
Visible light mediated synthesis of 4-aryl-1,2-dihydronaphthalene derivatives via single-electron oxidation or MHAT from methylenecyclopropanes†
Jiaxin
Liu
a,
Yin
Wei
 a and
Min
Shi
*ab
a and
Min
Shi
*ab
aState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@sioc.ac.cn
bShenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, Guangdong 518000, China
First published on 10th November 2020
Abstract
We report a direct single-electron oxidation of methylenecyclopropanes (MCPs) for the rapid construction of 4-aryl-1,2-dihydronaphthalene derivatives by merging visible light photoredox catalysis and cobalt catalysis. In MeCN with Et3N·3HF (1.0 equiv.), the fluorination of MCPs can be realized in the presence of 9-mesityl-10-methylacridinium perchlorate and Co(dmgH)2PyCl, affording fluorinated 4-aryl-1,2-dihydronaphthalene derivatives in moderate yields. In MeCN/HFIP (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), 4-aryl-1,2-dihydronaphthalene derivatives were obtained in good yields through a MHAT process under similar conditions.
:3), 4-aryl-1,2-dihydronaphthalene derivatives were obtained in good yields through a MHAT process under similar conditions.
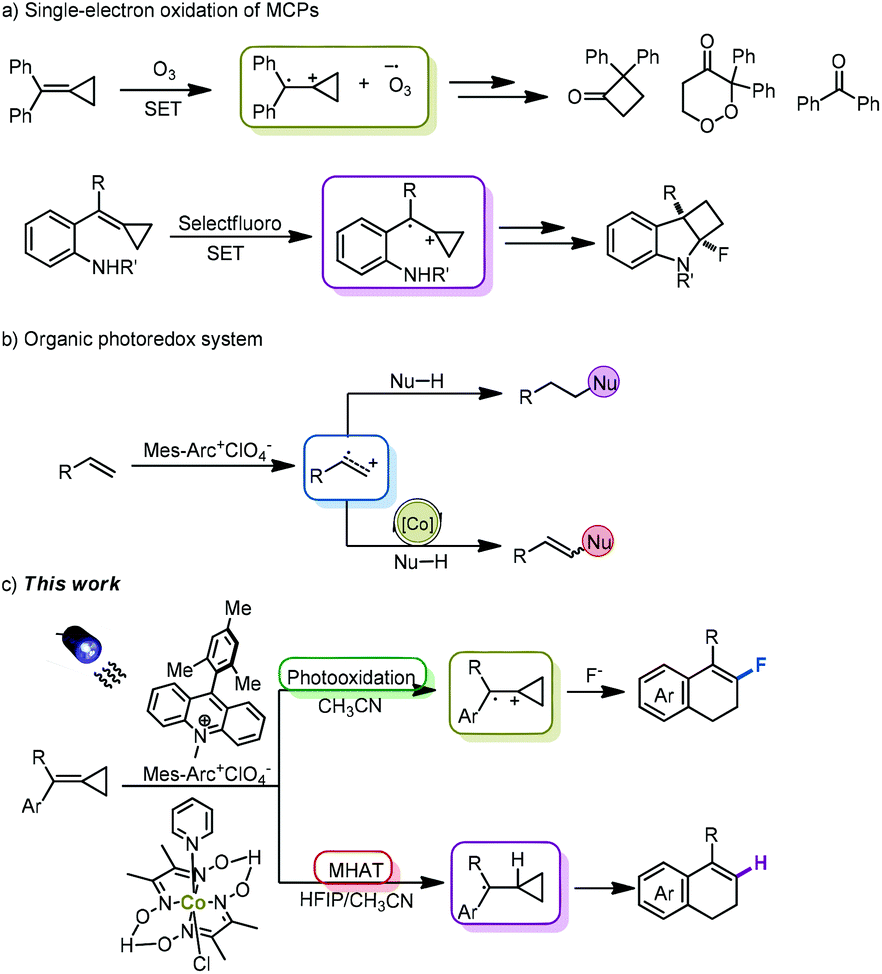
Methylenecyclopropanes (MCPs) are a class of strained small carbocycles that have drawn significant attention in the rapid construction of spiro-,1,2 hetero-3 and polycyclic4 compounds. As highly strained but readily accessible molecules,5 MCPs have also been often used as radical acceptors in reactions with a variety of radicals, furnishing the corresponding addition or cascade reaction products in traditional radical chemistry.6 However, the investigation on the reactivity of MCPs in other open-shell reaction pathways such as radical cation and triplet or singlet excited states has rarely been performed.
The direct oxidation of MCPs by ozone was first demonstrated by Beck in 2001 to afford cyclobutanone, peroxide and ketone derivatives, illustrating the possibility of a single-electron oxidation pathway with respect to MCPs (Scheme 1a).7 Another single-electron oxidation of MCPs with the employment of Selectfluor was reported by our group to forge fluorinated cyclobuta[b]indoline derivatives, rendering that the cationic radical species of MCPs could provide a new platform for the development of novel synthetic strategies in reactions with other nucleophiles (Scheme 1a).8 Therefore, we attempt to further exploit new transformations of MCPs through a single-electron oxidation process in this context.
 | ||
| Scheme 1 Our working hypothesis. | ||
Given the utility of visible light as a facile and robust alternative in photochemistry, photoredox catalysis has witnessed significant success recently and revealed fascinating potential in green chemistry.9 The renaissance of this photo-induced catalytic process depends on the utility of various of photoredox catalysts.10 Recently, Nicewicz's group developed an organic photoredox catalytic system with the utilization of 9-mesityl-10-methylacridinium perchlorate to expediently generate radical cationic species from various alkenes for reactions with a range of nucleophiles, furnishing the corresponding anti-Markovnikov addition products (Scheme 1b).11 Merging an organic photoredox catalytic system with a proton-reducing cobalt catalyst in the context of hydrogen evolution, Lei's group demonstrated various dehydrogenative cross-couplings of alkenes with a range of nucleophiles (Scheme 1b).12 Nowadays, this organic photoredox catalytic strategy has been also used by many groups to establish a variety of novel synthetic protocols under environment-benign conditions, respectively.13
Based on the aforementioned research circumstance, we could envisage a significant synthetic potential on integrating the single-electron oxidation of MCPs with an organic photoredox strategy owing to the close oxidation potential of ozone and Mes-Arc+ClO4−. Herein, we wish to report a novel synthetic method for the preparation of 4-aryl-1,2-dihydronaphthalene derivatives through the direct photooxidation of methylenecyclopropanes to radical cationic species upon visible light irradiation (Scheme 1c). The successful development of this new synthetic strategy can also enrich the chemistry of MCPs.
The preliminary investigation has been implemented by the utilization of Mes-Arc+ClO4− as a photocatalyst, which is known to catalyze a series of photooxidations of alkenes, and 4,4′-dinitrophenyl disulfide as the H atom transfer cocatalyst. On the basis of significant impact on fluorinated molecules in medicine chemistry and materials science,14 we attempted to introduce a fluorine atom into the desired 4-aryl-1,2-dihydronaphthalene derivatives in this chemical transformation. Thus, Et3N·3HF (1.0 equiv.) was employed as the fluorination reagent at the beginning, and MCP 1a was used as the substrate. Although the desired product 2a was acquired in a low yield, the formation of 2a indicated that the direct single-electron oxidation of methylenecyclopropane was feasible under mild conditions (Table 1, entry 1). It was speculated that the reduction of the in situ generated radical intermediate was the limiting process, which may not match with the generation of the H-atom transfer cocatalyst, leading to the production of 2a in a low yield.11a Thus, we turned our attention to pursue the proton-reducing cobalt catalyst, anticipating that it could facilitate this photoredox catalytic process. It was found that the yield of 2a increased to 44% upon the use of Co(dmgH)2PyCl as a replacement for the cocatalyst (Table 1, entry 2). The screening of various fluorination reagents revealed that only the use of Et3N·3HF could lead to the formation of the desired fluorinated product efficiently (Table 1, entries 3–5). By using a much stronger light source (100 W blue LEDs), the yield of 2a was improved to 55% (Table 1, entry 6). Next, a variety of solvents including DCM, 1,4-dioxane and toluene were employed in this reaction, but neither of them could provide 2a in a better yield (Table 1, entries 7–9). Meanwhile, the examination of a series of proton-reducing cobalt catalysts disclosed that Co(dmgH)2Cl2 and Co(dmgH)2(DMAP)Cl were less efficient than Co(dmgH)2PyCl (Table 1, entries 10 and 11). The screening of other photocatalysts revealed that cat II, cat III and Ir(dFCF3ppy)2(dtbbpy)PF6 could not catalyze the same reaction in as good efficiency as cat I could (Table 1, entries 12–14). The control experiments indicated that the photocatalyst, cocatalyst, and visible light irradiation were essential for this reaction (Table 1, entries 15–17) (for a more detailed examination, see Table S1 in the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Photocatalyst | F− source | Co catalyst | Solvent | Yield (%) |
| a Reactions were carried out with 1a (0.2 mmol), Et3N·3HF (1.0 equiv.), photocatalyst (3 mol%), and Co catalyst (3 mol%) in solvents (2 mL) at ambient temperature using 12 W blue LEDs irradiation for 12 hours. Yields were determined by isolated product. b Using 100 W blue LEDs. c Without photocatalyst. d Under dark conditions. | |||||
| 1 | Cat I | Et3N·3HF | 4,4′-Dinitrodiphenyl disulfide | CH3CN | 21 |
| 2 | Cat I | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | 41 |
| 3 | Cat I | CsF | Co(dmgH)2PyCl | CH3CN | — |
| 4 | Cat I | TBAF | Co(dmgH)2PyCl | CH3CN | — |
| 5 | Cat I | KHF2 | Co(dmgH)2PyCl | CH3CN | — |
| 6b | Cat I | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | 55 |
| 7b | Cat I | Et3N·3HF | Co(dmgH)2PyCl | DCM | 27 |
| 8b | Cat I | Et3N·3HF | Co(dmgH)2PyCl | 1,4-Dioxane | 14 |
| 9b | Cat I | Et3N·3HF | Co(dmgH)2PyCl | Toluene | Trace |
| 10b | Cat I | Et3N·3HF | Co(dmgH)2Cl2 | CH3CN | — |
| 11b | Cat I | Et3N·3HF | Co(dmgH)2(DMAP)Cl | CH3CN | 34 |
| 12b | Cat II | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | 27 |
| 13b | Cat III | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | 8 |
| 14b | Ir(dFCF3ppy)2(dtbbpy)PF6 | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | — |
| 15c | Cat I | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | — |
| 16b | Cat I | Et3N·3HF | — | CH3CN | Trace |
| 17d | Cat I | Et3N·3HF | Co(dmgH)2PyCl | CH3CN | — |

|
|||||

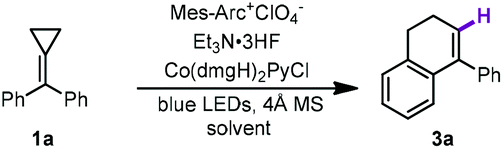
Moreover, during the examination of the solvent effects, protic solvents were also screened for probing the influence of the solvents in this reaction.11c Beyond our expectation, a hydrogen atom substituted product 3a was exclusively obtained while hexafluoroisopropanol (HFIP) was used as the cosolvent, suggesting that a different reaction pathway such as a metal-catalyzed hydrogen atom transfer process (MHAT) might have occurred under this condition.15 Therefore, the reaction conditions were also examined for this distinctly different process. The results are presented in Table 2. Using Mes-Arc+ClO4− and Co(dmgH)2PyCl as the photoredox catalytic system, 3a was obtained in 76% yield in the presence of Et3N·3HF (1.0 equiv.) with 4 Å MS in a mixed solvent of HFIP and CH3CN (Table 2, entry 1). The use of other protic solvents including trifluoroethanol (TFE) and AcOH showed lower efficiency than that of HFIP (Table 2, entries 2 and 3). The use of d4-acetic acid instead of AcOH gave 3a in a trace amount under the same conditions, suggesting that an acidic proton can affect the reaction outcome. Increasing the ratio of HFIP in the mixed solvent (CH3CN/HFIP = 1:1) gave 3a in 66% yield (Table 2, entry 4). Moreover, replacing Et3N·3HF with TFE (1.0 equiv.) or AcOH (1.0 equiv.) afforded 3a in 67% and 56% yields, respectively (Table 2, entries 5 and 6). In addition, the yield of 3a decreased to 57% in the absence of Et3N·3HF under otherwise identical conditions (Table 2, entry 7). The control experiments illustrated the requirement of a photocatalyst, Co catalyst and visible light irradiation in this new synthetic protocol (Table 2, entries 8–10).
|
|
|||
|---|---|---|---|
| Entry | Co catalyst | Solvent | Yield (%) |
| a Reactions were carried out with oven-dried 4 Å MS (50 mg), 1a (0.2 mmol), Et3N·3HF (1.0 equiv.), photocatalyst (3 mol%), and Co catalyst (3 mol%), in solvents (2 mL) at ambient temperature using 100 W blue LEDs irradiation for 12 hours. Yields were determined by isolated product. b Replacing Et3N·3HF with TFE (1.0 equiv.). c Replacing Et3N·3HF with AcOH (1.0 equiv.). d Without Et3N·3HF. e Without photocatalyst. f Under dark conditions. | |||
| 1 | Co(dmgH)2PyCl | CH3CN/HFIP (7:3) |
76 |
| 2 | Co(dmgH)2PyCl | CH3CN/TFE (7:3) |
32 |
| 3 | Co(dmgH)2PyCl | CH3CN/AcOH (7:3) |
16 |
| 4 | Co(dmgH)2PyCl | CH3CN/HFIP (1:1) |
66 |
| 5b | Co(dmgH)2PyCl | CH3CN/HFIP (7:3) |
67 |
| 6c | Co(dmgH)2PyCl | CH3CN/HFIP (7:3) |
56 |
| 7d | Co(dmgH)2PyCl | CH3CN/HFIP (7:3) |
57 |
| 8e | Co(dmgH)2PyCl | CH3CN/HFIP (7:3) |
Trace |
| 9 | — | CH3CN/HFIP (7:3) |
— |
| 10f | Co(dmgH)2Cl2 | CH3CN/HFIP (7:3) |
— |
With the optimized reaction conditions in hand, the generality for the production of fluorinated 4-aryl-1,2-dihydronaphthalene derivatives 2 was investigated using a series of substituted methylenecyclopropanes as substrates. The results are shown in Scheme 2. To our delight, using 4,4′-disubstituted bisaryl-MCPs as substrates, the reactions proceeded smoothly, affording the corresponding products 2a–2e in moderate yields whether the 4,4′-disubstituted bisaryl-MCPs had electron-donating or electron-withdrawing groups on the aromatic rings. With the use of 3,3′-dimethyl-substituted bisaryl-MCP as the substrate, two regioisomers 2f and 2f′ were afforded in 54% total yield in 4:1 ratio perhaps due to the steric effect. However, using 3,3′-dichloro-substituted bisaryl-MCP in the reaction only gave the desired product 2g in 37% yield presumably due to the steric and electronic effects. 3,3′-Dimethoxyl-substituted bisaryl-MCP was not tolerated without the formation of the desired product. The reaction also proceeded efficiently upon using 2,2′-dimethyl-substituted bisaryl-MCP 1i as the substrate, giving the desired product 2i in a moderate yield. In addition, as for monosubstituted aromatic MCPs, most of them afforded regioisomers in the ring-closing step, and the corresponding products 2j–2l and 2j′–2l′ were obtained in moderate total yields with different regioisomeric ratios (1.1:1, 1:1.3 and 2:1), respectively, due to the electronic effect. Only in the case of 2-chloro-substituted bisaryl-MCP 1m, the desired product 2m was acquired as one regioisomer, perhaps also due to the steric and electronic effects. Unfortunately, when 2-iodo-substituted bisaryl-MCP 1n was employed, none of the desired product was formed. Using monoaryl-substituted MCP as the substrate, no reaction occurred under the standard conditions. It should be mentioned here that benzophenones, derived from the oxidation of methylenecyclopropanes, and dihydronaphthalenes 3 were also formed at the same time, leading to the formation of 2 in moderate yields.
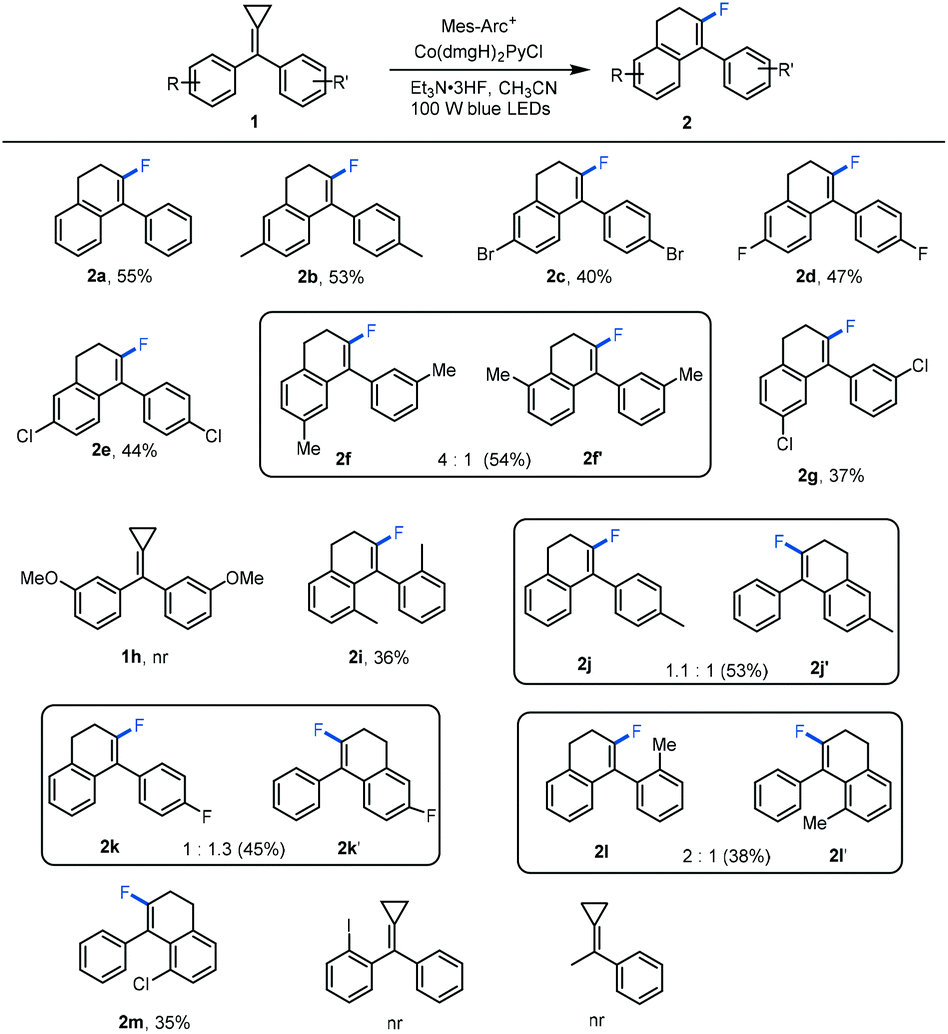 | ||
| Scheme 2 Substrate scope of fluorinated products. The reactions were carried out with 1 (0.2 mmol), a photocatalyst (3 mol%), and a Co catalyst (3 mol%) in solvents (2 mL) at ambient temperature using 100 W blue light irradiation for 12 hours. The yields were determined by isolated product. | ||
Subsequently, we investigated the substrate scope of this method to acquire 4-aryl-1,2-dihydronaphthalene. The results are shown in Scheme 3. As for 4,4′-disubstituted bisaryl-MCPs, regardless of whether an electron-donating or electron-withdrawing group was employed, the desired products 3a–3e could be obtained in moderate to good yields ranging from 54% to 76%. It should be noted that 3c and 3d were obtained as arylnaphthalene derivatives rather than 1,2-dihydronaphthalene derivatives due to the over oxidation during the reaction.16 Similarly, using 3,3′-dimethyl-substituted bisaryl-MCP 1f as the substrate, two regioisomers 3f and 3f′ were obtained in 73% total yield in 2:1 ratio perhaps due to the steric effect. However, in the cases of 3,3′-disubstituted bisaryl-MCPs 1g and 1h, only the corresponding single regioisomers 3g and 3h were obtained in 66% and 72% yields, respectively probably due to the radical property of this cyclization reaction. Moreover, the reaction also proceeded smoothly by using 2,2′-dimethyl-substituted bisaryl-MCP 1i as the substrate, affording the desired product 3i in 55% yield. Furthermore, 4-monosubstituted aromatic MCPs were examined as well, delivering the corresponding regioisomers 3j & 3j′ and 3k & 3k′ in 74% and 66% total yields with 1.1:1 and 1:2.5 ratios, respectively, due to the electronic effect. For 2-monosubstituted aromatic MCPs 1l and 1m, the corresponding products 3l and 3m were formed as only one regioisomer in 51% and 46% yields, respectively, presumably also due to the steric and electronic effects.
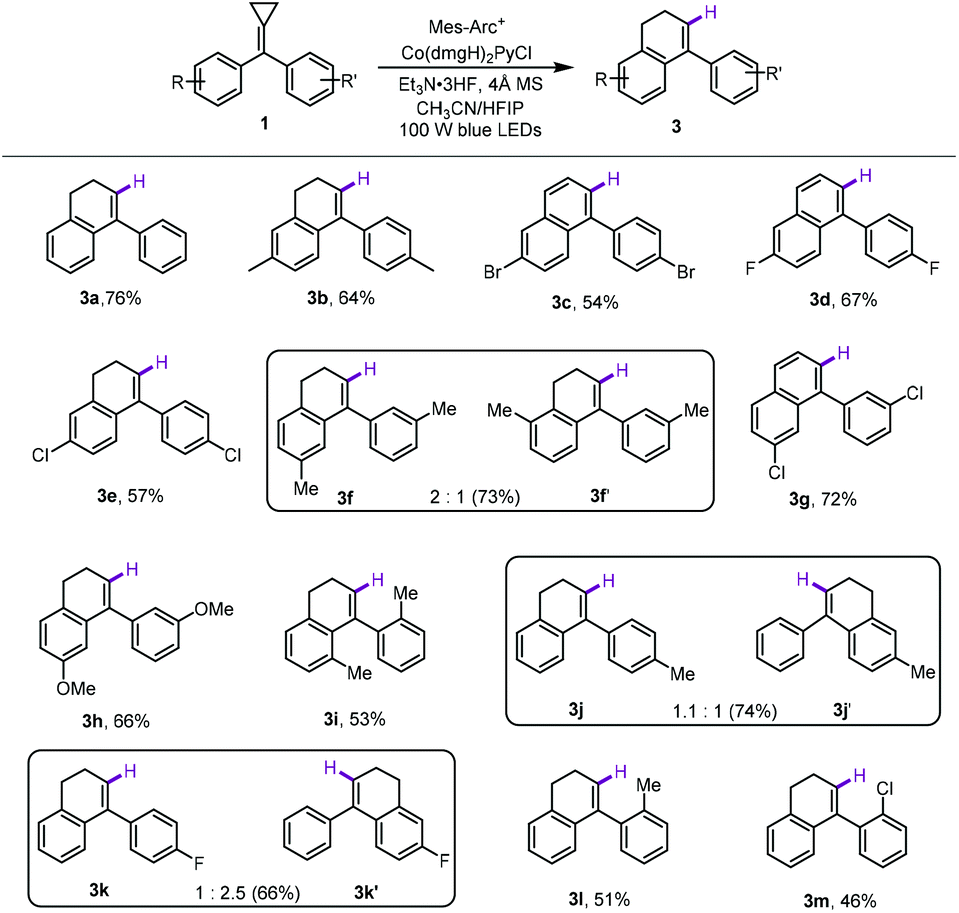 | ||
| Scheme 3 Substrate scope of the MHAT products. The reactions were carried out with oven-dried 4 Å MS (50 mg), 1 (0.2 mmol), a photocatalyst (3 mol%), and a Co catalyst (3 mol%) in 2 mL solvents (CH3CN/HFIP = 7:3) at ambient temperature using 100 W blue light irradiation for 12 hours. The yields were determined by isolated product. | ||
To understand the mechanistic insights of this reaction, a series of control experiments were carried out to investigate the reaction sequence. As shown in Scheme 4, the fluorescence quenching experiments of 9-mesityl-10-methylacridinium perchlorate with 1a and its Stern–Volmer analysis suggested that 1a could be oxidized by photocatalyst effectively through an SET process upon photo-irradiation. Namely, the fluorination reaction of MCPs was initiated by the direct single-electron photooxidation of MCPs with the generation of their radical cationic species, indicating that this visible light mediated catalytic system could provide a facile method to oxidize MCP to its radical cation via an open-shell pathway.
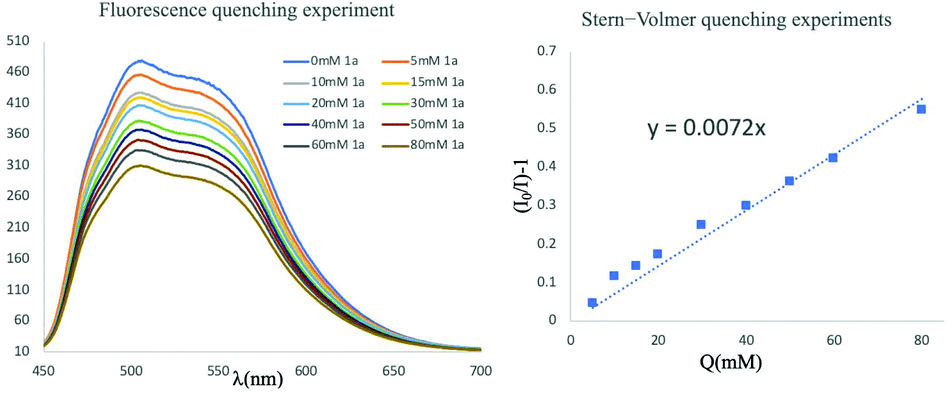 | ||
| Scheme 4 Fluorescence quenching experiments. | ||
On the basis of previous investigations12,13,15 and the above mechanistic study, two plausible mechanisms to illustrate the formation of two different products are shown in Schemes 5 and 6, respectively. For the fluorination of MCP,12 an SET process took place between substrate 1a and the PS* excited by visible light irradiation to generate a radical cation I and PS˙, which was attacked by a nucleophilic fluorine anion to generate a radical intermediate II. Meanwhile, the CoII species was reduced by PS˙ to generate CoI species, which captured a proton to generate CoIII–H species.15f The radical intermediate II underwent a ring-opening process and an intramolecular ring-closing process stepwise to give a radical intermediate IV, which was further oxidized by CoIII species to obtain a cationic intermediate V and regenerate CoII species. The desired product 2a was obtained by releasing a proton from V. The initial formation of CoII could be realized through an SET process between a CoIII catalyst and the in situ generated radical intermediate IV.12c
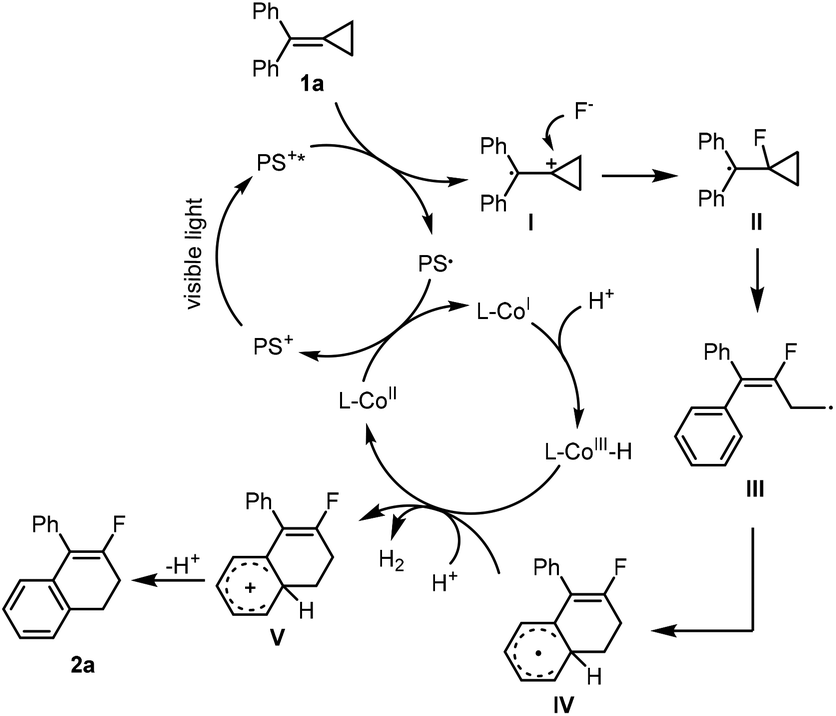 | ||
| Scheme 5 Mechanistic paradigm for the production of 2a. | ||
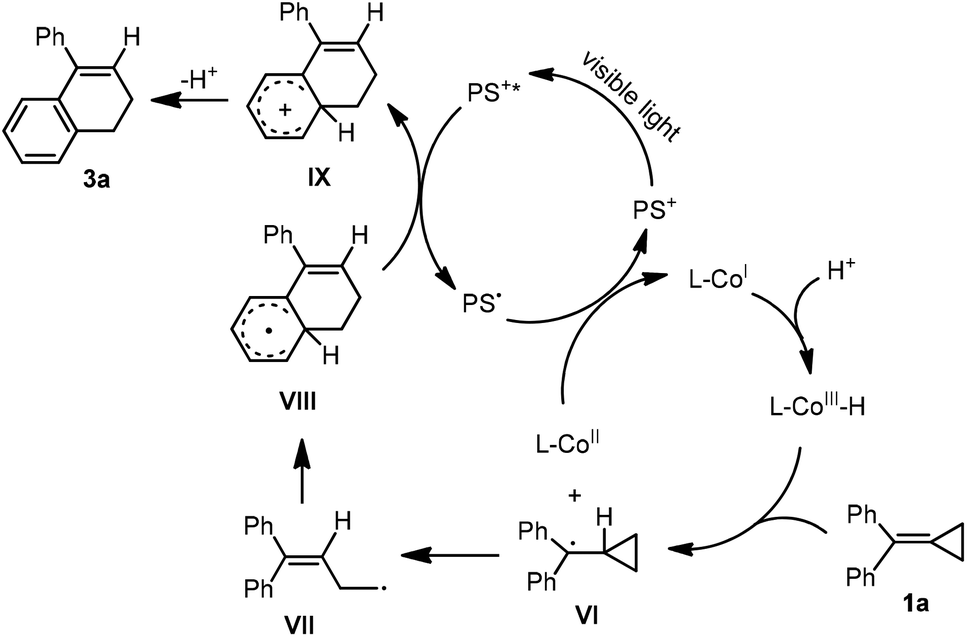 | ||
| Scheme 6 Proposed mechanism for the production of 3a. | ||
As for the formation of a hydrogen atom substituted product 3a, a MHAT process was suggested. Initially, the CoI species was generated by the above catalytic cycle. Subsequently, the CoI species was transformed to a CoIII–H species easily in a protic solvent.15f Then, a MHAT process took place between the CoIII–H species and 1a to generate a radical intermediate VI and CoII species.15g The generated CoII species was reduced by PS˙ to regenerate CoI species, and the radical intermediate VI underwent a ring-opening process and an intramolecular ring-closing process stepwise to give a radical intermediate VIII, which was further oxidized by PS* upon excitation by visible light irradiation to obtain a cationic intermediate IX. The release of a proton from intermediate IX afforded the desired product 3a.
In summary, we have accomplished the first example of a direct photooxidation and a MHAT process for the transformation of MCPs under mild conditions. These novel synthetic methods provide new protocols for the rapid construction of 4-aryl-1,2-dihydronaphthalene derivatives in moderate to good yields with a photoredox catalytic system induced by visible light irradiation, thereby demonstrating a robust synthetic strategy beyond photoredox catalysis to integrating a single-electron oxidation and a MHAT process into MCP chemistry. Further exploration of these synthetic methods from strained small rings is underway in our laboratory.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000) and sioczz201808, the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014 and 91956115) and the Shenzhen Nobel Prize Scientists Laboratory Project.Notes and references
- For reviews:
(a) J. Liu, R. Liu, Y. Wei and M. Shi, Recent Developments in Cyclopropane Cycloaddition Reactions, Trends Chem., 2019, 1, 779 CrossRef CAS
; (b) D.-H. Zhang, X.-Y. Tang and M. Shi, Gold-Catalyzed Tandem Reactions of Methylenecyclopropanes and Vinylidenecyclopropanes, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS
-
(a) F. Verdugo, L. Villarino, J. Durán, M. Gulías, J. L. Mascareñas and F. López, Enantioselective Palladium-Catalyzed [3C + 2C] and [4C + 3C] Intramolecular Cycloadditions of Alkylidenecyclopropanes, ACS Catal., 2018, 8, 6100 CrossRef CAS
-
(a) S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, Stereoselective Rhodium-Catalyzed [3 + 2 + 1] Carbocyclization of Alkenylidenecyclopropanes with Carbon Monoxide: Theoretical Evidence for a Trimethylenemethane Metallacycle Intermediate, J. Am. Chem. Soc., 2012, 134, 20569 CrossRef CAS
-
(a) K. Chen, X.-Y. Tang and M. Shi, Rh(II)-Catalyzed Formation of Pyrrolo[2,3-b]quinolines from Azide-methylenecyclopropanes and Isonitriles, Chem. Commun., 2016, 52, 1967 RSC
-
(a) C. A. Carsona and M. A. Kerr, Heterocycles from Cyclopropanes: Applications in Natural Product Synthesis, Chem. Soc. Rev., 2009, 38, 3051 RSC
-
(a) L.-Z. Yu, K. Chen, Z.-Z. Zhu and M. Shi, Recent Advances in the Chemical Transformations of Functionalized Alkylidenecyclopropanes (FACPs), Chem. Commun., 2017, 53, 5935 RSC
- R. F. Langler, R. K. Raheja, K. Schank and H. Beck, Ozonolysis of Allenes and Alkylidenecyclopropanes (Homoallenes), Helv. Chim. Acta, 2001, 84, 1943 CrossRef CAS
- X. Fan, Q. Wang, Y. Wei and M. Shi, Catalyst-Free Geminal Aminofluorination of Ortho-Sulfonamide-Tethered Alkylidenecyclopropanes via a Wagner-Meerwein Rearrangement, Chem. Commun., 2018, 54, 10503 RSC
-
(a) C. K. Prier, D. A. Rankic and D. W. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS
-
(a) D. A. Nicewicz and D. W. C. MacMillan, Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes, Science, 2008, 322, 77 CrossRef CAS
-
(a) D. S. Hamilton and D. A. Nicewicz, Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS
-
(a) G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C-H Thiolation for Constructing C–S Bonds, J. Am. Chem. Soc., 2015, 137, 9273 CrossRef CAS
-
(a) Q. Xu, B. Zheng, X. Zhou, L. Pan, Q. Liu and Y. Li, Photoinduced C(sp2)–H/C(sp2)–H Cross-Coupling of Alkenes: Direct Synthesis of 1,3-Dienes, Org. Lett., 2020, 22, 1692 CrossRef CAS
-
(a) K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881 CrossRef
-
(a) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins, Chem. Rev., 2016, 116, 8912 CrossRef CAS
-
(a) J. B. McManus, J. D. Griffin, A. R. White and D. A. Nicewicz, Homobenzylic Oxygenation Enabled by Dual Organic Photoredox and Cobalt Catalysis, J. Am. Chem. Soc., 2020, 142, 10325 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. See DOI: 10.1039/d0qo00853b |
| This journal is © the Partner Organisations 2021 |