neutral Ti3+–N vacancy associates were introduced into the TiO2 lattice by nitridation.
| Samples | t 1 (ps) | I 1 (%) | t 2 (ps) | I 2 (%) | t 3 (ps) | I 3 (%) |
|---|---|---|---|---|---|---|
| TiO2NTs | 174 | 60.5 | 288 | 34.5 | 8.3 | 5 |
| TiON2NTs | 178 | 57.4 | 297 | 35.4 | 10.4 | 7.2 |
| TiON4NTs | 181 | 55.2 | 301 | 36.6 | 12.1 | 8.9 |
| TiON6NTs | 188 | 52.9 | 311 | 37.9 | 13.5 | 9.2 |
The lifetime τ2 is larger than τ1, which implies that τ2 originated from positrons captured by defects of larger size. Thus, the τ2 of TiON2NTs (297 ps), TiON4NTs (301 ps), and TiON6NTs (311 ps) were greater than that of TiO2NTs (288 ps), implying the presence of larger-sized vacancy clusters attributed to the interaction among small neutral Ti3+–nitrogen-vacancy associates. The average electron density in larger-sized defects is lower than that in small-sized defects, which decreases the annihilation rate and increases the positron lifetime correspondingly. The longest lifetime (τ3) is plausibly attributed to the annihilation of orthopositronium atoms formed in the large voids in semiconductors. The τ3 of ammonia-annealed TiON2NTs (10.4 ps), TiON4NTs (12.1 ps), and TiON6NTs (13.5 ps) are higher than that of air annealed TiO2NTs (8.3 ps). This demonstrated the higher content of the few voids of N-vacancy associates on the nanoscale in ammonia-annealed samples.
In addition to the lifetimes of the positrons, their relative intensities provide information on the relative concentration of the defects. The relative intensities (I1) of TiON2NTs (57.4%), TiON4NTs (55.2%), and TiON6NTs (52.9%) were close to that of TiO2NTs (60.5%). The relative I2 of TiON2NTs (35.4%), TiON4NTs (36.6%), and TiON6NTs (37.9%) were slightly lower than that of TiO2NTs (34.5%). The I1 to I2 ratios of TiON2NTs (1.6), TiON4NTs (1.5), and TiON6NTs (1.39) were slightly lower than that of TiO2NTs (1.75). This demonstrates the decrease in the concentration of bulk defects (Cbd) to surface defects (Csd) of ammonia-annealed TiONxNTs relative to air-annealed TiO2NTs in addition to a higher concentration of monovacancies than the boundary-like defect. The relative intensities I3 of TiON2NTs (7.2%), TiON4NTs (8.9%), and TiON6NTs (9.2%) were higher than that of TiO2NTs (5%). This indicates the greater concentration of the voids of N-vacancy associates on the nanoscale in ammonia-annealed TiONxNTs as compared to air-annealed. The slight drop in the I3 originated from the sharp increase in I1 and I2. The summation of I1 + I2 + I3 should be equal to 100. Impressively, it is universally accepted that reducing the Cbd to Csd led to the retardation of the electron–hole recombination.37,39 As expected, the τ3 of TiON2NTs (10.4 ps), TiON4NTs (12.1 ps), and TiON6NTs (13.5 ps) were slightly higher than that of TiO2NTs (8.3 ps), demonstrating their higher content of the few voids of vacancy associates on the nanoscale. The relative intensities I3 of ammonia-annealed TiONxNTs were lower than that of TiO2NTs.
These results indicate that annealing under ammonia gas introduced different vacancy-type defects such as monovacancies, vacancy clusters, and a few voids inside the lattice structure of TiO2NTs. Such defects led to a significant reduction in the bandgap energy, retarding the electron–hole recombination and enhancing the UV-visible light absorption and photocatalytic water splitting. This is attributed to the substantial effect of the defects/vacancies on boosting the photocurrent in n-type TiO2.34,37,41 These defects form inter-bandgap states underneath the conduction band minimum and provide extra charge carriers under UV-light irradiation. Additionally, owing to the great electron trapping ability of defects, the lifetime of the photoelectrons increased significantly, resulting in supreme activity for solar-driven water splitting.
Conclusion
Herein, we rationally synthesized ultrathin, well-aligned, and uniform sub-100 nm multipodal TiO2NTs nanotubes (80 ± 2 nm length) via the electrochemical anodic oxidation of Ti-foil in formamide-based electrolytes. The as-formed TiO2NTs were annealed under ammonia for 2, 4, and 6 h to form TiON2NTs, TiON4NTs, and TiON6NTs, respectively. The N-content was ∼1.7, and 3.2, and 4.9%, in TiON2NTs, TiON4NTs, and TiON6NTs, respectively. The HRTEM, XPS, AES, and PALS warranted that a high concentration of vacancy-type defects was introduced inside the TiO2NTs lattice structure. The N-content and its subsequent defects inside the TiO2NTs lattice increased with the increment of the nitridation time that formed a mixture of Ti-nitride and oxynitride with various defect states. These kinds of defects decreased the bandgap energy to 2.4 eV, enhanced the visible-light response, IPCE, photocurrent density (by 8 times), and accelerated the charge carrier collection efficiency of ammonia-annealed TiONxNTs as compared to air-annealed TiO2NTs. The sub-100 nm nanotubes TiON6NTs outperformed the long compacted TiONxNTs with different lengths and TiO2 nanoparticles. This paper could pave the way towards optimizing the defect state of TiO2-based photocatalysts for solar-driven water splitting.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We greatly appreciate the Gas Processing Center, Qatar University, for supporting this work. Also, the SEM images were captured at the Core Labs of Hamad Bin Khalifa University, Doha, Qatar. This work was supported by the Qatar National Research Fund (QNRF, a member of the Qatar Foundation) through the National Priority Research Program Grant (NPRP) NPRP13S-0117-200095. Also, this publication was supported by Qatar University's internal grant IRCC-2021-015. Statements made herein are solely the responsibility of the authors.References
- F. Wu, K. Eid, A. M. Abdullah, W. Niu, C. Wang, Y. Lan, A. A. Elzatahry and G. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 31309–31318 CrossRef CAS PubMed.
- H. I. Abdu, K. Eid, A. M. Abdullah, Z. Han, M. H. Ibrahim, D. Shan, J. Chen, A. A. Elzatahry and X. Lu, Renewable Energy, 2020, 153, 998–1004 CrossRef CAS.
- K. Eid, M. H. Sliem, H. Al-Kandari, M. A. Sharaf and A. M. Abdullah, Langmuir, 2019, 35, 3421–3431 CrossRef CAS PubMed.
- K. Eid, M. H. Sliem and A. M. Abdullah, Nanoscale, 2019, 11, 11755–11764 RSC.
- K. Eid, M. H. Sliem, A. S. Eldesoky, H. Al-Kandari and A. M. Abdullah, Int. J. Hydrogen Energy, 2019, 44, 17943–17953 CrossRef CAS.
- Q. Lu, K. Eid, W. Li, A. M. Abdullah, G. Xu and R. S. Varma, Green Chem., 2021 10.1039/D1GC01303C.
- K. Eid, M. H. Sliem, K. Jlassi, A. S. Eldesoky, G. G. Abdo, S. Y. Al-Qaradawi, M. A. Sharaf, A. M. Abdullah and A. A. Elzatahry, Inorg. Chem. Commun., 2019, 107, 107460 CrossRef CAS.
- M. A. Ahsan, T. He, K. Eid, A. M. Abdullah, M. L. Curry, A. Du, A. R. Puente Santiago, L. Echegoyen and J. C. Noveron, J. Am. Chem. Soc., 2021, 143, 1203–1215 CrossRef CAS PubMed.
- A. K. Ipadeola, A. B. Haruna, L. Gaolatlhe, A. K. Lebechi, J. Meng, Q. Pang, K. Eid, A. Abdullah and K. I. Ozoemena, ChemElectroChem, 2021 DOI:10.1002/celc.202100574.
- K. Eid, H. Wang, P. He, K. Wang, T. Ahamad, S. M. Alshehri, Y. Yamauchi and L. Wang, Nanoscale, 2015, 7, 16860–16866 RSC.
- K. Eid, H. Wang, V. Malgras, Z. A. Alothman, Y. Yamauchi and L. Wang, J. Phys. Chem. C, 2015, 119, 19947–19953 CrossRef CAS.
- H. Wang, S. Yin, K. Eid, Y. Li, Y. Xu, X. Li, H. Xue and L. Wang, ACS Sustainable Chem. Eng., 2018, 6, 11768–11774 CrossRef CAS.
- A. Pancielejko, P. Mazierski, W. Lisowski, A. Zaleska-Medynska, K. Kosek and J. Łuczak, ACS Sustainable Chem. Eng., 2018, 6(11), 14510–14522 CrossRef CAS.
- V. Malgras, Y. Shirai, T. Takei and Y. Yamauchi, J. Am. Chem. Soc., 2020, 142, 15815–15822 CrossRef CAS PubMed.
- C. Zhao, H. Luo, F. Chen, P. Zhang, L. Yi and K. You, Energy Environ. Sci., 2014, 7, 1700–1707 RSC.
- R. Zazpe, H. Sopha, J. Prikryl, M. Krbal, J. Mistrik, F. Dvorak, L. Hromádko and J. Macak, Nanoscale, 2018, 10, 16601–16612 RSC.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 RSC.
- Z. Zhang and P. Wang, Energy Environ. Sci., 2012, 5, 6506–6512 RSC.
- A. E. R. Mohamed and S. Rohani, Energy Environ. Sci., 2011, 4, 1065–1086 RSC.
- T. Gakhar and A. Hazra, Nanoscale, 2020, 12, 9082–9093 RSC.
- Y. Alivov, V. Singh, Y. Ding, L. J. Cerkovnik and P. Nagpal, Nanoscale, 2014, 6, 10839–10849 RSC.
- H. Tsuchiya and P. Schmuki, Nanoscale, 2020, 12, 8119–8132 RSC.
- J. A. Seabold, K. Shankar, R. H. Wilke, M. Paulose, O. K. Varghese, C. A. Grimes and K.-S. Choi, Chem. Mater., 2008, 20, 5266–5273 CrossRef CAS.
- M. Marszewski, J. Marszewska, S. Pylypenko and M. Jaroniec, Chem. Mater., 2016, 28, 7878–7888 CrossRef CAS.
- M. Sluban, P. Umek, Z. Jagličić, J. E. Buh, P. Šmitek, A. Mrzel, C. Bittencourt, P. Guttmann, M.-H. Delville and D. Mihailović, ACS Nano, 2015, 9, 10133–10141 CrossRef CAS PubMed.
- Y.-J. Wei, C.-W. Peng, T.-M. Cheng, H.-K. Lin, Y.-L. Chen, C.-Y. Lee and H.-T. Chiu, ACS Appl. Mater. Interfaces, 2011, 3, 3804–3812 CrossRef CAS PubMed.
- Y. Zhao, X. Qiu and C. Burda, Chem. Mater., 2008, 20, 2629–2636 CrossRef CAS.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- E. Martínez-Ferrero, Y. Sakatani, C. Boissière, D. Grosso, A. Fuertes, J. Fraxedas and C. Sanchez, Adv. Funct. Mater., 2007, 17, 3348–3354 CrossRef.
- N. M. Nursam, X. Wang, J. Z. Tan and R. A. Caruso, ACS Appl. Mater. Interfaces, 2016, 8, 17194–17204 CrossRef CAS PubMed.
- D. Spanu, S. Recchia, S. Mohajernia, O. e. Tomanec, S. t. p. n. Kment, R. Zboril, P. Schmuki and M. Altomare, ACS Catal., 2018, 8, 5298–5305 CrossRef CAS.
- R. Vitiello, J. Macak, A. Ghicov, H. Tsuchiya, L. Dick and P. Schmuki, Electrochem. Commun., 2006, 8, 544–548 CrossRef CAS.
- D. Kim, S. Fujimoto, P. Schmuki and H. Tsuchiya, Electrochem. Commun., 2008, 10, 910–913 CrossRef CAS.
- Y. J. Jin, J. Linghu, J. Chai, C. S. Chua, L. M. Wong, Y. P. Feng, M. Yang and S. Wang, J. Phys. Chem. C, 2018, 122, 16600–16606 CrossRef CAS.
- Y. Liu, Q. Zhu, X. Li, G. Zhang, Y. Liu, S. Tang, E. Sharman, J. Jiang and Y. Luo, J. Phys. Chem. C, 2018, 122, 17221–17227 CrossRef CAS.
- M. D'Arienzo, N. Siedl, A. Sternig, R. Scotti, F. Morazzoni, J. Bernardi and O. Diwald, J. Phys. Chem. C, 2010, 114, 18067–18072 CrossRef.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
- F. Jin, X. Zhang, M. Wei, T. Chen, H. Ma and Y. Ma, J. Mater. Chem. A, 2020, 8, 20082–20090 RSC.
- X. Jiang, Y. Zhang, J. Jiang, Y. Rong, Y. Wang, Y. Wu and C. Pan, J. Phys. Chem. C, 2012, 116, 22619–22624 CrossRef CAS.
- A. Das, S. Deshagani, R. Kumar and M. Deepa, ACS Appl. Mater. Interfaces, 2018, 10, 35932–35945 CrossRef CAS PubMed.
- S. Ghosh, G. G. Khan, K. Mandal, A. Samanta and P. Nambissan, J. Phys. Chem. C, 2013, 117, 8458–8467 CrossRef CAS.
- K. Eid, K. A. Soliman, D. Abdulmalik, D. Mitoraj, M. H. Sleim, M. O. Liedke, H. A. El-Sayed, A. S. AlJaber, I. Y. Al-Qaradawi and O. M. Reyes, Catal. Sci. Technol., 2020, 10, 801–809 RSC.
- M. Samir, M. Salama and N. K. Allam, J. Mater. Chem. A, 2016, 4, 9375–9380 RSC.
- M. Wang, F. Zhang, X. Zhu, Z. Qi, B. Hong, J. Ding, J. Bao, S. Sun and C. Gao, Langmuir, 2015, 31, 1730–1736 CrossRef CAS PubMed.
- J. H. Han and J. H. Bang, J. Mater. Chem. A, 2014, 2, 10568–10576 RSC.
- J. B. Yoo, H. J. Yoo, H. J. Jung, H. S. Kim, S. Bang, J. Choi, H. Suh, J.-H. Lee, J.-G. Kim and N. H. Hur, J. Mater. Chem. A, 2016, 4, 869–876 RSC.
- J. S. Solomon and W. L. Baun, Surf. Sci., 1975, 51, 228–236 CrossRef CAS.
- K. G. Grigorov, G. I. Grigorov, L. Drajeva, D. Bouchier, R. Sporken and R. Caudano, Vacuum, 1998, 51, 153–155 CrossRef CAS.
- N. Finnegan, T. Y. Lee, R. T. Haasch, J. E. Greene and I. Petrov, Surf. Sci. Spectra, 2000, 7, 213–220 CrossRef CAS.
- R. T. Haasch, in Practical Materials Characterization, ed. M. Sardela, Springer New York, New York, NY, 2014, pp. 93–132, DOI: DOI:10.1007/978-1-4614-9281-8_3.
- O. Boese, W. E. S. Unger, E. Kemnitz and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2002, 4, 2824–2832 RSC.
- M. Fantauzzi, F. Secci, M. Sanna Angotzi, C. Passiu, C. Cannas and A. Rossi, RSC Adv., 2019, 9, 19171–19179 RSC.
- I. V. Chernyshova, S. Ponnurangam and P. Somasundaran, Phys. Chem. Chem. Phys., 2010, 12, 14045–14056 RSC.
- N. Saito and K. Ishizaki, J. Am. Ceram. Soc., 1996, 79, 1213–1217 CrossRef CAS.
- R. Pantel, D. Levy and D. Nicolas, J. Vac. Sci. Technol., A, 1988, 6, 2953–2956 CrossRef CAS.
- M. M. Soliman, M. H. Al Haron, M. Samir, S. A. Tolba, B. S. Shaheen, A. W. Amer, O. F. Mohammed and N. K. Allam, Phys. Chem. Chem. Phys., 2018, 20, 5975–5982 RSC.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989–2992 CrossRef CAS PubMed.
- S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2011, 12, 26–32 CrossRef PubMed.
- C.-T. Li, S.-R. Li, L.-Y. Chang, C.-P. Lee, P.-Y. Chen, S.-S. Sun, J.-J. Lin, R. Vittal and K.-C. Ho, J. Mater. Chem. A, 2015, 3, 4695–4705 RSC.
- M. M. Omar, S. M. Fawzy, A. B. El-Shabasy and N. K. Allam, J. Mater. Chem. A, 2017, 5, 23600–23611 RSC.
- J. Pan, D. Thierry and C. Leygraf, Electrochim. Acta, 1996, 41, 1143–1153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00274k |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |