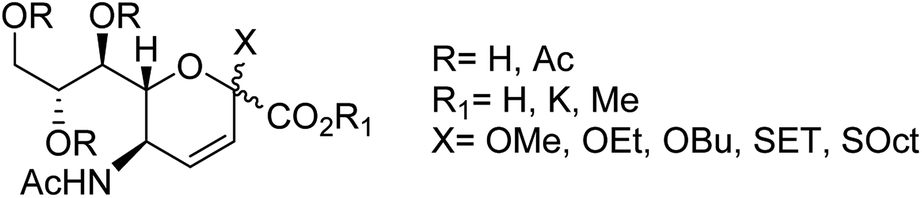DOI:
10.1039/D0RA07283D
(Review Article)
RSC Adv., 2021,
11, 1804-1840
Recent progress in chemical approaches for the development of novel neuraminidase inhibitors
Received
25th August 2020
, Accepted 22nd November 2020
First published on 6th January 2021
Abstract
Influenza virus is the main cause of an infectious disease called influenza affecting the respiratory system including the throat, nose and lungs. Neuraminidase inhibitors are reagents used to block the enzyme called neuraminidase to prevent the influenza infection from spreading. Neuraminidase inhibitors are widely used in the treatment of influenza infection, but still there is a need to develop more potent agents for the more effective treatment of influenza. Complications of the influenza disease lead to death, and one of these complications is drug resistance; hence, there is an urgent need to develop more effective agents. This review focuses on the recent advances in chemical synthesis pathways used for the development of new neuraminidase agents along with the medicinal aspects of chemically modified molecules, including the structure–activity relationship, which provides further rational designs of more active small molecules.
1. Introduction
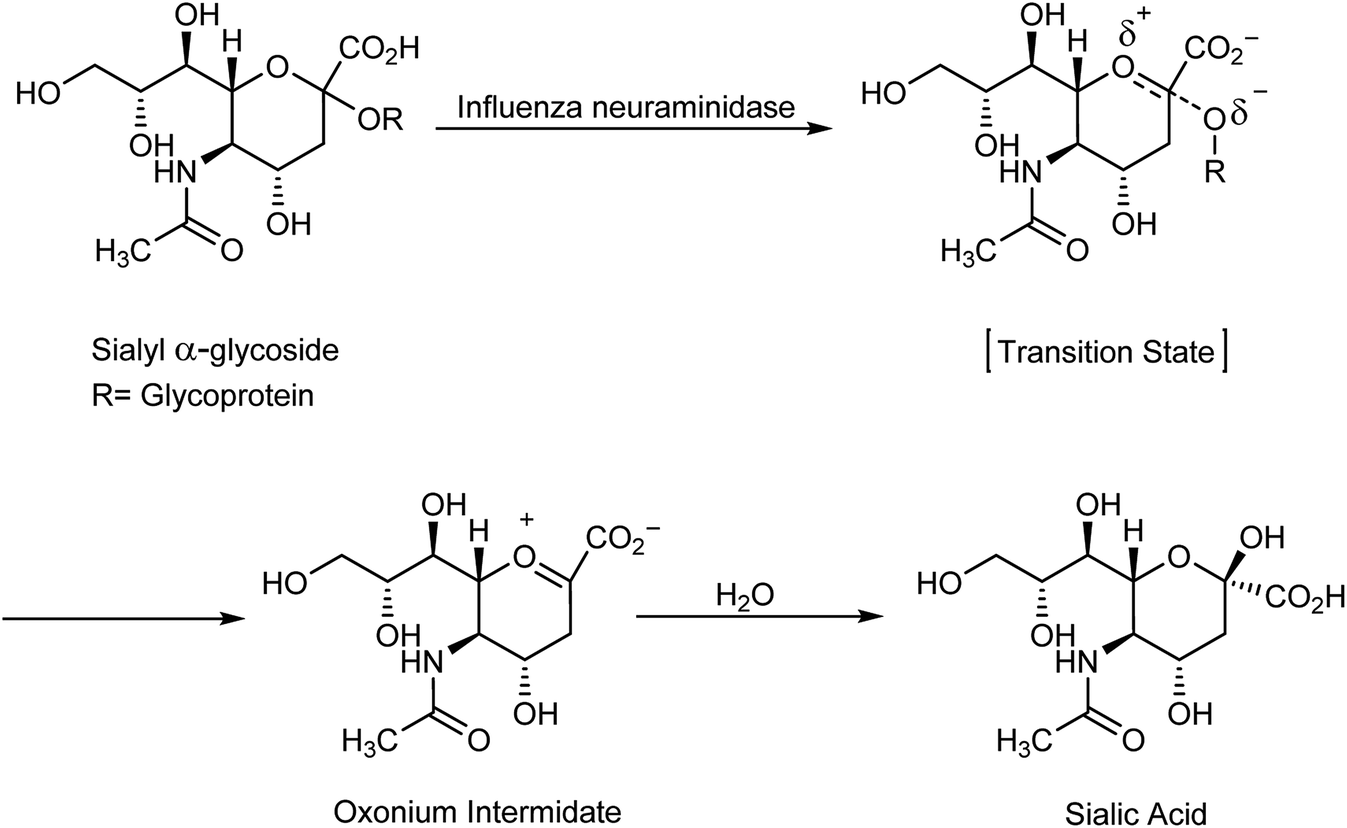
A highly infectious disease called influenza or flu is caused by influenza virus, and has symptoms such as fever, headache, muscle pains and runny nose, causing 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 500000 deaths.1 Importantly, there are three types of flu: A and B viruses are responsible for annual influenza epidemics, whereas C virus has less severe symptoms. The two classes of obtainable antiviral agents involve M2 inhibitors, which have potency only against virus A. Two classes of obtainable antiviral agents are involving the M2 inhibitors which have only potency against influenza A virus but with side effect including short time of effeteness and viral strain resistance2–7 and the neuraminidase inhibitors (NAIs) which have potency against influenza A and B viruses.8 Influenza is an enveloped RNA virus, which is a highly contagious pathogen worldwide and affects millions of people, animals and birds each year in seasonal epidemics.9,10 Neuraminidase inhibitors are drugs that inactivate viral neuraminidase (NA) protein. Blocking sialic acid receptors and inhibiting virus–host cell interactions have been the best possible way to control and prevent the infection. This stops the release of viruses and prevents new host cells from being infected.11 Therefore, inactivating NA using neuraminidase inhibitors offers the best possible method to inhibit sialic acid receptors. Oseltamivir and zanamivir are NA inhibitors, which inhibit all the subtypes of neuraminidase enzymes, and therefore, are effective against influenza viruses A and B. The role of neuraminidase inhibitors is important in the treatment of influenza, which act via blocking the release of viruses from the infected host cell and prevent the new host cells from infection. The role of antiviral agents is to block the neuraminidase enzymes from being more effective against A and B viruses. Neuraminidase inhibitors act by preventing the cleavage of sialic acid by a glycoprotein enzyme called neuraminidase and inhibiting the distribution of viruses to neighboring cells12,13 (Scheme 1).
000 to 500000 deaths.1 Importantly, there are three types of flu: A and B viruses are responsible for annual influenza epidemics, whereas C virus has less severe symptoms. The two classes of obtainable antiviral agents involve M2 inhibitors, which have potency only against virus A. Two classes of obtainable antiviral agents are involving the M2 inhibitors which have only potency against influenza A virus but with side effect including short time of effeteness and viral strain resistance2–7 and the neuraminidase inhibitors (NAIs) which have potency against influenza A and B viruses.8 Influenza is an enveloped RNA virus, which is a highly contagious pathogen worldwide and affects millions of people, animals and birds each year in seasonal epidemics.9,10 Neuraminidase inhibitors are drugs that inactivate viral neuraminidase (NA) protein. Blocking sialic acid receptors and inhibiting virus–host cell interactions have been the best possible way to control and prevent the infection. This stops the release of viruses and prevents new host cells from being infected.11 Therefore, inactivating NA using neuraminidase inhibitors offers the best possible method to inhibit sialic acid receptors. Oseltamivir and zanamivir are NA inhibitors, which inhibit all the subtypes of neuraminidase enzymes, and therefore, are effective against influenza viruses A and B. The role of neuraminidase inhibitors is important in the treatment of influenza, which act via blocking the release of viruses from the infected host cell and prevent the new host cells from infection. The role of antiviral agents is to block the neuraminidase enzymes from being more effective against A and B viruses. Neuraminidase inhibitors act by preventing the cleavage of sialic acid by a glycoprotein enzyme called neuraminidase and inhibiting the distribution of viruses to neighboring cells12,13 (Scheme 1).
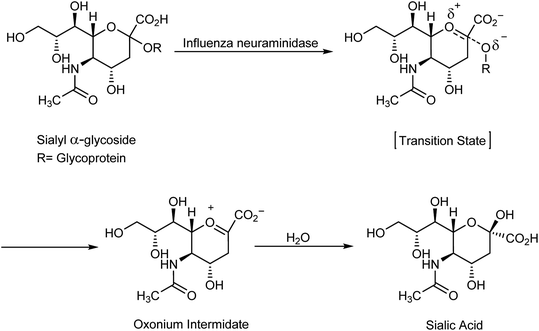 |
| | Scheme 1 Hydrolysis step of sialylglycosides. | |
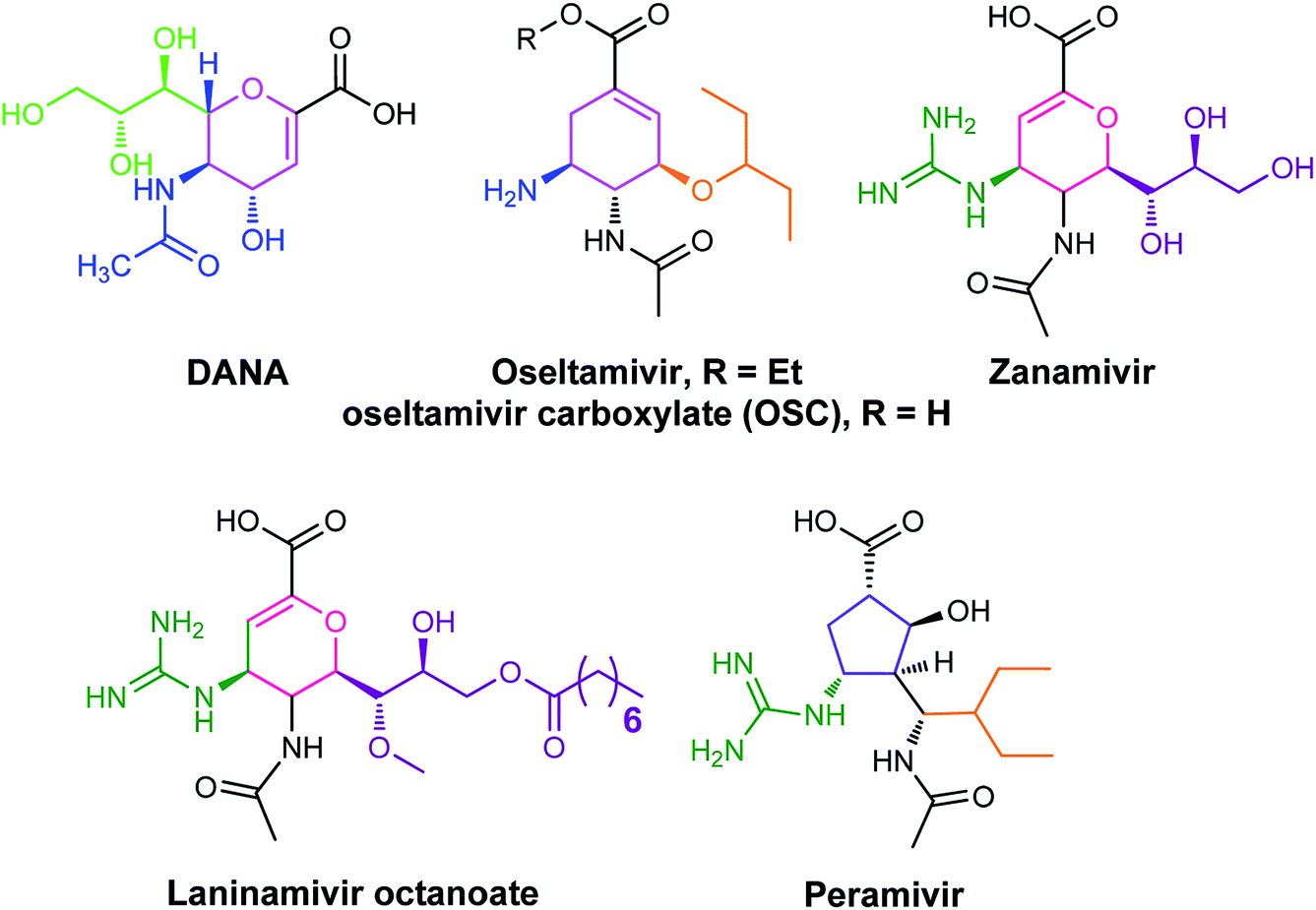
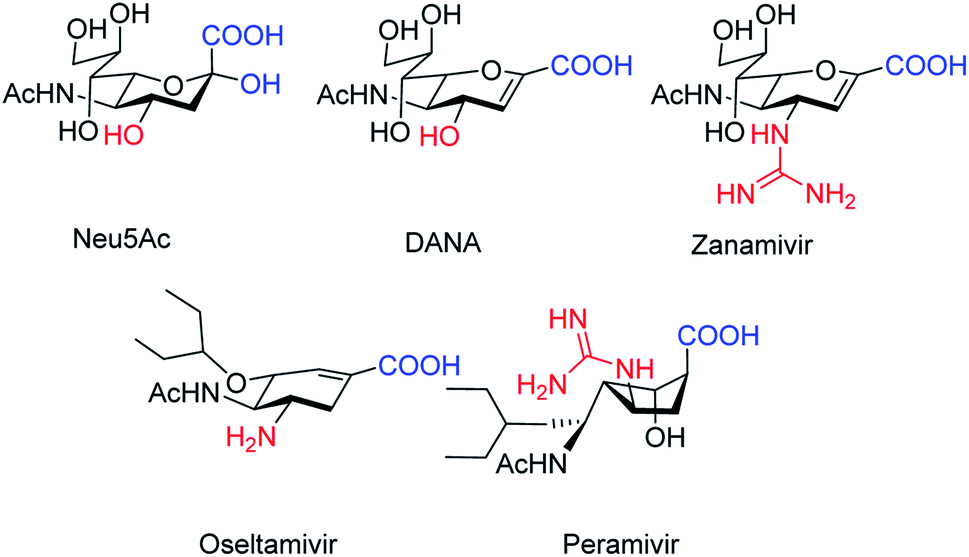
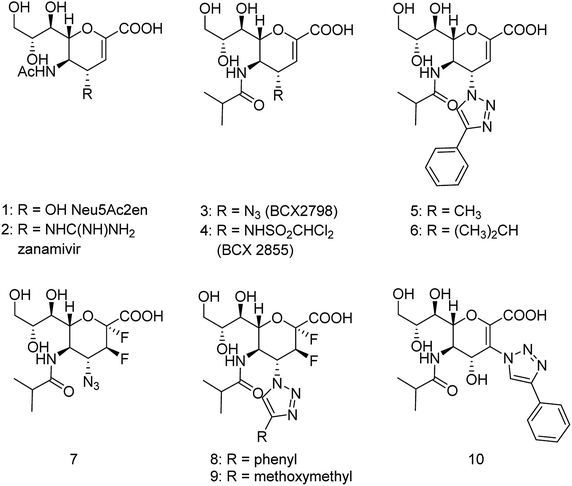
The antiviral agents are considered to be alternative choices of the use of vaccines since there are many disadvantages including the time of production storage and effective time limitation. There are many approved neuraminidase drugs in the market worldwide including 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA), zanamivir (Relenza),14 oseltamivir phosphate (Tamiflu),15,16 laninamivir17 and peramivir,18 as shown in Fig. 1.
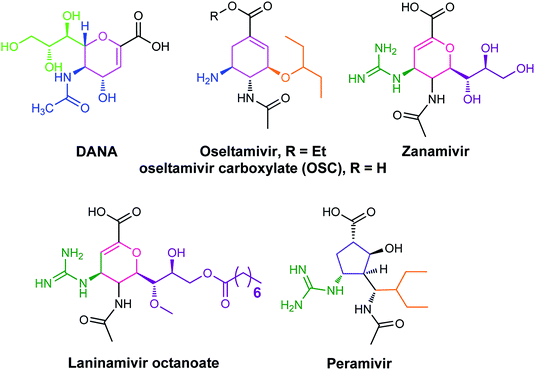 |
| | Fig. 1 DANA, zanamivir and oseltamivir phosphate structures. | |
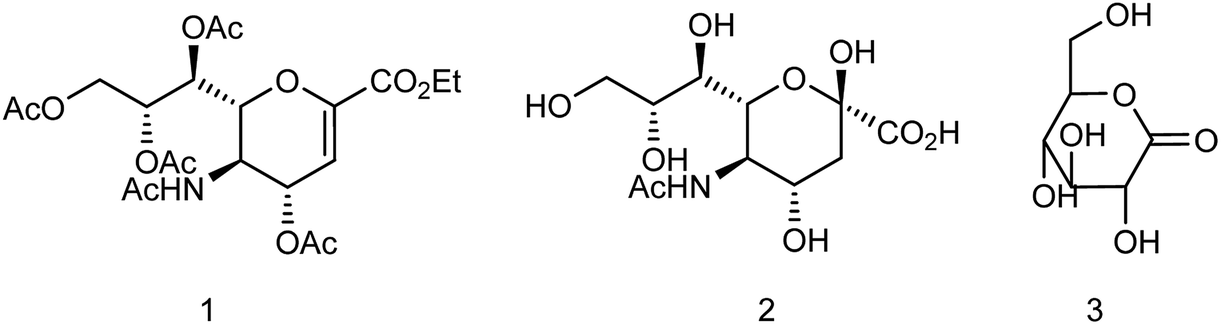
Zanamivir has low bioavailability and can be administrated by inhalation and was first synthesized from methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-2,6-anhydro-3,5-dideoxy-D-glycero-D-galacto-non-2-enonate (1),19–22 N-acetylneuraminic acid (2, NANA),23 N-acetylneuraminic acid 2 (ref. 24) and D-glucono-δ-lactone 3 (ref. 25) (Fig. 2).
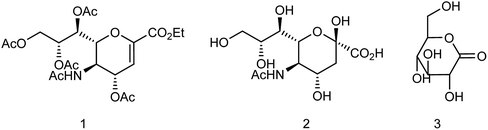 |
| | Fig. 2 Structures of starting materials for the synthesis of zanamivir. | |
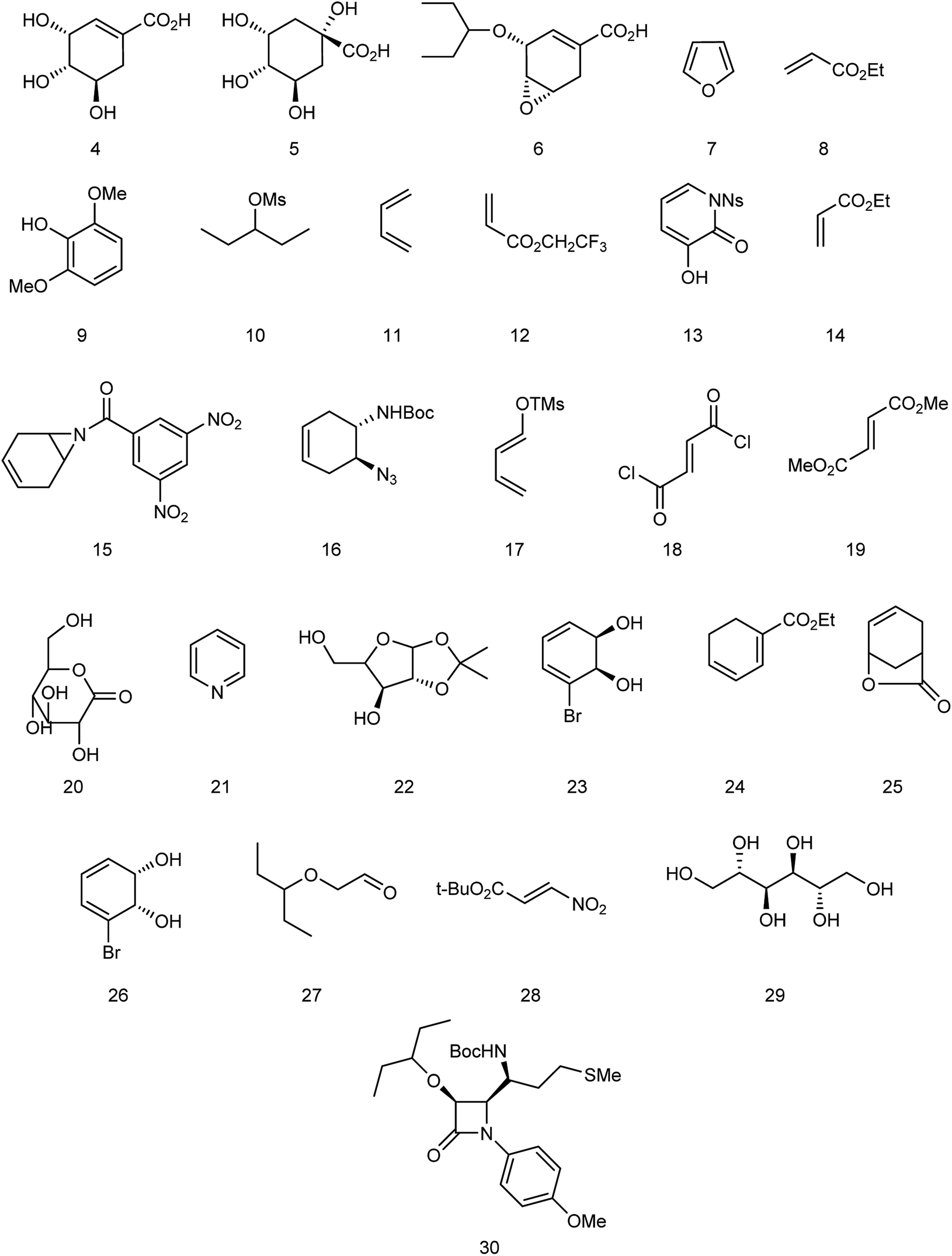
Oseltamivir phosphate is an oral active prodrug that requires ester hydrolysis to form active oseltamivir carboxylate as an inhibitor against influenza virus. Many strategies were described for the synthesis of this inhibitor including Gilead starting from (−)-shikimic acid (4)26 and (−)-quinic acid (5),27 F. Hoffmann-La Roche Ltd starting from (−)-shikimic acid (4) and (−)-quinic acid (5),28 epoxide (6),29 furan (7) and ethyl acrylate (8),30 2,6-dimethoxyphenol (9) and mesylate (10),30 Roche Colorado Corporation starting from epoxide (6),31 Corey starting from 1,3-butadiene (11) and 2,2,2-trifluoroethyl acrylate (12),32 Okamura starting from N-nosyl-3-hydroxy-2-pyridone (13) and ethyl acrylate (14),33 Shibasaki starting form N-3,5-dinitrobenzoylaziridine (15),34,35 azide (16),35 1-(trimethylsiloxy)-1,3-butadiene (17) and fumaryl chloride (18),36 1-(trimethylsiloxy)-1,3-butadiene (17) and dimethyl fumarate (19),37 Yao starting from D-glucono-δ-lactone (20),38 Fukuyama starting from pyridine (21),39,40 Fang starting from D-xylose (22)41 and cis-1,2-dihydrodiol (23),42 Kann starting from cyclohexadienoic acid ethyl ester (24),43 Trost starting from lactone (25),44 Banwell starting from cis-1,2-dihydrodiol (26),45 Shi starting from (−)-shikimic acid (4),46,47 Hayashi starting from aldehyde (27) and nitroalkene (28),48 Mandai starting from D-mannitol (29)49 and L-methionine derivative (30)50 (Fig. 3).
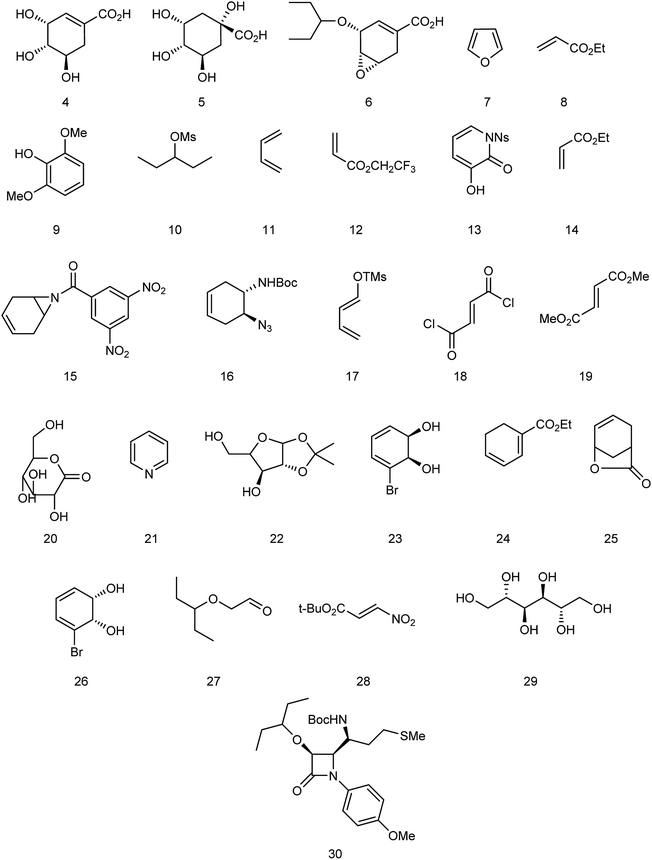 |
| | Fig. 3 Structures of starting materials for the synthesis of oseltamivir phosphate. | |
2. Types of influenza virus
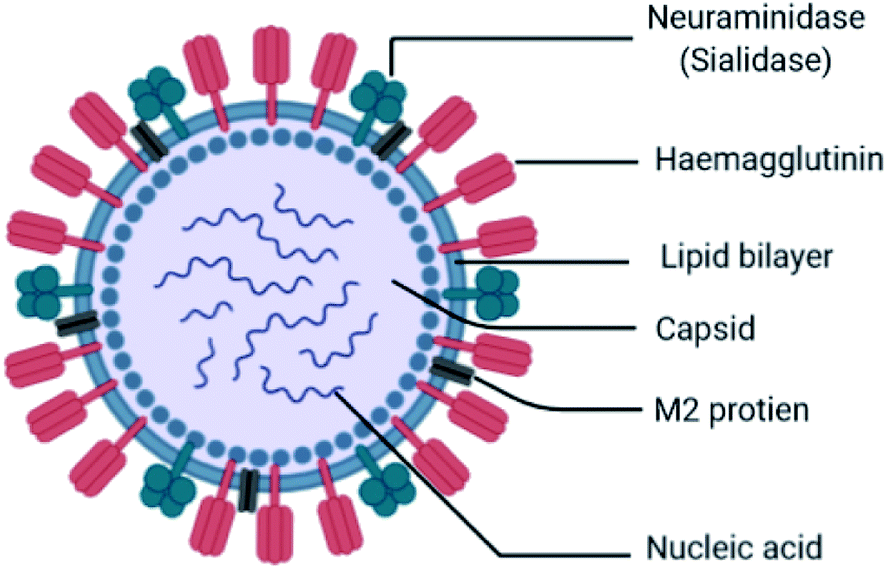
Influenza is an RNA virus that exists mostly in spherical shape 80–120 nm in diameter. They belong to the Orthomyxoviridae family of viruses, which contain a negative-stranded RNA genome made up of eight RNA segments that encode ten viral proteins. It is an enveloped virus (lipoprotein of host). The envelope contains two different varieties of glycoproteins: HA (hemagglutinin attachment to the host) and NA (neuraminidase for release, Fig. 4).
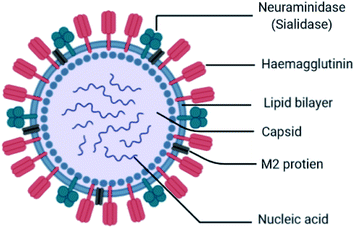 |
| | Fig. 4 Structure of influenza virus. | |
These are the main exposed segments of the virus responsible for virus entry. Inside the envelope, the matrix protein is present, which gives additional support to the envelope. Inside the matrix, the influenza genome is encapsidated. The influenza genome consists of 8 strands of RNA. These are the genetic materials combined with the capsid called nucleocapsid. The doted mark is known as RNA polymerase, which replicates RNA. The virus can survive on surfaces for few hours. It may enter into the body through mouth and nose by inhaling the droplets containing viruses from the sneeze and cough of an infected person within a range of 6 ft. When it enters the body, it binds sialic acid sugars of epithelial cells of the upper respiratory tract using hemagglutinin.
The influenza viruses are classified into four types (A, B, C, and D) based on the host predilections, ability to undergo genetic reassortment, and the severity of the disease they cause.51 Influenza A virus has several variants based on the presence of different HA (1–18) and NA (1–11) types. The virus undergoes major genetic variations (antigenic shift) and adapts itself in different hosts including humans, pigs, horses, birds, reptiles, and others resulting in pandemics. The influenza type B viruses are exclusively seen in humans and cause mild to moderate illness. They cause seasonal diseases, undergo minor genetic changes (antigenic drift) and are responsible for epidemic flu-like illnesses. The type C influenza viruses usually cause infections in pigs. They are not known to undergo genetic changes and are rarely reported in humans. Various types of influenza viruses and their infection types are listed in Table 1 for better understanding.51–65
Table 1 Types of influenza virus
| Types |
Name |
Year of finding |
Origin |
Key hosts |
Infection type |
Ref. |
| XType-A (8 RNA segments) (antigenic shift) |
H1N1 |
1918, 1976 (swine), 1977 (Russian flu), 2009 |
Spanish flu, Alberta, Canada |
Mutated avian influenza virus, duck |
Most common, most severe and can mutate |
51–54 |
| Highly pathogenic |
| Infects humans and animals |
| H2N2 |
1957 Asian flu |
Singapore |
Birds and pigs |
Pathogenic to humans |
54 and 57 |
| H3N2 |
1968 |
America |
Human |
Most common, most severe and can mutate |
54 |
| H3N2v |
2011 |
America |
Pigs |
Infects humans and animals |
|
| H5N1 |
1997 (avian) (bird flu) |
China |
Geese, poultry |
Pathogenic to humans |
54 |
| H5N5 |
2008 |
China |
Ducks, poultry |
Zoonotic |
56 and 58 |
| H5N8 |
2010 |
China |
Poultry |
Zoonotic |
56 |
| H7N7 |
2003 |
Europe |
Poultry |
Zoonotic |
54 and 59 |
| H7N9 |
2013 (avian) |
China novel avian influenza a ducks, poultry, and wild birds |
Poultry |
Pathogenic to humans |
55 |
| Humans and birds |
| H7N3 |
2003 (bird flu) |
Taiwan, British Columbia, North America |
Migratory birds, poultry |
Zoonotic |
60 |
| H9N2 |
1966 (bird flu) |
America, Turkey |
Chicken, duck, pigeon |
Potentially pathogenic to humans |
61 |
| H10N8 |
2012 (bird flu) |
China |
Ducks |
Potentially pathogenic to humans |
62 |
| Type-B (8 RNA segments) (antigenic drift) |
Have HA and NA, two types |
1988 |
Yamagata, Japan |
Humans |
Less common and doesn't mutate as often |
63 |
| Yamagata and Victoria |
Infects only humans |
| Type-C (7 RNA segments) (antigenic drift) |
No HA, and NA, but have hemagglutinin-esterase fusion (HEF) glycoprotein |
1982 |
Sao Paulo, Brazil |
Pigs, cows |
Least common and least likely to mutate |
64 |
| Infects children and pigs |
| Type-D (7 RNA segments) (antigenic drift) |
No HA, and NA, but have hemagglutinin-esterase fusion (HEF) glycoprotein |
2011 |
America |
Pigs, cattle |
No human infections reported |
65 |
3. Symptoms
The common symptoms are acute-onset fever, headache, runny nose, sore throat, and dry cough. Other symptoms noted during the flu include myalgia, fatigue/weakness, vomiting, and diarrhea. The incubation period ranges from 1 to 4 days and uncomplicated infection resolves within 1 week. The complications are acute otitis media, bronchiolitis, croup, sinusitis, pneumonia, myocarditis, and secondary microbial infections. Young children, pregnant women, adults above 65 years and persons having heart or lung disease are at higher risk of developing complications.66–68
4. Diagnosis
Influenza viral infections are diagnosed based on the clinical symptoms as discussed earlier. Virus can be cultured from nasal, nasopharyngeal, and throat swabs taken from the infected and suspected persons. Laboratory diagnosis by enzyme-linked immunosorbent assay (ELISA) can be performed for detecting the IgM antibodies in acute infections, and IgG antibodies from the convalescent persons. Diagnosis of flu can also be done by the complement fixation test, hemagglutination, and hemagglutination inhibition tests. Rapid influenza diagnostic tests (detect type does not strain) and polymerase chain reaction (detects viral RNA) are preferred for preliminary diagnosis and confirmation of the disease, respectively.69–71
5. Mechanism
5.1. Virus entry mechanism
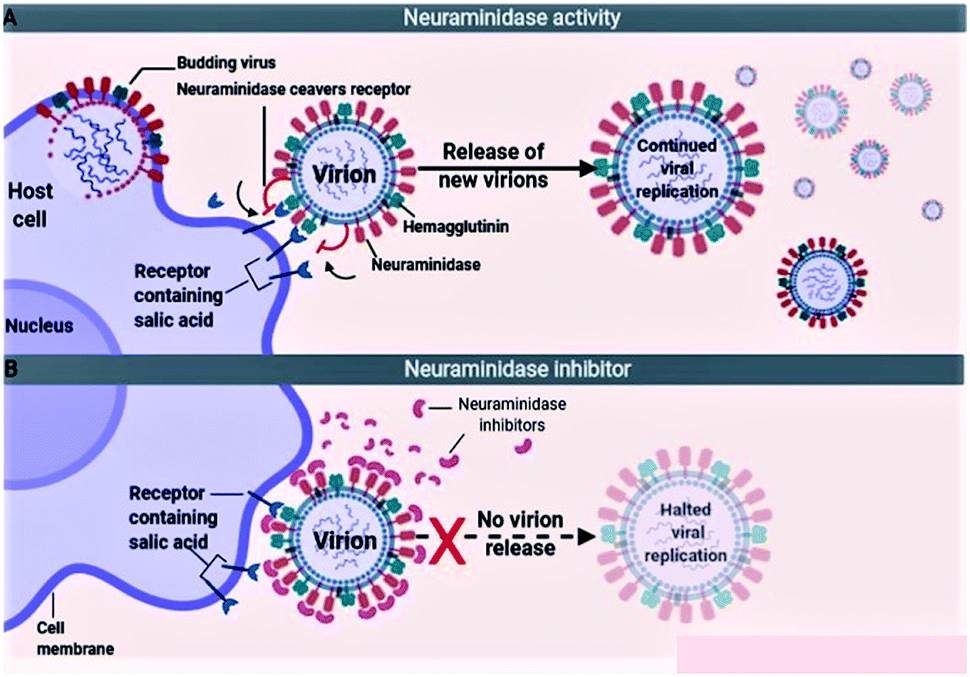
The influenza viruses adhere to the host cells with the help of spike-like projections protruding from the viral envelope called HA (Fig. 5A). Later, the viruses identify appropriate sites on the host cells, known as N-acetylneuraminic acid (sialic acid) residue-containing areas to initiate binding/attachment. The terminal α-sialic acid residues are oligosaccharide chains of N- and O-linked glycoproteins and lipids, which bind to the underlying galactose by a-2,3 or a-2,6 glycosidic linkages. This linkage and the resulting structural consequences influence how well the virus can bind to its receptor. After successfully attaching to the host cell, the virus enters the cell by using the clathrin-dependent endocytosis or the micropinocytosis. The host factors like the C-type lectins, Annexin V, 6-Sulfo sialyl Lewis X, dynamin, actin, clathrin, espin-1, EGFR, c-met kinase, and PLC-γ1 facilitate the successful attachment, and internalization of virus to the host cell. After entering into the host cell, the endosome fuses with the viral HA and increases the pH (acidity) within the virion. This causes release of the viral nucleic acid into the host cell cytoplasm. The host cell factors including Rab5, Rab7, PKC βII, Cullin 3, HDAC8, vATPase, CD81, and ITCH facilitate the endosomal trafficking, acidification, fusion, and uncoating of the viral nucleic acid. The viral nucleic acid then uses the host cell nuclear import pathways and importin-α, and importin-β proteins, to enter the nucleosome. The host factors that contribute to the import of viral RNA into the nucleosome include karyopherin (α1; α3; α5), Ran, p 10, and CSE1.72,73
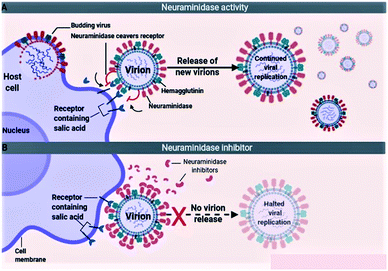 |
| | Fig. 5 Mechanism of virus entry (A) and neuraminidase inhibitors (B). | |
5.2. Neuraminidase inhibition mechanism
Because of the significance of sialic acid receptors in the attachment and entry of viruses into the host cells, blocking these receptors and inhibiting virus–host cell interactions have been the best possible way to control and prevent the infection. For the virus to successfully attach to the host cell, sialic acid must first be cleaved to expose the site of attachment for the virus. This function is performed by the NA proteins present on the virus envelope as projections similar to HAs. Therefore, inactivating NA using the neuraminidase inhibitors offers the best possible method to inhibit sialic acid receptors (Fig. 5B). The most efficient drugs in the treatment of flu caused by influenza A and pathogenic influenza B virus including oseltamivir and zanamivir are NA inhibitors.74–76
6. Neuraminidase inhibitors (NAIs)
6.1. Synthetic neuraminidase inhibitors
Several aryl and alkyl substituted 3-hydroxypyridin(1H)-2-ones were studied by LaVoie and co-workers77 for their ability to inhibit IAV endonuclease activity. Their endonuclease inhibitory activities are provided. The effect of para-substituents on the phenyl group substituted at position 5 was assessed, which indicates the importance of having an acidic functional group at this site. A number of 2,3-difluorosialic acids and their derivatives (Fig. 6) were synthesized chemoenzymatically by Chen and co-workers.78 PmAldolase was found to catalyze the formation of a C5-azido analogue of 3-fluoro(a)-sialic acid, whereas its Escherichia coli homologue (EcAldolase) did not. Both EcAldolase and PmAldolase could catalyze the synthesis of 3-fluoro(a/e)-sialic acids and their C-9 analogues, but PmAldolase was more efficient.
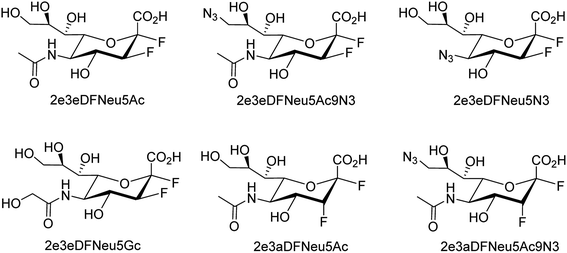 |
| | Fig. 6 Structures of 2,3-difluorosialic acid analogues. | |
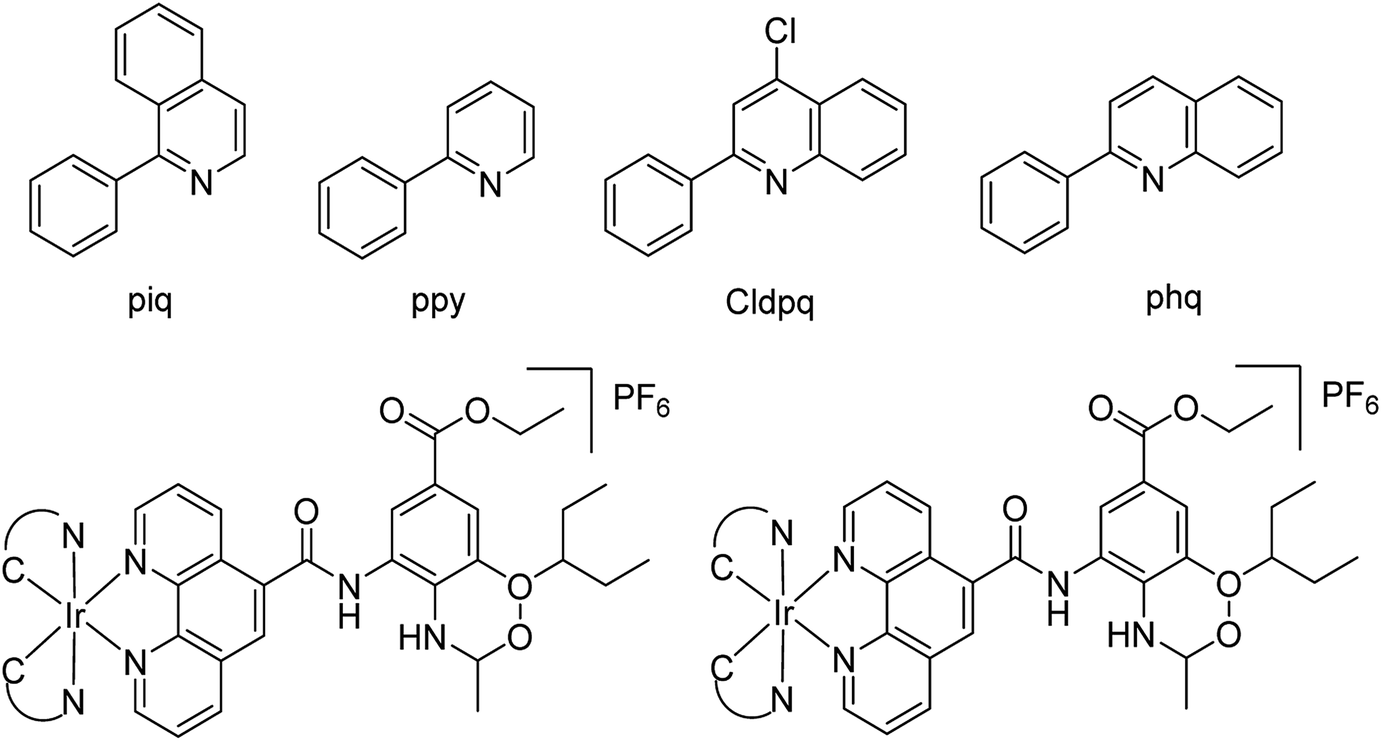
Ma and co-workers79 have designed a dual-functional luminescent probe and NA inhibitor, a key influenza target (Fig. 7). The lead iridium(III) compound displayed appreciated inhibition against NA compared to oseltamivir (FDA-approved drug), and could detect NA even in the presence of an auto-fluorescent background.
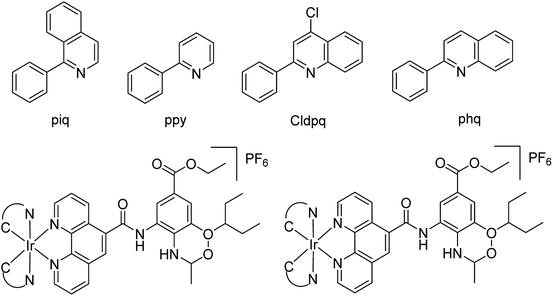 |
| | Fig. 7 Structures of iridium(III) complexes. | |
Hu and co-workers80 have designed twenty-seven novel chalcone derivatives (Fig. 8), and their inhibitory activities were evaluated against NA of influenza A virus in vitro. As per the study, the derivatives having a pyran ring displayed better NA inhibitory activity than those without a pyran ring. The molecular docking studies revealed that some compounds are in the good binding mode with zanamivir binding sites. Moreover, the structure–activity relationship was also reported.
 |
| | Fig. 8 Structures of chalcone derivatives. | |
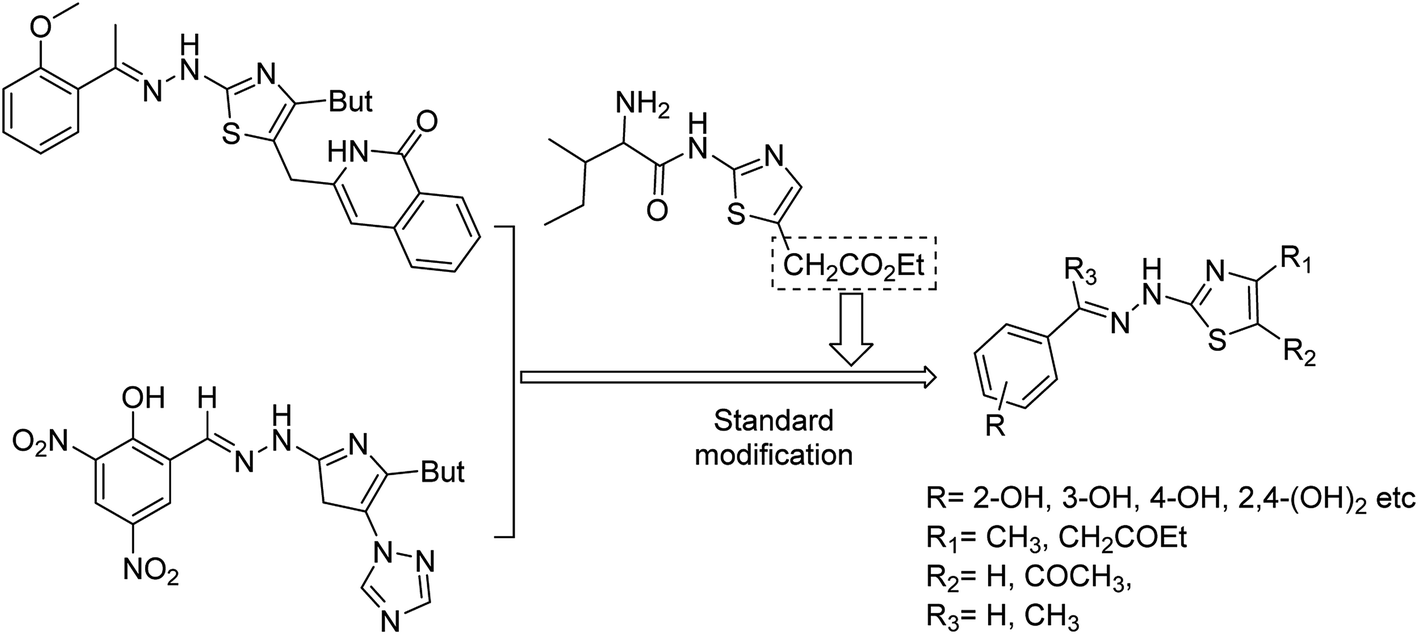
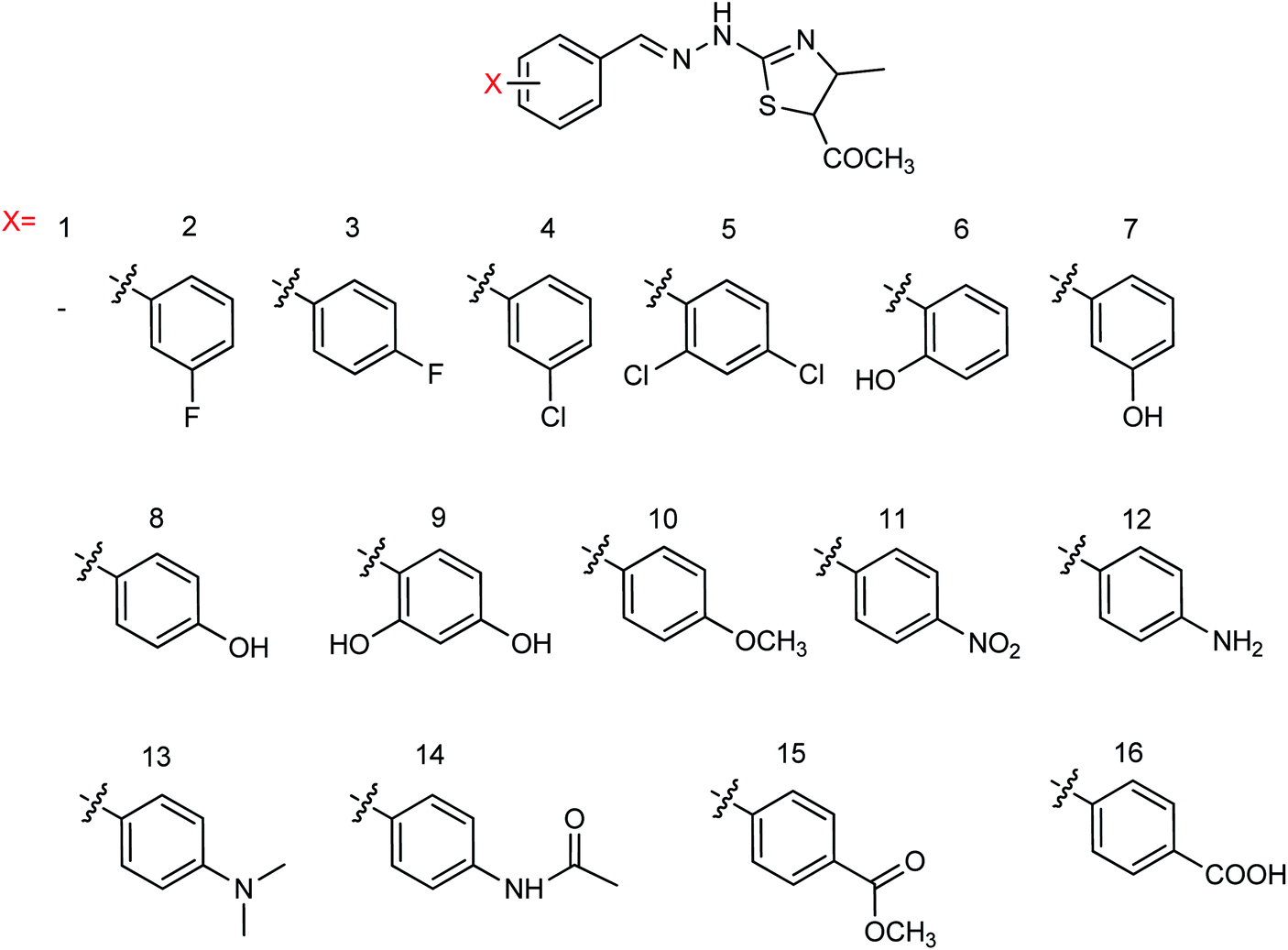
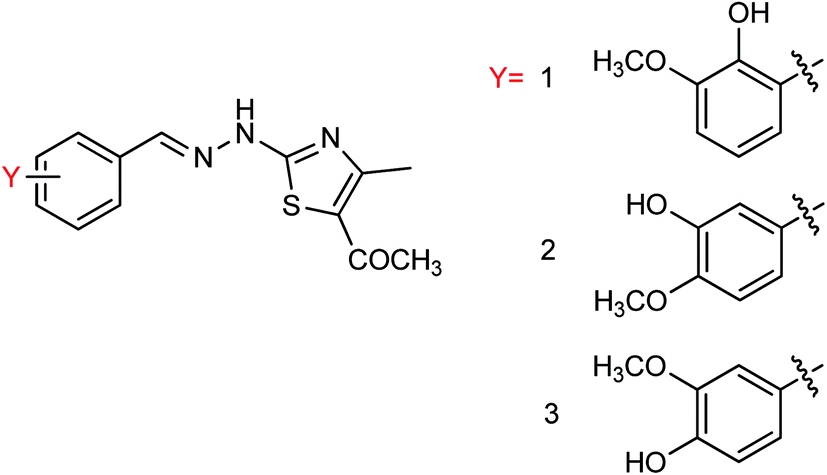
Hu and co-workers81 have evaluated the neuraminidase (NA) inhibitory activity of a number of thiazolylhydrazone derivatives (Fig. 9) against H1N1 (influenza virus) in vitro. Most of the compounds exhibited moderate-to-good inhibitory activity. The NA inhibitory activity of the most active compound was found to be IC50 = 7.12 μg mL−1. Furthermore, a molecular docking study was performed to understand the possible interactions between the compound and the active site of NA. In addition, DFT calculations and SAR were derived and discussed.
 |
| | Fig. 9 Design and synthesis of 2-thiazolylhydrazone derivatives. | |
hPIV causes acute respiratory tract infections and becomes life-threatening to young children and immunocompromised individuals. As there are no anti-hPIV drugs, it is important to develop specific antiviral therapies to decrease the mortality. HN is the hPIV surface protein playing critical roles in binding and cleavage. von Itzstein and co-workers82 have discussed the structural features of the HN protein that are being exploited for structure-guided inhibitor designs (Fig. 10), which will be very helpful for antiviral drug design.
 |
| | Fig. 10 Structures of Neu5Ac2en-derived hPIV-3 HN inhibitors. | |
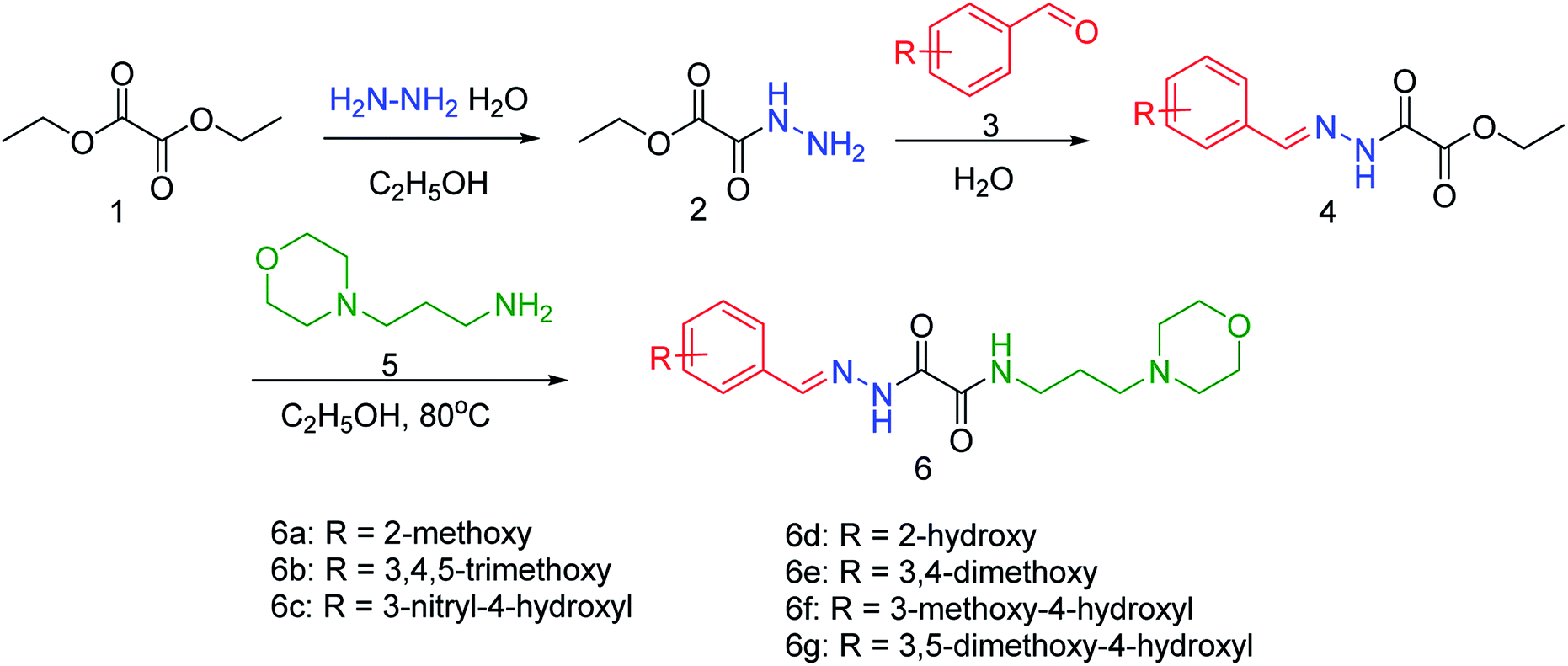
NA is used for the treatment of influenza A and B infections, as it plays a vital role in the replication and transmission of the virus. Wu and co-workers83 have discovered a new lead neuraminidase inhibitor by receptor-based virtual screening, ligand-based virtual screening, MD simulation, and bioassay validation. A series of acylhydrazone NA inhibitors (Fig. 11) have also been synthesized based on the lead compound. The compound 6e exerted IC50 = 2.37 ± 0.5 mM (lower than OC) against NA.
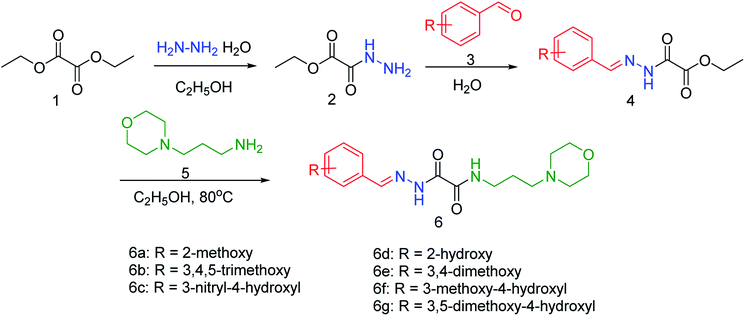 |
| | Fig. 11 Synthesis of acylhydrazone NA inhibitors. | |
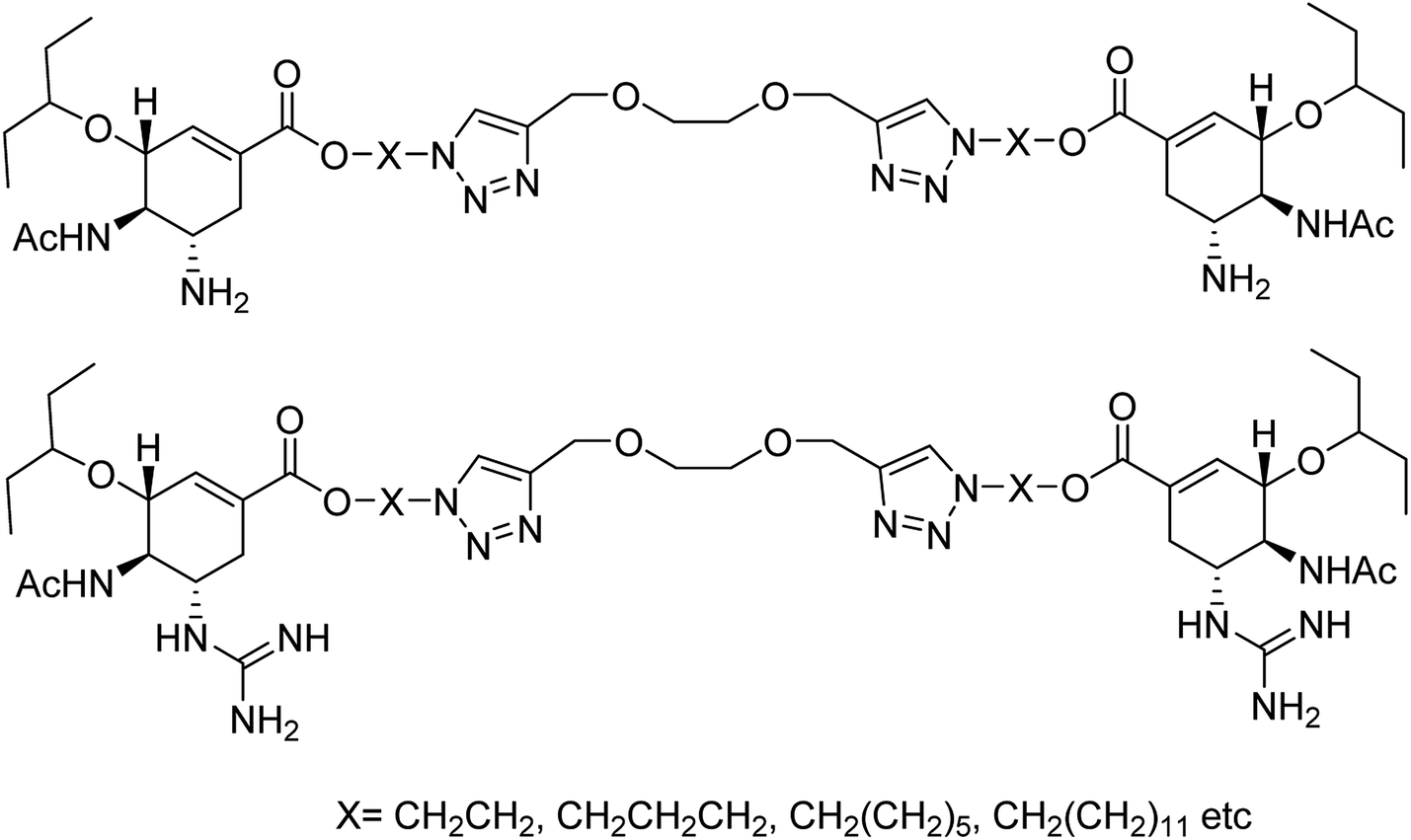
Yang and co-workers84 have prepared a series of divalent guanidino oseltamivir and oseltamivir analogues via a click reaction (Fig. 12). The SAR study suggested an appropriate distance between two oseltamivir monomers, which result in highly effective NA inhibitors against three strains of influenza virus. This study provides a basis for the multivalent modification on oseltamivir.
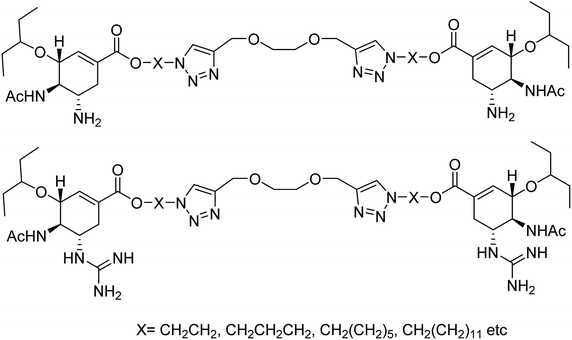 |
| | Fig. 12 Structures of divalent oseltamivir analogues. | |
The combined therapy improves the treatment outcomes and reduces the emergence of drug-resistant variants. Kiso and co-workers85 have infected immunocompromised nude mice with influenza A virus and treated them with viral polymerase (favipiravir) and/or neuraminidase (oseltamivir and laninamivir) inhibitors. The combination therapy (for 28 days) increased the survival times compared with monotherapy, but the mice died after the treatment was terminated. This study explained that combination therapy increases survival times in immunocompromised hosts, but does not suppress the emergence of neuraminidase inhibitor-resistant variants. The designing of novel neuraminidase inhibitors is very essential, which may withstand the challenges of resistance. Malbari and co-workers86 have designed (Fig. 13) five series of scaffolds (aurones, chalcones, cinnamic acid analogues, pyrimidine analogues and cinnamic acid linkages) based on virtual screening against H1N1 virus. The molecular modelling studies revealed that the reported compounds occupied 430-loop cavity of neuraminidase. The favourable compounds have synthesized and evaluated for their cytotoxicity and cytopathic effect inhibition by H1N1 virus. The study indicated that aurones will be treated as potential neuraminidase inhibitors against the pandemic H1N1 virus.
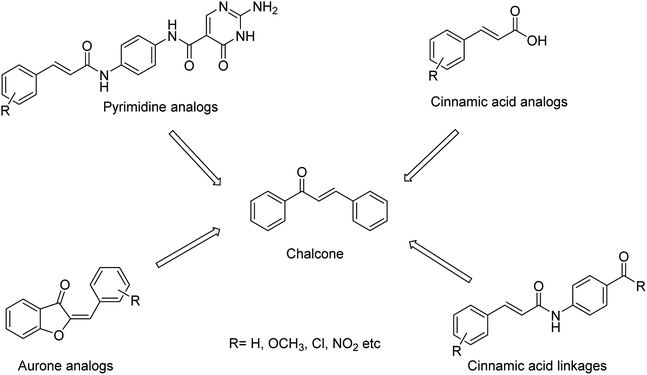 |
| | Fig. 13 Structures of aurones, chalcones, cinnamic acid analogues, pyrimidine analogues and cinnamic acid linkages. | |
Prasath and co-workers87 have analysed the anti-influenza agent LGN using FT-Raman and FT-IR spectra. The experimental results were compared with DFT calculations, which are in good agreement with the computational one. The suitability of a drug candidate was evaluated for human intake by ADMET properties. According to the study, LGN molecules exhibited good bioactive score and less toxicity. Moreover, molecular docking study was carried out to determine the binding affinity in the active site. The correct configuration in sialosides at C2 is a major challenge due to the absence of anomeric hydrogen. Therefore, the correct configuration of 3,4-unsaturated derivatives of Neu5Ac could be assigned as per the reported 3,4-unsaturated 2O-methyl sialosides. In this context, Anastasia and co-workers88 have tried to resolve this issue through the synthesis of the corresponding unreported unsaturated 1,7-lactones (Fig. 14).
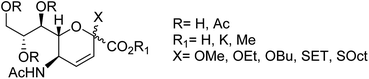 |
| | Fig. 14 Structures of 3,4-unsaturated Neu5Ac derivatives. | |
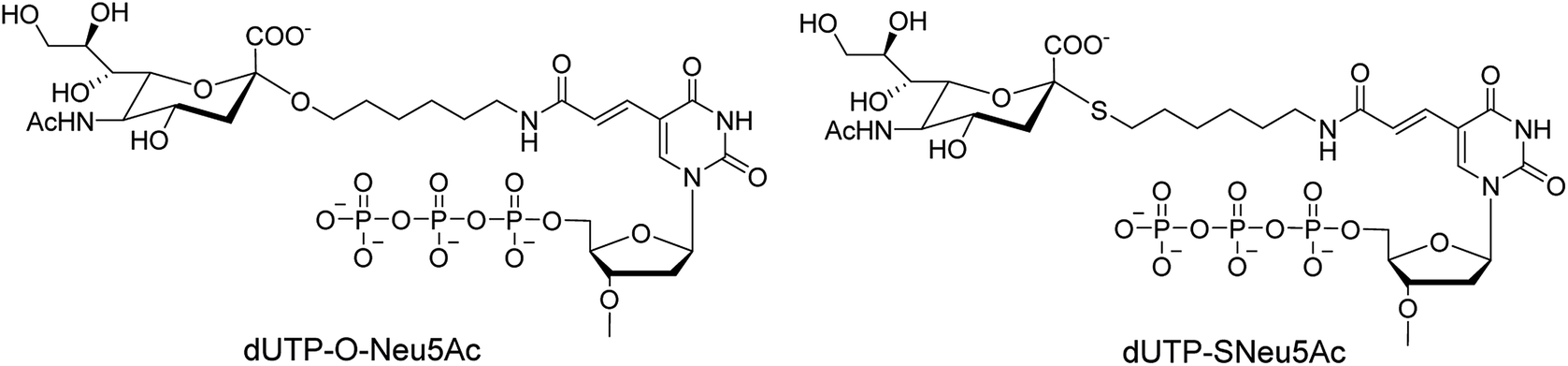
Ebaraa and co-workers89 have synthesized (Fig. 15) and modified NA-resistant sialoside (having unnatural S-glycoside bonds) on a 3WJ DNA to display complementary distribution to its binding sites on a HA trimer. This modified sialoside maintained high binding affinity and displayed certain NA resistance. Most importantly, this study may help in the development of NA-resistant sialoside derivatives for a broad spectrum of viruses.
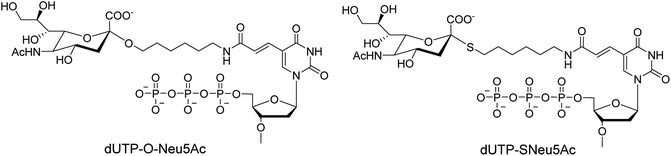 |
| | Fig. 15 Structures of dUTP-ONeu5Ac and dUTP-SNeu5Ac derivatives. | |
We all know that influenza viruses are causing severe upper respiratory illness in humans. Moreover, the current IAV strains have developed resistance to neuraminidase inhibitors. Manicassamy and co-workers90 have summarized the existing antiviral strategies for combating influenza viruses. This report may provide ample references for the treatment of influenza virus disease and the development of antiviral resistance. Fang and co-workers91 have reported the anti-influenza and NA inhibition activity of some OC, GOC (by replacing 5-amino group with a guanidino group) and their carboxyl bioisosteres (Fig. 16). The GOC congeners showed better anti-influenza and NA inhibition activity than the corresponding OC congeners. The GOC sulfonic acid congener (EC50 = 2.2 nM) is the most potential influenza agent against the wild-type H1N1 virus.
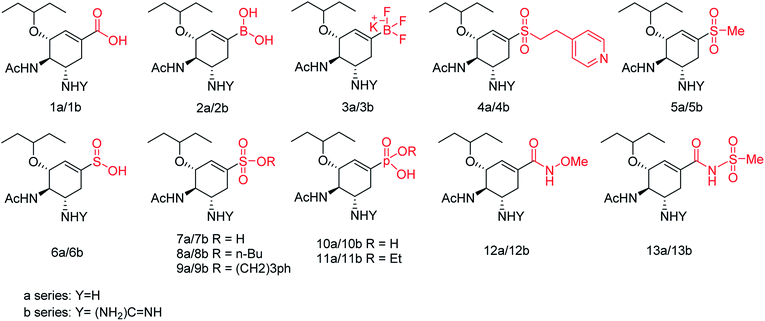 |
| | Fig. 16 Structures of oseltamivir carboxylic acid (1a, OC), guanidino OC (1b, GOC) and their carboxyl bioisosteres. | |
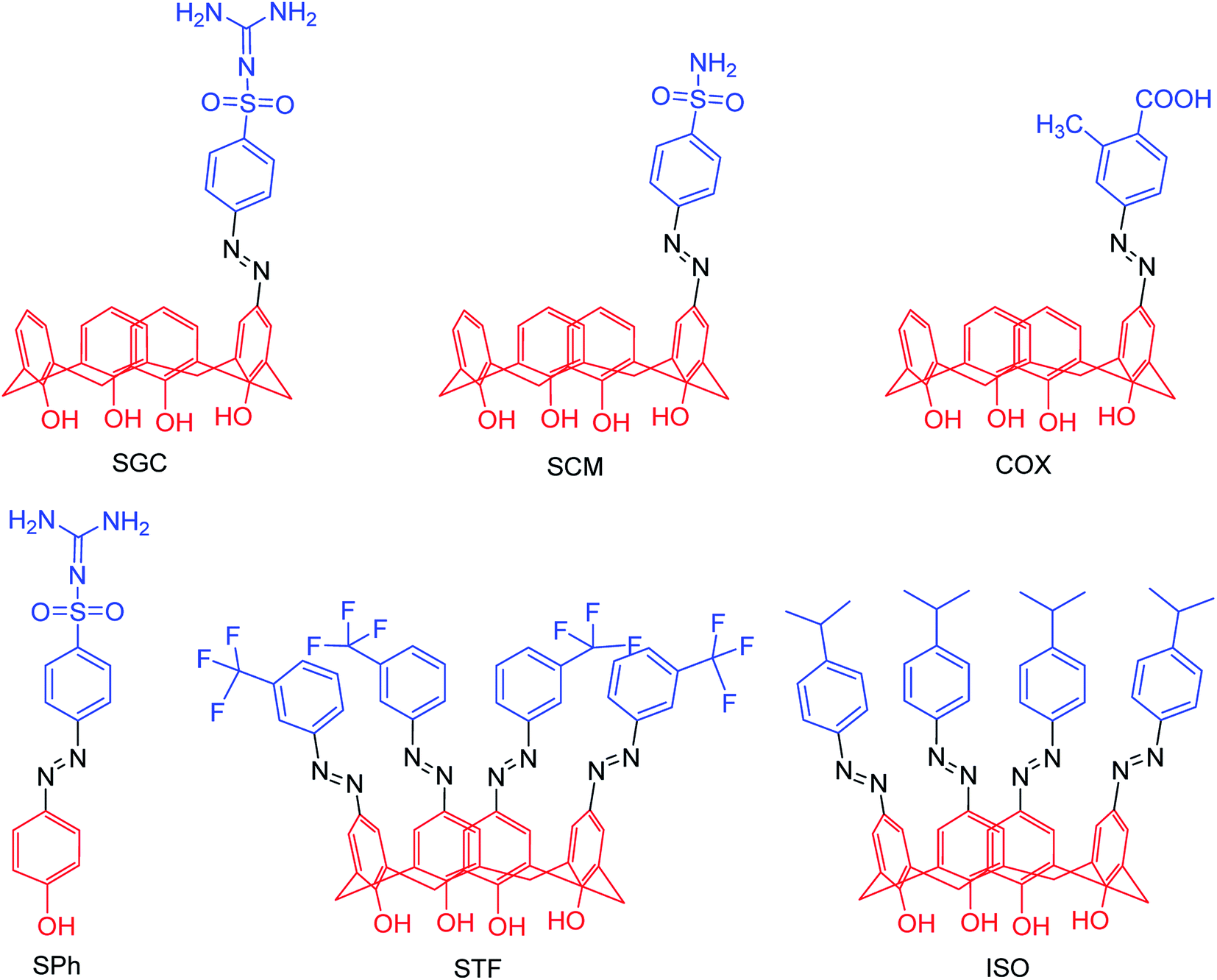
Due to the potential therapeutic properties of calixarene derivatives, Hamid and co-workers92 have reported the antibacterial and antiviral potentials of some azo-based derivatives of calix[4]arene (Fig. 17). The mono azo products of sulfaguanidine, sulphanilamide, and 2-methyl-4-aminobenzoic acid displayed good activity against some selected bacterial strains. The use of sodium acetate trihydrate and calix[4]arene in 3:1 molar ratio helped in partial substitution. Moreover, a molecular docking study was carried out to understand the interactions with bacterial and viral receptors.
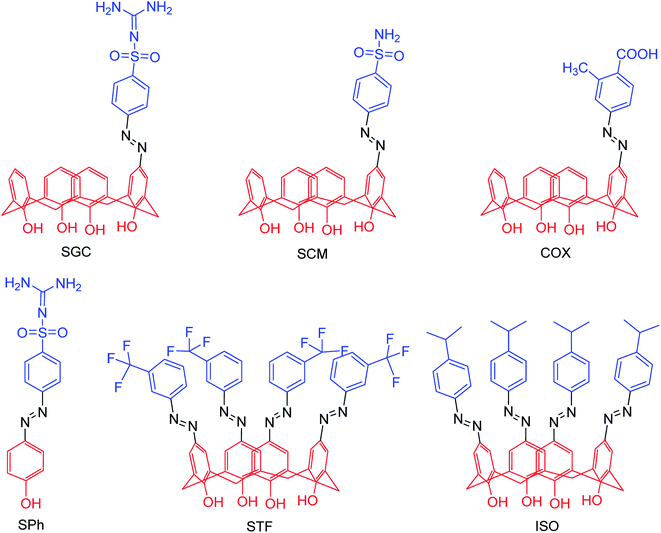 |
| | Fig. 17 Structures of azo derivatives. | |
Abdalsalam93 has reported virtual screening and docking analysis of several compounds obtained from the ZINC database against the H1N13TI6 protein (Fig. 18) by using Autodock Vina.
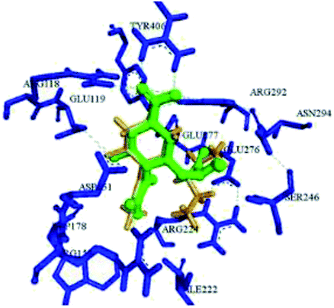 |
| | Fig. 18 Superimposition between the docked conformation (yellow) and the crystal structure (green) of H1N1 3TI6. | |
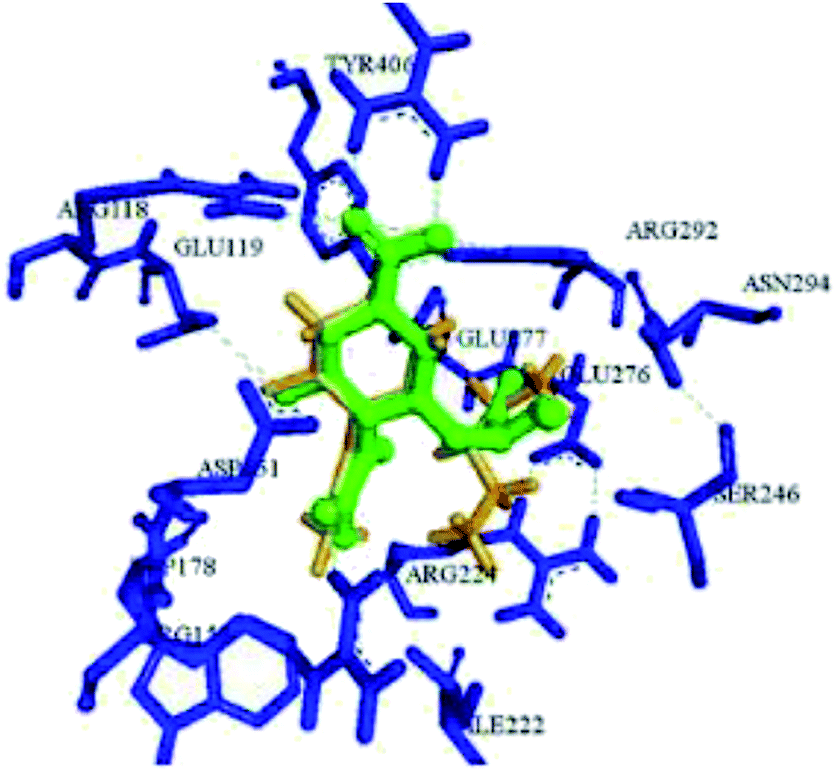
Here, the exhibited data are compared with the co-crystallized drug oseltamivir. The novel compounds were successfully identified, which could serve as a potential lead compound for the development of a new anti-neuraminidase drug. Oseltamivir resistance against H1N1 influenza virus has been reported lately. Tambunan and co-workers94 have reported the docking simulation of disulfide cyclic peptide ligands (DNY, LRL, NNT) (Fig. 19) along with zanamivir and oseltamivir as the standard ligands using the MOE 2008.10 software. The study showed that all disulfide cyclic peptide ligands have lower ΔG binding than the standard ligands, with the lowest for DNY (ΔG binding = −7.8544 kcal mol−1). Furthermore, these ligands had better interactions with neuraminidase than the standards, hence may be used as potential neuraminidase H1N1 inhibitors.
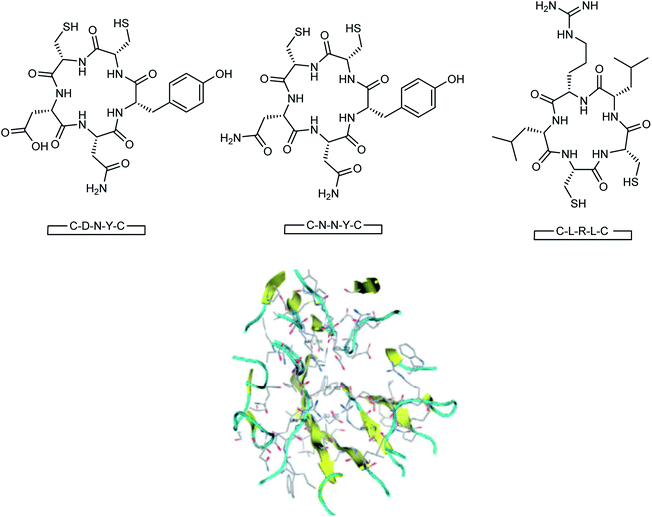 |
| | Fig. 19 Structures of cyclic peptides and the visualization of the catalytic site of influenza A (H1N1) neuraminidase. | |
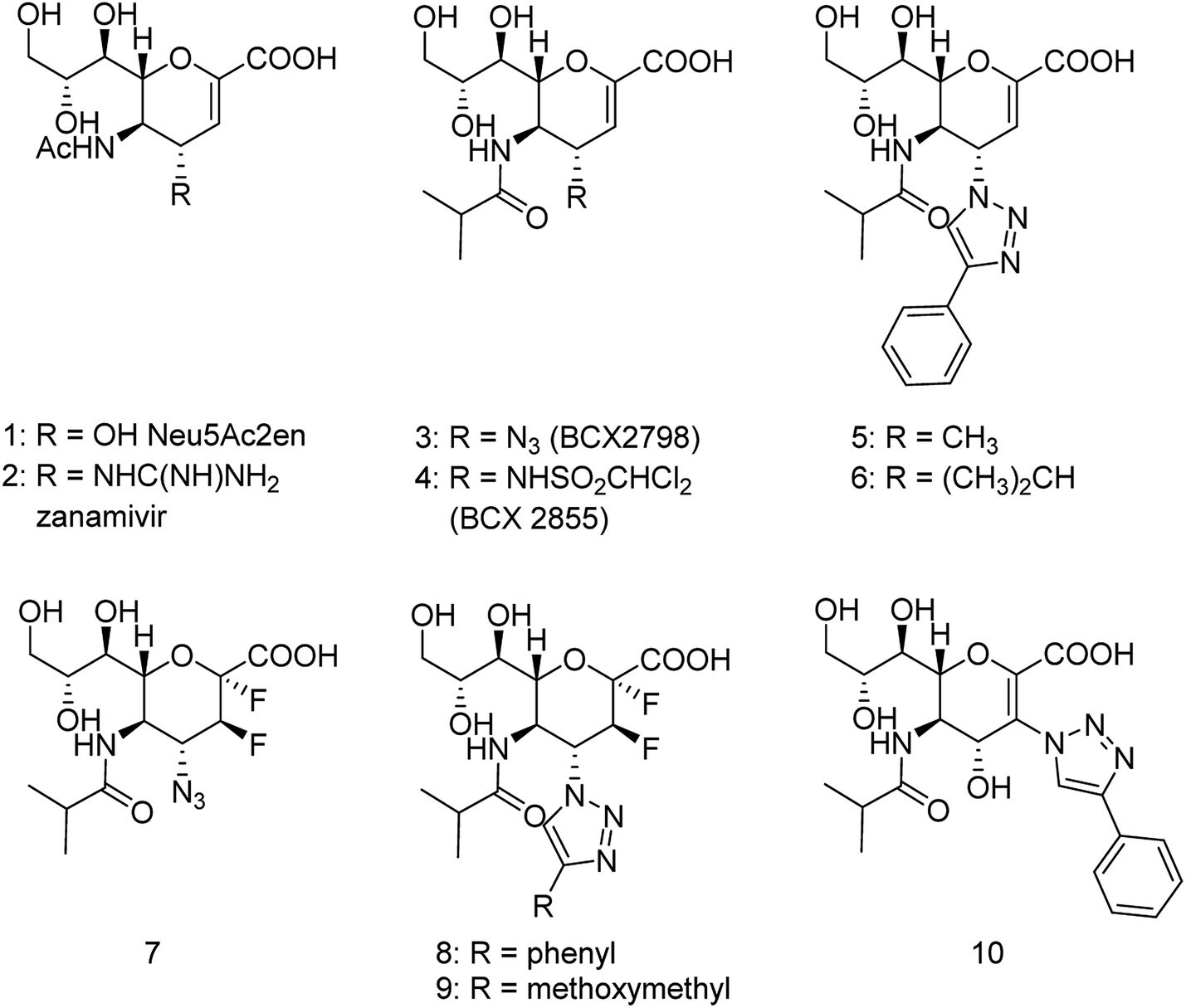
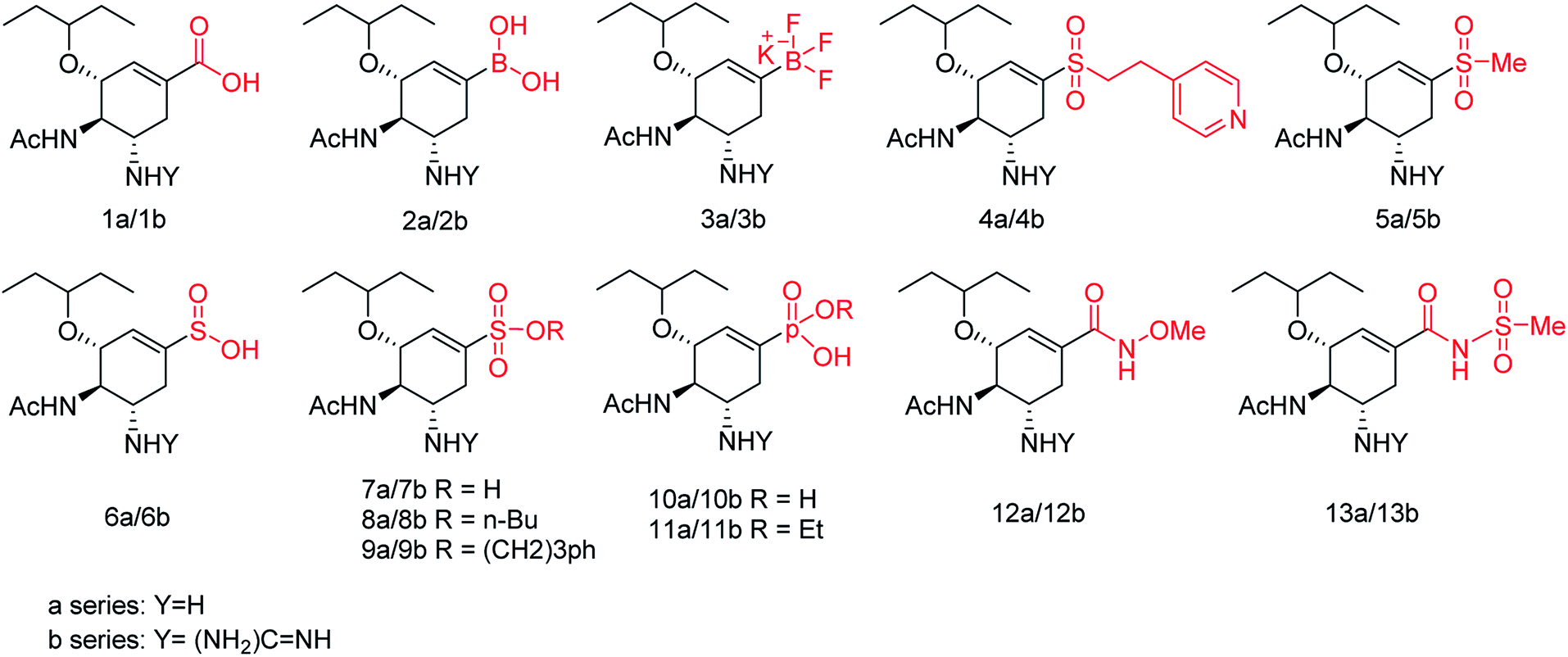
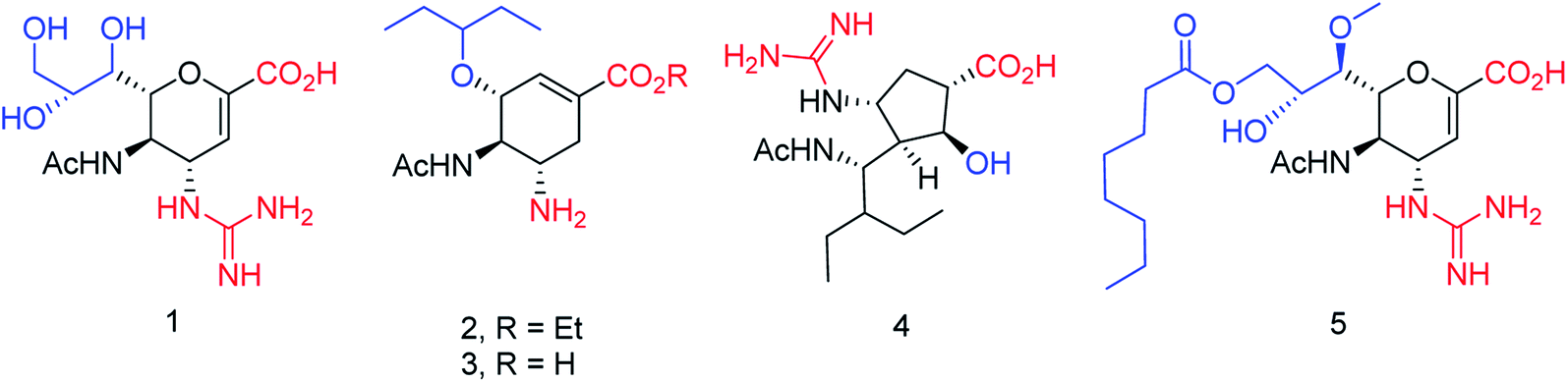
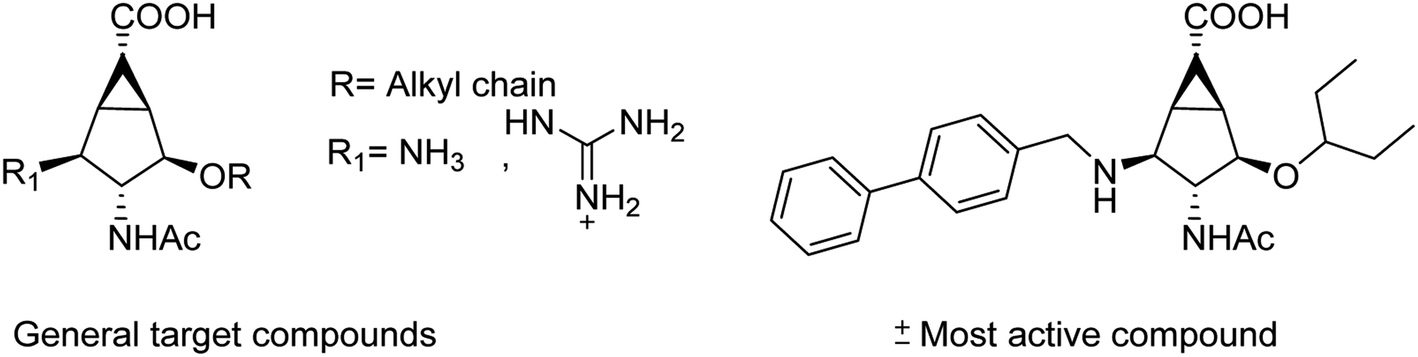
The new strains of influenza virus, resistant to current inhibitors (oseltamivir, amantadine, etc.), are making serious threats to the public health. Hence, it is essential to develop better and more effective vaccines and therapeutics. With this in view, Yassine and co-workers95 have designed a computational study to identify potential flavonoid inhibitors that bind to the contact epitopes of the HA stem that are targeted by bNAb. The QSAR study was performed for 100 natural compounds along with molecular docking analysis. According to the study, 18 lead flavonoids have strong binding abilities to bNAb epitopes of various HA subtypes. Cairo and co-workers96 have reported a library of modified DANA analogues (Fig. 20) and evaluated them against four human neuraminidase isoenzymes (NEU1-4). The best NEU1 inhibitor was C5-hexanamido–C9-acetamido–DANA having 340-fold selectivity (Ki = 53 ± 5 nM). Moreover, it was demonstrated that C5-modifications combined with a C4-guandino group provided the most potent NEU2 inhibitor.
 |
| | Fig. 20 Synthesis of C5-modified DANA analogues. | |
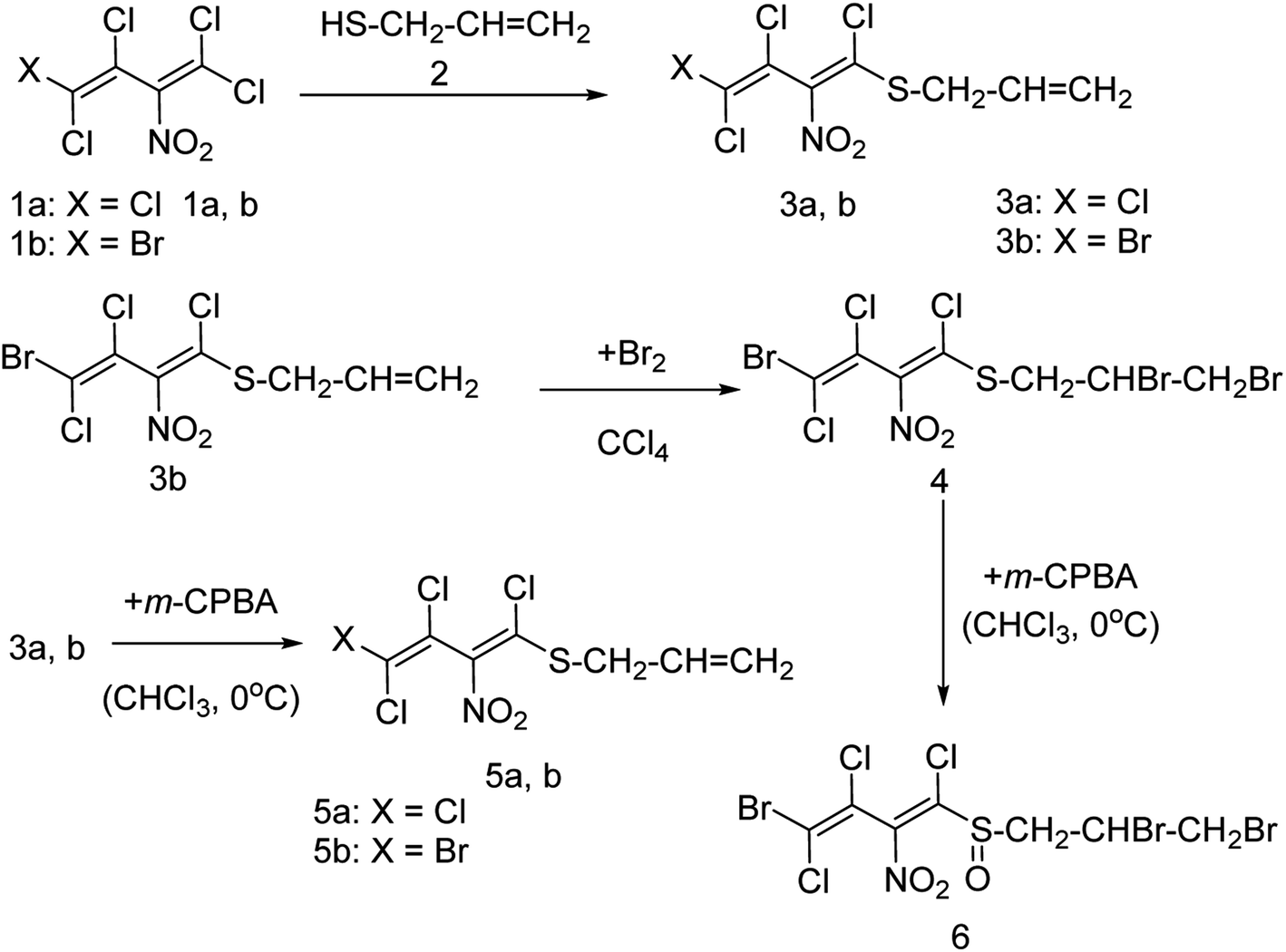
Onul and co-workers97 have reported the synthesis of S-substituted perhalo-2-nitrobuta-1,3-dienes (3a,b) from the reaction of allyl mercaptan with polyhalo-2-nitrobuta-1,3-dienes (1a,b). The addition of bromine to S-substituted polyhalo-2-nitrobuta-1,3-diene (3b) in CCl4 yielded 1-(2,3-dibromopropanethio)-4-bromo-1,3,4-trichloro-2-nitrobuta-1,3-diene (4). The reaction of thiosubstituted polyhalonitrobutadienes (3a,b, and 4) with m-CPBA in CHCl3 yielded sulfoxides (5a,b, and 6). The structures of these newly reported compounds were confirmed by 1H NMR, 13C NMR, FTIR and MS data. These reported compounds (Fig. 21) exhibited antixanthine oxidase, antielastase, antityrosinase, and antineuraminidase activities.
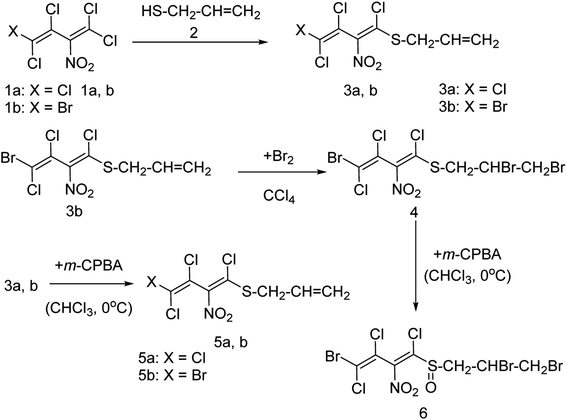 |
| | Fig. 21 Synthesis of S-substituted perhalonitrobuta-1,3-diene derivatives. | |
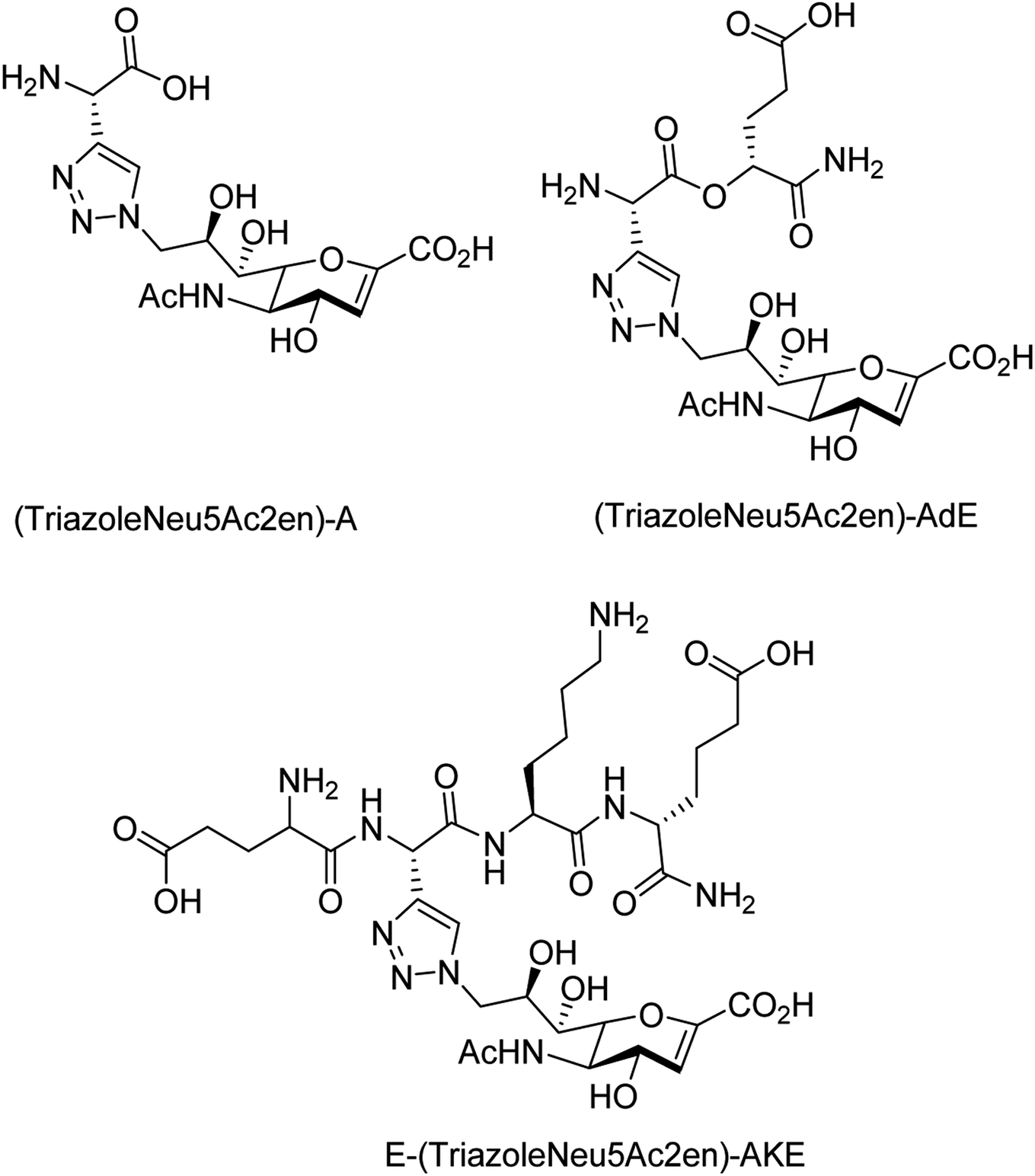
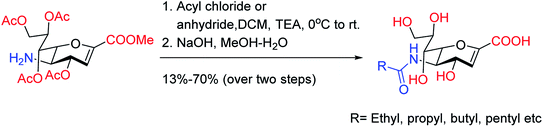
To overcome the drug–resistance activity, the biological nanoparticles are playing significant rules in the development of novel anti-influenza drugs. In this context, selenium nanoparticles with low toxicity have attracted increasing attention for biomedical intervention. Zhu and co-workers98 have designed the surface decoration of selenium NPs by amantadine. Se@AM has less toxicity and inhibited the ability of H1N1 to infect host cells through suppression of neuraminidase activity. Moreover, Se@AM could prevent H1N1 from infecting the MDCK cell line. In another work, Zhu and co-workers99 have also reported the synthesis of oseltamivir (OTV) surface-modified SeNPs with superior antiviral properties. Se@OTV had less toxicity and inhibited H1N1 infection. Se@OTV interfered with H1N1 virus to host cells through suppression of neuraminidase and hemagglutinin activity. The study demonstrated that Se@OTV is a promising efficient antiviral pharmaceutical for H1N1. Cheng and co-workers100 have studied a series of zanamivir derivatives as potential NA inhibitors by using molecular docking, 3D-QSAR, and MD simulation techniques. Out of which, two selected compounds were synthesized. Both the compounds were biologically evaluated by virus inhibition and NA inhibition assays. One compound exhibited excellent antiviral activity (IC50 = 0.670 μM, SI > 149) against A/WSN/33H1N1, superior to zanamivir (IC50 = 0.873 μM, SI > 115). This study may provide significant contribution for the development of new anti-influenza drugs. Sialidases and neuraminidases are becoming the main targets for drug development as they are playing important roles in bacterial and viral infections. Several derivatives of Neu5Ac2en (DANA) have been designed and synthesized by Chen and co-workers101 (Fig. 22) as triazole-linked transition state analogs. The inhibition studies confirmed that E-(TriazoleNeu5Ac2en)-AKE and (TriazoleNeu5Ac2en)-A were selective inhibitors against V. cholerae sialidase, while (TriazoleNeu5Ac2en)-AdE selectively inhibited V. cholerae and A. ureafaciens sialidases.
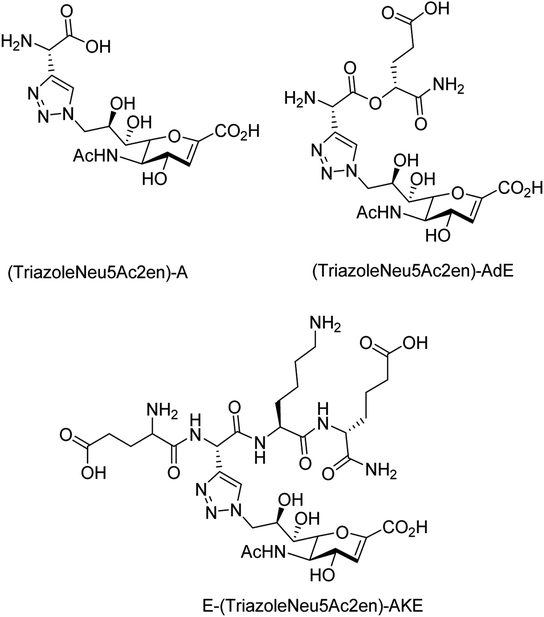 |
| | Fig. 22 Structures of (TriazoleNeu5Ac2en)-A, (TriazoleNeu5Ac2en)-AdE and E-(TriazoleNeu5Ac2en)-AKE. | |
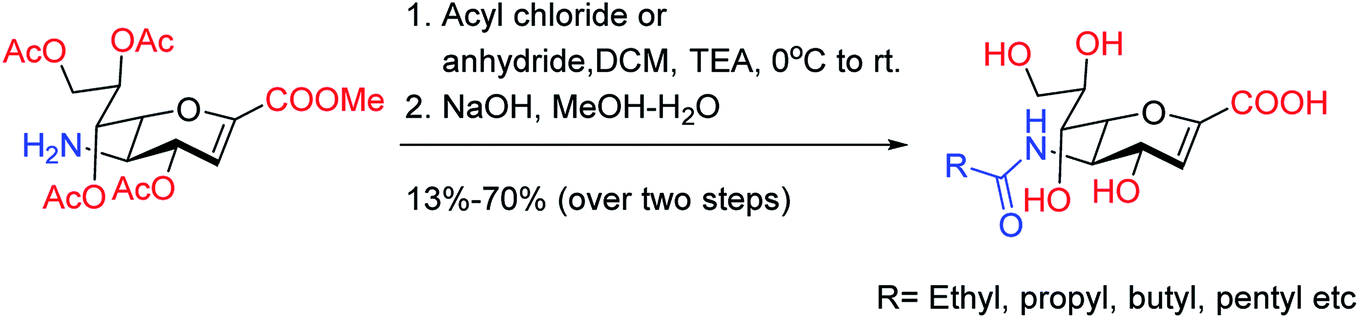
On the basis of molecular docking study, Pratama and co-workers102 have reported the relationship between artemisinins and neuraminidase (NA). The docking study was performed using AutoDock 4.2.3 toward NA in complexes with oseltamivir as a co-crystal ligand. Artesunate provided most negative free ΔG (−9.55 kcal mol−1) and lowest Ki (100.66 nM) toward NA. Artesunate showed higher affinity than oseltamivir with interactions between artesunate and amino acids at position 246, which had important influences on artesunate affinity toward NA from H5N1. The study indicated that artesunate could be considered as a NA inhibitor and should have potential to be developed as an anti-influenza drug particularly to H5N1 with oseltamivir resistance. OSMIP@silica gel was prepared by Yang and co-workers103 to improve the chromatographic performance. NIP@silica gel has been made for comparison. These were characterized through FT-IR, SEM, SSAA and porosity measurements. The OSMIP@silica gel column displayed good affinity and selectivity for template OS and peramivir, and hence, it could be employed to search more active neuraminidase inhibitor analogues from traditional herbs. Li and co-workers104 have studied the inhibitory activities of a series of carboxyl-modified oseltamivir analogs against neuraminidase from H5N1 virus. One compound has shown potent inhibitory activity (IC50 = 1.30 ± 0.23 μM) and is more lipophilic (logD = −0.12) than oseltamivir carboxylate (logD = −1.69) at pH of 7.4. There results may be helpful in identifying neuraminidase inhibitors in the exploration of 430-cavity with optimal lipophilicity. The active ingredients of four neuraminidase inhibitors are shown in Fig. 23.
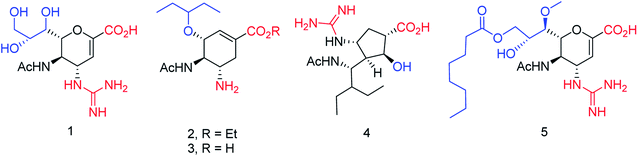 |
| | Fig. 23 Structures of carboxyl-modified oseltamivir analogues. | |
Igdoura and co-workers105 have evaluated the effect of sialidase NEU1 on atherogenesis. They have generated ApoE-deficient mice and found that hypomorphic NEU1 expression in male mice reduces serum levels of VLDL and LDL cholesterol. The reduction decreases atherosclerosis via its significant effects on lipid metabolism and inflammation. From the study, it was concluded that NEU1 may represent a promising target for managing atherosclerosis. Colombo and co-workers106 have reported a mini library of new bicyclic compounds (Fig. 24) and characterized them by enzyme inhibition assays. None of the synthesized compounds presented fair activity against influenza A neuraminidases at concentrations less than 2 mM. According to them, choice and positioning of the functional groups still need to be properly tuned.
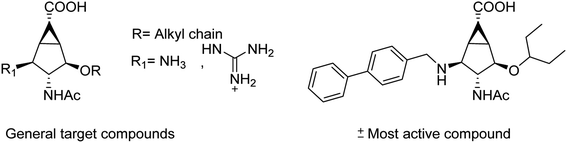 |
| | Fig. 24 Structures of bicyclo[3.1.0]hexane analogues of sialic acid. | |
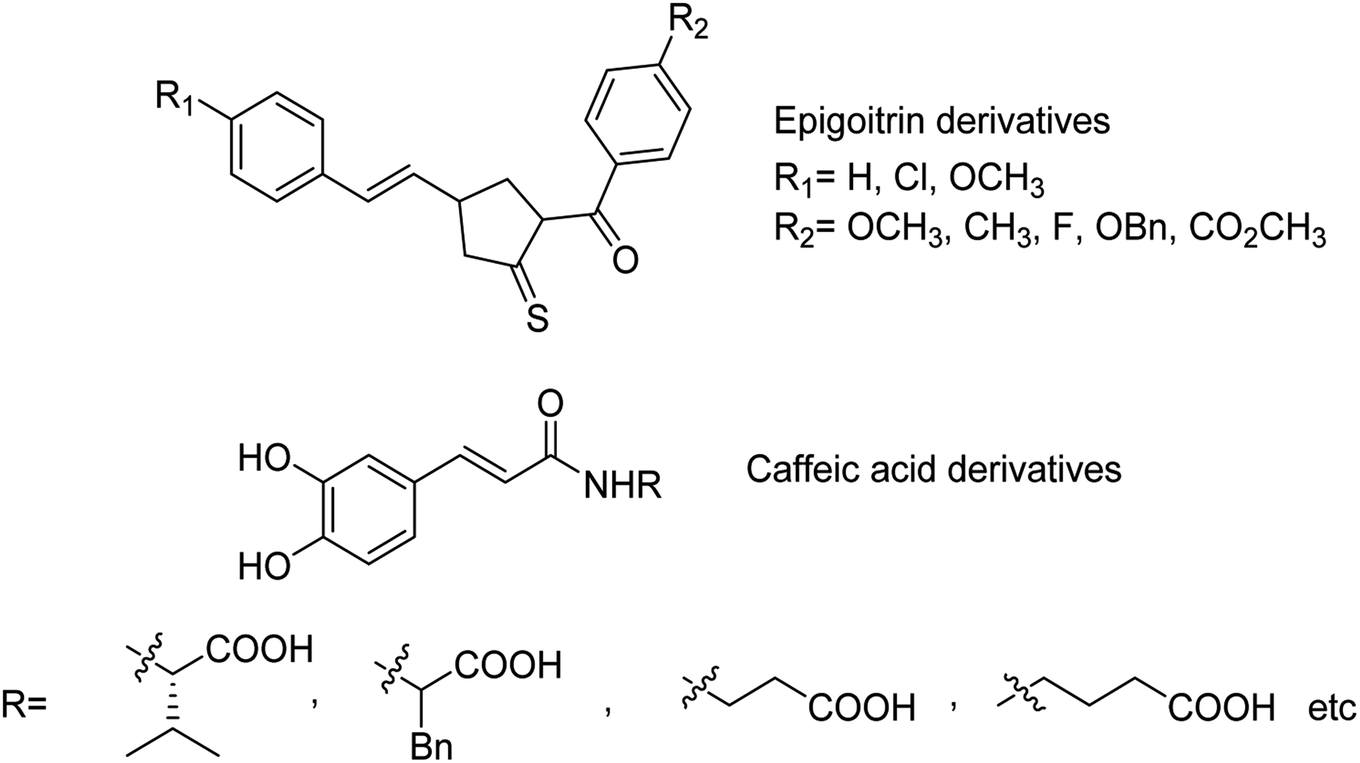
The sialidase's activity was correlated with the pathogenicity of many pathogenic microorganisms. Sialidases efficiently and specifically cleave the glycosidic bond in sialo-glycoconjugates. Xing and co-workers107 have reported a sialic acid-coated TPE4S with good stability, excellent hydrophilicity, high sensitivity and unique selectivity towards sialidases. Moreover, TPE4S was conveniently employed for the screening of sialidase inhibitors and diagnosis of bacterial vaginosis. Molecular docking and 3D-QSAR methods were used by Yin co-workers108 to explore the SAR with caffeic acid derivatives109 (Fig. 25). Ten epigoitrin derivatives have been outlined on the basis of computational results and analysed. The computational studies governed the designation of epigoitrin derivatives as novel neuraminidase inhibitors.
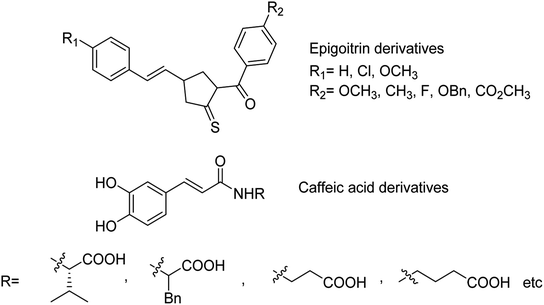 |
| | Fig. 25 Structures of caffeic acid and epigoitrin derivatives. | |
Lee and co-workers110 have reported some potential agents using turmeric and curcumin analogues for treating bacterial infections caused by S. pneumoniae Nan A. The SAR studies were described, and the synthesised derivatives (Fig. 26) were potent inhibitors of S. pneumoniae sialidase (IC50 = 0.2 ± 0.1 μM). The most potent compound displayed a 3.0-fold improvement in inhibitory activity over that of curcumin. This study suggested that the curcumin derivatives may be used for the treatment of sepsis by bacterial infections.
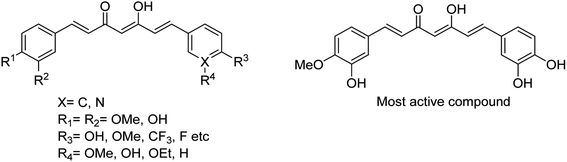 |
| | Fig. 26 Structures of curcumin analogues. | |
Mistry and co-workers111 have analyzed the biological activities of substituted piperazine-based berberine analogues conjugated through a pentyloxy side chain. All the analogs have been screened for their in vitro antiviral activity against some influenza strains by using SRB and CPE assays. Moreover, the cytotoxicity was evaluated using MDCK cell lines. In addition, the anti-influenza activities of BPD were examined. As per the study, these compounds were found to be potential neuraminidase inhibitors that merge to the active site of neuraminidase. Smith and co-workers112 have described the recent progress of the biotin/(strept)avidin self-assembly system and the modern design rules for creating synthetic mimics. Moreover, a series of case studies have been given on synthetic derivatives of cucurbiturils, cyclodextrins and various organic cyclophanes (calixarenes, pillararenes, tetralactams and deep cavitands).
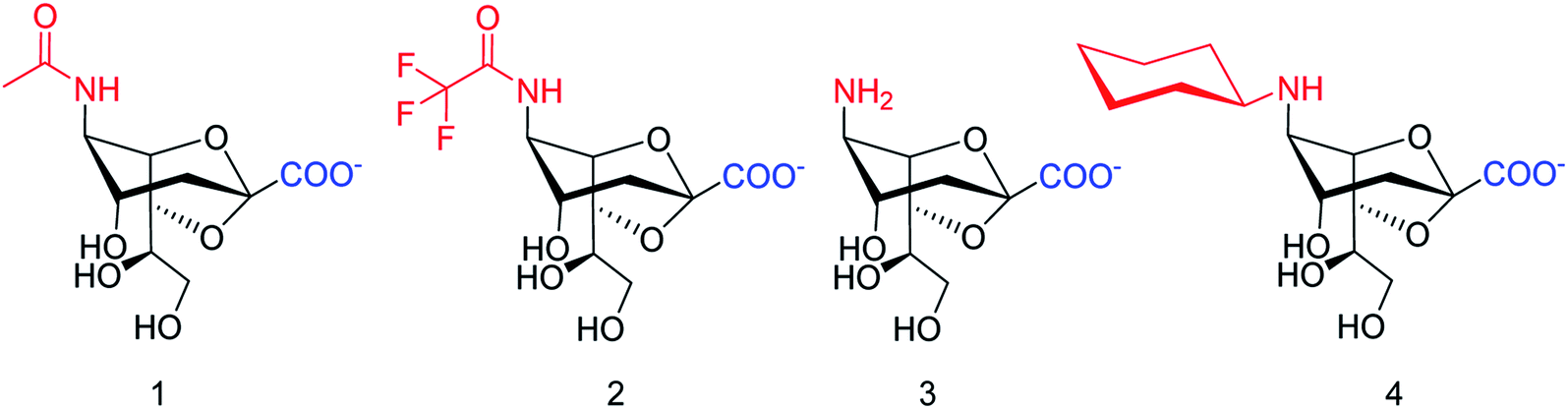
SpNanB is responsible for streptococcal infection of the upper and lower respiratory tract. Chen and co-workers113 have reported the development of a potent OPME system for synthesizing 2,7-anhydro-Neu5Ac and its derivatives (Fig. 27). An N-cyclohexyl derivative of 2,7-anhydro-neuraminic acid was synthesized and found to be a selective inhibitor against SpNanB and SpNanC. The study demonstrated a unique strategy of synthesizing 2,7-anhydro-sialic acids and potential applications of their derivatives as selective sialidase inhibitors.
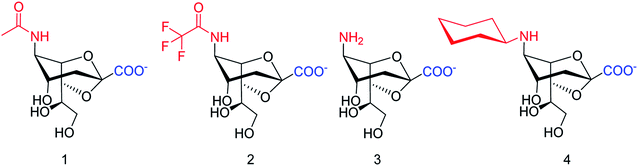 |
| | Fig. 27 Structures of enzymatically synthesized 2,7-anhydro-Neu5Ac (1) and its chemoenzymatically synthesized derivatives 2–4. | |
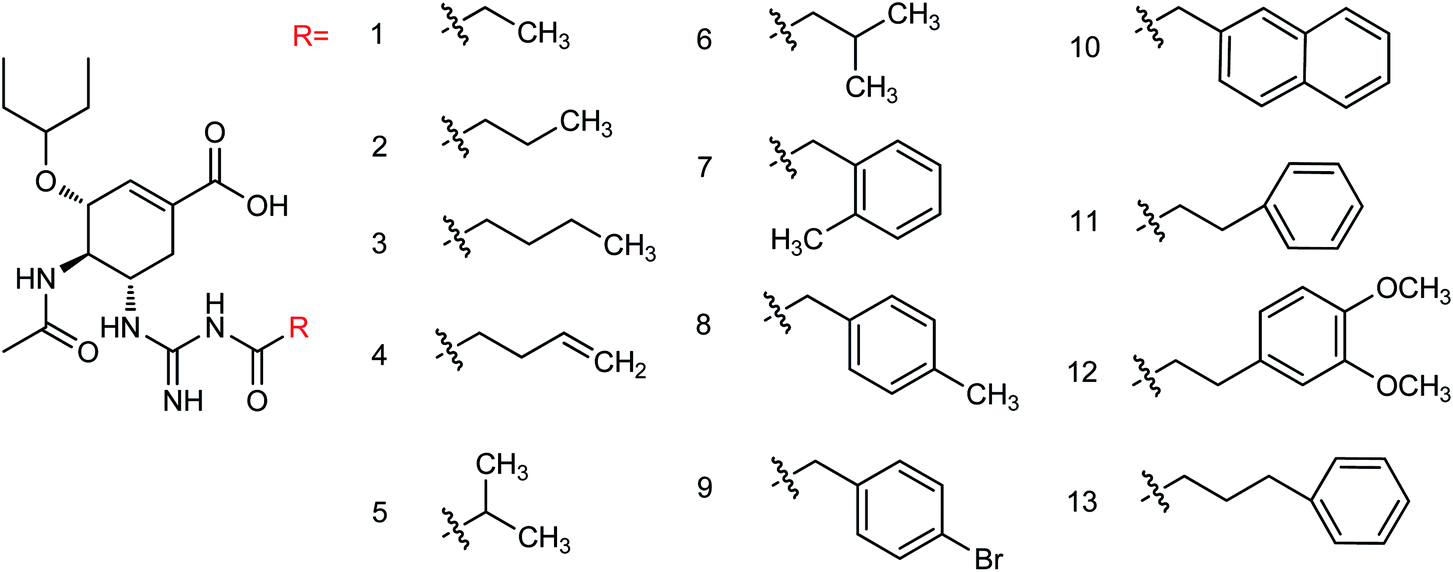
Liu and co-workers114 have reported some potent oseltamivir derivatives on the basis of their earlier discovery of N1-selective inhibitors (Fig. 28). Some of the synthesized derivatives were exceptionally active against group-1 and group-2 NAs. These compounds displayed greater inhibitory activity than OSC towards E119V and H274Y variant. Moreover, they showed significant potency toward viruses (H5N2, H5N1, H5N6, and H5N8) in cellular assays. Furthermore, molecular dynamics and computational modeling studies have also been discussed to understand the role of R group. This study will be an important breakthrough in the development of more potent anti-influenza agents.
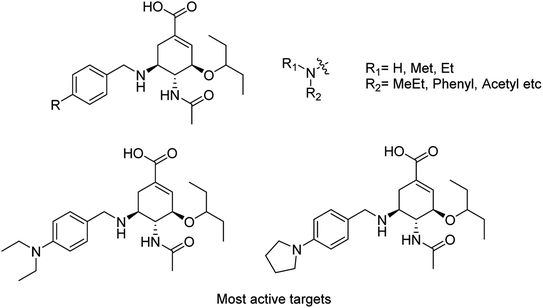 |
| | Fig. 28 Structures of N-substituted oseltamivir derivatives. | |
Cairo and co-workers115 have tested the inhibitory activity of DANA, oseltamivir, zanamivir, and peramivir against the human isoenzymes (Fig. 29). These compounds showed poor activity against the human neuraminidase enzymes except DANA and zanamivir. Ordered water is important for inhibitor design according to molecular docking simulation of NEU2 and NEU3.
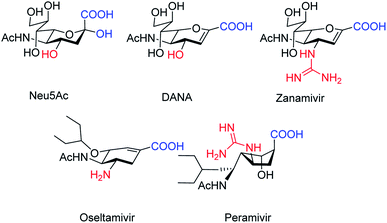 |
| | Fig. 29 Structures of tested compounds. | |
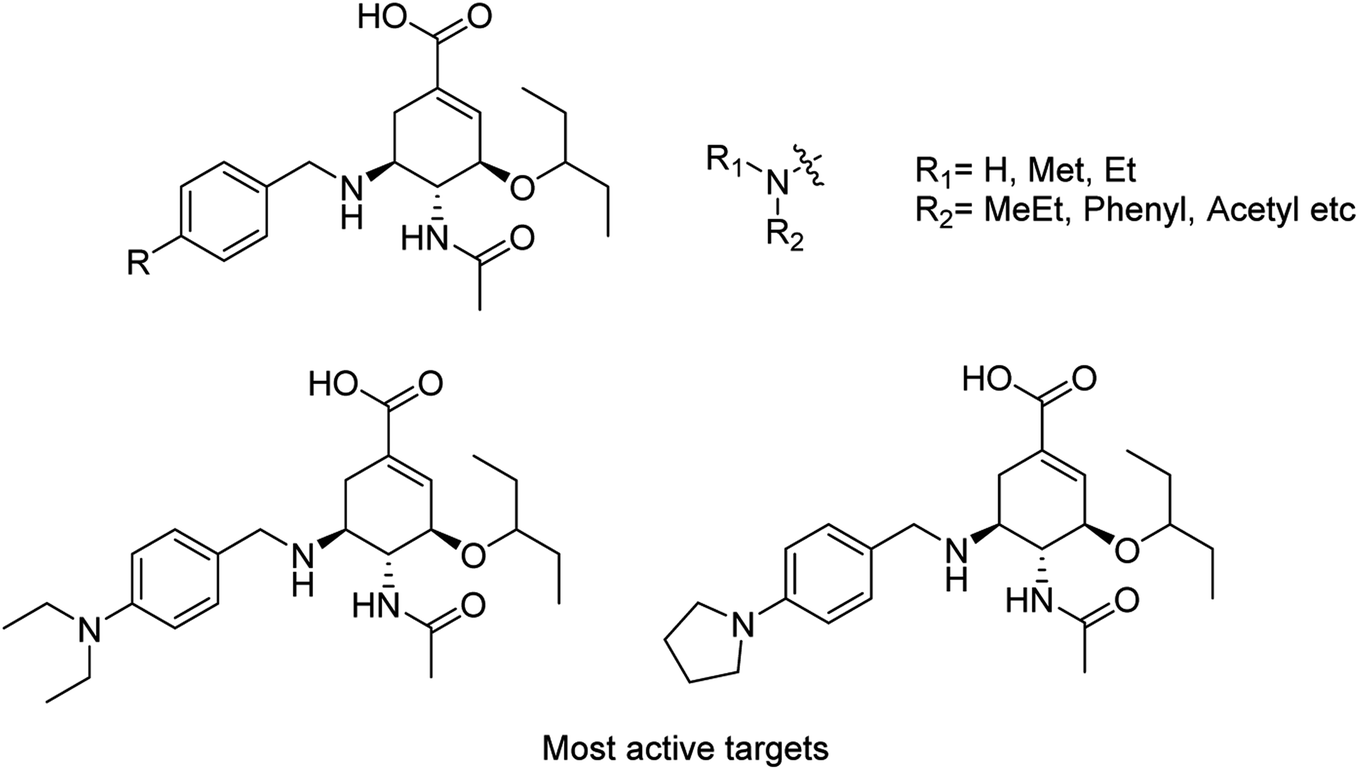
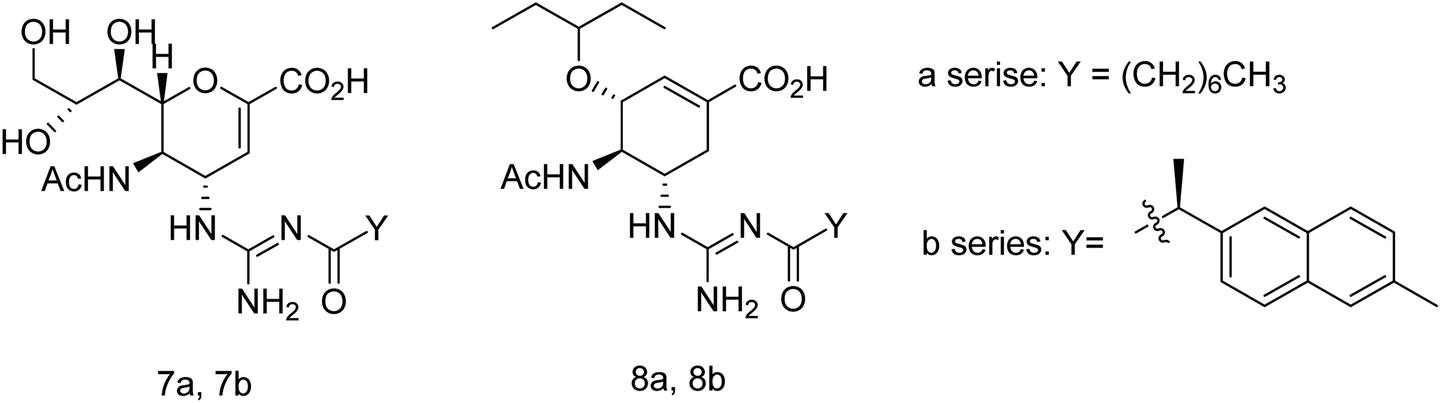
Toshimaa and co-workers116 have chemically synthesized a series of fucoidan derivatives to bind HAs of influenza virus and inhibit the hemagglutination activity. Moreover, a synthesized ligand with three sulfated oligofucoside moieties showed attractive hemagglutination inhibition activity. One of the reported compounds effectively inhibited influenza virus infection. Zanamivir and GOC are the potent inhibitors against influenza neuraminidase. Fang and co-workers117 have investigated the use of zanamivir and GOC acylguanidine derivatives (Fig. 30) as possible oral prodrugs. The derivatives have synthesized by coupling with either (S)-naproxen or n-octanoic acid. According to molecular docking studies, the straight octanoyl chain could extend to 150-cavity and 430-cavity of NA to gain extra hydrophobic interactions. Mice taking a zanamivir octanoylguanidine derivative survived better than those treated with zanamivir. Moreover, a GOC octanoylguanidine derivative could be orally administrated to treat mice with efficacy equal to oseltamivir. This study may be helpful for the development of oral drugs for influenza therapy.
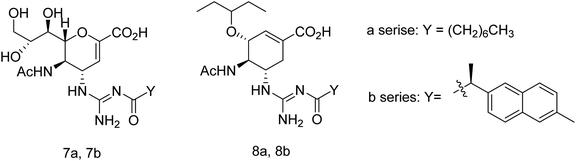 |
| | Fig. 30 Structures of acylguanidine derivatives. | |
Yin and co-workers118 have synthesized a series of 1-amino-2-alkanols. According to the study, (E)-1-amino-4-phenylbut-3-en-2-ol had better inhibitory activities than 2-amino-1-arylethan-1-ol derivatives. The sulfonation of (E)-1-amino-4-phenylbut-3-en-2-ol with 4-methoxybenzenesulfonyl chloride afforded more active inhibitor II. Moreover, molecular docking and SAR analysis also aided valuable information towards the discovery of (E)-1-amino-4-phenylbut-3-en-2-ol derivatives as potent and novel NA inhibitors. Kobayashi and co-workers119 have reported the MGO suppressed influenza A virus replication in a strain-independent manner. In this study, the anti-influenza activity of methylglyoxal against influenza B strains by using MDCK cells was evaluated. The susceptibilities of influenza A and B viruses were compared. This study confirmed that methylglyoxal has potent inhibitory activity against influenza B viruses. A novel approach to hPIV-3 inhibitor design (Fig. 31) has been evaluated by von Itzstein and co-workers120 with the introduction of nitrogen-based functionalities on the neuraminidase inhibitor template N-acyl-Neu2en. Due to introduction of triazole substituents, the compounds showed a 48-fold improved potency over the corresponding C-3 unsubstituted analogue. However, C-3-triazole N-acyl-Neu2en derivatives were less active against the virus' haemagglutinin function and showed insignificant in vitro antiviral activity.
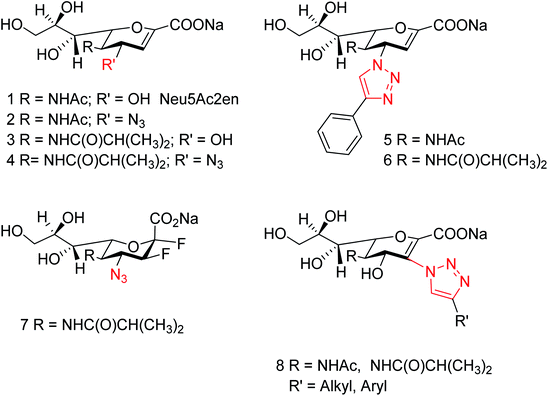 |
| | Fig. 31 Structures of reference hPIV-3 HN inhibitors (1–7) and the potential new target inhibitors (8). | |
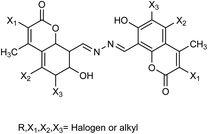
To enhance the therapeutic effects, the azo (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) linkage is very much useful for linking two bioactive moieties. Based on this, Narasimhan and co-workers121 have discussed the recent developments on diazenyl derivatives as antimicrobial agents during the last five years. This study has provided useful information to develop novel antimicrobial agents by slight modifications in active derivatives. Due to poor solubility in saline, baicalin has limited clinical use. In this context, Dou and co-workers122 have prepared sodium baicalin (Fig. 32), which showed more solubility in saline. It was evaluated against oseltamivir-resistant mutant H1N1-H275Y in vitro and in vivo. The treatment alleviated lung injury and body weight loss. This study confirmed that sodium baicalin is effective on wild-type and oseltamivir-resistant viral strains.
N) linkage is very much useful for linking two bioactive moieties. Based on this, Narasimhan and co-workers121 have discussed the recent developments on diazenyl derivatives as antimicrobial agents during the last five years. This study has provided useful information to develop novel antimicrobial agents by slight modifications in active derivatives. Due to poor solubility in saline, baicalin has limited clinical use. In this context, Dou and co-workers122 have prepared sodium baicalin (Fig. 32), which showed more solubility in saline. It was evaluated against oseltamivir-resistant mutant H1N1-H275Y in vitro and in vivo. The treatment alleviated lung injury and body weight loss. This study confirmed that sodium baicalin is effective on wild-type and oseltamivir-resistant viral strains.
 |
| | Fig. 32 Synthesis of sodium baicalin. | |
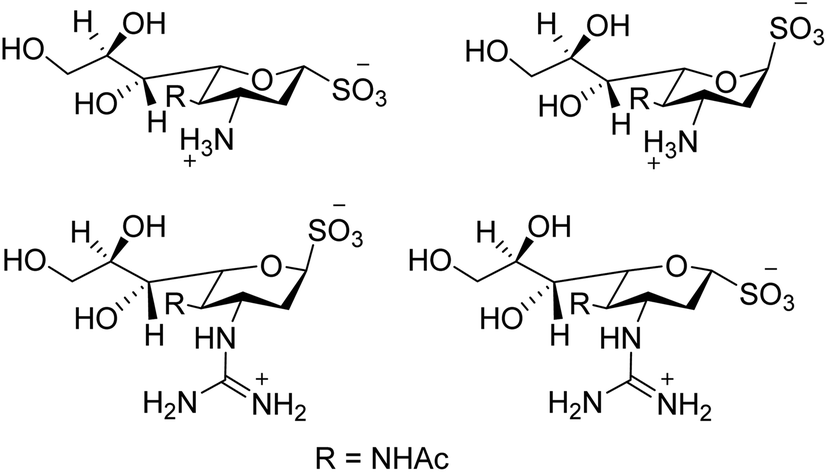
Swine flu is caused by influenza A virus in both swine and human. Neuraminidase plays a significant role in the infection of progeny virions inside the respiratory tract of the host body. Hafeez and co-workers123 have predicted possible neuraminidase inhibitors from the online available tools using a bioinformatics software. The active site has been predicted and then allowed for docking studies. According to docking results, indacaterol have minimum free energy (−8.2 kcal mol−1). An extremely potent inhibitor of influenza virus sialidase (Fig. 33) was identified by von Itzstein and co-workers.124 The synthesised 3-guanidino sialosyl α-sulfonate inhibits virus replication in vitro comparable to that of zanamivir. As per protein X-ray crystallography, the sialosyl α-sulfonate template binds inside the sialidase active site in 1C4 chair conformation. This sulfonozanamivir analogue will bring a fruitful step towards the anti-influenza drug development.
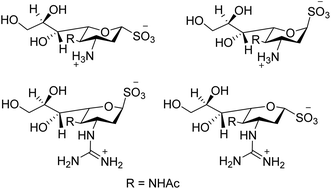 |
| | Fig. 33 Structures of 3-amino sialosyl sulfonates and 3-guanidino sialosyl α-sulfonates. | |
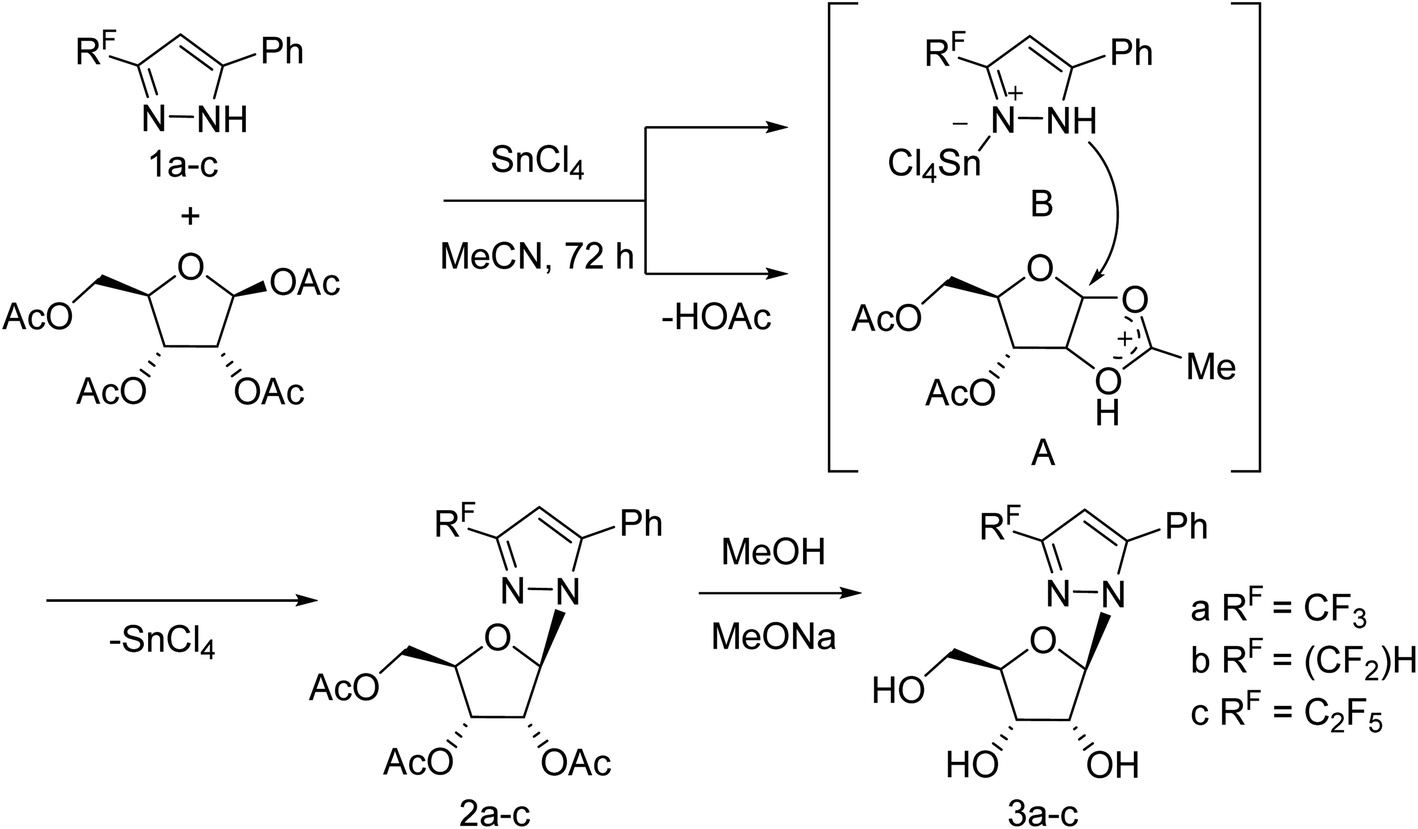
Jia and co-workers125 have reported the progress of small molecular inhibitors such as HA inhibitors, NA inhibitors, RdRp inhibitors and M2 ion channel protein inhibitors acting as antiviral agents. They also summarized some newly reported potential targets and strategies to develop new anti-influenza drugs. Ivanova and co-workers126 have reported the anti-influenza activity of some pyrazole derivatives (Fig. 34). In this study, 1-(β-d-ribofuranosyl)-3-polyfluoroalkyl-5-phenyl-1H-pyrazoles were synthesized from regiospecific ribosylation of 3-polyfluoroalkyl-5-phenylpyrazoles and 1,2,3,5-tetra-O-acetyl-b-d-ribofuranose in the presence of SnCl4 followed by deacetylation.
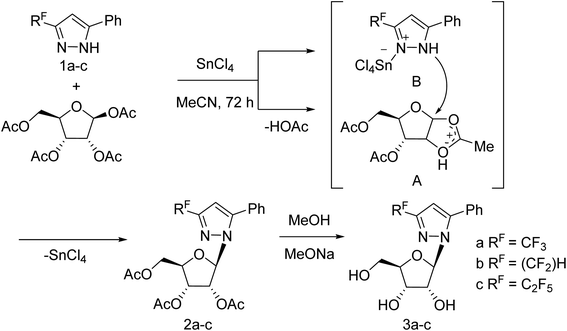 |
| | Fig. 34 Synthesis of 1-(β-d-ribofuranosyl)-3-polyfluoroalkyl-5-phenyl-1H-pyrazoles. | |
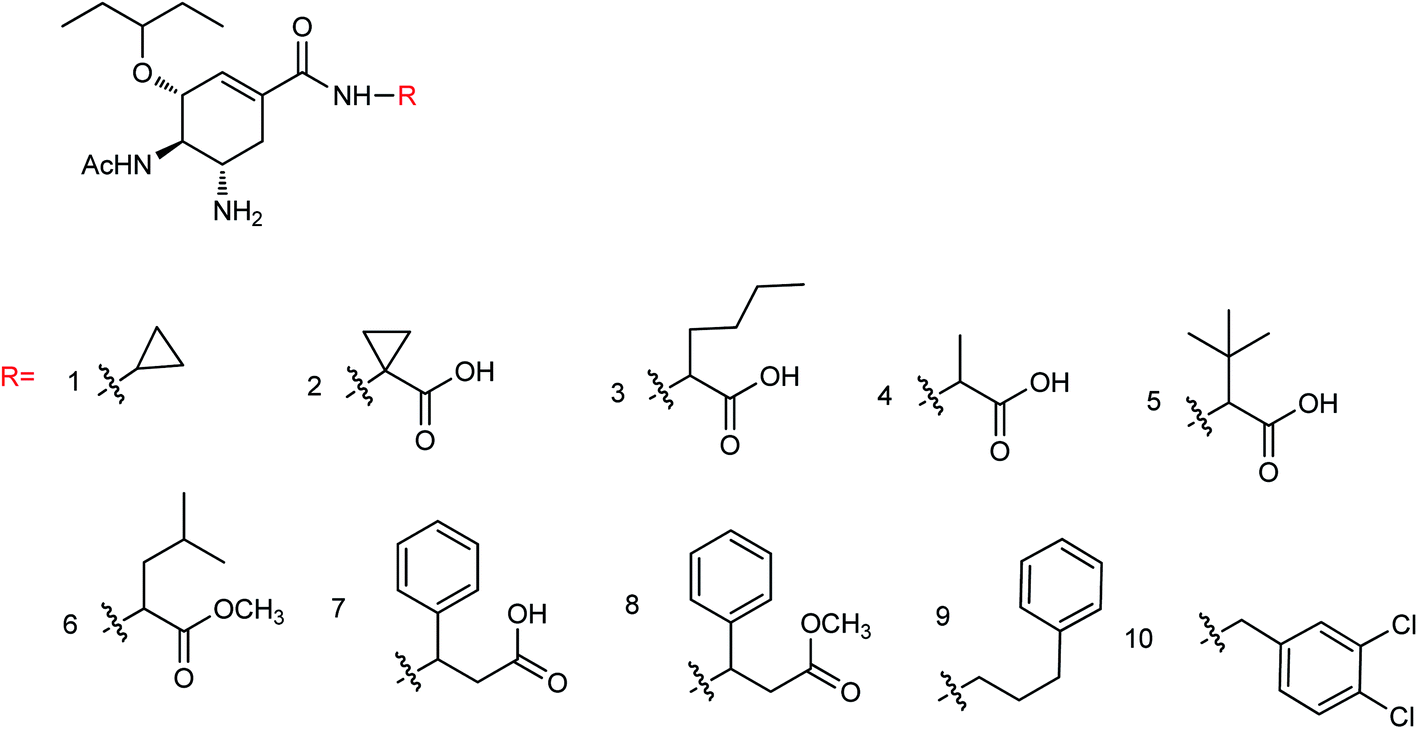
Liu and co-workers127 have synthesized a group of novel oseltamivir derivatives with C-1 modification, targeting the 430-cavity. These reported compounds (Fig. 35) showed robust anti-influenza potencies against H5N1 and H5N6 viruses. Moreover, the molecular modeling study has also been carried out. The study will be helpful in the discovery of potent inhibitors against group-1 and group-2 NAs.
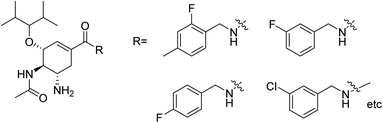 |
| | Fig. 35 Structures of C-1-modified oseltamivir derivatives. | |
Cairo and co-workers128 have designed a series of DANA analogues (Fig. 36) with modifications at C4 and C9 positions. They discovered selective inhibitors targeting the human NEU3 isoenzyme. The most selective inhibitor (Ki = 320 ± 40 nM) has a 15-fold selectivity over other human neuraminidase isoenzymes. It blocks the glycolipid processing by NEU3 in vitro. Some esters were also synthesized from the best inhibitors. Few synthesized compounds displayed selective inhibition of NEU orthologues from the murine brain.
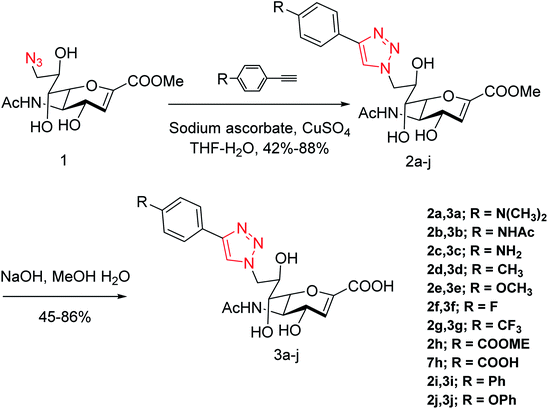 |
| | Fig. 36 Synthetic route to C9-phenyltriazole DANA analogues. | |
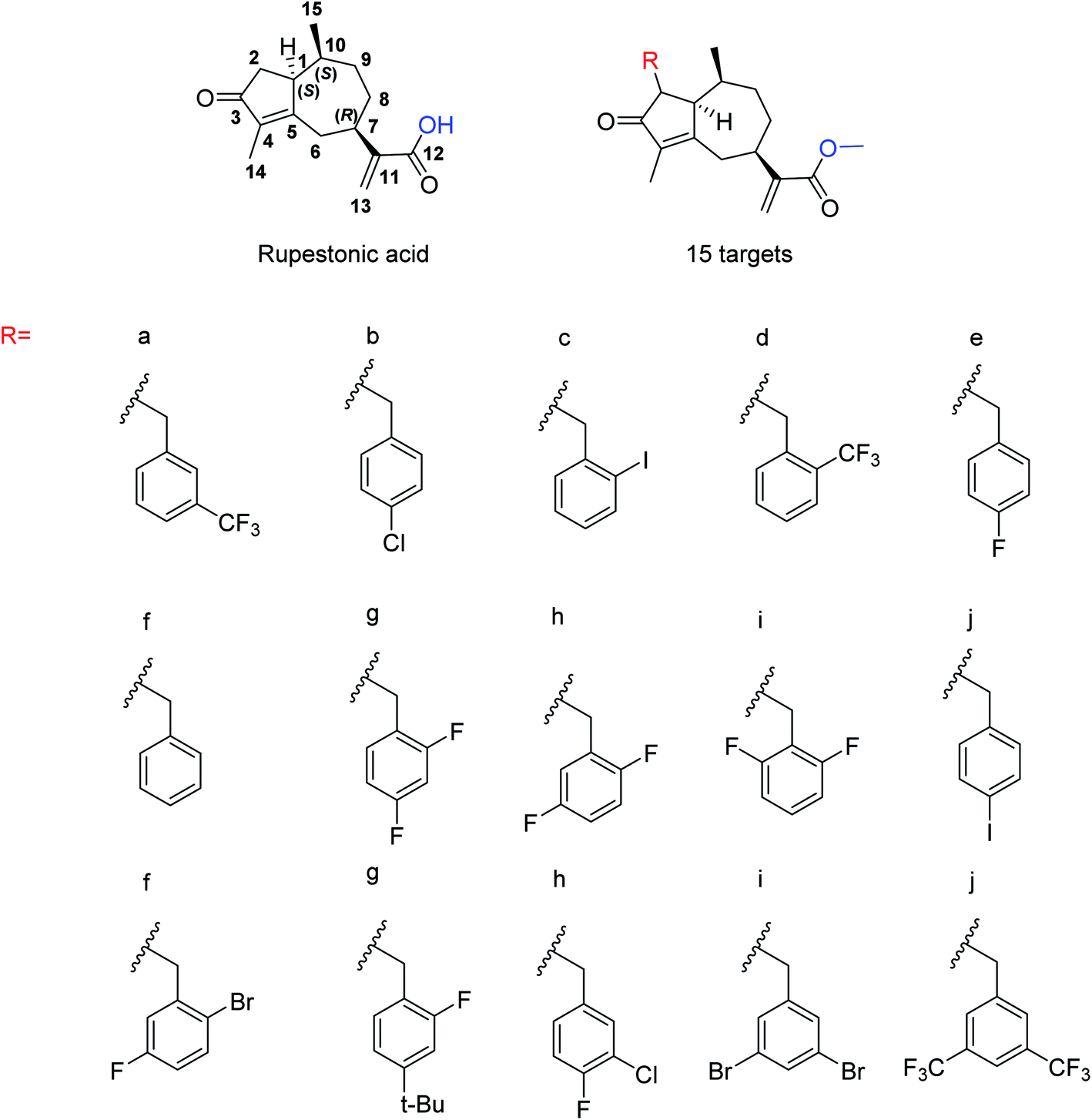
Due to the drug-resistant activity, it is an essential and urgent need to discover some novel antiviral drugs. Artemisia rupestris L. is a folk medicine used for the treatment of cold. Huang and co-workers129 have modified rupestonic acid (Fig. 37), a component of A. rupestris, to synthesize a series of fifteen 2-substituted rupestonic acid methyl esters. The compounds were characterized by using several spectral tools. These compounds (3b and 3c) displayed potent activities against H1N1 and may be used for the treatment of influenza.
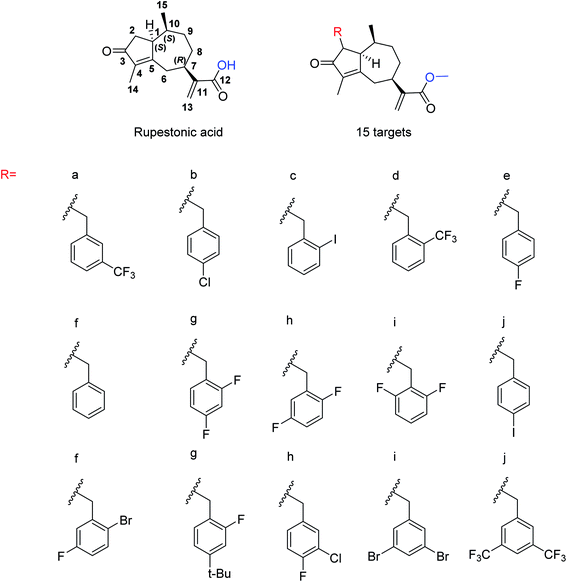 |
| | Fig. 37 Structures of isolated rupestonic acid and rupestonic acid derivatives. | |
6.2. Natural neuraminidase inhibitors
The Laggera pterodonta herb may inhibit influenza virus infection but its anti-influenza components remain unknown. Yang and co-workers130 have isolated the components (Fig. 38) from the plant by column chromatography. NA inhibition was determined by chemiluminescence assay. The anti-inflammation and anti-virus effects were measured by dual-luciferase reporter assay, quantitative real-time PCR, immunofluorescence, and luminex assay. It was suggested that pterodontic acid (isolated from L. pterodonta) might be a potential antiviral agent against influenza A virus.
 |
| | Fig. 38 Structure of pterodontic acid. | |
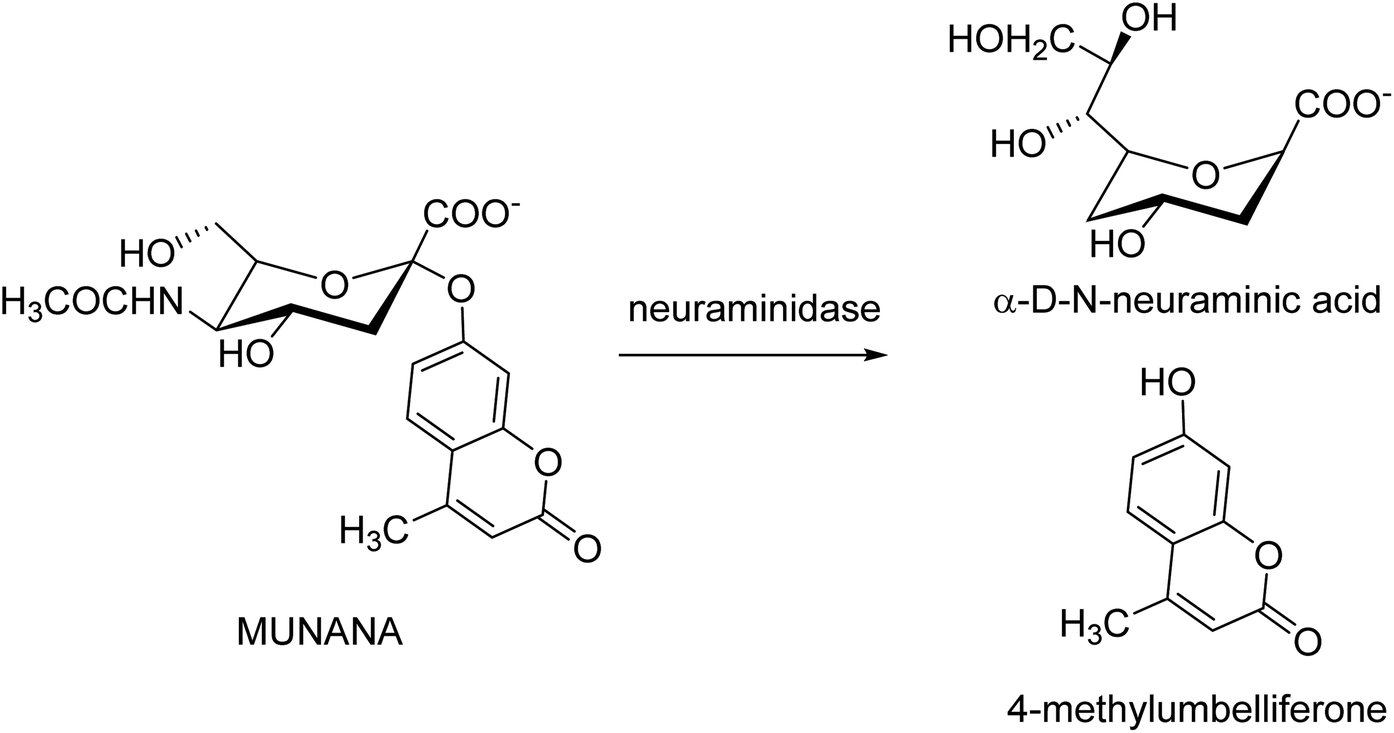
Kolodziej and co-workers131 have assessed the inhibitory activity of plant tannins (Fig. 39) using a MUNANA-based activity assay on both bacterial and viral model neuraminidase. The evaluated potency was compared with OC and zanamivir against H1N1 and VCNA. All the tested polyphenols presented a weak inhibition of the viral enzyme but higher potency on the bacterial neuraminidase. In addition, SAR analyses were performed to understand which groups are crucial for inhibitory activity.
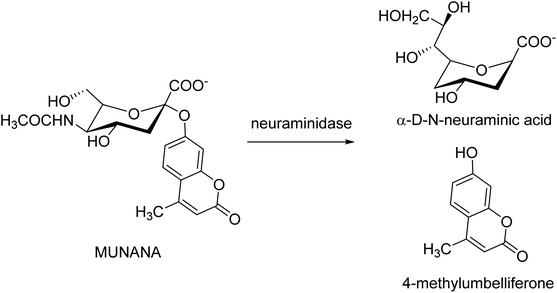 |
| | Fig. 39 Neuraminidase-catalyzed hydrolysis of 20-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (MUNANA). | |
Marine microbials have attracted tremendous attention in the last two decades. Wang and co-workers132 have reported simpterpenoid A (Fig. 40) containing a highly functionalized cyclohexadiene moiety (ring C), identified from mangrove-derived Penicillium simplicissimum MA-332. The X-ray crystallographic analysis was also reported to understand the unambiguous assignment of the planar structure and steric configuration. Simpterpenoid A showed inhibitory activities against influenza neuraminidase in nanomolar quantities.
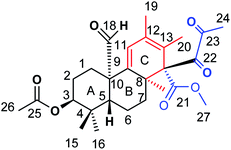 |
| | Fig. 40 Structure of simpterpenoid A. | |
Kanyalkar and co-workers133 have designed a series of chalcone derivatives based on isoliquiritigenin (natural NA inhibitor). Molecular modeling studies suggested that isoliquiritigenin and its analogs occupied a 430-loop cavity of NA. Moreover, the favorable compounds were synthesized and evaluated. The inhibitory effect was also quantified by H1N1-NA inhibition and HA assay. The marine-derived microbes are biologically active natural products. Keeping this in mind, Wang and co-workers134 have reported ten citrinin analogues from the EtOAc extracts of a coculture of two marine algal-derived endophytic fungal strains: Penicillium citrinum (EN-535) and Aspergillus sydowii (EN-534). The structures were confirmed by NMR, ECDs, HRESIMS, optical rotation, and XRD data. The antimicrobial and influenza neuraminidase inhibitory activities of these compounds were also assessed. Due to the drug resistance activity of unfamiliar strains to the available drugs, there is a rapid demand for the discovery of antiviral drugs. Neuraminidase is a crucial enzyme for viral replication and is the most promising target for the new drugs. Chandra and co-workers135 have reported the update on neuraminidase enzyme highlighting its structure, function, mechanism and inhibition by natural products, which will be helpful in the discovery of antiviral agents in the future. Some experimental studies have reported that tea catechins may inhibit influenza viral adsorption and suppressed replication and neuraminidase activity. In addition, tea catechins may be helpful to enhance immunity against viral infection. Moreover, regular consumption of green tea decreases the infection rates of some cold symptoms, and gargling with tea catechin may protect against influenza infection. Yamada and co-workers136 have briefly discussed the effect of tea catechins on common cold and influenza infection with a focus on clinical studies. Park and co-workers137 have reported the isolation of phenolic metabolites (Fig. 41) from the methanol extract of Usnea longissima by phytochemical investigation. All these compounds were characterized by several spectral techniques. Most of the compounds displayed dose-dependent inhibition when assessed against BNA. The compounds 2 and 3 (IC50 = 7.8, 8.2 μM) had three-fold greater potency than quercetin (IC50 = 21.4 μM). Furthermore, compounds 2 and 3 showed reversible and mixed type-I inhibitory behaviors (Ki = 6.8 and 7.2 μM, respectively).
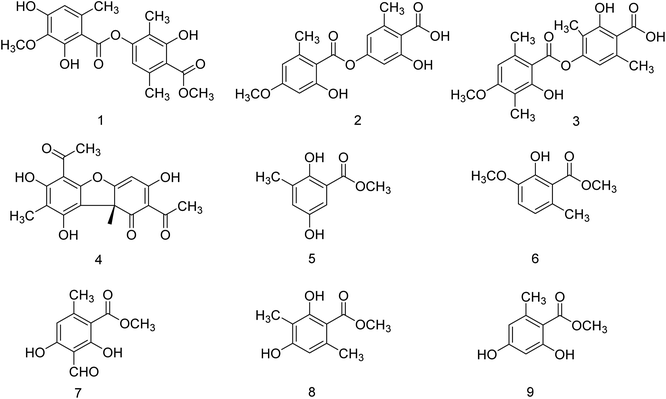 |
| | Fig. 41 Structures of isolated compounds from U. longissima. | |
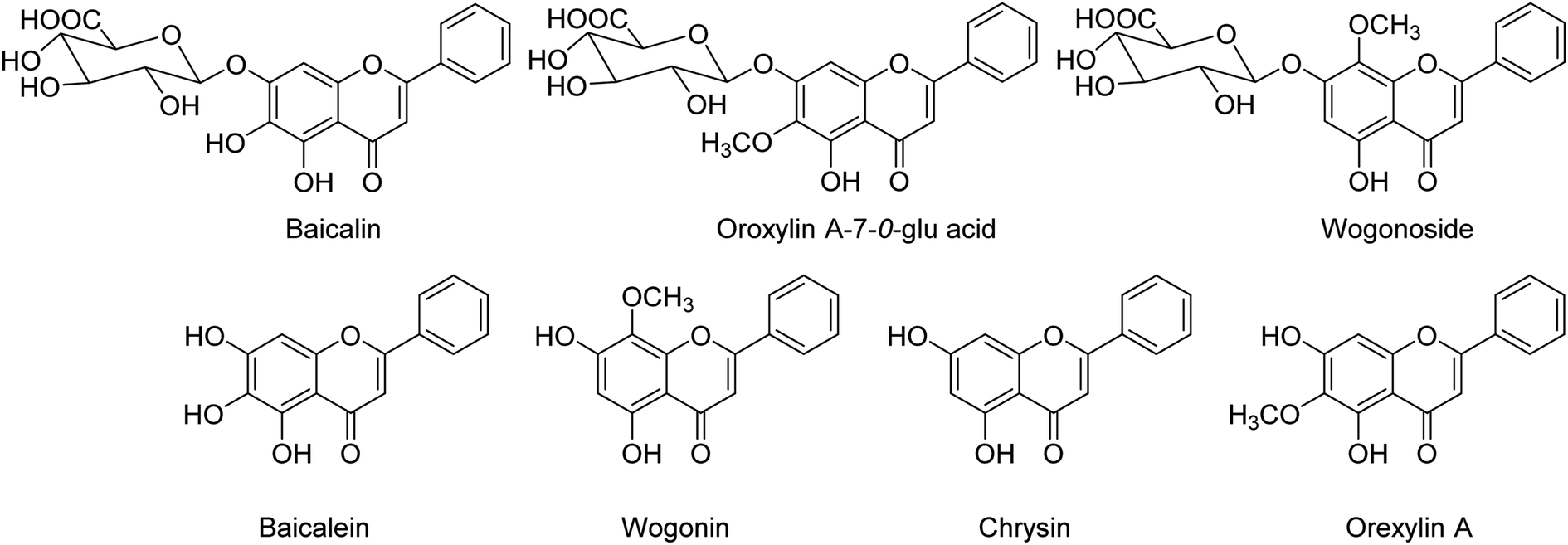
BF-30 (peptide) isolated from the venom of Bungarus fasciatus (snake) is suggested to have antitumor and antibacterial activities. Dou and co-workers138 have suggested that BF-30 could inhibit influenza virus strains (H1N1 and H3N2) and oseltamivir-resistant H1N1, in vitro. Mice treated with BF-30 exhibited 50% survival at a dosage of 4 μM, with an approximately 2log viral titer decrease in the lung. According to the study, peptide BF-30 showed an effective inhibitory activity against influenza A virus, and hence, it will be a promising candidate for influenza therapy. Flavonoids containing Scutellaria baicalensis root are traditionally used to treat against fever, common cold, and influenza. The anti-influenza and anti-inflammatory activities of FESR and the main flavonoids (Fig. 42) were assessed by Chen and co-workers139 in vitro. Baicalin (BG) was given as a reference control. According to the assays, FESR has a great potential against ALI induced by IAV. Moreover, FESR exhibited more potent therapeutic effects than BG.
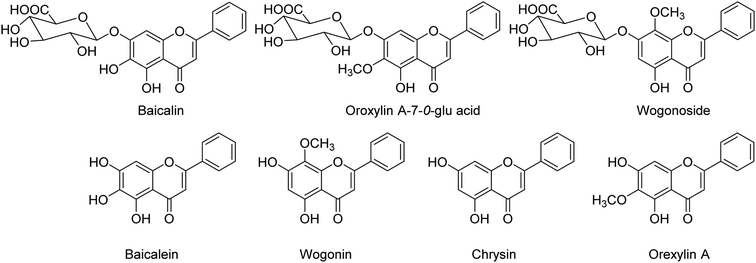 |
| | Fig. 42 Structures of baicalin, oroxylin A-7-O-glu acid, wogonoside, baicalein, wogonin, chrysin and oroxylin A. | |
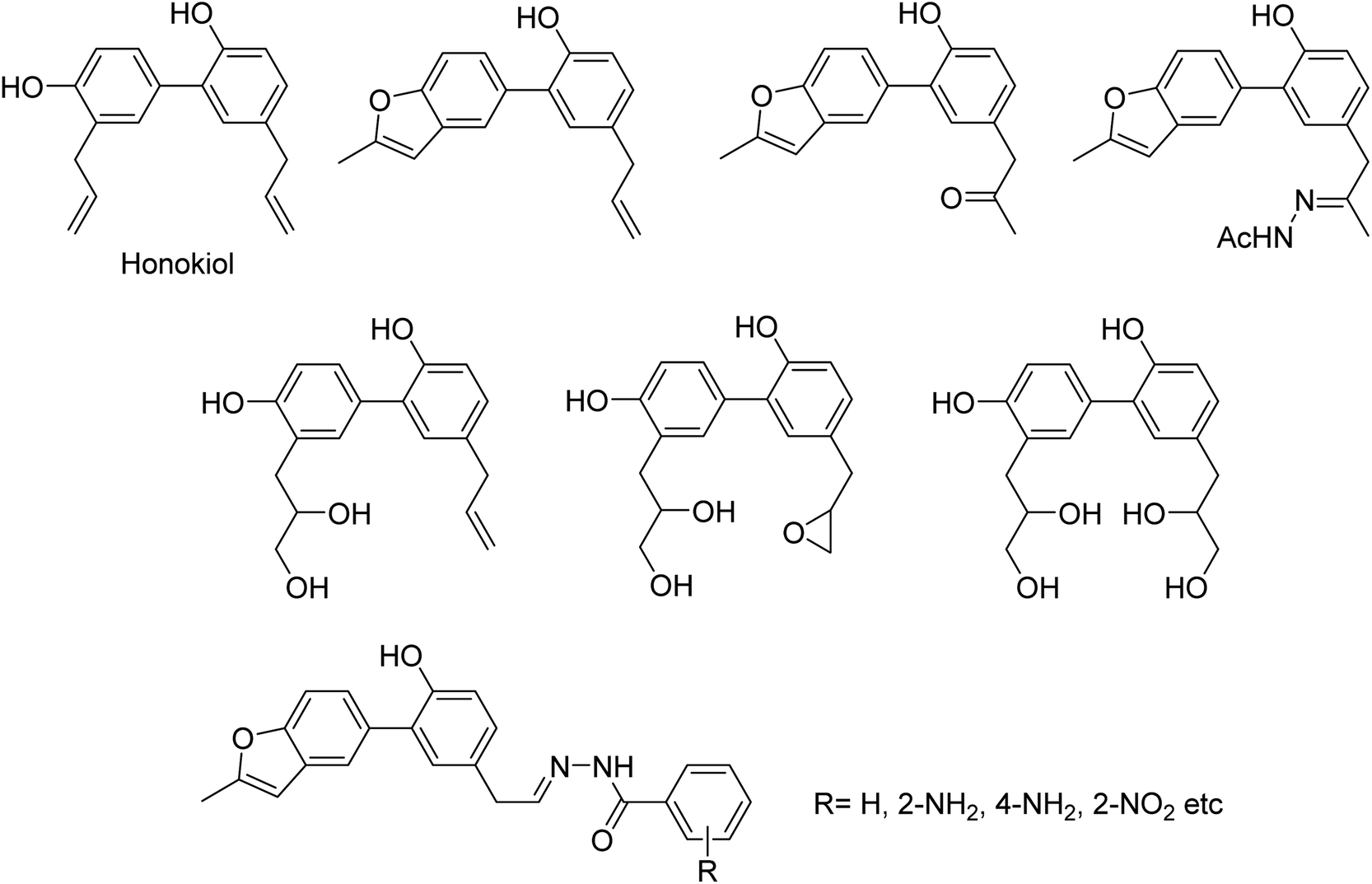
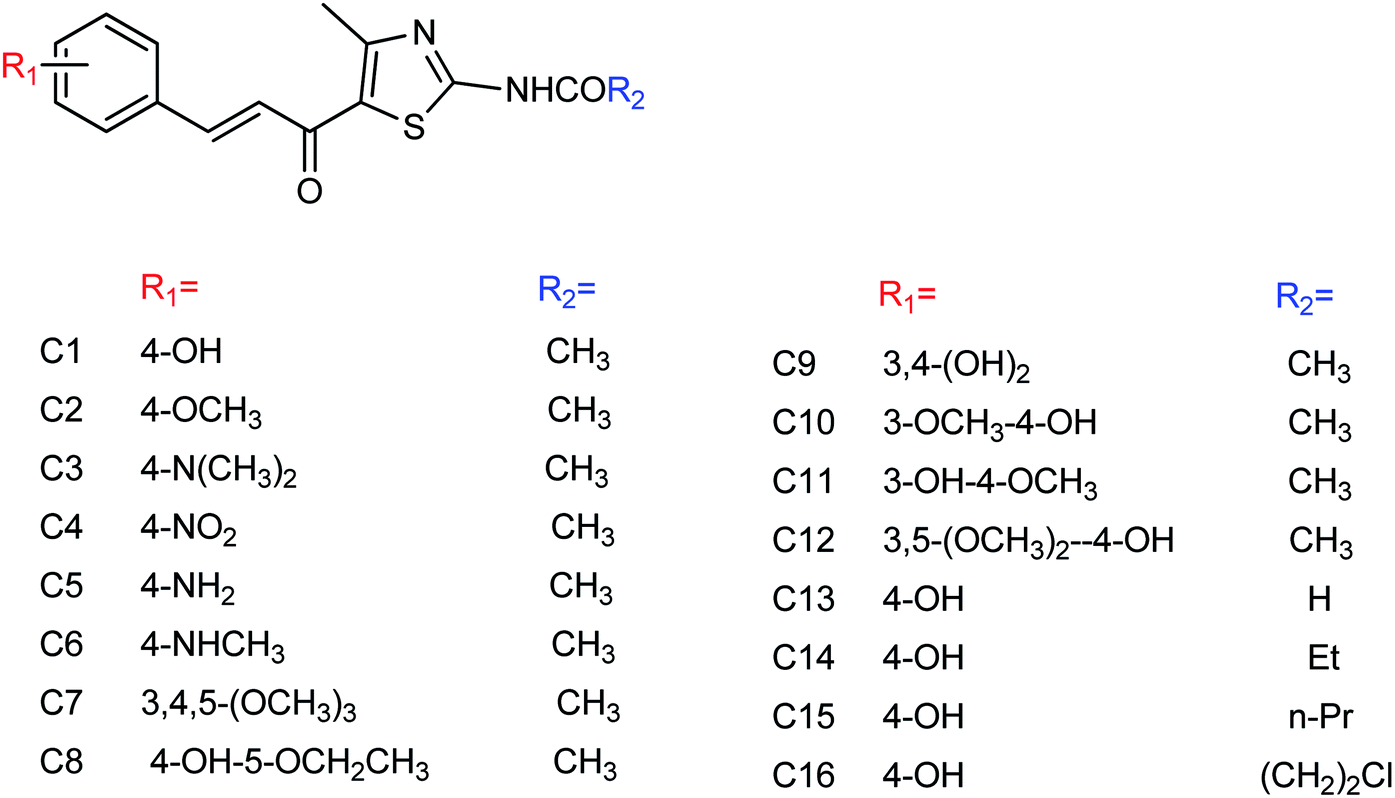
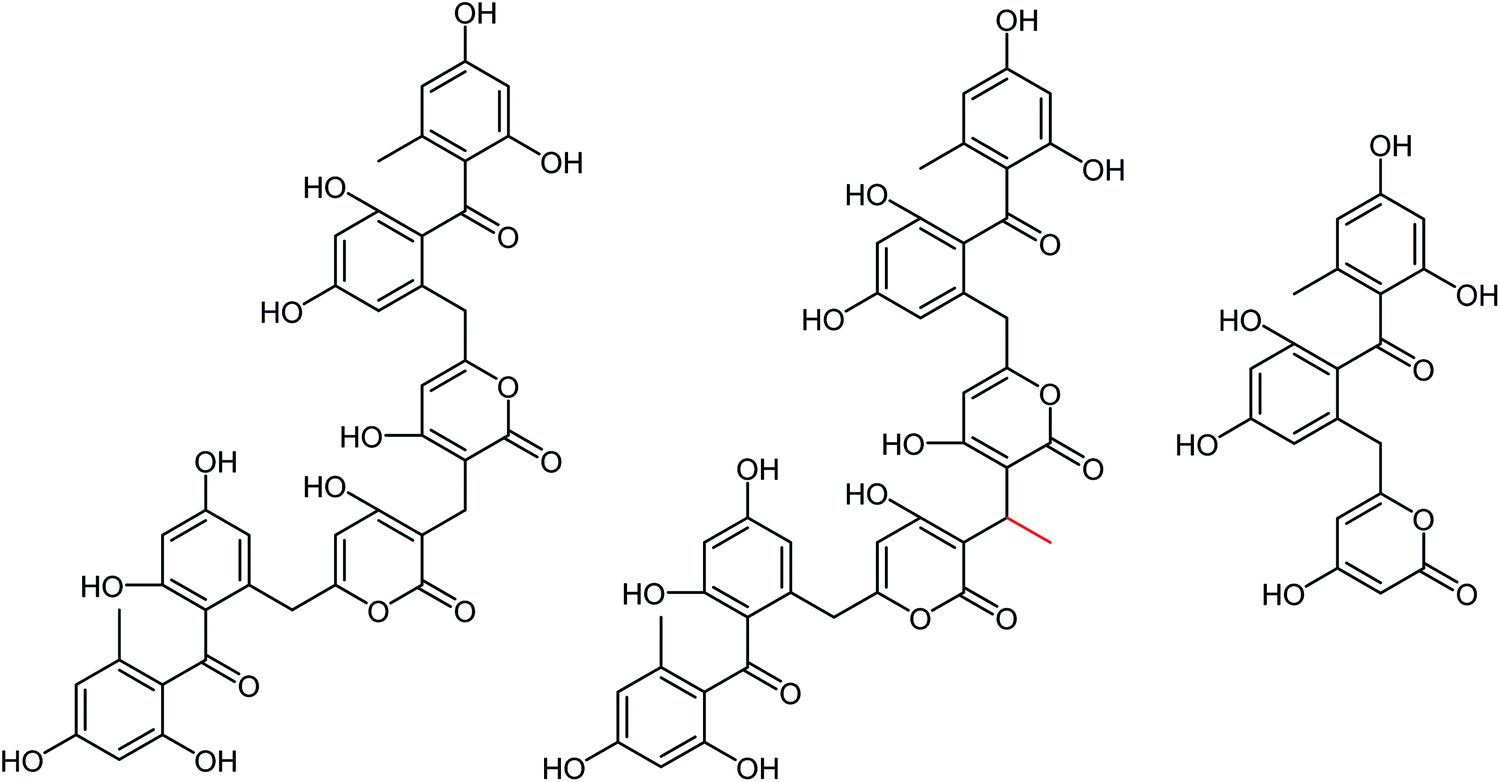
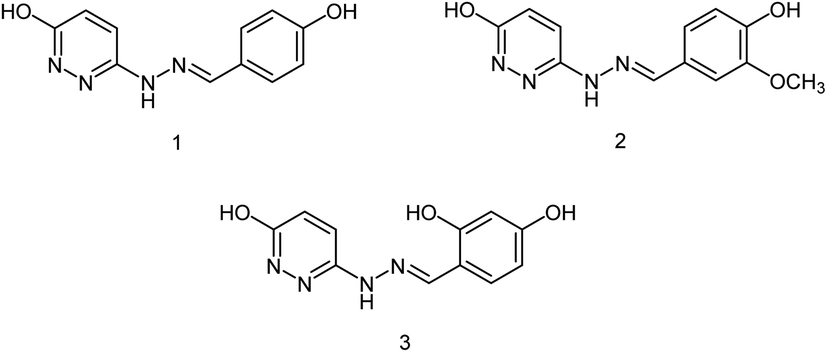
DS is a traditional herb available in various Asian countries. Nile and co-workers140 have reported the extraction of bioactive compounds and their biological activities from the EA, Bu and DW extracts of Dianthus superbus. The antiviral, antioxidant and anticancer activities were determined. EA extracts exhibited strong anticancer activities on NCL-H1299, SKOV, and Caski cancer cell lines. The Bu extracts showed the strongest antiviral activity with respect to both influenza A and B viruses. The isorhamnetin 3-glucoside and quercetin 3-rutinoside exhibited higher neuraminidase inhibition activity in a dose-dependent manner. This study may provide vital information for the effective utilization of DS for food, medicinal and therapeutic purposes. Geniposide extracted from Gardenia jasminoides Ellis (GJ) fruit, has antiviral and anti-inflammation activities. Ganzhu and co-workers141 have reported the antiviral and cytotoxicity activity of geniposide by an MTT assay. The influenza respiratory tract infection murine model was studied with A/Jiangsu/1/2009 (H1N1) virus. Geniposide substantially inhibited the virus-induced alveolar wall changes. The study confirmed that geniposide effectively inhibited the cell damage caused by A/Jiangsu/1/2009 (H1N1) virus and mitigated virus-induced acute inflammation. A natural polyphenol (Honokiol) having NA inhibitory activity was structurally modified by Hu and co-workers1 (Fig. 43). The authors have reported 23 compounds with their ortho-effects in epoxidation and hydrolyzation reactions. The benzoylhydrazone derivatives exhibited much better anti-NA activity than honokiol. According to SAR analysis, polyphenols showed better anti-NA activity than monophenols and biphenols. Moreover, the most active compound had potent anti-influenza virus activity.
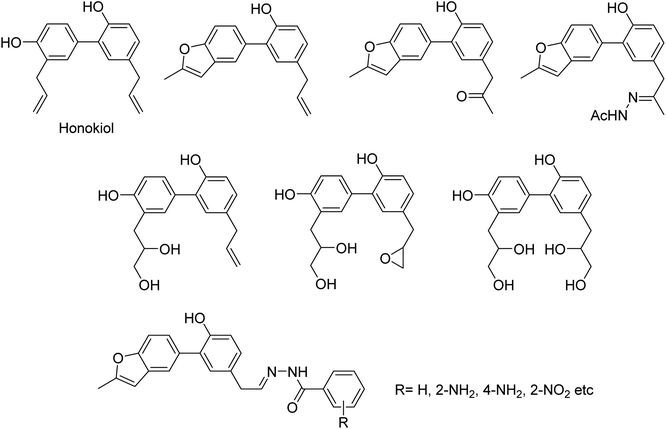 |
| | Fig. 43 Structures of honokiol and honokiol derivatives. | |
The chromenone derivatives (Fig. 44) obtained from Flemingia philippinensis exhibit significant inhibition against bacterial NA and play an important role in the pathogenesis of bacterial infection.142 As per the kinetic study, 8,8-diprenyl compounds showed a competitive inhibitory mode. Moreover, inhibitors (1 and 2) were simple, reversible and slow-binding against bacterial neuraminidase.
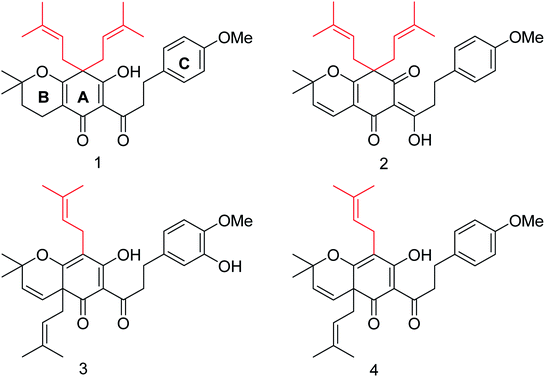 |
| | Fig. 44 Structures of isolated chromenone derivatives from Flemingia philippinensis. | |
The inhibition of tyrosinase and melanogenesis are two skin-whitening activities of artocarpin (Fig. 45), an isoprenyl flavone from Artocarpus species. It protects UVB-induced skin damage. Besides this, artocarpin possesses other pharmacological properties such as antimicrobial, anti-inflammatory, antioxidant, antiplasmodial, antitubercular, cytotoxic, and neuraminidase inhibitory activities. Chan and co-workers143 have reported the chemistry and pharmacology of artocarpin.
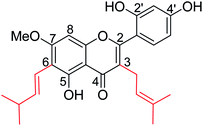 |
| | Fig. 45 Structure of artocarpin. | |
6.3. Patents study
Ye and co-workers144 have synthesized three novel compounds (Fig. 46) from 4-dimethylaminobenzaldehyde with the yields ranging from 59 to 89%. These compounds could inhibit neuraminidase to varying degrees. The anti-influenza virus aminotransferase activity of these compounds was detected in the enzyme reaction system.
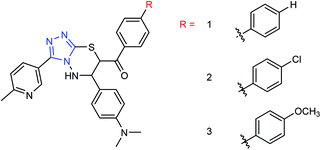 |
| | Fig. 46 Structures of 4-dimethylaminobenzaldehyde derivatives. | |
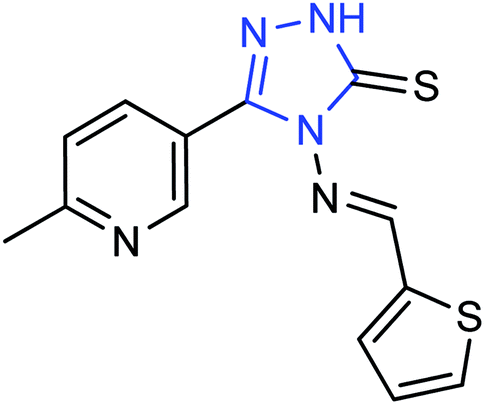
A new compound (Fig. 47), 3-(6-methylpyridine-3-yl)-4-[(thiophene-2-yl) methylene amino]-1H-1,2,4-triazol-5(4H)-thione, was synthesized from 6-methylnicotinic acid methyl ester by Ye and co-workers145 with a yield of 73%. At a concentration of 40 μg mL−1, the inhibition rate of the compound was 42.19%. It was proved that the compound has promising activities as an anti-influenza neuraminidase inhibitor and can be used for further preparation of neuraminidase inhibitors.
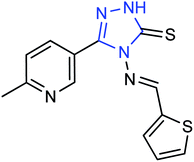 |
| | Fig. 47 Structure of 3-(6-methylpyridine-3-yl)-4-[(thiophene-2-yl) methylene amino]-H-1,2,4-triazol-5(4H)-thione. | |
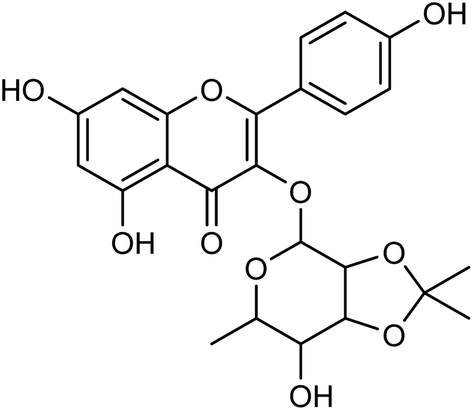
Cui and co-workers146 have used the branches, leaves or fruits of Cassia trees as raw materials, and separated a kind of flavonoid compounds (Fig. 48) by organic solvent ultrasonic extraction, organic solvent extraction, silica gel column chromatography and liquid chromatography. The molecular formula of the flavonoid is C24H24O10, named kaempferol-3-0-[(4′′5′′-0-isoprotylidene)-a-L-rhamnopyranoside]. The results indicated that the compound exhibited good neuraminidase inhibitory activity with an IC50 value of 187.40 μML−1. It can be used as a leading compound of anti-influenza drugs.
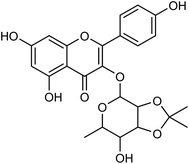 |
| | Fig. 48 Structure of kaempferol-3-0-[(4′′5′′-0-isoprotylidene)-a-L-rhamnopyranoside. | |
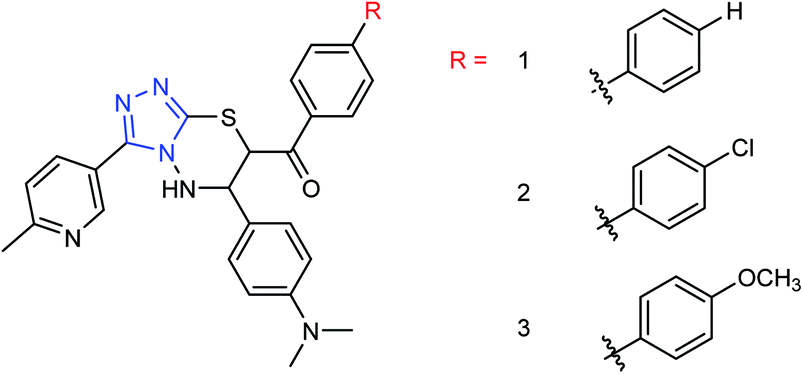
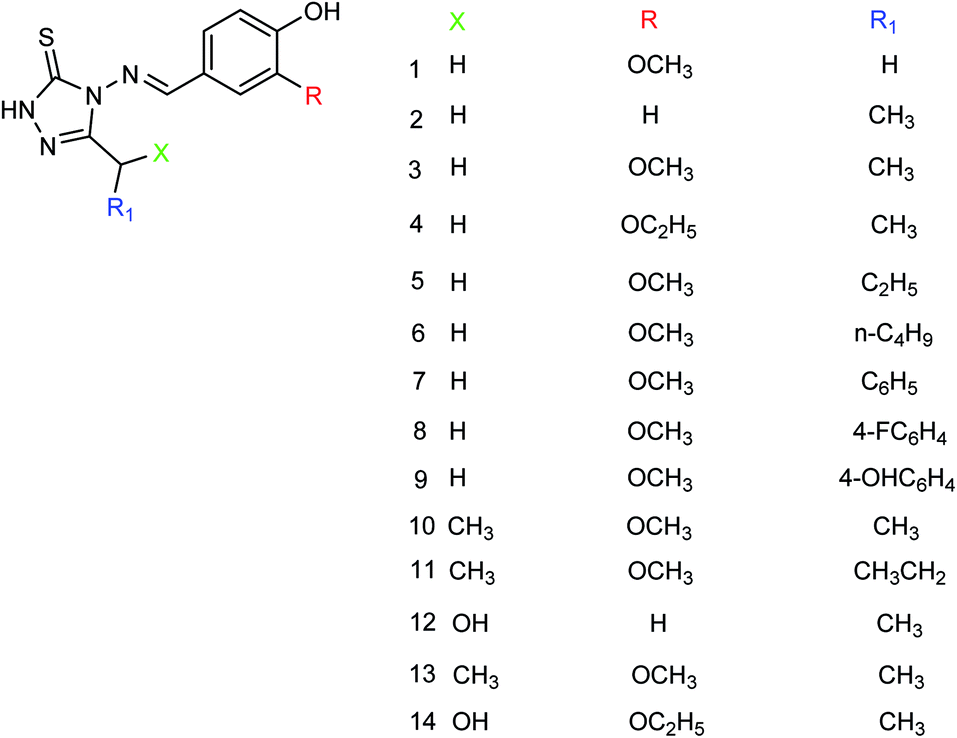
Peptide inhibitors are widely used in the prevention and treatment of viral infection. Ning and co-workers147 have used molecular docking to screen small molecules, which can dock with neuraminidase from a small molecular library. According to the docking score, the best 20 protein sequences were selected, and the method of increasing or reducing 2–4 amino acids was used to obtain peptide inhibitors with a higher antiviral bioactivity. In vitro experiments have investigated that the screened 10 peptides showed low IC50 values against influenza virus (H1N1). Among them, two peptides showed good inhibitory activities against H1N1 with IC50 values of 2.24 and 2.99 μM, and the results indicated a potent strong inhibitory effect against virus-infected cells. Their protein sequences were errKPAQP and hsFHHKPAK respectively, and the non-toxic effect of peptide inhibitors on HEK 293T was investigated. Three derivatives of 4-(arylimino)-3-alkyl-1H-1,2,4-triazol-5(4H)-thioketone (Fig. 49) were designed and synthesized by Yan and co-workers148 with the yields ranging from 60 to 75%. In vitro experiments indicated that the synthesized compound showed good activity against influenza virus neuraminidase, which can be used to prepare further neuraminidase inhibitors of influenza virus. The activity of compound 3 is the best, the IC50 value is 19 μg mL−1, and the inhibition rate of compound 3 to neuraminidase H1N1 is 70%.
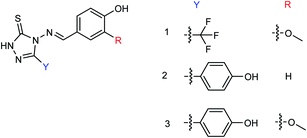 |
| | Fig. 49 Structure of 4-(arylimino)-3-alkyl-1H-1,2,4-triazol-5(4H)-thioketone derivatives. | |
Hu and co-workers149 designed and prepared a series of 4-(4-hydroxyphenyl-methyleneamino)-1,2,4-triazol-5-thione derivatives (Fig. 50) with the yields ranging from 55 to 82%. The most potent derivative was compound 3 with an IC50 value of 14.68 μg mL−1.
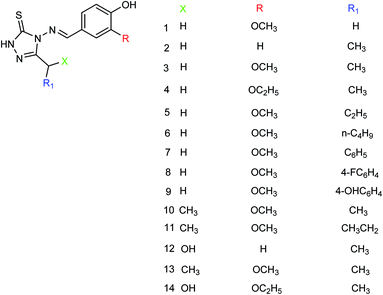 |
| | Fig. 50 Structures of 4-(4-hydroxyphenylmethyleneamino)-1,2,4-triazol-5-thione derivatives. | |
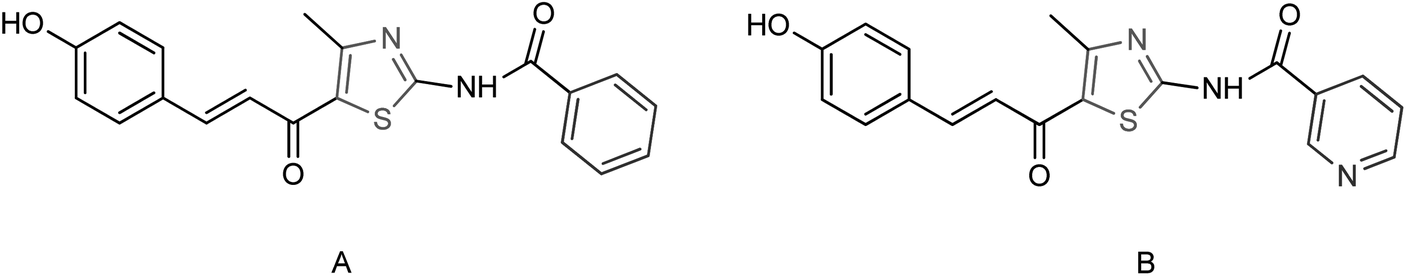
Ye and co-workers150 designed and synthesized two N-thiazolylpyridine formamide derivatives A and B as neuraminidase inhibitors (Fig. 51) with yields of 67% for both of them. Compounds A and B showed good inhibition rates of 82.7 and 83.6% respectively at a concentration of 40.0 μg mL−1. Compounds A and B showed potent inhibitory activities with IC50 values of 44.7 μM and 33.9 μM respectively.
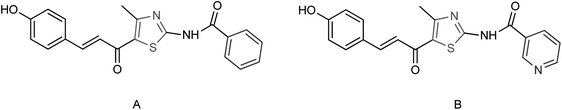 |
| | Fig. 51 The synthesized N-thiazolylpyridine formamide derivatives. | |
Two oxazolamide derivatives were synthesized (Fig. 52) by Ye and co-workers151 with the yields of 43.5 to 63.8% respectively. In vitro results showed that the inhibition rates of the compounds against A/PR/8/34 (H1N1) are 72.5% and 69.8% respectively at a concentration of 40.0 μg mL−1. The compounds showed inhibitory activity with IC50 values of 67.9 and 73.3 μM respectively. 4-Alkyl-5-(3-phenylacryloyl)-2-alkanoyl-aminoxazole showed good anti-influenza activity, which can be used to prepare potent neuraminidase inhibitors.
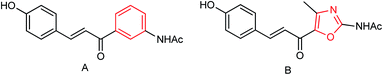 |
| | Fig. 52 Structures of oxazolamide derivatives. | |
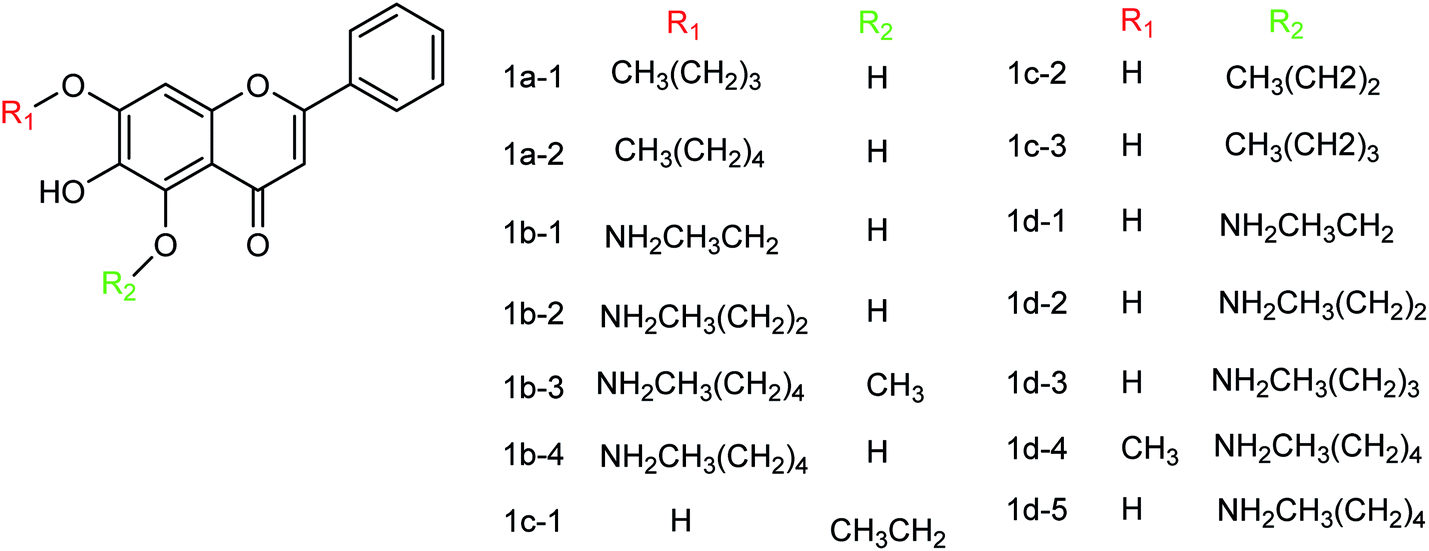
Baicalein and its derivatives including flavonoid have significant inhibitory effects on influenza virus; therefore, Li and co-workers152 designed and synthesized a series of baicalein derivatives (Fig. 53) bearing a novel structure with the yields ranging from 25 to 40%. In order to play the role of prevention and treatment to influenza virus H1N1, type I neuraminidase was chosen as the target. The compounds showed better inhibitory activity than baicalein as a positive control (the half inhibitory concentration of type I neuraminidase was lower than the baicalein control). In vitro results indicated good inhibitory activity of compound IB-3 with IC50 4.4 μg mL−1. Compound IB-3 showed good inhibitory activity with an IC50 value of 4.4 μg mL−1 and in general baicalein phenol hydroxyl esterified derivatives showed good activity and can play an important pharmacological role against H1N1 influenza virus for the treatment of influenza A. The synthesis process is simple and easy to implement and the reaction conditions are stable, controllable and easy to promote and apply.
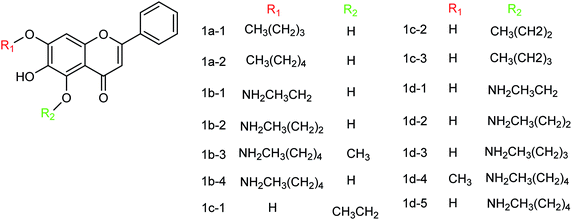 |
| | Fig. 53 Structures of baicalein derivatives. | |
Another study of Li and co-workers153 was to establish a small library of baicalein phenol hydroxyl etherified derivatives (Fig. 54) and a series of eighty-two derivatives were synthesized with the yields ranging from 20.2 to 41.4%. In vitro study showed the best activity of compound id-5 with IC50 of 4.5 μg mL−1 and the inhibitory activity was the same as the positive drug zanamivir. In addition, the activity of all compounds was better than baicalin, which has good inhibitory effects on type I neuraminidase.
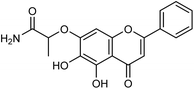 |
| | Fig. 54 Structure of compound Id-5. | |
Nine derivatives of 5-(3-phenylacryloyl)-2-benzoylaminothiazole (Fig. 55) were synthesized by Ye and co-workers154 with the yields ranging from 44 to 88%. Compound 4-methyl-5-acetyl-2-(3-methyl-4-nitrobenzoylamino)thiazole was selected by the national drug screening center of the Institute of Medicine, Chinese Academy of Medical Sciences as a positive drug for in vitro experiment. Compound J showed potent inhibitory activities among all of the synthesized compounds with an IC50 value of 14.5 μM at a concentration of 40.0 μg mL−1.
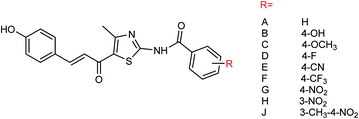 |
| | Fig. 55 Structures of 5-(3-phenylacryloyl)-2-benzoylaminothiazole derivatives. | |
There are many advantages in the preparation of neuraminidase compounds by microbial fermentation because there is no reduction of species and no pollution. Li and co-workers155 used ethyl acetate to extract the fermentation products of the marine fungus Aspergillus WENTII and purified them by gradient elution with reduced pressure silica gel column chromatography (Fig. 56). Four new diterpenoids with significant neuraminidase inhibitory activities were found. Compounds 1–4 showed significant inhibitory activities with IC50 values of 18.2, 10.9, 14.2 and 16.7 μM respectively.
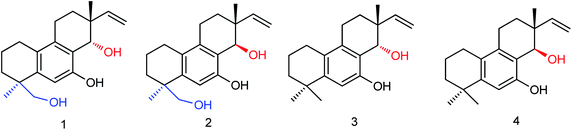 |
| | Fig. 56 Structures of diterpenoids. | |
Thirteen derivatives of 2-(5-nitrothiazol-2-yl)-imino-4-thiazolinone (Fig. 57) were designed and synthesized by Ye and co-workers156 with the yields ranging from 52 to 89%. Most of the compounds have good inhibitory activities at a concentration of 40.0 μg mL−1. Among them, 2-(4-tert-butyl-5-nitrothiazole-2-yl)imino-4-thiazoline showed the best activity with an IC50 value of 12.50 μg mL−1.
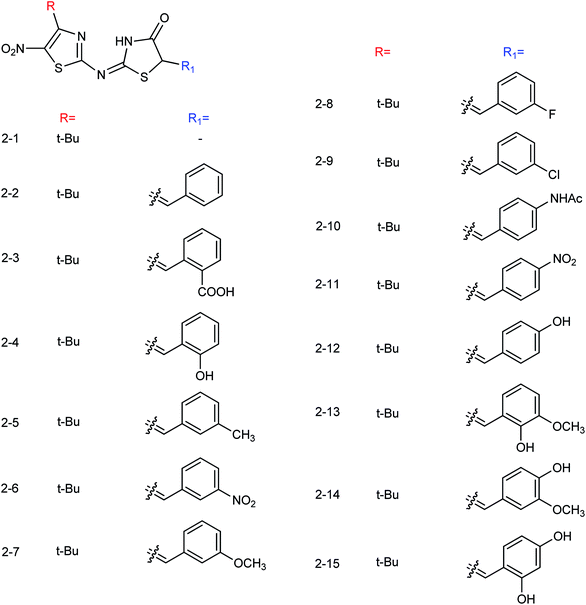 |
| | Fig. 57 Structures of 2-(5-nitrothiazol-2-yl)-imino-4-thiazolinone derivatives. | |
Ding and co-workers157 have extracted Chinese gallnut with ethyl acetate and n-butanol to obtain the corresponding ester and alcohol extracts. Gallnut possesses good inhibitory activity against H1N1 influenza virus, effectively controls the infectivity of the virus, down regulates the expression of inflammatory cytokines TNF-α, and improves the lung inflammation of influenza virus-infected mice. In order to further clarify the active parts of gallnut against influenza virus, the authors have carried out in vitro experiments on the components of gallnut. The alcohol extract and gallate extract of gallnut showed different degrees of inhibition, while the effect of the gallate extract was particularly obvious with the inhibition rate up to 83%. There are sesquiterpenes, chromogenic ketones, phenylpropanoids, alkaloids and other components in traditional Chinese medicine, which have a wide range of pharmacological activities. Qin and co-workers158 have studied the inhibitory activity of the extracts from dried Flos Farfarae which is also known as Kuandonghua in China. The results of pharmacodynamic experiments showed that the n-butanol extract of Flos Farfarae exhibited significant inhibitory activities. Among them, the n-butanol part (d) showed a strong inhibitory activity with IC50 = 27.64 ± 0.184 μg mL−1, indicating that the n-butanol part (d) is the effective part to inhibit H1N1 neuraminidase, which can be used to prepare further drugs for the prevention and treatment of H1N1 influenza. Jia and co-workers159 have synthesized a series of 5-(3-phenylacryloyl)thiazole derivatives (Fig. 58) with the yields ranging from 52.2 to 89.7%. Compound 5-(3-phenylacryloyl)thiazole derivatives showed good inhibitory activities and compound C1 showed the best activity with an IC50 value of 27.1 ± 0.1 μM. At the same time, the author compared the anti-influenza virus neuraminidase activity of reference compounds A and B (Institute of medicine, Chinese Academy of Medical Sciences) with compound C1. The results indicated that the inhibition rates of compounds A and B were 62.59% and 79.25% at a concentration of 40 μg mL−1, while the inhibition rate of compound C1 was 92.32%.
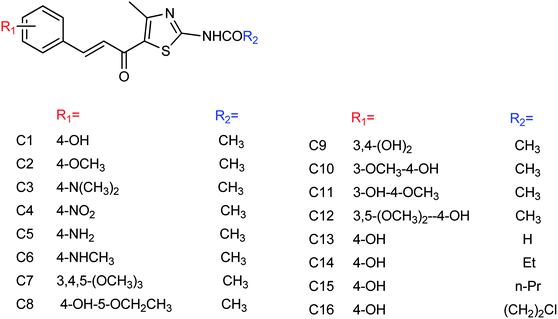 |
| | Fig. 58 Structures of 5-(3-phenylacryloyl)thiazole derivatives. | |
Two furan formamide derivatives 1 and 2 (Fig. 59) were designed and synthesized by Li and co-workers160 with yields of 67.7% and 57.7% respectively. The bioactivity of the two compounds was subsequently evaluated. The two compounds have a certain anti-influenza virus neuraminidase activity with IC50 values of 44.7 μM and 33.5 μM respectively.
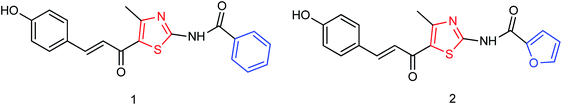 |
| | Fig. 59 Structures of furan formamide derivatives. | |
Ye and co-workers161 prepared three (4-alkyl-5-acyl-2-thiazolyl)hydrazone derivatives (Fig. 60) from acetylacetone with yields ranging from 61.1 to 81.7%. In vitro study showed that these compounds displayed good inhibitory activity on the A/PR/8/34 (H1N1) cell line. Among them, compound 2 showed the best activity with an IC50 value of 21.07 μg mL−1 and an inhibition rate of 71.68%.
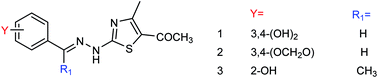 |
| | Fig. 60 Structures of (4-alkyl-5-acyl-2-thiazolyl)hydrazone derivatives. | |
Neuraminidase is one of the targets of anti-influenza drugs. In order to find a new neuraminidase inhibitor, Li and co-workers162 designed and synthesized three novel hydrazide neuraminidase inhibitors (Fig. 61) based on the structure of neuraminidase using computer virtual screening technology and computer-aided drug design. Three compounds showed good neuraminidase inhibitory activity with IC50 values of 0.075 μM, 0.46 μM and 0.21 μM respectively, which have higher activity than the OS group (IC50 = 33.75 μM).
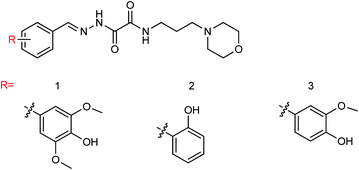 |
| | Fig. 61 Structures of hydrazide neuraminidase inhibitors. | |
The emergence of oseltamivir resistant strains, such as H274Y, I117V, E119A, R292K and other neuraminidase mutants, has decreased the effectiveness of neuraminidase inhibitors; therefore, it is urgent to develop new neuraminidase inhibitors. Huang and co-workers163 designed and synthesized thirteen new oseltamivir derivatives (Fig. 62) with yields ranging from 27 to 69%. In vitro study showed good inhibitory activity of the synthesized compounds against H1N1, H3N2 and H259Y (H1N1) influenza viruses. Among them, compound 12 showed potent inhibitory activities with IC50 values of 1.43 μM (H1N1), 3.64 μM (H3N2) and 14.47 μM (H259Y) respectively.
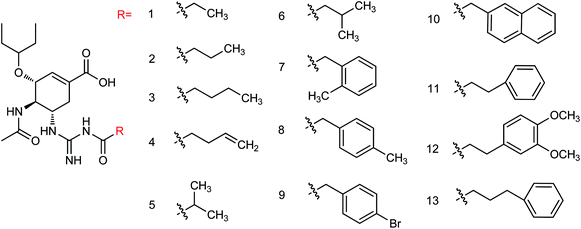 |
| | Fig. 62 Structures of oseltamivir derivatives. | |
The secondary metabolites of marine microorganisms are rich in structural diversity and significant biological activity, which are important sources of drug leading compounds. The mixed terpenes with austin skeleton structure are a kind of bioactive compounds produced by fungi which have certain inhibitory activities on aquatic pathogenic fungi, bacteria and Artemia. Li and co-workers164 isolated and purified three new terpenoids (Fig. 63) from the fermentation products of marine fungus Aspergillus ustus. The molecular formulas of compounds 1–3 are C33H34O12, C32H38O11, and C25H28O8. In vitro bioactivity experiments showed significant inhibitory activities for these terpenoids with IC50 values of 5.28 μM, 8.95 μM and 10.72 μM respectively, which are expected to become new potential anti-influenza drugs.
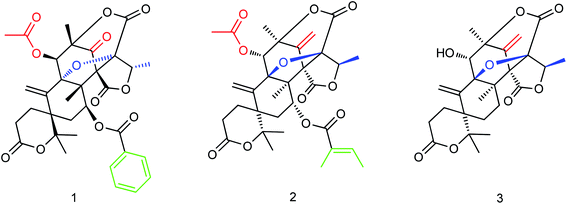 |
| | Fig. 63 Structures of terpenoids. | |
Ma and co-workers165 found that the neuraminidase inhibitors oseltamivir phosphate and zanamivir displayed effective therapeutic effects on myocarditis model mice. After treatment with oseltamivir phosphate or zanamivir, the serum cTnI concentration was significantly reduced and the degree of myocardial injury was improved. The score of myocardial cases was significantly reduced and the degree of myocardial lesion was relieved; therefore, oseltamivir phosphate and zanamivir can be used to prepare drugs for the treatment of myocarditis. Ten novel neuraminidase inhibitors (Fig. 64) were prepared by the modification of ethyl carboxylate at the C-1 position of oseltamivir by Li and co-workers166 with the yields ranging from 60.4 to 85.3%. The results indicated good inhibitory activities against neuraminic acid A/Anhui/1/2005 (H5N1) for all derivatives and compounds 1–5 showed strong inhibitory activity. Compound 5 showed potent inhibitory activities among all the synthesized derivatives with an inhibition rate of 90.5%.
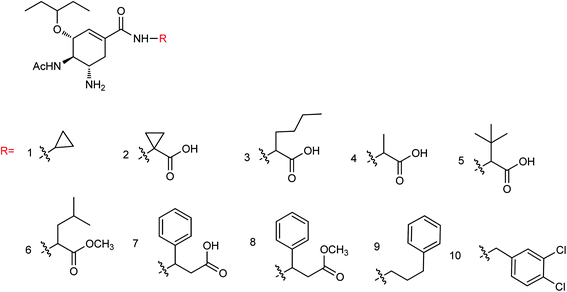 |
| | Fig. 64 Structures of oseltamivir derivatives. | |
The most effective way to obtain effective antiviral drugs is from direct extraction of antiviral drugs from natural products or chemical modification of the extracts. Coumarins have excellent antiviral, antitumor and anticoagulant properties. Li167 has used 4-methyl-7-hydroxy-8-formylcoumarin as a starting material reacting with hydrazine (NH2–NH2) at room temperature to yield N,N′-bis(7-hydroxycoumarin-8-methylene)hydrazine (Fig. 65). The synthesis was carried out in ethanol as a solvent without any catalyst at room temperature. In vitro study showed that the inhibition rate was 42.3% for N,N′-bis(4-methyl-7-hydroxycoumarin-8-methylene)hydrazine at a concentration of 30.0 μg mL−1. The results indicated that N,N′-bis(4-methyl-7-hydroxycoumarin-8-methylene)hydrazine showed good inhibition activity and could be used to prepare further anti-influenza drugs.
 |
| | Fig. 65 Structures of N,N′-bis(4-methyl-7-hydroxycoumarin-8-methylene)hydrazine derivatives. | |
Sixteen 2-(2-benzylhydrazide)-5-acylthiazole derivatives (Fig. 66) were designed and synthesized by Jia and co-workers168 with the yields ranging from 52.4 to 92.1%. Compound 15 showed potent inhibitory activities with IC50 values of 8.62 (H1N1) and 15.69 (H3N2) μg mL−1 at a concentration of 40.0 μg mL−1.
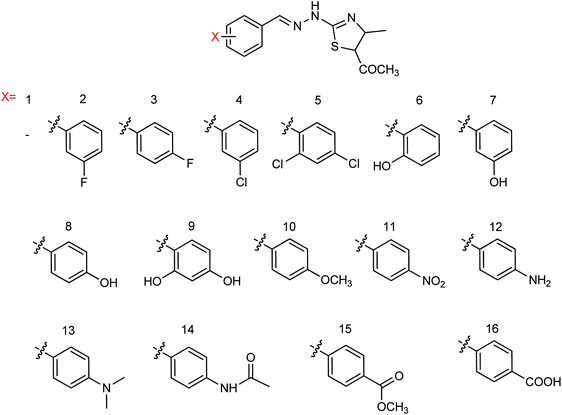 |
| | Fig. 66 Structures of 2-(2-benzylhydrazide)-5-acylthiazole derivatives. | |
Based on vanillin and its isomers, three novel 2-(2-benzylhydrazinyl)-5-acylthiazole derivatives (Fig. 67) bearing hydroxy and methoxy groups were designed and synthesized from acetylacetone by Wei and co-workers169 with the yields ranging from 51.2 to 83.3%. The inhibition rate of compounds was determined at a concentration of 40.0 μg mL−1. The results indicated that compound 3 has the best activity with an IC50 value of 7.13 μg mL−1 and an inhibition rate up to 93.33%.
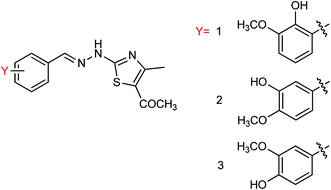 |
| | Fig. 67 Structures of 2-(2-benzylhydrazinyl)-5-acylthiazole derivatives. | |
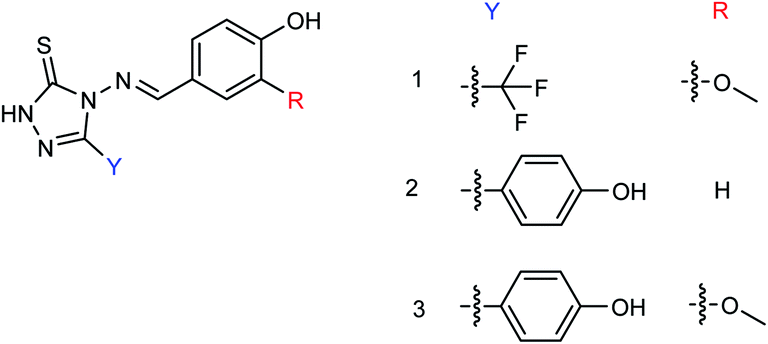
Tiliroside (Fig. 68), a flavonoid isolated and purified from traditional Chinese medicine Duchesnea indica, has a wide range of anti-inflammatory and antioxidant activities. However, there are few studies on the anti-influenza virus activity. Sheng co-workers170 have obtained 4 mg tiliroside from 1 kg of Duchesnea indica plant. An in vitro study revealed that it can inhibit the oseltamivir-sensitive and drug-resistant influenza virus strains and reduce the replication of oseltamivir-sensitive and drug-resistant influenza virus strains. Tiroside showed inhibitory activity against oseltamivir-sensitive (197.9 μM L−1) and drug-resistant neuraminidase (125.4 μM L−1). At doses of 40 μM and 200 μM, the replication of oseltamivir sensitive influenza virus strain PR8 and resistant influenza virus strain H274Y was reduced respectively. At the same time, it was found that tiliroside did not produce cytotoxicity to MDCK at a dose of 1 mM. Tiliroside can be used as a safe and effective natural product to provide a theoretical basis for the preparation of influenza prevention and treatment drugs. It is expected to be promoted and used as a new neuraminidase inhibitor.
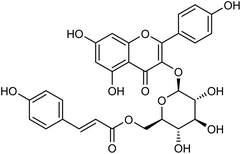 |
| | Fig. 68 Structure of tiliroside. | |
Fifteen new anti-influenza compounds (Fig. 69) were designed and synthesized by Chen and co-workers.171 The synthesized derivatives showed good inhibitory activity against influenza virus (N2 (H3N2, E119V)) resistant to zanamivir. The fat water partition coefficient experiment showed that the lipid water partition coefficient of these compounds was significantly higher in comparison to zanamivir. It is expected to significantly improve the pharmacokinetics of zanamivir to be good potential agents.
 |
| | Fig. 69 Structures of anti-influenza derivatives. | |
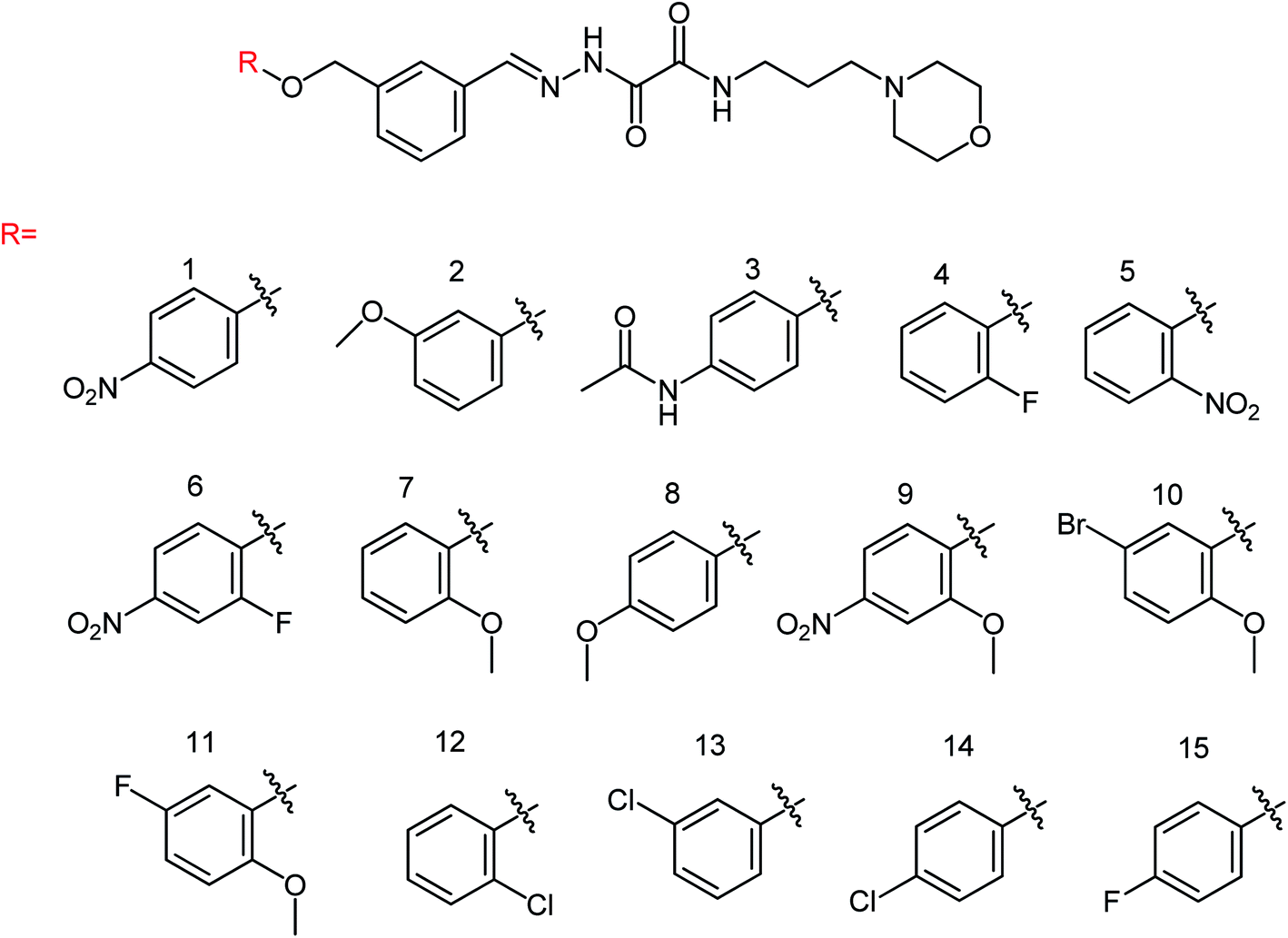
In order to solve the problem of drug resistance caused by virus variants, Xiao and co-workers172 designed and synthesized 15 novel acylhydrazone neuraminidase inhibitors (Fig. 70) with the yields ranging from 86 to 93%. In vitro study showed the best inhibitory activity for the compounds L8, L10, L14 and L15 with IC50 values of 0.60 μM, 7.31 μM, 6.20 μM and 5.29 μM respectively, which were significantly higher than oseltamivir acid (IC50 = 17.00 μM). SAR study has proved the importance of substitution for the improvement of the activity by introducing the same substituent at different positions; therefore, the activity of position 4 was higher than that of the substituent at position 2 such as compounds L1 and L5, compounds L4 and L15, compounds L7 and L8 and compounds L12 and L14 due to the binding mode and the number of hydrogen bonds formed between inhibitors and substrate amino acids.
 |
| | Fig. 70 Structures of acylhydrazone derivatives. | |
Naturally occurring anti-influenza compounds have received much attention in recent years due to their applications in medicinal chemistry and drug discovery. Actinomycetes produce active secondary metabolites with a novel structure and a unique mechanism due to their special living environment. Therefore, researchers pay more attention to find active ingredients from actinomycetes to prevent and treat the influenza disease. Streptomyces, the largest genus of actinomycetes, has attracted much attention due to their abundant secondary metabolites with significant bioactivities. A class of sek15 polyketides with neuraminidase inhibitory activities were isolated from Streptomyces sp. by Liu and co-workers.173 These three compounds (Srepolyketide B, Srepolyketide C and SEK15) showed inhibitory activities with IC50 values of 85.6 μM, 58.2 μM and 51.3 μM respectively (Fig. 71).
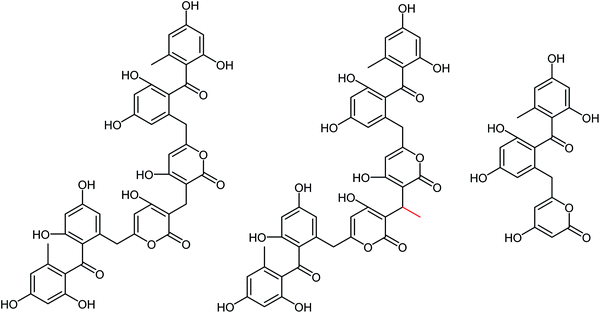 |
| | Fig. 71 Structures of Srepolyketide B, Srepolyketide C and SEK15. | |
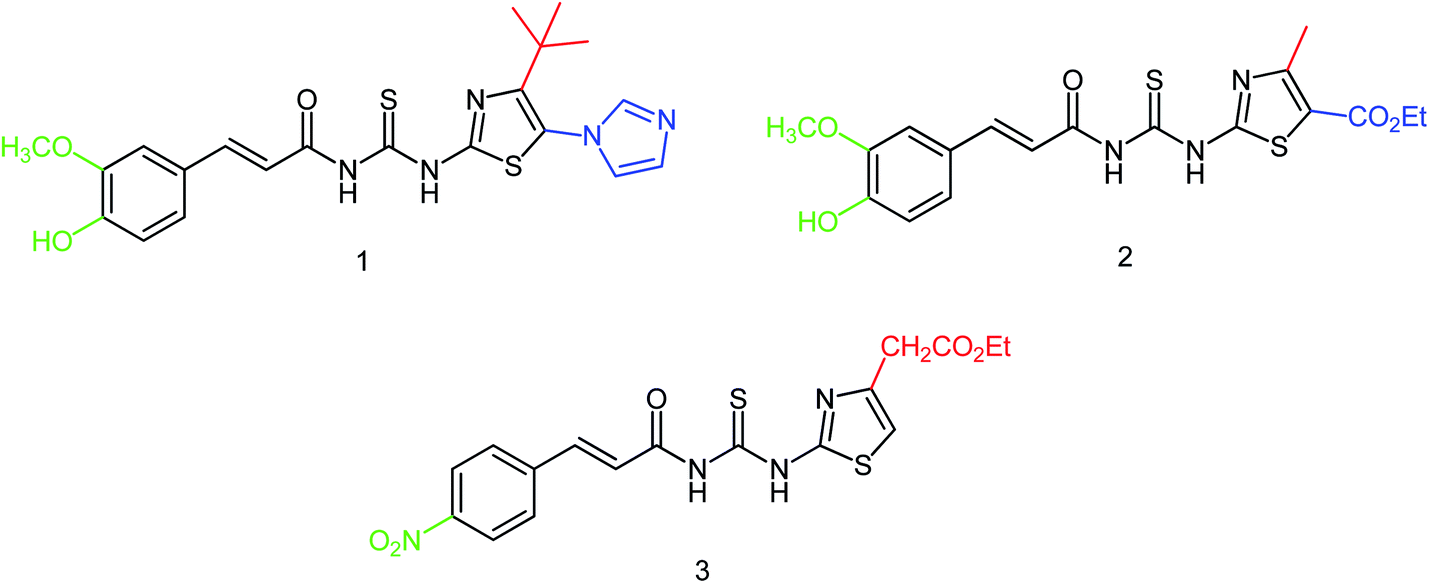
It has been proved that thiourea compounds have inhibitory activities against influenza virus. On this basis, Hu and co-workers174 have designed and synthesized three thiourea derivatives (Fig. 72) and determined their bioactivity against influenza virus (H1N1). The inhibitory rate of these three compounds were 70.13 ± 7.05, 68.3 ± 5.66 and 56.59 ± 6.21% respectively with the IC50 values of 22.78 ± 3.73, 21.76 ± 5.22 and 32.69 ± 10.46 μM respectively.
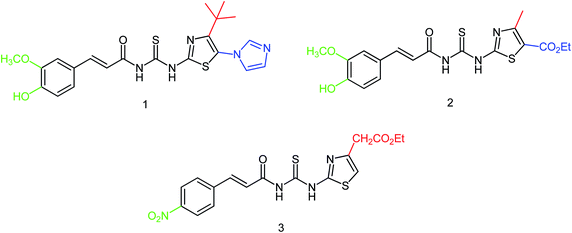 |
| | Fig. 72 Structures of thiourea derivatives. | |
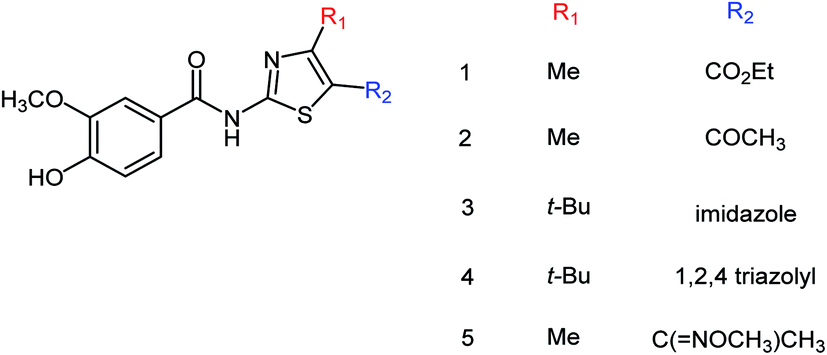
Five vanillin derivatives (Fig. 73) were designed and synthesized by Hu and co-workers175 with the yields ranging from 79.6 to 95.0%. The synthesized derivatives showed potent inhibitory activities and may be used to prepare further inhibitors. Compound 5 showed the best activity with an IC50 value of 12.33 ± 3.01 μM.
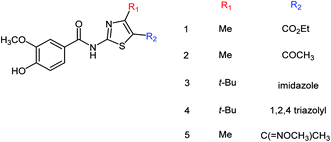 |
| | Fig. 73 Structures of vanillin derivatives. | |
In another study, Hu and co-workers176 have designed and synthesized three pyridazine hydrazone derivatives (Fig. 74), and in vitro determination of their anti-influenza H1N1 activity has been investigated. The yields were ranging from 71.9 to 86.9%. Compound 3 showed significant inhibitory activities with the inhibition rate of 84.52 ± 1.32% and an IC50 value of 11.17 ± 0.34 μM.
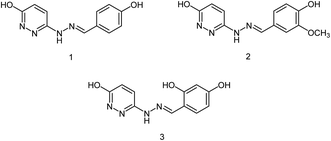 |
| | Fig. 74 Structures of pyridazine hydrazone derivatives. | |
7. Conclusions
Influenza virus (IFV) is a respiratory pathogen that can cause life-threatening diseases such as pneumonia and encephalitis when aggravated. Due to influenza infections, occasional, seasonal, and pandemic influenza transmission occurs in humans as well as animals and spreads easily via droplets and contact. Neuraminidase inhibitors are widely used in the treatment of influenza infection, and there is an urgent need to develop more potent agents due to drug resistance activities. This review has discussed various developments in the field of neuraminidase inhibitors along with their medicinal properties. It also focuses on the recent advances of chemical synthesis pathways used for the development of new neuraminidase agents along with medicinal aspects of chemically modified molecules. Hence, this review presents a strong intellectual background in the said field, which gives an in-depth insight into several aspects of this global pandemic and will provide ample references for the researchers, which may be useful for the ongoing development of therapeutic agents and vaccines.
Abbreviations
| ALI | Acute lung injury |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| ApoE | Apolipoprotein E |
| BNA | Bacterial neuraminidase |
| BPD | Berberine-piperazine derivatives |
| BCCAO | Bilateral common carotid artery occlusion |
| bNAb | Broadly neutralizing antibodies |
| Bu | butanol |
| DFT | Density functional theory |
| DANA | 2-Deoxy-2,3-didehydro-N-acetylneuraminic acid |
| DNA | Deoxyribonucleic acid |
| DS | Dianthus superbus |
| DW | Distilled water |
| FESR | Flavonoids-enriched extract of S. baicalensis |
| GOC | Guanidino OC |
| GOC | Guanidino-oseltamivir carboxylic acid |
| HN | Haemagglutinin-neuraminidase |
| HA | Hemagglutinin |
| HEK 293T | Human embryonic kidney cells 293 |
| hPIV | Human para influenza virus |
| hPIV-3 | Human parainfluenza virus type-3 |
| IAV | Influenza A virus |
| LGN | Liquiritigenin |
| MDCK | Madin–Darby canine kidney |
| MGO | Methylglyoxal |
| MUNANA | 20-(4-Methylumbelliferyl)-α-D-N-acetylneuraminic acid |
| MD | Molecular dynamics |
| MIP | Molecularly imprinted polymer |
| M2 | Matrix protein 2 |
| Neu5Ac | N-Acetylneuraminic acid |
| N-Acyl-Neu2en | N-Acyl-2,3-dehydro-2-deoxy-neuraminic acid |
| NPs | Nanoparticles |
| NA | Neuraminidase |
| NEU1 | Neuraminidase 1 |
| NEU2 | Neuraminidase 2 |
| NEU3 | Neuraminidase 3 |
| NIP@silica gel | Non-imprinted polymer with silica gel |
| OPME | One-pot multienzyme |
| OS | Oseltamivir |
| OTV | Oseltamivir |
| OC | Oseltamivir carboxylate |
| OSMIP | Oseltamivir molecularly imprinted polymer |
| Se@OTV | OTV decoration of SeNPs |
| PmAldolase | Pasteurella multocida sialic acid aldolase |
| RNA | Ribonucleic acid |
| RdRp | RNA-dependent RNA polymerase |
| SEM | Scanning electron microscopy |
| OSMIP@silica gel | Silica gel coated with an MIP layer for OS |
| SSAA | Specific surface area analysis |
| SpNanB | Streptococcus pneumoniae sialidase |
| SAR | Structure–activity relationship |
| TPE4S | Tetraphenylethene luminogen |
| 3WJ | Three–way junction |
| VLDL | Very-low-density lipoprotein |
| VCNA | Vibrio cholerae neuraminidase |
Conflicts of interest
The authors declare that they have no conflict of interests in this work.
Acknowledgements
Ahmed Mahal acknowledges the Chinese Academy of Sciences (CAS) for financial support through the CAS President's International Fellowship Initiative (2016PM032) and Cihan University as well. This work was supported by Natural Science Foundation of Guangdong Province, China (2017A030313775, 2019A1515011822). RKM is also thankful to Government College of Engineering, Keonjhar, for his help and facilities.
References
- D. Lin, Y.-J. Yi, M.-W. Xiao, J. Chen, J. Ye, A.-X. Hu, W.-W. Lian, A.-L. Liu and G.-H. Du, J. Asian Nat. Prod. Res., 2019, 21, 1052–1067, DOI:10.1080/10286020.2018.1509854.
- A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith, EMBO J., 1985, 4, 3021–3024, DOI:10.1002/j.1460-2075.1985.tb04038.x.
- A. J. Hay, Semin. Virol., 1992, 3, 21–30 CAS.
- F. G. Hayden, A. Minocha, D. A. Spyker and H. E. Hoffman, Antimicrob. Agents Chemother., 1985, 28, 216–221, DOI:10.1128/AAC.28.2.216.
- F. G. Hayden and R. B. Couch, Rev. Med. Virol., 1992, 2, 89–96, DOI:10.1002/rmv.1980020205.
- F. G. Hayden, E. B. Belshe, R. D. Clover, A. J. Hay, M. G. Oakes and W. Soo, N. Engl. J. Med., 1989, 321, 1696–1702, DOI:10.1056/NEJM198912213212502.
- S. Grambas, M. S. Bennett and A. J. Hay, Virology, 1992, 191, 541–549, DOI:10.1016/0042-6822(92)90229-i.
- J. L. McKimm-Breschkin, Influenza Other Respir, Viruses, 2012, 7, 25–36, DOI:10.1111/irv.12047.
- Y. Li, Z. Lin, M. Zhao, T. Xu, C. Wang, L. Hua, H. Wang, H. Xia and B. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 24385–24393 CrossRef CAS.
- C.-Z. Wang, H.-H. Han, X.-Y. Tang, D.-M. Zhou, C. Wu, G.-R. Chen, X.-P. He and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 25164–25170 CrossRef CAS.
- A. Moscona, N. Engl. J. Med., 2005, 353, 1363–1373 CrossRef CAS.
- A. F. Abdel-Magid, C. A. Maryanoff and S. J. Mehrman, Curr. Opin. Drug Discovery Dev., 2001, 4, 776–791 CAS.
- V. Y. Glanz, V. A. Myasoedova, A. V. Grechko and A. N. Orekhov, Drug Des., Dev. Ther., 2018, 12, 3431–3437, DOI:10.2147/DDDT.S176220.
- J. Magano, Chem. Rev., 2009, 109, 4398–4438, DOI:10.1021/cr800449m.
- V. Farina and J. D. Brown, Angew. Chem., Int. Ed., 2006, 45, 7330–7334, DOI:10.1002/anie.200602623.
- M. Shibasaki and M. Kanai, Eur. J. Org. Chem., 2008, 1839–1850, DOI:10.1002/ejoc.200800033.
- N. Anuwongcharoen, W. Shoombuatong, T. Tantimongcolwat, V. Prachayasittikul and C. Nantasenamat, PeerJ, 2016, 4, e1958, DOI:10.7717/peerj.1958.
- A. Moscona, N. Engl. J. Med., 2009, 360, 953–956, DOI:10.1056/NEJMp0900648.
- M. Itzstein, W.-Y. Wu and B. Jin, Carbohydr. Res., 1994, 259, 301–305, DOI:10.1016/0008-6215(94)84065-2.
- M. Itzstein, B. Jin, W.-Y. Wu and M. Chandler, Carbohydr. Res., 1993, 244, 181–185, DOI:10.1016/0008-6215(93)80014-6.
- E. Schreiner, E. Zbiral, R. G. Kleineidam and R. Schauer, Liebigs Ann. Chem., 1991, 1991, 129–134, DOI:10.1002/jlac.199119910124.
- K. Kim, Y.-T. Lin and H. S. Mosher, Tetrahedron Lett., 1988, 29, 3183–3186, DOI:10.1016/0040-4039(88)85116-5.
- J. Scheigetz, R. Zamboni, M. A. Bernstein and B. Roy, Org. Prep. Proced. Int., 1995, 27, 637–644, DOI:10.1080/00304949509458521.
- M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright and C. Williamson, J. Chem. Soc., Perkin Trans. 1, 1995, 1173–1180, 10.1039/P19950001173.
- K.-G. Liu, S. Yan, Y.-L. Wu and Z.-J. Yao, Org. Lett., 2004, 6, 2269–2272, DOI:10.1021/ol0491890.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690, DOI:10.1021/ja963036t.
- J. C. Rohloff, K. M. Kent, M. J. Postich, M. W. Becker, H. H. Chapman, D. E. Kelly, W. Lew, M. S. Louie, L. R. McGee, E. J. Prisbe, L. M. Schultze, R. H. Yu and L. Zhang, J. Org. Chem., 1998, 63, 4545–4550, DOI:10.1021/jo980330q.
- M. Federspiel, R. Fischer, M. Hennig, H.-J. Mair, T. Oberhauser, G. Rimmler, T. Albiez, J. Bruhin, H. Estermann, C. Gandert, V. Göckel, S. Götzö, U. Hoffmann, G. Huber, G. Janatsch, S. Lauper, O. Röckel-Stäbler, R. Trussardi and A. G. Zwahlen, Org. Process Res. Dev., 1999, 34, 266–274, DOI:10.1021/op9900176.
- M. Karpf and R. Trussardi, J. Org. Chem., 2001, 66, 2044–2051, DOI:10.1021/jo005702l.
- S. Abrecht, P. Harrington, H. Iding, M. Karpf, R. Trussardi, B. Wirz and U. Zutter, Chimia, 2004, 58, 621–629, DOI:10.2533/000942904777677605.
- P. J. Harrington, J. D. Brown, T. Foderaro and R. C. Hughes, Org. Process Res. Dev., 2004, 8, 86–91, DOI:10.1021/op0302107.
- Y. S. Hong and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 6310–6311, DOI:10.1021/ja0616433.
- N. T. Kipassa, H. Okamura, K. Kina, T. Hamada and T. Iwagawa, Org. Lett., 2008, 105, 815–816, DOI:10.1021/ol7029646.
- Y. Fukuta, T. Mita, N. Fukuda, M. Kanai and M. D. Shibasaki, J. Am. Chem. Soc., 2006, 128, 6312–6313, DOI:10.1021/ja061696k.
- T. Mita, N. Fukuda, F. X. Roca, M. Kanai and M. Shibasaki, Org. Lett., 2007, 9, 259–262, DOI:10.1021/ol062663c.
- K. Yamatsugu, S. Kamijo, Y. Suto, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2007, 48, 1403–1406, DOI:10.1016/j.tetlet.2006.12.093.
- K. Yamatsugu, L. Yin, S. Kamijo, Y. Kimura, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 1070–1076, DOI:10.1002/anie.200804777.
- X. Cong and Z.-J. Yao, J. Org. Chem., 2006, 71, 5365–5368, DOI:10.1021/jo060633h.
- N. Satoh, T. Akiba, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2007, 46, 5734–5736, DOI:10.1002/anie.200701754.
- N. Satoh, T. Akiba, S. Yokoshima and T. Fukuyama, Tetrahedron, 2009, 65, 3239–3245, DOI:10.1016/j.tet.2008.09.103.
- J.-J. Shie, J.-M. Fang, S.-Y. Wang, K.-C. Tsai, Y.-S. E. Cheng, A.-S. Yang, S.-C. Hsiao, C.-Y. Su and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 11892–11893, DOI:10.1021/ja073992i.
- J.-J. Shie, J.-M. Fang and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 5788–5791, DOI:10.1002/anie.200801959.
- K. M. Bromfield, H. Graden, D. P. Hagberg, T. Olsson and N. Kann, Chem. Commun., 2007, 30, 3183–3185, 10.1039/B703295A.
- B. M. Trost and T. Zhang, Angew. Chem., Int. Ed., 2008, 47, 3759–3761, DOI:10.1002/anie.200800282.
- M. Matveenko, M. G. Banwell and A. C. Willis, Tetrahedron Lett., 2008, 49, 7018–7020, DOI:10.1016/j.tetlet.2008.09.130.
- L.-D. Nie and X.-X. Shi, Tetrahedron: Asymmetry, 2009, 20, 124–129, DOI:10.1016/j.tetasy.2008.11.027.
- L.-D. Nie, X.-X. Shi, K. H. Ko and W.-D. Lu, J. Org. Chem., 2009, 74, 3970–3973, DOI:10.1021/jo900218k.
- H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304–1307, DOI:10.1002/anie.200804883.
- T. Mandai and T. Oshitari, Synlett, 2009, 783–786, DOI:10.1055/s-0028-1087941.
- T. Oshitari and T. Mandai, Synlett, 2009, 787–789, DOI:10.1055/s-0028-1087940.
- F. Krammer, G. J. D. Smith, R. A. M. Fouchier, M. Peiris, K. Kedzierska, P. C. Doherty, P. Palese, M. L. Shaw, J. Treanor, R. G. Webster and A. García-Sastre, Influenza, Nat Rev Dis Primers., 2018, 4, 3, DOI:10.1038/s41572-018-0002-y.
- M. Billings, The Influenza Pandemic of 1918, 2005, http://virus.stanford.edu/uda/ Search PubMed.
- Centers for Disease Control and Prevention (CDC), 2009 H1N1 Pandemic (H1N1pdm09 virus), 2019, https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html Search PubMed.
- European Centre for Disease Prevention and Control, Expert opinion on neuraminidase inhibitors for the prevention and treatment of influenza – review of recent systematic reviews and meta-analyses, ECDC, Stockholm, 2017, https://www.ecdc.europa.eu/sites/portal/files/documents/Scientific-advice-neuraminidase-inhibitors-2017.pdf Search PubMed.
- E. Chen, Y. Chen, L. Fu, Z. Chen, Z. Gong, H. Mao, D. Wang, M. Y. Ni, P. Wu, Z. Yu, T. He, Z. Li, J. Gao, S. Liu, Y. Shu, B. J. Cowling, S. Xia and H. Yu, Eurosurveillance, 2013, 18, 20616 CrossRef.
- K. Zhao, M. Gu, L. Zhong, Z. Duan, Y. Zhang, Y. Zhu, G. Zhao, M. Zhao, Z. Chen, S. Hu, W. Liu, X. Liu, D. Peng and X. Liu, Vet. Microbiol., 2013, 163, 351–357, DOI:10.1016/j.vetmic.2012.12.025.
- U. Joseph, M. Linster, Y. Suzuki, S. Krauss, R. A. Halpin, D. Vijaykrishna, T. P. Fabrizio, T. M. Bestebroer, S. Maurer-Stroh, R. J. Webby, D. E. Wentworth, R. A. M. Fouchier, J. Bahl and G. J. D. Smith, J. Virol., 2015, 89, 2442–2447, DOI:10.1128/JVI.02590-14.
- M. Gu, W. Liu, Y. Cao, D. Peng, X. Wang, H. Wan, G. Zhao, Q. Xu, W. Zhang, Q. Song, Y. Li and X. Liu, Emerging Infect. Dis., 2011, 17, 1060–1063, DOI:10.3201/eid/1706.101406.
- H. Liu, C. Xiong, J. Chen, G. Chen, J. Zhang, Y. Li, Y. Xiong, R. Wang, Y. Cao, Q. Chen, D. Liu, H. Wang and J. Chen, Emerging Microbes Infect., 2018, 7, 62, DOI:10.1038/s41426-018-0064-7.
- N. Beerens, R. Heutink, F. Harders, A. Bossers, G. Koch and B. Peeters, J. Virol., 2020, 94, e01818–19, DOI:10.1128/JVI.01818-19.
- T. H. P. Peacock, J. James, J. E. Sealy and M. Iqbal, Viruses, 2019, 11, 620, DOI:10.3390/v11070620.
- E. Montomoli and C. M. Trombetta, Pathog. Global Health, 2014, 108, 213, DOI:10.1179/2047772414Z.000000000215.
- P. A. Rota, T. R. Wallis, M. W. Harmon, J. S. Rota, A. P. Kendal and K. Nerome, Virology, 1990, 175, 59–68, DOI:10.1016/0042-6822(90)90186-U.
- G. Yuanji, J. Fengen, W. Ping, W. Min and Z. Jiming, J. Gen. Virol., 1983, 64, 177–182 CrossRef.
- B. M. Hause, M. Ducatez, E. A. Collin, Z. Ran, R. Liu, Z. Sheng, A. Armien, B. Kaplan, S. Chakravarty, A. D. Hoppe, R. J. Webby, R. R. Simonson and F. Li, PLoS Pathog., 2013, 9, e1003176, DOI:10.1371/journal.ppat.1003176.
- Flu Symptoms & Complications, https://www.cdc.gov/flu/symptoms/symptoms.htm Search PubMed.
- S. W. Boktor and J. W. Hafner, Influenza. [Updated 2020 May 26], in StatPearls [Internet]. Treasure Island (FL), StatPearls Publishing; 2020 Jan, https://www.ncbi.nlm.nih.gov/books/NBK459363/ Search PubMed.
- V. Kumar, Indian J. Pediatr., 2017, 84, 139–143, DOI:10.1007/s12098-016-2232-x.
- V. K. Trombetta, Y. L. Chan and M. J. Bankowski, Hawaii J. Med. Public Health, 2018, 77, 226–230 Search PubMed.
- E. Egilmezer, G. J. Walker, P. Bakthavathsalam, J. R. Peterson, J. J. Gooding, W. Rawlinson and S. Stelzer-Braid, Rev. Med. Virol., 2018, 28, e1995, DOI:10.1002/rmv.1995.
- S. H. Choi, J. W. Chung, T. Kim, K. H. Park, M. S. Lee and Y. G. Kwak, Korean J. Intern. Med., 2018, 33, 391–396, DOI:10.3904/kjim.2016.226.
- D. Dou, R. Revol, H. Östbye, H. Wang and R. Daniels, Front. Immunol., 2018, 9, 1581, DOI:10.3389/fimmu.2018.01581.
- T. O. Edinger, M. O. Pohl and S. Stertz, J. Gen. Virol., 2014, 95, 263–277, DOI:10.1099/vir.0.059477-0.
- Y. A. Shtyrya, L. V. Mochalova and N. V. Bovin, Acta Naturae, 2009, 1, 26–32 CrossRef CAS.
- A. G. Winquist, K. Fukuda, C. B. Bridges and N. J. Cox, Morb. Mortal. Wkly. Rep., 1999, 48, 1–9 Search PubMed.
- A. J. Burnham, T. Baranovich and E. A. Govorkova, Antiviral Res., 2013, 100, 520–534, DOI:10.1016/j.antiviral.2013.08.023.
- E. J. LaVoie, H. Y. Sagong, J. D. Bauman, A. Nogales, L. Martínez-Sobrido and E. Arnold, ChemMedChem, 2019, 14, 1204–1223, DOI:10.1002/cmdc.201900084.
- W. Li, A. Santra, H. Yu, T. J. Slack, M. M. Muthana, D. Shi, Y. Liu and X. Chen, J. Org. Chem., 2019, 84, 6697–6708, DOI:10.1021/acs.joc.9b00385.
- C. Wu, K.-J. Wu, J.-B. Liu, X.-M. Zhou, C.-H. Leung and D.-L. Ma, Chem. Commun., 2019, 55, 6353–6356, 10.1039/c9cc02189b.
- C. Aiyu, L. Yongdong, Y. Jiao, H. Aixi, L. Wenwen, L. Ailin and D. Guanhua, Chem. Res. Chin. Univ., 2019, 35, 395–402, DOI:10.1007/s40242-019-8346-8.
- M.-Y. Cui, J.-X. Nie, Z.-Z. Yan, M.-W. Xiao, D. Lin, J. Ye and A.-X. Hu, Med. Chem. Res., 2019, 28, 938–947, DOI:10.1007/s00044-019-02343-3.
- V. P. Chibanga, L. Dirr, P. Guillon, I. M. El-Deeb, B. Bailly, R. J. Thomson and M. von Itzstein, Antiviral Res., 2019, 167, 89–97, DOI:10.1016/j.antiviral.2019.04.001.
- Z. X. Zhao, L. P. Cheng, M. Li, W. Pang and F. H. Wu, Eur. J. Med. Chem., 2019, 173, 305–313, DOI:10.1016/j.ejmech.2019.04.006.
- Z.-L. Yan, A.-Y. Liu, X.-X. Wei, Z. Zhang, L. Qin, Q. Yu, P. Yu, K. Lu and Y. Yang, Carbohydr. Res., 2019, 477, 32–38, DOI:10.1016/j.carres.2019.03.012.
- M. Kiso, T. J. S. Lopes, S. Yamayoshi, M. Ito, M. Yamashita, N. Nakajima, H. Hasegawa, G. Neumann and Y. Kawaoka, J. Infect. Dis., 2018, 217, 887–896, DOI:10.1093/infdis/jix606.
- K. D. Malbari, A. S. Chintakrindi, L. R. Ganji, D. J. Gohil, S. T. Kothari, M. V. Joshi and M. A. Kanyalkar, Mol. Diversity, 2019, 23, 927–951, DOI:10.1007/s11030-019-09919-6.
- B. Sathya and M. Prasath, Res. Chem. Intermed., 2019, 45, 2135–2166, DOI:10.1007/s11164-018-03727-7.
- P. L. Rocca, P. Rota, M. Piccoli, F. Cirillo, M. Orioli, A. Ravelli, P. Allevi and L. Anastasia, J. Org. Chem., 2019, 84, 5460–5470, DOI:10.1021/acs.joc.9b00431.
- M. Yamabea, A. Fujitaa, K. Kaihatsub and Y. Ebara, Carbohydr. Res., 2019, 474, 43–50, DOI:10.1016/j.carres.2019.01.008.
- J. Han, J. Perez, A. Schafer, H. Cheng, N. Peet, L. Rong and B. Manicassamy, Curr. Med. Chem., 2018, 25, 5115–5127 CrossRef CAS.
- B.-T. Hong, Y.-S. E. Cheng, T.-J. Cheng and J.-M. Fang, Eur. J. Med. Chem., 2019, 163, 710–721, DOI:10.1016/j.ejmech.2018.12.027.
- Y. Ali, N. M. Bunnori, D. Susanti, A. M. Alhassan and S. A. Hamid, Front. Chem., 2018, 6, 210, DOI:10.3389/fchem.2018.00210.
- A. A. A. Abdalsalam, J. Appl. Pharm. Sci., 2017, 7, 024–033 CAS.
- R. P. Putra, R. Imaniastuti, M. A. F. Nasution, D. Kerami and U. S. F. Tambunan, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 349, 012052, DOI:10.1088/1757-899X/349/1/012052.
- S. Mathew, A. A. Al-Thani and H. M. Yassine, PLoS One, 2018, 13, e0203148, DOI:10.1371/journal.pone.0203148.
- T. Guo, R. Héon-Roberts, C. Zou, R. Zheng, A. V. Pshezhetsky and C. W. Cairo, J. Med. Chem., 2018, 61, 11261–11279 CrossRef CAS.
- N. Onul, O. Ertik, N. G. Mermer and R. Yanardag, J. Chem., 2018, 4386031, DOI:10.1155/2018/4386031.
- Y. Li, Z. Lin, M. Guo, M. Zhao, Y. Xia, C. Wang, T. Xu and B. Zhu, Int. J. Nanomed., 2018, 13, 2005–2016 CrossRef CAS.
- Y. Li, Z. Lin, M. Guo, Y. Xia, M. Zhao, C. Wang, T. Xu, T. Chen and B. Zhu, Int. J. Nanomed., 2017, 12, 5733–5743 CrossRef CAS.
- L. P. Cheng, T. C. Wang, R. Yu, M. Li and J. W. Huang, Bioorg. Med. Chem. Lett., 2018, 28, 23–24, DOI:10.1016/j.bmcl.2018.10.040.
- T. J. Slack, W. Li, D. Shi, J. B. McArthur, G. Zhao, Y. Li, A. Xiao, Z. Khedri, H. Yu, Y. Liu and X. Chen, Bioorg. Med. Chem., 2018, 26, 5751–5757, DOI:10.1016/j.bmc.2018.10.028.
- M. R. F. Pratama and T. Gusdinar, Asian J. Pharm. Clin. Res., 2017, 10, 304–308, DOI:10.22159/ajpcr.2017.v10i8.18667.
- Y.-J. Yang, X.-W. Liu, X.-J. Kong, Z. Qin, Z.-H. Jiao, S.-H. Li and J.-Y. Li, Molecules, 2018, 23, 1881, DOI:10.3390/molecules23081881.
- B. Wang, K. Wang, P. Meng, Y. Hu, F. Yang, K. Liu, Z. Lei, B. Chen and Y. Tian, Bioorg. Med. Chem. Lett., 2018, 26, 5751–5757, DOI:10.1016/j.bmcl.2018.09.014.
- E. J. White, G. Gyulay, S. Lhoták, M. M. Szewczyk, T. Chong, M. T. Fuller, O. Dadoo, A. E. Fox-Robichaud, X. R. C. Austin, B. L. Trigatti and S. A. Igdoura, J. Biol. Chem., 2018, 293, 14689–14706 CrossRef CAS.
- C. Colombo, Č. Podlipnik, L. Lo Presti, M. Niikura, A. J. Bennet and A. Bernardi, PLoS One, 2018, 13, e0193623, DOI:10.1371/journal.pone.0193623..
- G. Liu, B. Wang, Y. Zhang, G. Xing, X. Yang and S. Wang, Chem. Commun., 2018, 54, 10691–10694, 10.1039/C8CC06300A.
- F. Meng, Y. Yin, C. Lu, Y. Duan, Y. Zhu, C. Huang, Y. Zhou, X. Xu and F. Wu, Monatsh. Chem., 2018, 149, 2037–2046, DOI:10.1007/s00706-018-2245-4.
- Y.-C. Xie, B. Huang, K.-X. Yu, F.-Y. Shi, T.-Q. Liu and W.-F. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 3556–3560, DOI:10.1016/j.bmcl.2013.04.033.
- B. R. Kim, J.-Y. Park, H. J. Jeong, H.-J. Kwon, S.-J. Park, I.-C. Lee, Y. B. Ryu and W. S. Lee, J. Enzyme Inhib. Med. Chem., 2018, 33, 1256–1265, DOI:10.1080/14756366.2018.1488695.
- G. Enkhtaivan, D. H. Kim, G. S. Park, M. Pandurangan, D. A. Nicholas, S. H. Moon, A. A. Kadam, R. V. Patel, H.-S. Shin and B. M. Mistry, Int. J. Biol. Macromol., 2018, 119, 1204–1210, DOI:10.1016/j.ijbiomac.2018.08.047.
- W. Liu, S. K. Samanta, B. D. Smith and L. Isaacs, Chem. Soc. Rev., 2017, 46, 2391–2403, 10.1039/c7cs00011a.
- A. Xiao, T. J. Slack, Y. Li, D. Shi, H. Yu, W. Li, Y. Liu and X. Chen, J. Org. Chem., 2018, 83, 10798–10804 CrossRef CAS.
- J. Zhang, V. Poongavanam, D. Kang, C. Bertagnin, H. Lu, X. Kong, H. Ju, X. Lu, P. Gao, Y. Tian, H. Jia, S. Desta, X. Ding, L. Sun, Z. Fang, B. Huang, X. Liang, R. Jia, X. Ma, W. Xu, N. A. Murugan, A. Loregian, B. Huang, P. Zhan and X. Liu, J. Med. Chem., 2018, 61, 6379–6397 CrossRef CAS.
- M. R. Richards, T. Guo, C. D. Hunter and C. W. Cairo, Bioorg. Med. Chem., 2018, 26, 5349–5358 CrossRef CAS.
- S. Kosono, A. Kasai, S. Komaba, T. Matsubara, T. Sato, D. Takahashi and K. Toshima, Chem. Commun., 2018, 54, 7467–7470, 10.1039/C8CC03865A.
- P.-H. Hsu, D.-C. Chiu, K.-L. Wu, P.-S. Lee, J.-T. Jan, Y.-S. E. Cheng, K.-C. Tsai, T.-J. Cheng and J.-M. Fang, Eur. J. Med. Chem., 2018, 154, 314–323, DOI:10.1016/j.ejmech.2018.05.030.
- C. Lu, Y. Yin, F. Meng, Y. Dun, K. Pei, C. Wang, X. Xu and F. Wu, Bioorg. Med. Chem. Lett., 2018, 28, 2003–2007 CrossRef CAS.
- S. Charyasriwong, T. Haruyama and N. Kobayashi, Drug Discoveries Ther., 2016, 10, 201–210 CrossRef CAS.
- M. Pascolutti, L. Dirr, P. Guillon, A. V. DenBergh, T. Ve, R. J. Thomson and M. von Itzstein, ACS Chem. Biol., 2018, 13, 1544–1550, DOI:10.1021/acschembio.8b00150.
- H. Kaur and B. Narasimhan, Curr. Top. Med. Chem., 2018, 18, 3–21 CrossRef CAS.
- J. Jin, Y. Chen, D. Wang, L. Ma, M. Guo, C. Zhou and J. Dou, Arch. Pharmacal Res., 2018, 41, 664–676, DOI:10.1007/s12272-018-1022-6.
- A. Hafeez, M. Roy, P. K. Pal, R. Kumar, P. Gangwar and A. Mohan, Int. J. Biol., Pharm. Allied Sci., 2015, 4, 5277–5283 CAS.
- Á. Hadházi, L. Li, B. Bailly, A. Maggioni, G. Martin, L. Dirr, J. Dyason, R. Thomson, G. Gao, A. Borbás, T. Ve, M. Pascolutti and M. von Itzstein, ChemMedChem, 2018, 13, 785–789, DOI:10.1002/cmdc.201800092.
- X. Wu, X. Wu, Q. Sun, C. Zhang, S. Y. Yang, L. Li and Z. Jia, Theranostics, 2017, 7, 826–845, DOI:10.7150/thno.17071.
- A. E. Ivanova, Y. V. Burgart, V. I. Saloutin, Y. R. Orshanskaya and V. V. Zarubaev, Mendeleev Commun., 2018, 28, 52–54 CrossRef CAS.
- H. Ju, J. Zhang, Z. Sun, Z. Huang, W. Qi, B. Huang, P. Zhan and X. Liu, Eur. J. Med. Chem., 2018, 146, 220–231 CrossRef CAS.
- T. Guo, P. Datwyler, E. Demina, M. R. Richards, P. Ge, C. Zou, R. Zheng, A. Fougerat, A. V. Pshezhetsky, B. Ernst and C. W. Cairo, J. Med. Chem., 2018, 61, 1990–2008 CrossRef CAS.
- M. Obul, X. Wang, J. Zhao, G. Li, H. A. Aisa and G. Huang, Mol. Diversity, 2019, 23, 1–9, DOI:10.1007/s11030-018-9840-5.
- W. Guan, J. Li, Q. Chen, Z. Jiang, R. Zhang, X. Wang, Z. Yang and X. Pan, Molecules, 2017, 22, 1738, DOI:10.3390/molecules22101738.
- S. Quosdorf, A. Schuetz and H. Kolodziej, Molecules, 2017, 22, 1989, DOI:10.3390/molecules22111989.
- H.-L. Li, R. Xu, X.-M. Li, S.-Q. Yang, L.-H. Meng and B.-G. Wang, Org. Lett., 2018, 20, 1465–1468 CrossRef CAS.
- A. S. Chintakrindi, D. J. Gohil, S. T. Kothari, A. S. Chowdhary and M. A. Kanyalkar, Med. Chem. Res., 2018, 27, 1013–1025, DOI:10.1007/s00044-017-2124-2.
- S.-Q. Yang, X.-M. Li, X. Li, H.-L. Li, L.-H. Meng and B.-G. Wang, Phytochem. Lett., 2018, 25, 191–195 CrossRef CAS.
- N. Singh, N. Anjum and R. Chandra, Phytochem. Rev., 2019, 18, 69–107, DOI:10.1007/s11101-018-9581-1.
- D. Furushima, K. Ide and H. Yamada, Molecules, 2018, 23, 1795, DOI:10.3390/molecules23071795.
- M. Ullah, Z. Uddin, Y. H. Song, Z. P. Li, J. Y. Kim, Y. J. Ban and K. H. Park, S. Afr. J. Bot., 2018, 120, 326–330, DOI:10.1016/j.sajb.2018.10.020.
- J. Xu, S. Chen, J. Jin, L. Ma, M. Guo, C. Zhou and J. Dou, Peptides, 2019, 112, 14–22, DOI:10.1016/j.peptides.2018.10.004.
- H.-J. Zhi, H.-Y. Zhu, Y.-Y. Zhang, Y. Lu, H. Li and D.-F. Chen, Phytomedicine, 2018, 57, 105–116, DOI:10.1016/j.phymed.2018.12.009.
- D. H. Kim, G. S. Park, A. S. Nile, Y. D. Kwon, G. Enkhtaivan and S. H. Nile, Food Chem. Toxicol., 2019, 125, 313–321, DOI:10.1016/j.fct.2019.01.013.
- Z. Yunshi, Y. Jing, Q. Xian, L. Xing, L. Xieqin and F. Ganzhu, Antiviral Ther., 2017, 22, 599–611, DOI:10.3851/IMP3152.
- Y. Wang, J. Y. Kim, Y. H. Song, Z. P. Li, S. H. Yoon, Z. Uddin, Y. J. Ban, K. W. Lee and K. H. Park, Int. J. Biol. Macromol., 2019, 128, 149–157, DOI:10.1016/j.ijbiomac.2019.01.105.
- E. W. C. Chan, S. K. Wong, J. Tangah and H. T. Chan, Syst. Rev. Pharm., 2018, 9, 58–63 CrossRef CAS.
- A.-X. Hu, Q.-Y. Tao, Y.-K. Yuan and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 109305979 A 20190205, 2019.
- A.-X. Hu, K.-Y. Yuan and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 109293644 A 20190201, 2019.
- X.-M. Gao, H. Zhu, S.-S. Wang, Z. Zhang and D. Cui, Faming Zhuanli Shenqing, Design Patent, CN 109265503 A 20190125, 2019.
- J.-M. Chen, X.-Y. An, G. Gao, J.-K. Zhang, X.-Y. Huang, J.-L. Ge, Y. Zhang and X.-H. Ning, Faming Zhuanli Shenqing, Design Patent, CN 109160937 A 20190108, 2019.
- A.-X. Hu, L. Liu, J. Ye and Y. Zhu, Faming Zhuanli Shenqing, Design Patent, CN 109053607 A 20181221. 2018.
- J. Ye, M. He, L. Liu, Y. Zhu and A.-X. Hu, Faming Zhuanli Shenqing, Design Patent, CN 109053606 A 20181221, 2018.
- A.-X. Hu, Z.-Z. Yan, M. Zhang and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 108864073 A 20181123, 2018.
- A.-X. Hu, Y.-J. Yi, M. Zhang, X.-Q. Wei and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 108863972 A 20181123, 2018.
- Y.-L. Zhao, H.-T. Li, X.-L. Zhou, S.-C. Wei, J.-Y. Li and K. Li, Faming Zhuanli Shenqing, Design Patent, CN 108794441 A 20181113, 2018.
- Y.-L. Zhao, H.-T. Li, X.-L. Zhou, S.-C. Wei, J.-Y. Li and K. Li, Faming Zhuanli Shenqing, Design Patent, CN 108794442 A 20181113, 2018.
- A.-X. Hu, Y.-J. Yi, M. Zhang and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 108774193 A 20181109, 2018.
- B.-G. Wang, X. Li, X.-D. Li and X.-M. Li, Faming Zhuanli Shenqing, Design Patent, CN 108774115 A 20181109, 2018.
- A.-L. Liu, A.-X. Hu, W.-W. Lian, M.-W. Xiao and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 108689961 A 20181023, 2018.
- L. Li, F.-G. Lu, H.-H. Huang, L.-J. Chen, K. Wei, Y. Ning, J. Hu and H. Ding, Faming Zhuanli Shenqing, Design Patent, CN 108578439 A 20180928, 2018.
- Z.-Y. Li, S.-T. Yu, G.-H. Du, A.-L. Liu and X.-M. Qin, Faming Zhuanli Shenqing, Design Patent, CN 108578458 A 20180928, 2018.
- A.-X. Hu, M. Zhang, X.-Q. Wei, J. Ye, A.-L. Liu and H. Jia, Faming Zhuanli Shenqing, Design Patent, CN 108546254 A 20180918, 2018.
- Y.-W. Li, M. Zhang, A.-X. Hu and W.-S. Li, Faming Zhuanli Shenqing, Design Patent, CN 108530439 A 20180914, 2018.
- A.-X. Hu, W. Chen, J.-X. Nie, M.-Y. Cui and J. Ye, Faming Zhuanli Shenqing, Design Patent, CN 108503604 A 20180907, 2018.
- L.-P. Cheng, Z.-X. Zhao, W. Pang, R. Ye and Y.-P. Li, Faming Zhuanli Shenqing, Design Patent, CN 108383805 A 20180810, 2018.
- W. Xiao, Z.-L. Li, Y.-C. Meng, Z.-Q. Meng, G. Ding, Z.-Z. Wang and W.-Z. Huang, Faming Zhuanli Shenqing, Design Patent, CN 108299246 A 20180720, 2018.
- B.-G. Wang, L. Li and X.-M. Li, Faming Zhuanli Shenqing, Design Patent, CN 108299462 A 20180720, 2018.
- L.-W. Qi, L. Zhang, X.-J. Yin, J. Li and G.-X. Ma, Faming Zhuanli Shenqing, Design Patent, CN 108125942 A 20180608, 2018.
- Y.-S. Tian, K.-L. Wang, B.-Y. Wang, L.-J. Li, D.-Y. Liu and G.-G. Li, Faming Zhuanli Shenqing, Design Patent, CN 108101804 A 20180601, 2018.
- H.-P. Li, Faming Zhuanli Shenqing, Design Patent, CN 108078980 A 20180529, 2018.
- A.-X. Hu, J.-X. Nie, J. Ye, A.-L. Liu and H. Jia, Faming Zhuanli Shenqing, Design Patent, CN 108047160 A 20180518, 2018.
- A.-X. Hu, W.-X. Xie, J.-X. Nie and X.-Q. Wei, Faming Zhuanli Shenqing, Design Patent, CN 107987033 A 20180504, 2018.
- H.-Y. Tian, Z.-J. Jiang, S.-F. Luo, C.-F. Li and C.-M. Shen, Faming Zhuanli Shenqing, Design Patent, CN 111265539 A 20200612, 2020.
- X.-B. Li, X. Lv, P.-F. Wang and J.-Z. Chen, Faming Zhuanli Shenqing, Design Patent, CN 111233962 A 20200605, 2020.
- L.-P. Chen, M. Li, Z.-J. Zhong, W. Yu, L.-P. Shi, W. Pang and X.-Z. Xiao, Faming Zhuanli Shenqing, Design Patent, CN 111233790 A 20200605, 2020.
- Z.-J. Ma, Y.-J. Jiang and M.-X. Liu, Faming Zhuanli Shenqing, Design Patent, CN 111205252 A 20200529, 2020.
- Z.-L. Wu, M.-Y. Cui and A.-X. Hu, Faming Zhuanli Shenqing, Design Patent, CN 111153898 A 20200515, 2020.
- P. Chen, M.-Y. Cui and A.-X. Hu, Faming Zhuanli Shenqing, Design Patent, CN 111138377 A 20200512, 2020.
- Z.-L. Wu, A.-Y. Chen and A.-X. Hu, Faming Zhuanli Shenqing, Design Patent, CN 111100074 A 20200505, 2020.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abl,
Meitao Duancd,
Dhafer S. Zinad
*abl,
Meitao Duancd,
Dhafer S. Zinad