
DAP derived fatty acid amide organogelators as novel carrier for drug incorporation and pH-responsive release†
Eqvinshi
Yadav
,
Anil Kumar
Khatana
,
Sharol
Sebastian
and
Manoj K.
Gupta
 *
*
Department of Chemistry, School of Basic Sciences, Central University of Haryana, Mahendergarh-123 031, Haryana, India. E-mail: mkgupta@cuh.ac.in; Tel: +91 9728739075
First published on 26th November 2020
Abstract
Inflammation is associated with many different class of diseases and NSAIDs (non-steroidal anti-inflammatory drugs) are mostly preferred for long-term use. Although they are safe to use, some serious side effects are associated with these class of compounds; therefore, local drug delivery is an option to minimize the side effects. In this study, we have designed a new gel formulation for topical and transdermal applications of the NSAIDs with enhanced properties. For this purpose, low molecular mass DAP (2,6-diaminopyridine) derived fatty acid amides with varying alkyl chain lengths are synthesized. These fatty acid amides form stable self-assembled aggregates in organic solvents as well as in organic and aqueous solvent mixtures affording organogels and bigels, respectively. The minimum gelation concentration (MGC) of the organic gel is 0.5% w/v, which behaves as a super gelator. The various functionality present in the DAP-derived fatty acid amide gelators play an important role in the self-aggregation such as pyridine moiety stack through π–π and alkyl chain via van der Waals interactions resulting in the formation of stable organo and bigels network. The prepared organogel emulsions with these fatty acid amides are capable to encapsulate and release the drug molecule ibuprofen at room temperature without altering its structure and activity. Therefore, these analogues can be successfully utilized in pharmaceutical industries as a novel drug delivery carrier.
Introduction
A large number of pharmaceutical drugs have poor therapeutic efficacy due to its low solubility in water.1 Therefore tremendous effort has been made to improve the low water solubility of the pharmaceutical drugs.2 Due to low membrane permeability and low bioavailability of pharmacology, it is still in high demand to investigate new techniques for rapid administration of active principles towards targeted drug delivery systems. In the recent years, research efforts have been directed toward the application of self-assembled composites as ‘‘transporters’’ to deliver molecular ‘‘cargo’’ to the targets in a sustained way,3 in which the most lucrative plan to execute these condition is the entanglement of the drugs into gels.4Gels have a tendency to trap large amount of solvents without being dissolved and produce three-dimensional cross-linked network structure with an immobilized external solvent phase. Hydrogels trap a large amount of water, whereas organogels trap organic solvents.5 Recently, some new classes of gels such as emulgels and bigels have been reported in the literature.6 The interactions among the gels are via hydrogen bonds, π–π stacking and van der Waals forces.7–9
There are various applications of hydrogels in the pharmaceutical and other fields, as reviewed by Peppas et al.10 while organogels received the attention of scientific community from late 1990s.11,12 Organogel comprises numerous applications in fat-free food products,13 drug delivery systems,14 self-healing materials,15 pollutant removal,16 analysis and purification-related systems.17 However, bigels have the advantages of both phases18 being together with and better properties than either of the single gel.19 It has a tendency to deliver both hydrophilic and hydrophobic agents,20 cooling and moisturizing effect,21 spreadability,22 washability after application,21 better permeability of drugs through the skin21 and better stability at room temperature.23 All these properties of bigels make them suitable for various applications such as pharmaceutical, cosmetics and aqueous phase, and have been extensively studied by researchers in the past decade, particularly for drug delivery applications.24,25
A number of fatty acid amides of naturally occuring amino acids forming gels having a potential for drug delivery have already been reported in the literature.26–31 However, due to the burst release of drug, the applications of these gelators in drug delivery systems are limited. To avoid this issue, Hu et al. designed new formulation compositions of amino acids (L-alanine acid, L-glutamic acids and L-serine acid) derived gelators.32 It was reported that the mechanical strength and thermal stabilities of the gel forming ability from fatty acid amides of amino acid gelators improved with the increase in chain length of the fatty acids.33a However, this procedure is not fascinating in terms of its large scale production due to the non availabilty of natural amino acids. Recently, the self-assembled amino acid biometallo hydrogels have also drawn attention to local drug delivery, long-term drug effects, drug dose reduction, and mechanical properties associated with these class of hydrogels.33b–d
Recently DAP (2,6-diaminopyridine) has gained considerable attention due to its unique symmetrical charaterisation such as their hydrogen bonding motifs34 and macrocyclic synthetic receptors.35 Although the self-assembled derivatives of 2,6-diaminopyridine forming various nanostructures such as nanotubes, nanosheets and nanofibres with a number of applications in medicine, material science and biochemistry are already reported in various literatures,36,37 no report is available for the study of gel behavior of 2,6-diaminopyridine derived fatty acid amides with varying chain lengths.
Considering the numerous applications of organic and bigels, we focused on the synthesis of a scaffold that can easily self-assemble into various solvents and provide substitutations to the naturally occuring lecithin like gelators, which are expensive and not available for large-scale production. The unique feature of these derivatives is its straight forward one-step synthesis and operational simplicity that makes this process an exciting one.
For the first time, we herewith report the DAP derived fatty acid amide derivatives as organogelators (biocompatible solvent DMSO is used as a solvent for gelation) for drug encapsulation and pH responsive release. Organogels's propensity for the delivery of NSAID was studied using ibuprofen as a model drug because it could interact with the gel not only by van der Waals forces but because of the carboxylic acids moiety, hydrogen bonding can be retained in the gel. In addition, ibuprofen is UV-active, which makes subsequent studies easier by UV spectroscopy. Moreover, the study of drug loading and its release in bigels continues.
Experimental section
Materials
Ibuprofen and analytical grade solvents were purchased from Sigma Aldrich. 2,6-Diaminopyridine, lauric acid, palmitoyl chloride, oleic acid, arachidic acid, behenic acid, oxalyl chloride and triethyl amine were purchased from Central Drug House and used as such. The distillation of solvents was done before its use.Methods
Nuclear magnetic resonance (NMR) spectra (1H and 13C) were recorded on Bruker 400 MHz and 101 MHz spectrometer, respectively. The samples were made in chloroform-d or DMSO-d6 using tetramethylsilane (TMS) as the internal standard and chemical shift values were measured in ppm on a scale downfield from TMS and the coupling constant J are in Hz. The molecular mass of the synthesized compounds were obtained by high resolution LC-MS (Agilent) instrument. The UV-visible absorption spectra were obtained with a Shimadzu UV Visible spectrophotometer. The IR spectra were obtained with FT-IR Bruker Instrument.General procedure for the syntheses of DAP derived fatty acid amide gelators
A series of DAP derived fatty acid amides were synthesized (Table 1) by reacting 2,6-diaminopyridine (1 mmol) with the corresponding acyl chloride (1 mmol) in the presence of anhyd. triethylamine (2 mmol) and anhydrous THF (10 mL). Progress of reaction was monitored with the help of TLC. After completion of the reaction, water (1 mL) was added to quench the reaction and solvents were dried under reduced pressure. The crude mass was dissolved in ethyl acetate (10 mL) and H2O (10 mL) and extracted with ethyl acetate (3 × 10 mL). The assorted organic extracts were dried over anhydrous Na2SO4, filtered and solvent was removed under reduced pressure. The native product was eluted by column chromatography using ethyl acetate–hexane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) as an eluent. The purity of the products was examined by obtaining its spectral data.
:7) as an eluent. The purity of the products was examined by obtaining its spectral data.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 157.1 (Ar-C), 149.9 (Ar-C), 140.2 (Ar-C), 104.1 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.5, 29.4, 29.2, 29.2, 25.5, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C21H37N3O: 348.3009 (M + H)+. Found 348.3012.
O), 157.0 (Ar-C), 149.9 (Ar-C), 140.2 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.6, 29.5, 29.4, 29.4, 29.3, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C17H29N3O: 292.2383 (M + H)+. Found 292.2384.
CHCH2–), 1.60 (p, J = 7.4 Hz, 2H; –CH2CH2CO–), 1.22–1.19 {m, 20H; (CH2)n–}, 0.80 (t, J = 6.6 Hz, 3H; –CH3); 13C NMR (101 MHz, chloroform-d) δ 171.7 (NHCO), 157.0 (Ar-C), 149.8 (Ar-C), 140.3 (Ar-C), 130.0 (Olefinic-C), 129.7 (Olefinic-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.8, 29.7, 29.6, 29.5, 29.3, 29.3, 29.2, 29.1, 27.2, 27.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C23H39N3O: 373.3166 (M + H)+. Found 374.3167.
O), 156.7 (Ar-C), 149.6 (Ar-C), 140.6 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.4, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1; HRMS (ESI) calcd for C25H45N3O: 404.3635 (M + H)+. Found 404.3636.
O), 156.7 (Ar-C), 149.6 (Ar-C), 140.7 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.2, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C27H49N3O: 432.3948 (M + H)+. Found 432.3952.
O), 149.5 (Ar-C), 140.8 (Ar-C), 109.4 (Ar-C), 37.8 (–COCH–), 31.8, 29.5, 29.4, 29.3, 29.2, 29.1, 25.3, 22.6 {–(CH2)n–}, 14.0 (–CH3); HRMS (ESI): calcd for C29H52N3O2: 474.4042 (M + H)+. Found: 474.4059.
O), 157.1 (Ar-C), 149.9 (Ar-C), 140.2 (Ar-C), 104.1 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.5, 29.4, 29.2, 29.2, 25.5, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C21H37N3O: 348.3009 (M + H)+. Found 348.3012.
O), 157.0 (Ar-C), 149.9 (Ar-C), 140.2 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.6, 29.5, 29.4, 29.4, 29.3, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C17H29N3O: 292.2383 (M + H)+. Found 292.2384.
CHCH2–), 1.60 (p, J = 7.4 Hz, 2H; –CH2CH2CO–), 1.22–1.19 {m, 20H; (CH2)n–}, 0.80 (t, J = 6.6 Hz, 3H; –CH3); 13C NMR (101 MHz, chloroform-d) δ 171.7 (NHCO), 157.0 (Ar-C), 149.8 (Ar-C), 140.3 (Ar-C), 130.0 (Olefinic-C), 129.7 (Olefinic-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.8, 29.7, 29.6, 29.5, 29.3, 29.3, 29.2, 29.1, 27.2, 27.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C23H39N3O: 373.3166 (M + H)+. Found 374.3167.
O), 156.7 (Ar-C), 149.6 (Ar-C), 140.6 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.4, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1; HRMS (ESI) calcd for C25H45N3O: 404.3635 (M + H)+. Found 404.3636.
O), 156.7 (Ar-C), 149.6 (Ar-C), 140.7 (Ar-C), 104.2 (Ar-C), 103.3 (Ar-C), 37.8 (–COCH–), 31.9, 29.7, 29.7, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.2, 29.2, 25.4, 22.7 {(–CH2)n–}, 14.1 (–CH3); HRMS (ESI) calcd for C27H49N3O: 432.3948 (M + H)+. Found 432.3952.
O), 149.5 (Ar-C), 140.8 (Ar-C), 109.4 (Ar-C), 37.8 (–COCH–), 31.8, 29.5, 29.4, 29.3, 29.2, 29.1, 25.3, 22.6 {–(CH2)n–}, 14.0 (–CH3); HRMS (ESI): calcd for C29H52N3O2: 474.4042 (M + H)+. Found: 474.4059.
Organogel preparation
A fixed amount of gelator was taken in a sample vial and added in 1 mL of solvent. The vial was heated up to complete dissolution of the gelator. The solution was then allowed to cool at room temperature for appropriate time (Table 2). The state of gel was examined by inverting the glass sample vial.| Gelator | Solvent (v/v: 1 mL) | Gel | Gelation time | MGC (mg mL−1) | T gel (K) |
|---|---|---|---|---|---|
| a Tube Inversion Method. b Semi-transparent gel. c Opaque gel; MCG = minimum gelation concentration; Tgel = Gel-to-sol phase transition temperature. | |||||
| 16DAP | DMSO | No | — | — | — |
| CH3OH:H2O (1:1) |
Yes | <5 min | 10 | 338 | |
| C2H5OH:H2O |
Yes | 5 min | 10 | 337 | |
| DMF:water |
No | — | — | — | |
| DMSO:water |
Yes | 10 min | 10 | 334 | |
| DCM:water |
No | — | — | — | |
| Ethylacetate:water |
No | — | — | — | |
| Acetonitrile:water |
Yes | 10 min | 10 | 336 | |
| Hexane:water |
No | — | — | — | |
| 12DAP | CH3OH:H2O (1:1) |
Yes | 24 h | 10 | 300 |
| DMSO | No | — | — | — | |
| 20DAP | CH3OH:H2O (1:1) |
Yes | <5 min | 10 | 350 |
| DMSO | No | — | — | — | |
| CH3OH | Yes | 30–45 min | 10 | 302 | |
| 22DAP | CH3OH:H2O (1:1) |
Yes | <5 min | 10 | 356 |
| DMSO | Yesb | 15–30 min | 5 | 309 | |
| CH3CN | Yesc | 30 min | 5 | 310 | |
| CH3OH | Yesc | 30 min | 5 | 313 | |
| 18UDAP | CH3OH:H2O (1:1) |
Yes | 3–4 days | 10 | — |
| DMSO | No | — | — | — | |
| 18UDAP | CH3OH:H2O (1:1) |
No | — | — | — |
| DMSO | No | — | — | — | |
Bigel preparation
A particular amount of gelator was dissolved in 0.5 mL solvent in a sample vial and heated up to complete dissolution of the gelator and then immediately added 0.5 mL distilled water to the vial. The solution was then allowed to cool at room temperature for appropriate time (Table 2). The minimum gelation concentration (MGC) was obtained by calculating minimum amount of gelator required for the formation of stable gel at room temperature. The “stable to inversion of a test tube” method was used to determine the gel state.Thermal stability
Gel-to-sol phase transition temperature (Tgel) was evaluated with a tube inversion method. The gel was taken in a glass sample vial and kept in temperature controlled oil bath at 20 °C. The temperature of the hot plate was increased by 1 °C after every 1 min. The temperature at which the viscous gel breaks down was esteemed as Tgel.Scanning electronic microscopy (SEM)
The bigels and organogels were deep freeze dried under lyoflization and morphology analysis was carried out with the SEM EVO18 Zeiss model.Drug loading
A fixed amount of gelator (22DAP) was added to 1 mL of solvent (DMSO) at their MGC and heated to form a clear solution. Then a weighed amount of ibuprofen was added to the clear gelator solution at 70 °C. The solution was mixed thoroughly and allowed to cool at temperature 20 °C for 1 h. The amount of ibuprofen was increased till stable gel formation achieved and drug loading was calculated by using the following equation.38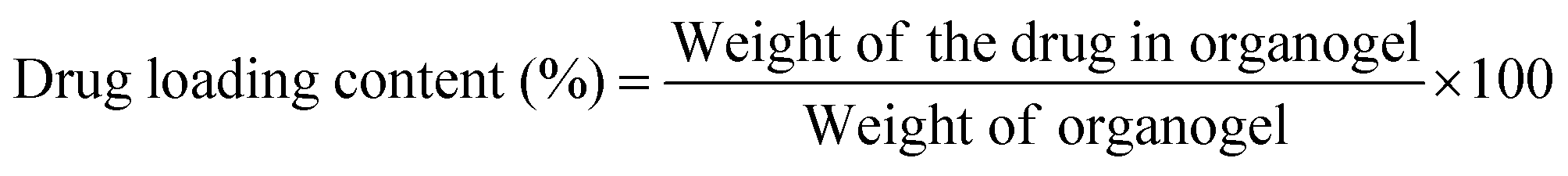 | (1) |
Drug release study
pH-Responsive drug release from the ibuprofen composite gel was studied at pH 5.5 and 7.4. Accordingly, ibuprofen encapsulated gel was placed at room temperature for 2 h to attain the equilibrium, and then the buffer solution of pH 5.5 was added and mixed thoroughly. The degradation pattern of the gel was examined at pH 5.5 by obtaining the absorbance at 264 nm in the kinetic absorption study. In the same way, the degradation pattern of the gel was examined at pH 7.4.Results and discussion
We have synthesized five derivatives of fatty acid amide gelators (12DAP, 16DAP, 18UDAP, 20DAP and 22DAP) for this study using DAP as a linker and alkyl chains of varying lengths. The products were obtained in good to excellent yields (Table 1). It was observed that when the length of alkyl chain of fatty acids increases, selective mono-acylation occurs and corresponding products were exclusively obtained (entries 4 and 5, Table 1).Gelation of the above fatty acid amide gelators was studied in various solvent system by the heating–cooling method. Initially, efforts were made to solubilize the fatty acid amide (16DAP) in water, but it was insoluble even upon heating. Then we have tried to dissolve 16DAP in polar solvents such as methanol and DMSO, and observed that it appeared as a clear solution at room temperature. However, it was heated and then cooled to room temperature, which did not afford an organogel. We next focused our study towards screening of the co-solvent system and a methanol and water mixture was used in 1:1 ratio (10 mg mL−1) and heated to 50 °C to obtain a clear solution. The resulting clear solution was allowed to cool to room temperature under ambient conditions for 5–10 min, which afforded white solid aggregates at the bottom of the sample vial. Bigel formation was confirmed by the stable-to-inversion of the test tube method (Fig. 1).38
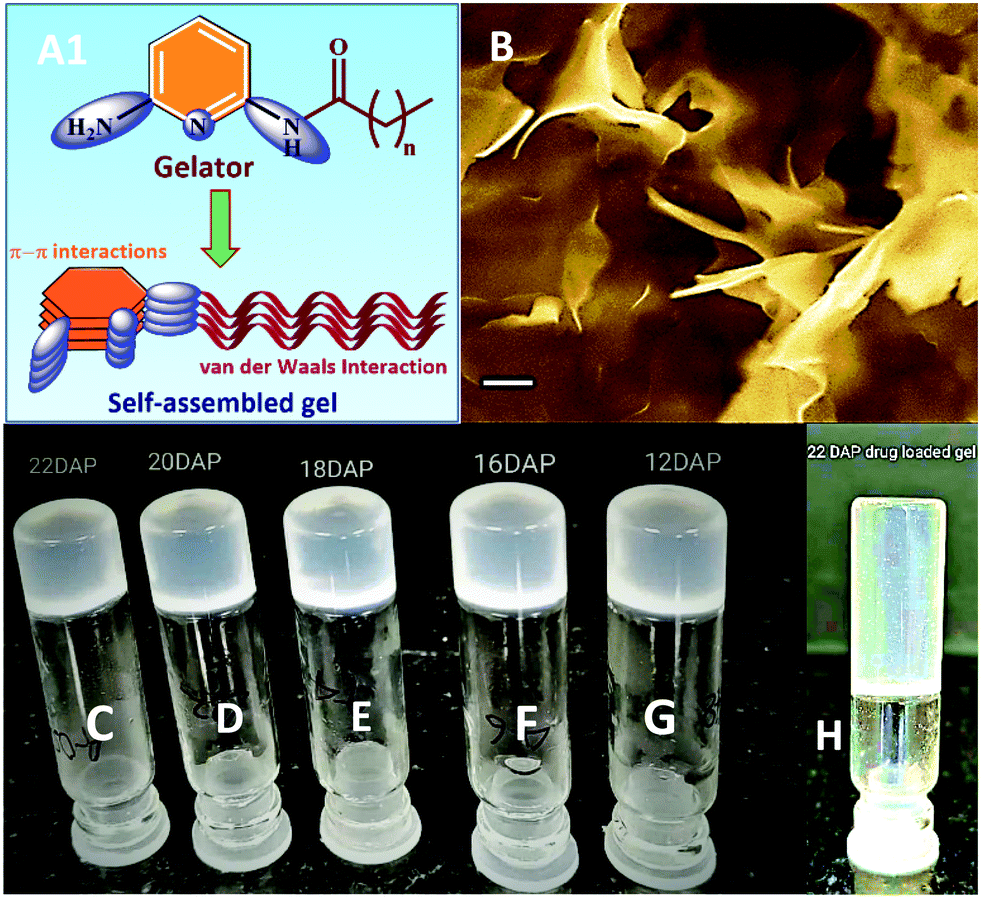 | ||
| Fig. 1 (A1) Proposed packing model for the self-assembled gel.36a (B) SEM image of bigel of 16DAP in 1:1 MeOH–H2O mixture (image recorded for xerogel; scale bar = 1 μM); photograph of vials showing gelation of fatty acid amides gelators; bigel in 1:1 MeOH–H2O mixture (C–G). C = 22DAP; D = 20DAP; E = 16DAP; F = 12DAP; G = 18UDAP; H = 22DAP (organogel formed in DMSO). | ||
Further a relative study of 16DAP gelator was performed in various solvent–water mixtures (1:1 ratio) and the obtained results are summarized in Table 2. This study gave a clear evidence that methanol–water combination should be a good choice for the self-assembling of the fatty acid amides for aggregation.
After establishing the optimal gelating conditions, next we were interested in exploring the effect of alkyl chains on the gelation behaviour. Accordingly, the length of fatty acid alkyl chain was reduced from sixteen (16DAP) to twelve carbon (12DAP), but did not yield any promising results and bigel formation occurred only after cooling the vial for 24 h at ambient temperature. However, upon increasing the length of fatty acid amides (20DAP and 22DAP), the bigel formation happened instantly, which demonstrates that the stability of bigels are directly dependent on the length of the fatty acid alkyl chains because of the increasing strength of the van der Waals forces.29 Furthermore, fatty acid amides having unsaturation (18UDAP) were screened for its gelation ability, which hardly produced bigel after 3–4 days at 20 °C. This phenomenon can be explained by the fact that the saturated fatty acid tails pack closely together, causing them to be solid at room temperature, whereas unsaturated fatty acids cannot pack closely together resulting in relatively low melting points and turned up liquid at room temperature (Fig. 1).
We further screened the gelation ability of bis fatty acid amide (12BDAP) under the optimized procedure, which failed to give any such promising results and produced a sol rather than a gel.27
Finally we have studied the concentration ratio of MeOH–H2O mixture at which the bigel formation (16DAP) occurs and results are presented in Table 3.
| Gelator (10 mg mL−1) | Methanol water 1 mL, (v/v) | Gel | Gelation time |
|---|---|---|---|
| 16GAP | 10:90 |
No | — |
| 20:80 |
Yes | 15–20 min | |
| 30:70 |
Yes | <5 min | |
| 40:60 |
Yes | <5 min | |
| 50:50 |
Yes | <5 min | |
| 60:40 |
Yes | <5 min | |
| 70:30 |
Yes | 10–15 min | |
| 80:20 |
Yes | 10–20 min | |
| 90:10 |
Yes | 30–45 min |
With the ability of these fatty acid amides to produce bigels, we expanded our investigation to screen the ability of organogel formation of longer saturated fatty acid amides in various polar organic solvents such as MeOH, DMSO and acetonitrile. Surprisingly 20DAP and 22DAP afforded organogel with MeOH (opaque gel); however, only 22DAP produced semi-transparent gel in DMSO. A detailed study of the solvents systems for the study of bi- and organogelation ability is presented in Table 2.
Bigels are more stable as compared to the organogels and its thermal stability Tgel (K) was determined by the inverted-tube method.40 In relative study, the Tgel of 12DAP, 16DAP, 20DAP and 22DAP was measured at 1% w/v of gelator and results are presented in Table 2, which suggests that with the increase of the aliphatic side chain of the fatty acid amide gelators, thermal stability of the gels simultaneously got enhanced due to the increased strength of the van der Waals forces.39 These bigels are stable for more than six months at 25–30 °C.
The 22DAP organogel (DMSO) was stable at room temperature but not at a higher temperature. Further it was also observed that the 22DAP organogel was deformed on heating and reversibly formed again on cooling, and the stability was checked by the tube inversion method (Table 2).40
Morphology of xerogels
The mechanism and the forces that drive the formation of self-assembled morphologies is not clearly understood. FAA (fatty acid amide) gelators possess the DAP moiety and it is likely that the π–π interactions between the pyridine groups provide the required rigidity and play a major role in directing the aggregation.The morphology of self-assembled freeze dried (xerogel) bigels and organogels were characterized by the scanning electron microscopy (SEM) technique. Xerogels were prepared by the removal of water and solvent from the bigels using a freeze-drying method. The morphologies of the xerogels (22DAP, 20DAP and 16DAP) are given in Fig. 2. The SEM images revealed that the bigels were self-assembled into a collection of flower bunch and vein like structures (Fig. 2).
 | ||
| Fig. 2 SEM images of xerogels: 22DAP (1 and 2; sacle bar 10 μM and 2 μM); 20DAP (3 and 4; scale bar = 2 μM); 16DAP (5 and 6; sacle bar 10 μM and 2 μM). | ||
Drug incorporation and pH-responsive release
Earlier methods reported that self-assembly derived from the chemically modified prodrug gelators and drug loaded thermo-responsive hydrogels were involved in the drug delivery processes.40 Since prodrugs were prepared by the covalent linkage of the drug molecules with the gelators, there could be a high possibility of losing original activity of the drug during chemical modification of the gelator while its release makes this process limited for drug delivery applications. Diaminopyridine derivatives are known to have a lower tendency to dimerise and can recognize and bind to ligands through hydrogen bonding.41 These results indicate that drug (ibuprofen) encapsulation in the organogels can occur through the hydrogen bond network and not by any chemical modification that may be released by the action of a pH dependent degradation method.In this study, ibuprofen, a non-steroidal anti-inflammatory drug (NSAID), was taken as the model drug, which is known to be a best-quality hydrophobic drug comprising a large hydrophobic part comprised of an aromatic ring and a carbon tail along with a small hydrophilic head where the oxygen groups are located.
The drug composite gel was found to be transparent and the amount of entrapped ibuprofen in the 22DAP organogel was 6.78% (percentage of drug loading was calculated by equation 1) in 0.5% w/v, which indicates high degree of encapsulation. The SEM morphologies of the xerogel (22DAP) containing ibuprofen (B and C) are given in Fig. 3.
 | ||
| Fig. 3 Photograph of drug loaded gel (7); SEM images of xerogel 22DAP (organogel in DMSO) contained ibuprofen (8 and 9) scale bar (8) = 2 μM; scale bar (9) = 20 μM. | ||
Fourier transmission infrared spectroscopy (FT-IR) analysis
The FT-IR spectra of ibuprofen (a); xerogel (b); and ibuprofen drug composite xerogel (c) are given in Fig. 4. In the spectrum of native xerogel (b), there are characteristic peaks at 3288 and 3164 cm−1, which are attributed to the NH bond stretching of NH2. The peaks at 2918 and 2850 cm−1 correspond to the CH stretching of the alkyl chains, whereas the peak at 1673 cm−1 corresponds to the stretching vibration of the amide carbonyl >CO and other characteristic peaks of xerogel are 2375, 2313, 716 and 710 cm−1. Ibuprofen drug comprise a strong peak at 1714 cm−1, which corresponds to the stretching vibration frequency of acid carbonyl >CO functionality while other characteristic peaks are at 2912, 2860, 1459, 1415, 1371, 864 and 514 cm−1 (a). FT-IR spectra of the drug composite xerogel (c) had bands at 1701 and 1660 cm−1, relative to the stretching vibration of the acid carbonyl >CO of ibuprofen and stretching vibration of the amide carbonyl CO of native xerogel, respectively. The other peaks are at 2912, 2860, 1459, 1415, 1371, 864 and 514 cm−1, which are relative to the characteristic peaks of ibuprofen drug while the peaks at 2375, 2313, 716 and 710 cm−1 are relative to the peaks of xerogel.
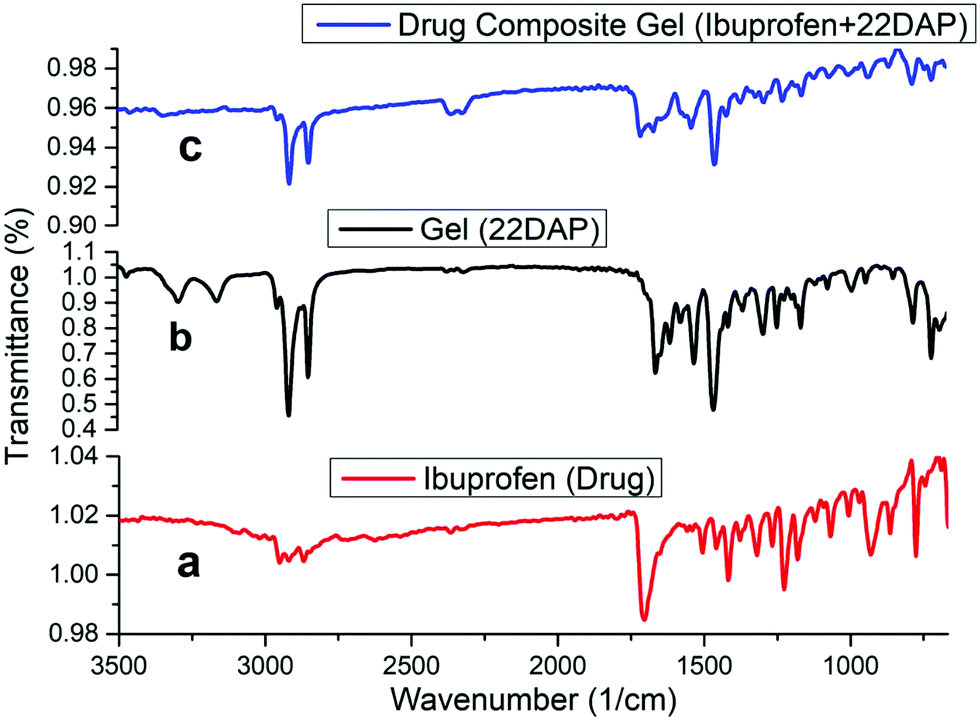 | ||
| Fig. 4 FT-IR spectra (a) ibuprofen drug; (b) organogel (22DAP); (c) ibuprofen drug composite organogel. | ||
From Fig. 4, it is clear that the drug composite xerogel contains the characteristic peaks of both the drug and native xerogel, while the absence of peaks at 3289 and 3169 cm−1, which are attributed to the NH bond stretching frequencies of the NH2 support indicates that NH2 is involved in the H-bonding in the drug composite gel. It has been observed that during the gel preparation, 22DAP forms a stable gel at neutral pH 7.4 on standing while it immediately gets precipitated at acidic pH 5.5 even on standing.
This behaviour shows that the terminal amine group plays an important role in the self-assembling process by the formation of hydrogen-bonding networks. The pH effect on the degradation of gel could be observed by the naked eye.
Comparative pH-responsive: kinetics of ibuprofen drug from organogel
The release kinetics of drug composite organogel was systematically investigated using hydrophobic ibuprofen at pH 5.5 and 7.4.3a The pH responsive gel degradation of the 22DAP gelator was useful to encapsulate the drug in the organogel and later release it by changing the pH of the medium.42 The release behavior of ibuprofen was initially investigated on gelator's MGC at pH 5.5. At a particular interval of time, 100 μL of the supernatant solution was taken out and diluted to 3 mL and then filtered. The absorbance at 264 nm was measured for each sample and the concentration of ibuprofen that was released from the organogel was obtained. The kinetic degradation profile of the drug composite organogel was recorded by UV absorption spectroscopy (Fig. 5).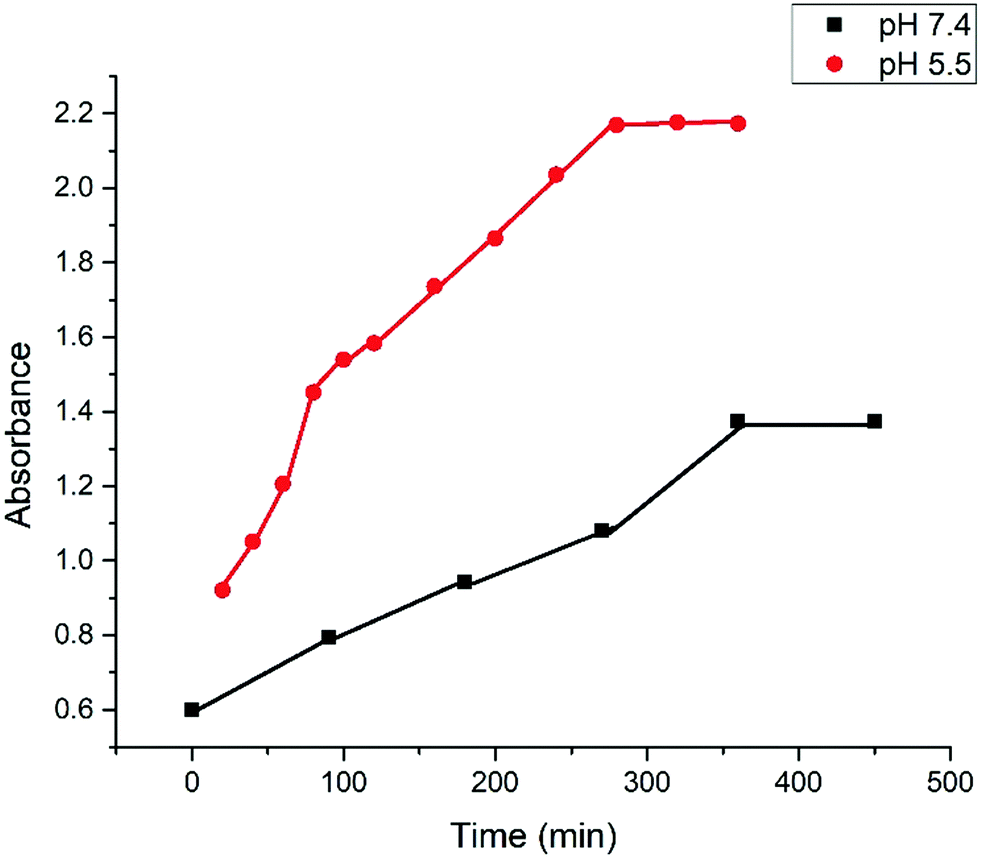 | ||
| Fig. 5 pH-Responsive cumulative release of ibuprofen from the ibuprofen composite organogels at pH 5.5 and pH 7.4. | ||
The pH-responsive cumulative release of ibuprofen from the ibuprofen composite organogel is shown in Fig. 5, which indicates that there was no more release of the drug observed after 280 min.
In case of pH 7.4, only 35% drug was released from the drug composite gel in 360 min and no more release was observed after 360 min as ibuprofen has a bulky molecular structure, leading to slower transmission through the porous channel of gel at neutral pH (7.4). However, in the case of acidic pH (5.5), the release percentage of ibuprofen was increased up to 57% in 280 min and no more release was observed afterwards. In the case of acidic pH (5.5) both the release amount and release rate were increased because of the weakening of the H-bonding interactions. These results were reproduced twice.
Conclusions
In summary, DAP-derived low molecular weight fatty acid amides were synthesized using standard organic procedures. Further, these gelators self-assembled in a water–solvent mixture and polar organic solvents and formed bi and organogel, respectively. The analogues having saturated long aliphatic chains formed stable bigels while the unsaturated analogues hardly afforded bigels. Stability of the bigels increases with increase in the length of the saturated fatty acid alkyl chain. Incubation technique concluded that the bigels are thermally more stable compared to the organogels. The minimum gelation concentration of the organic gel is 0.5% w/v, which behaves like a super gelator. The prepared gel emulsion encapsulated and released the drug molecule ibuprofen at ambient temperature without altering the structure and activity. The results reported here offer an opportunity in the field of pharmaceuticals to use these analogues as drug carrier vehicles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The SAIF facility from All India Institute of Medical Sciences (AIIMS), New Delhi is highly acknowledged for providing SEM images. EY and AKK thank Council of Scientific and Industrial Research (CSIR), New Delhi and SS thanks University Grants Commission (UGC), New Delhi for the award of research fellowships. The research leading to these results has received from the University Grants Commission, New Delhi under Early Career Start-up Research Grant [Ref. No. 30-367/2017(BSR)].References
- (a) S. Song, B. Tian, F. Chen, W. Zhang, Y. Pan, Q. Zhang, X. Yang and W. Pan, Drug Dev. Ind. Pharm., 2015, 41, 51–62 CrossRef CAS; (b) M. Z. Ahmad, A. A. Mohammed and M. M. Ibrahim, Pharm. Dev. Technol., 2016, 7450, 1–10 Search PubMed; (c) M. Z. Ahmad, S. Akhter, N. Mohsin, B. A. Abdel-Wahab, J. Ahmad, M. H. Warsi, M. Rahman, N. Mallick and F. J. Ahmad, Curr. Drug Discovery Technol., 2014, 11, 197–213 CrossRef CAS.
- (a) R. E. Kohman, S. S. Cha, H.-Y. Man and X. Han, Nano Lett., 2016, 16, 2781–2785 CrossRef CAS; (b) S. Young, M. Wong, Y. Tabata and A. G. Mikos, J. Controlled Release, 2005, 109, 256–274 CrossRef CAS; (c) S. V. Bhosale and S. V. Bhosale, Sci. Rep., 2013, 2013(3), 1982 CrossRef; (d) H. A. Strobel, E. I. Qendro, E. Alsberg and M. W. Rolle, Front. Pharmacol., 2018, 9, 1229 CrossRef.
- (a) E.-B. Lim, T. A. Vy and S.-W. Lee, J. Mater. Chem. B, 2020, 8, 2096–2106 RSC; (b) B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC; (c) C. M. Dias, H. Li, H. Valkenier, L. E. Karagiannidis, P. A. Gale, D. N. Sheppard and A. P. Davis, Org. Biomol. Chem., 2018, 16, 1083–1087 RSC; (d) W. Cui, J. Li and G. Decher, Adv. Mater., 2016, 28, 1302–1311 CrossRef CAS; (e) A. Fuertes, M. Juanes, J. R. Granja and J. Montenegro, Chem. Commun., 2017, 53, 7861–7871 RSC; (f) F. Din, W. Aman, I. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef; (g) N. Machinaga, G. W. Ashley, R. Reid, A. Yamasaki, K. Tanaka, K. Nakamura, Y. Yabe, Y. Yoshigae and D. V. Santi, TVST, 2018, 7, 21 CrossRef; (h) H. Li, H. Valkenier, A. G. Thorne, C. M. Dias, J. A. Cooper, M. Kieffer, N. Busschaert, P. A. Gale, D. N. Sheppard and A. P. Davis, Chem. Sci., 2019, 10, 9663–9672 RSC.
- (a) J. Mayr, C. Saldías and D. D. Díaz, Chem. Soc. Rev., 2018, 47, 1484–1515 RSC; (b) S. Lange, J. Unsleber, P. Drücker, H. Galla, M. P. Waller and B. J. Ravoo, Org. Biomol. Chem., 2015, 13, 561–569 RSC; (c) L. Liu, W. D. Yao, Y. F. Rao, X. Y. Lu and J. Q. Gao, Drug Delivery, 2017, 24, 569–581 CrossRef CAS; (d) S. Roy, M. Maiti, S. Das and A. Roy, Chem. Papers, 2020, 74, 183–196 CrossRef CAS; (e) K. P. C. Sekhar, D. K. Swain, S. A. Holey, S. Bojja and R. R. Nayak, Langmuir, 2020, 36, 3080–3088 CrossRef CAS; (f) K. P. C. Sekhar, H. Adicherla and R. R. Nayak, Langmuir, 2018, 34, 8875–8886 CrossRef CAS.
- E. D. Co and A. G. Marangoni, J. Am. Oil Chem. Soc., 2012, 89, 749–780 CrossRef.
- K. Rehman and M. H. Zulfakar, Drug Dev. Ind. Pharm., 2014, 40, 433–440 CrossRef CAS.
- (a) P. McNeice, Y. Zhao, J. Wang, G. F. Donnelly and P. C. Marr, Green Chem., 2017, 19, 4690–4697 RSC; (b) C. S. Hawes, A. D. Lynes, K. Byrne, W. Schmitt, G. Ryan, M. E. Mobius and T. Gunnlaugsson, Chem. Commun., 2017, 53, 5989–5992 RSC; (c) Z. Li, Y. Huang, D. Fan, H. Li, S. Liu and L. Wang, Front. Chem. Sci. Eng., 2016, 10, 552–561 CrossRef CAS; (d) D. Kong, Y. Xia, D. Li and R. Hou, Supramol. Chem., 2017, 29, 102–110 CrossRef CAS; (e) C.-W. Chu and B. J. Ravoo, Chem. Commun., 2017, 53, 12450–12453 RSC; (f) Y. G. Jia, J. Jin, S. Liu, L. Ren, J. Luo and X. X. Zhu, Biomacromolecules, 2018, 19, 626–632 CrossRef CAS.
- (a) C. Tsitsilianis, G. Serras, C.-H. Ko, F. Jung, C. M. Papadakis, M. Rikkou-Kalourkoti, C. S. Patrickios, R. Schweins and C. Chassenieux, Macromolecules, 2018, 51, 2169–2179 CrossRef CAS; (b) Y. Zhang, J. Ji, H. Li, N. Du, S. Song and W. Hou, Soft Matter, 2018, 51, 2169–2179 Search PubMed; (c) Z. Li, H. Bai, S. Zhang, W. Wang, P. Ma and W. Dong, New J. Chem., 2018, 42, 13453–13460 RSC.
- (a) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS; (b) K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS; (b) W. L. Hinze, I. Uemasu, F. Dai and J. M. Braun, Curr. Opin. Colloid Interface Sci., 1996, 1, 502–513 CrossRef CAS.
- (a) S. Murdan, B. Ven den Bergh, G. Gregoriadis and A. T. Florence, J. Pharm. Sci., 1999, 88, 615–619 CrossRef CAS; (b) J. H. Van Esch and B. L. Feringa, Angew. Chem., Int. Ed. Engl., 2000, 39, 2263–2266 CrossRef CAS; (c) H. Willimann, P. Walde, P. L. Luisi, A. Gazzaniga and F. Stroppolo, J. Pharm. Sci., 1992, 81, 871–874 CrossRef CAS.
- (a) A. R. Patel, P. S. Rajarethinem, A. Gredowska, O. Turhan, A. Lesaffer, W. H. De Vos, D. Van de Walle and K. Dewettinck, Food Funct., 2014, 5, 645–652 RSC; (b) A. R. Patel, N. Cludts, M. D. B. Sintang, A. Lesaffer and K. Dewettinck, Food Funct., 2014, 5, 2833–2841 RSC.
- B. Behera, S. S. Sagiri, K. Pal and A. Srivastava, J. Appl. Polym. Sci., 2013, 127, 4910–4917 CrossRef CAS.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- W. L. Hinze, I. Uemasu, F. Dai and J. M. Braun, Curr. Opin. Colloid Interface Sci., 1996, 1, 502–513 CrossRef CAS.
- V. K. Singh, I. Banerjee, T. Agarwal, K. Pramanik, M. K. Bhattacharya and K. Pal, Colloids Surf., B, 2014, 123, 582–592 CrossRef CAS.
- A. Blumlein and J. J. McManus, J. Mater. Chem. B, 2015, 3, 3429–3435 RSC.
- B. Behera, S. S. Sagiri, K. Pal, K. Pramanik, U. A. Rana, I. Shakir and A. Anis, Polym.-Plast. Technol. Eng., 2015, 54, 837–850 CrossRef CAS.
- I. Almeida, A. Fernandes, L. Fernandes, M. Pena Ferreira, P. Costa and M. Bahia, Pharm. Dev. Technol., 2008, 13, 487–494 CrossRef CAS.
- B. Behera, S. S. Sagiri, V. K. Singh, K. Pal and A. Anis, Starch-Starke., 2014, 66, 865–879 CrossRef CAS.
- L. Di Michele, D. Fiocco, F. Varrato, S. Sastry, E. Eiser and G. Foffi, Soft Matter, 2014, 10, 3633–3648 RSC.
- M. N. Lee and A. Mohraz, Adv. Mater., 2010, 22, 4836–4841 CrossRef CAS.
- V. K. Singh, A. Anis, S. Al-Zahrani, D. K. Pradhan and K. Pal, Int. J. Electrochem. Sci., 2014, 9, 5049–5060 Search PubMed.
- S. Gupta, R. P. Singh, A. Sarkar, H. Panchai and D. Pandey, Pharm. Globale, 2011, 5 Search PubMed.
- G. Tarun, B. Ajay, K. Bhawna, K. Sunil and J. Ravi, Int. Res. Pharm., 2011, 2, 15–21 Search PubMed.
- B. Hu, W. Wang and Y. Wang, Mater. Sci. Eng., C, 2018, 82, 80–90 CrossRef CAS.
- Z. Li, J. Cao and B. Hu, Drug Dev. Ind. Pharm., 2016, 42, 1732–1741 CrossRef CAS.
- Z. Li, J. Cao and H. Li, Drug Delivery, 2016, 23, 3168–3178 CrossRef CAS.
- B. Hu, W. Sun and H. Li, Int. J. Pharm., 2018, 547, 637–647 CrossRef CAS.
- B. Hu, H. Yan, Y. Sun, X. Chen, Y. Sun, S. Li, Y. Jing and H. Li, Artif. Cells, Nanomed., Biotechnol., 2020, 48, 266–275 CrossRef CAS.
- (a) A. Pal, Y. K. Ghosh and S. Bhattacharya, Tetrahedron, 2007, 63, 7334–7348 CrossRef CAS; (b) J. Song, C. Yan, T. Jiao, R. Xing, M. Yang, D. J. Adams and X. Yan, Small, 2020, 16, 1907309 CrossRef CAS; (c) R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS; (d) L. Ge, M. Zhang, R. Wang, N. Li, L. Zhang, S. Liu and T. Jiao, RSC Adv., 2020, 10, 15091–15097 RSC.
- M. Mazik, D. Blaser and R. Boese, Tetrahedron, 1999, 55, 12771 CrossRef CAS.
- (a) S. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318 CrossRef CAS; (b) S. Chang, D. Van Engen, E. Fan and A. D. Hamilton, J. Am. Chem. Soc., 1991, 113, 7640 CrossRef CAS.
- (a) J. S. Yadav, M. K. Gupta, I. Prathap, M. P. Bhadra, P. K. Mohan and B. Jagannadh, Chem. Commun., 2007, 3832–3834 RSC; (b) J. S. Yadav and M. K. Gupta, Int. J. Pharm. Sci. Res., 2012, 3, 4822–4826 CAS.
- M. F. Hassan and A. Rauf, J Nanostruct. Chem., 2014, 4, 83–93 CrossRef.
- B. A. Kumar and R. R. Nayak, New J. Chem., 2019, 43, 5559–5567 RSC.
- G. John, M. Mason, P. M. Ajayan and J. S. Dordick, J. Am. Chem. Soc., 2004, 126, 15012–15013 CrossRef CAS.
- K. J. C. van Bommel, M. C. Stuart, B. L. Feringa and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 RSC.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Koojman and A. Spek, J. Org. Chem., 1996, 61, 6371–6380 CrossRef CAS.
- (a) K. Lalitha, Y. S. Prasad, C. U. Maheswari, V. Sridharan, G. John and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5560–5568 RSC; (b) J. Liu, C. Detrembleur, A. Debuigne, M. C. De Pauw-Gillet, S. Mornet, L. Vander Elst, S. Laurant, E. Duguet and C. Jerome, J. Mater. Chem. B, 2014, 2, 1009–1023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04611f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |