
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotolysis of the BODIPY dye activated by pillar[5]arene†
Haifan
Zhang
 a,
Long
Wang
a,
Puyang
Dong
a,
Shuqiang
Mao
a,
Pu
Mao
*a and
Guoxing
Liu
*ab
a,
Long
Wang
a,
Puyang
Dong
a,
Shuqiang
Mao
a,
Pu
Mao
*a and
Guoxing
Liu
*ab
aCollege of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450001, P. R. China. E-mail: gxliu@henau.edu.cn; maopu@haut.edu.cn
bCollege of Science, Henan Agricultural University, Zhengzhou, 450002, China
First published on 15th February 2021
Abstract
Here, a pseudo[3]rotaxane comprising a fluorescent BODIPY derivative and pillar[5]arene was conveniently fabricated via host–guest complexation. Importantly, in this system, the efficient photodecomposition of the BODIPY derivative in the presence of pillar[5]arene was witnessed upon irradiation at 311 nm light, which was demonstrated via UV-Vis absorption, fluorescence emission, NMR and HR-MS spectroscopy techniques, but the only BODIPY dye in the absence of pillar[5]arene couldn't undergo photodegradation. We demonstrated that pillar[5]arene could act as an activator to trigger the photodegradation reaction of BODIPY derivatives via free radical reactions even without supramolecular interactions. The present results provide a new strategy for the efficient photolysis of organic dyes.
Superfluous organic dyes are one of the most serious contamination sources that are discharged at will, which is a cause for concern. The efficient elimination of organic dyes is a hugely challenging problem that needs and remains to be solved. Most approaches, such as membrane separation,1 activated carbon adsorption,2 ion exchange on synthetic adsorbent resins3 and coagulation by chemical agents,4 were adopted to dispose of the organic dye pollution. However, these superfluous organic dyes cannot be thoroughly degraded via these conventional methods. Light is considered one of the most desirable stimuli because of its non-invasiveness, easy controllability, low cost, and ubiquity.5 Owing to the above-mentioned advantages, light is often utilized as an effective strategy to manage responsive materials, which have been applied in numerous research fields, such as separation technology,6 photolithography,7 photocontrolled singlet oxygen generation,8 optical memory storage,9 and photomodulated control-release of anions.10 Among numerous stimuli-responsive systems, photolyzable matrices that can irreversibly decompose after irradiation have aroused wide research interests in the design and construction of photodegradable materials, controlled drug release systems and tissue engineering systems.11 The supramolecular method was alternatively used to develop photodegradable materials.12 Recently, Liu et al. developed a photoresponsive amphiphilic supramolecular assembly that achieved efficient photolysis of anthracene derivatives via calixarene-induced aggregation.13 Subsequently, they employed several multi-charged macrocycles such as water-soluble cyclodextrins, calixarenes, crown ethers and cucurbiturils as platforms for the rapid and broad spectral photodecomposition of aromatic dyes.14 Although there are a few reports on the construction of photodegradable supramolecular assemblies, the fabrication of photoresponsive supramolecular systems equipped with photolysis remains highly unexplored. Consequently, it is still necessary to develop novel high-efficiency photolyzable supramolecular materials. Pillararenes (particularly pillar[5]arene and pillar[6]arene), as a new type of supramolecular macrocycles, have been used as a host to construct photodegradable materials.15 For instance, Wang et al. prepared a bola-type supra-amphiphilic photoresponsive supramolecular material based on water-soluble pillar[5]arene, achieving the photodecomposition of the 9,10-dialkoxyanthracene group.16
Due to excellent photophysical and photochemical properties such as good photostability and fluorescence quantum yield,17 BODIPY dyes have been generally applied in bioimaging and fluorescent sensing,18 photodynamic therapy,19 advanced optical materials,20 etc. However, the elimination of surplus and used BODIPY dyes and other fluorescent dyes has gradually become a major issue to be addressed due to use and waste of large amounts of organic dyes applied in living matter. Furthermore, some BODIPY derivatives have high cytotoxicity.21 Consequently, it is crucial to seek a simple and effective method for dealing with excess and useless organic pollutants. Herein, to the best of our knowledge, we first put forward a new way for the efficient photolysis of BODIPY dyes via free radical reaction triggered by P5.
The BODIPY dye G-BODIPY and pillar[5]arene (P5) acted as the guest and host, respectively. Their synthetic routes were prepared by simple steps, as shown in Scheme 1. 3,4-Dihydroxybenzaldehyde (1) reacted with propargyl bromide to obtain compound (2). Subsequently, the reaction of (2) and pyrrole catalyzed by TFA produced substance (3). Then, (3) encountered with DDQ, Et3N and BF3·OEt2 in succession to give compound (4). Finally, the click reaction of (4) and (5) by the catalyzation of CuSO4·5H2O and sodium ascorbate prepared G-BODIPY. Macrocyclic host P5 was synthesized by 1,4-dimethoxybenzene by the catalyzation of FeCl3.
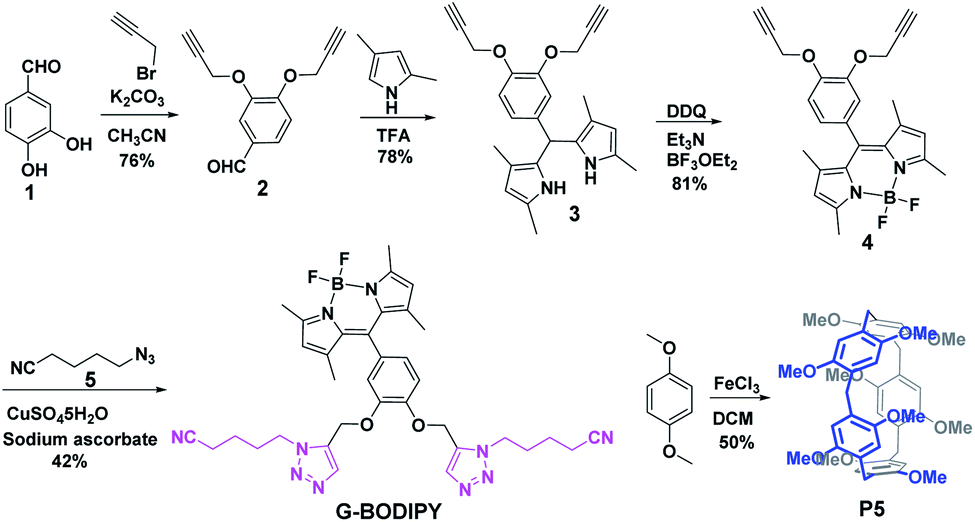 | ||
| Scheme 1 The synthesis route of G-BODIPY and P5. | ||
It is well-known that a strong binding ability (Ka = 1.2 × 104 M−1) exists between P5 and the neutral guests bearing two or three short alkyl chains with a triazole site and a cyano site at either end in chloroform.22 Therefore, we speculated that G-BODIPY would assemble with P5 by an equivalent proportion of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (guest/host) and strong complexation. As displayed in Fig. S5,† an NMR titration experiment was performed. From the NMR titration spectroscopy, an apparent upfield shift of the resonance for methylene in the guest (Ha–d) was observed, and their integral areas remained unchanged with the continuous addition of 2 equiv. P5, indicating the 2:1 host–guest binding stoichiometry. The complex stability constant (KS) was determined as 5.66 × 106 M−2 using the 1H NMR single point method (see Fig. S5 and computational method in ESI†). Clearly, the assembled model of G-BODIPY and P5 are manifested in Scheme 2. The corresponding proof was provided by the 1H NMR spectroscopic contrast, indicating the apparent upfield shift for the resonance of methylene (Ha–d) of the guest, which implies the occurrence of an assembling behavior and the formation of a new supramolecular assembly G-BODIPY⊂P52 (Fig. 1). Furthermore, as shown in Fig. S6 and S7,† the absorbance (at 504 nm) and fluorescence intensity (at 517 nm) of G-BODIPY increased slightly with the addition of P5, implying the formation of the host-guest complex G-BODIPY⊂P52. Therefore, G-BODIPY and P5 self-assembled to a [3]pseudorotaxane G-BODIPY⊂P52 in chloroform (Scheme 2).
:2 (guest/host) and strong complexation. As displayed in Fig. S5,† an NMR titration experiment was performed. From the NMR titration spectroscopy, an apparent upfield shift of the resonance for methylene in the guest (Ha–d) was observed, and their integral areas remained unchanged with the continuous addition of 2 equiv. P5, indicating the 2:1 host–guest binding stoichiometry. The complex stability constant (KS) was determined as 5.66 × 106 M−2 using the 1H NMR single point method (see Fig. S5 and computational method in ESI†). Clearly, the assembled model of G-BODIPY and P5 are manifested in Scheme 2. The corresponding proof was provided by the 1H NMR spectroscopic contrast, indicating the apparent upfield shift for the resonance of methylene (Ha–d) of the guest, which implies the occurrence of an assembling behavior and the formation of a new supramolecular assembly G-BODIPY⊂P52 (Fig. 1). Furthermore, as shown in Fig. S6 and S7,† the absorbance (at 504 nm) and fluorescence intensity (at 517 nm) of G-BODIPY increased slightly with the addition of P5, implying the formation of the host-guest complex G-BODIPY⊂P52. Therefore, G-BODIPY and P5 self-assembled to a [3]pseudorotaxane G-BODIPY⊂P52 in chloroform (Scheme 2).
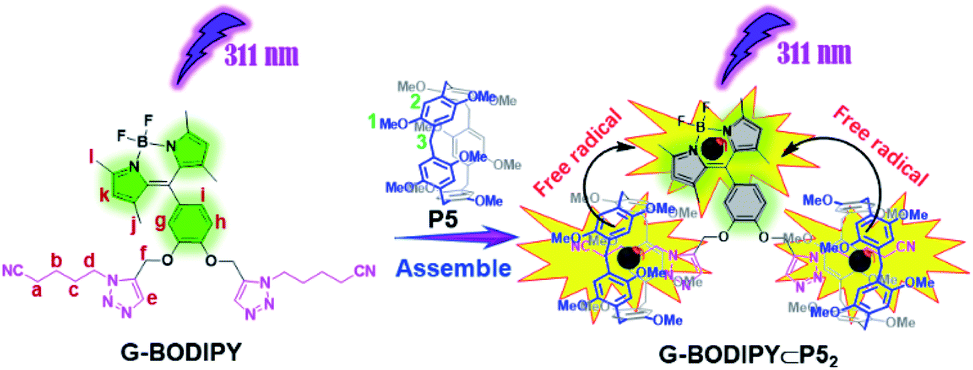 | ||
| Scheme 2 Schematic illustration of formation pattern of assembly G-BODIPY⊂P52 and photolysis of the BODIPY guest induced by P5. | ||
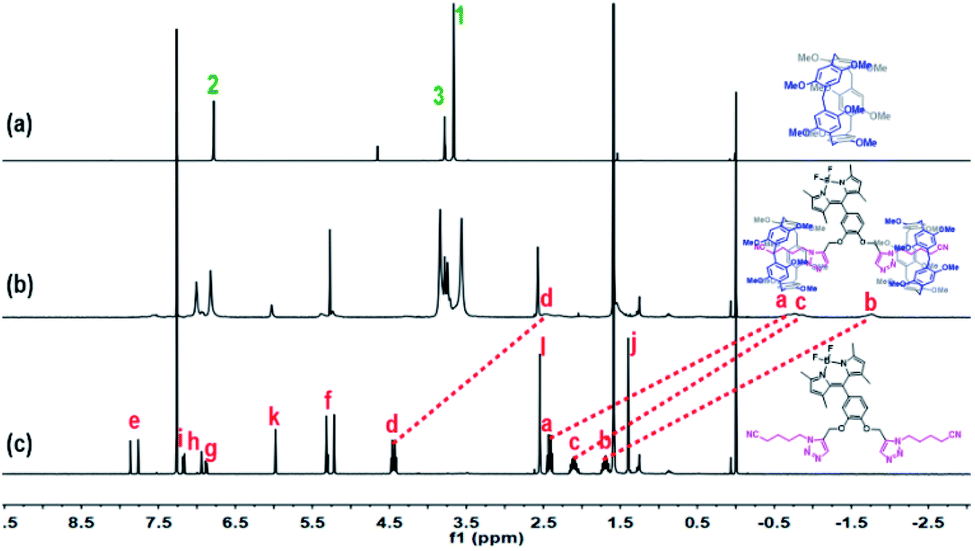 | ||
| Fig. 1 1H NMR (400 MHz, CDCl3) spectra of (a) P5, (b) assembly G-BODIPY⊂P52 and (c) guest G-BODIPY. [P5] = 2[G-BODIPY] = 6 × 10−3 mol L−1. | ||
To our surprise, as illustrated in Fig. 2a, when the sample containing G-BODIPY⊂P52 was irradiated using 311 nm light, the absorbance at 295 nm and 503 nm (assigned to BODIPY unit) significantly declined by 83% (503 nm) with four isosbestic points at 286 nm, 308 nm, 458 nm and 518 nm, respectively, implying that the BODIPY unit of G-BODIPY was dramatically destroyed. The photochemical quantum yield (311 nm light irradiation) of the pseudo[3]rotaxane was determined to be 0.34, and the test method was added in the ESI.† However, in the control experiment, only the guest G-BODIPY expressed no obvious change under the same condition, indicating that its structure exhibited invariability upon irradiation at 311 nm. Furthermore, we selected p-phenyl dimethyl ether (PDME), which is 5 equivalents to the host (P5), to evaluate photodegradation performance. As shown in Fig. 2c, the absorbance of G-BODIPY/PDME at 503 nm decreased by only 43%, suggesting that PDME also influenced the photodegradation of the guest to some extent but unsatisfactory. More visually, the variation curve of the absorbance at 503 nm belonged to the BODIPY skeleton over irradiation time at 311 nm light for only G-BODIPY, G-BODIPY⊂P52 and G-BODIPY/PDME, as presented in Fig. 2d. Moreover, the color change of the samples containing G-BODIPY, G-BODIPY⊂P52 and G-BODIPY/PDME in the above irradiation process is visually shown in Fig. 2b, a and 2c (inset), which reveal that the sample containing G-BODIPY⊂P52 turned from yellow to almost colorless, while the others did not display a wide variation. The aforementioned investigation clearly illustrated that the intervention of P5 caused the best photolysis efficiency of G-BODIPY.
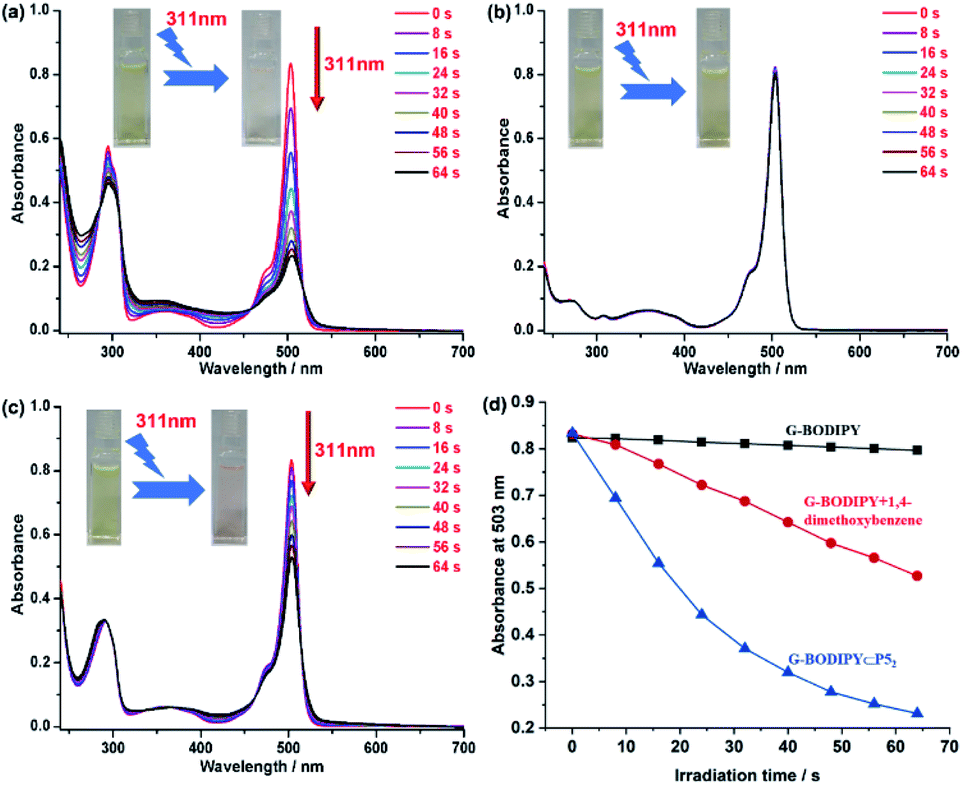 | ||
| Fig. 2 (a) The variation of UV-Vis absorption spectra of G-BODIPY with P5 upon continuous irradiation of 311 nm. (b) The variation of the UV-Vis absorption spectra of G-BODIPY upon continuous irradiation of 311 nm. (c) The variation of UV-Vis absorption spectra of G-BODIPY with 10 eq. PDME upon continuous irradiation of 311 nm. (d) The variation of absorbance of G-BODIPY, G-BODIPY with 10 eq. PDME and G-BODIPY with P5 at 503 nm upon continuous irradiation of 311 nm. [G-BODIPY] = 1.0 × 10−5 mol L−1. | ||
In addition, we employed a fluorescence spectrometer to confirm if P5 played a vital role in the photolysis of G-BODIPY. As expected, the fluorescence of G-BODIPY⊂P52 at 516 nm was quenched by 77%, which was consistent with the absorbance change (Fig. 3a). Intuitively, strong green fluorescence turned out to be very weak and nearly invisible to our eyes in the irradiation process (Fig. 3a, inset). On the contrary, no apparent variation of only G-BODIPY was observed in the fluorescence spectrum upon irradiation at 311 nm (Fig. 3b). Meanwhile, no fluorescent color and intensity changes were observed from photographs before and after irradiation (Fig. 3b, inset). Moreover, with regards to BODIPY/PDME, approximately 51% of fluorescence intensity was decreased, and its photodegradation efficiency was inferior to that of G-BODIPY⊂P52 (Fig. 3c). More visually, G-BODIPY⊂P52 manifested the highest fluorescence quenching efficiency (Fig. 3d), suggesting that P5 induced high-efficiency photolysis of the BODIPY dye.
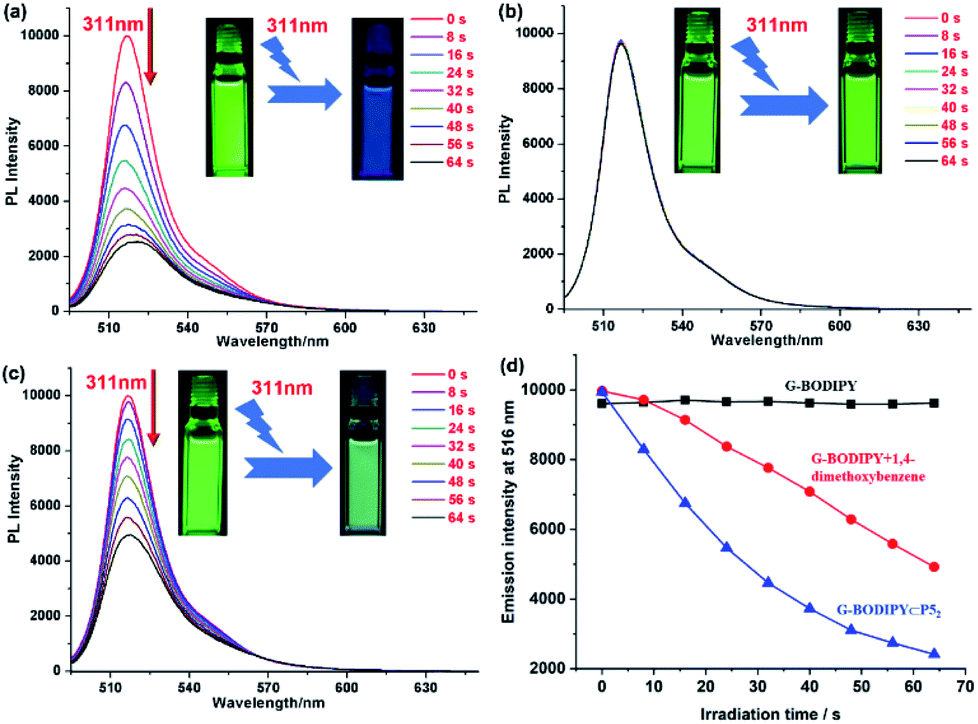 | ||
| Fig. 3 (a) The variation of the fluorescence spectra of G-BODIPY with P5 upon continuous irradiation of 311 nm. (b) The variation of the fluorescence spectra of G-BODIPY upon continuous irradiation of 311 nm. (c) The variation of the fluorescence spectra of G-BODIPY with 10 eq. PDME upon continuous irradiation of 311 nm. (d) The variation of the fluorescence intensity of G-BODIPY, G-BODIPY with 10 eq. PDME and G-BODIPY with P5 at 516 nm upon continuous irradiation of 311 nm. [G-BODIPY] = 1.0 × 10−5 mol L−1; excitation at 480 nm; slit = 1, 2.5. | ||
Subsequently, to further verify the aforementioned consequence, we performed an NMR irradiation experiment for the sample containing G-BODIPY⊂P52 in CDCl3. As shown in Fig. S8,† the resonance of the protons on the BODIPY group (Hk) in the assembly gradually disappeared with irradiation at 311 nm light for 3 h, indicating that the BODIPY group in G-BODIPY⊂P52 was almost completely damaged. In addition, we measured the HR-MS spectra of P5 and G-BODIPY⊂P52 before and after the irradiation of 311 nm. As shown in Fig. S9–S12,† the HR-MS spectra of P5 and G-BODIPY⊂P52 after irradiation showed the peaks at 768.3761 (assigned to P5 + NH4+) and 681.3404 (belonged to G-BODIPY) both thoroughly disappeared and only some small molecular weight peaks were observed, revealing that P5 and G-BODIPY⊂P52 were both broken into pieces. In contrast, when the sample containing only G-BODIPY was irradiated by 311 nm light, the resonance of the protons on the BODIPY group in the guest did not manifest any change (Fig. S13†). In addition, we conducted photoirradiation experiments of P5 and PDME via UV-Vis absorption spectra upon irradiation at 311 nm, showing that photodecomposition reactions of P5 and PDME occurred (Fig. S14 and S15†).
From the aforementioned investigation, the photolysis of G-BODIPY was efficiently activated by P5. We presumed that the three investigated manners of degradation were as follow: (a) energy transfer (as shown in Fig. S16,† P5 and PDME with a specific absorbance at 311 nm can absorb 311 nm UV light into the solution containing G-BODIPY and deliver the absorbed light energy to G-BODIPY), (b) reactive oxygen species (photoreaction of P5 and PDME caused by reactive oxygen species further triggered the photodecomposition of G-BODIPY), and (c) free radical species (photoreaction of P5 and PDME caused by the free radical-activated photolysis of G-BODIPY). To clarify the credible manner, we subsequently performed a series of control experiments. As displayed in Fig. S17†, we directly employed a 500 nm wavelength light (which could also generate heat) on free G-BODIPY and complex G-BODIPY⊂P52 through the variation of UV-Vis absorption and emission spectra, and no changes were observed. The results implied that the degradation reaction was not caused by heat. Furthermore, the spectral overlap of UV-Vis absorption of G-BODIPY and fluorescence emission of P5 (Fig. S18†) were performed and displayed almost very little degree of overlap (Fig. S19†). Thus, we could rule out the photolysis of the BODIPY dye caused by energy transfer from P5 to G-BODIPY. Subsequently, we also investigated the photoirradiation experiments of P5 and G-BODIPY⊂P52 under both aerobic and anaerobic conditions. The manifested results in the photoreactions mentioned above were not photooxidation reactions caused by oxygen (Fig. S20 and S21†). Then, we speculated that the photoreaction of P5 caused by free radicals upon irradiation at 311 nm light could further trigger the photodecomposition reaction of G-BODIPY. To verify our speculation, we selected TEMPO as a free radical scavenger to perform some control experiments. As displayed in Fig. S22b,† the absorption spectrum of G-BODIPY⊂P52 in the presence of TEMPO showed no apparent change in comparison to that of G-BODIPY⊂P52 in the absence of TEMPO (Fig. S22a†) upon irradiation at 311 nm light. Furthermore, a striking contrast for the absorbance variation of the pseudo[3]rotaxane at 503 nm in the presence and absence of TEMPO is presented in Fig. S22c.† Therefore, the above investigations demonstrated that photoreaction of P5 caused by free radicals further triggered photodecomposition of G-BODIPY upon irradiation at 311 nm light. In the control experiments, we selected compound (4) that lacked the valeronitrile binding site to investigate its photoirradiation experiments in the presence and absence of P5 using UV-Vis absorption and fluorescence emission spectra. As shown in Fig. S23 and S24,† the photolysis of compound (4) in the presence of P5 without the host–guest complexation was also achieved upon irradiation at 311 nm light, which further implied that these photodecomposition reactions were caused by free radicals and not related to supramolecular interactions.
Conclusion
In summary, we synthesized a valeronitrile-modified BODIPY derivative via the “Click” reaction as a guest. Holosymmetric methoxyl-pillar[5]arene, as the host was prepared, and assembled with the BODIPY guest to form a pseudo[3]rotaxane. Importantly, pillar[5]arene as an activator could activate the high-efficiency photolysis of the BODIPY dye via free radical reactions, and was verified via UV-Vis absorption, fluorescence emission, NMR and HR-MS spectroscopy techniques. Through a series of control experiments, we also demonstrated that the photodecomposition reactions caused by free radicals were not related to supramolecular interactions. The study provides a new strategy to induce the efficient photodegradation of residual and useless organic dyes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21801063), the Science and Technology Foundation of Henan Province (No. 192102210039), the Colleges and Universities Key Research Program Foundation of Henan Province (No. 19A150022) and China Postdoctoral Science Foundation (No. 2018M642767) for their financial support.References
- (a) S. P. Dharupaneedi, S. K. Nataraj, M. Nadagouda, K. R. Reddy, S. S. Shukla and T. M. Aminabhavi, Sep. Purif. Technol., 2019, 210, 850–866 CrossRef CAS; (b) P. S. Goh and A. F. Ismail, Desalination, 2018, 434, 60–80 CrossRef CAS.
- (a) N. Kannan and M. M. Sundaram, Dyes Pigm., 2001, 51, 25–40 CrossRef CAS; (b) P. K. Malik, J. Hazard. Mater., 2004, 113, 81–88 CrossRef CAS.
- S. S. Beheraa, S. Dasb, P. K. Parhia, S. K. Tripathyb, R. K. Mohapatrac and M. Debatac, Desalin. Water Treat., 2017, 60, 249–260 CrossRef.
- (a) G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS; (b) V. K. Gupta and Suhas, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS.
- (a) M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS; (b) G. Liu, Y. M. Zhang, X. Xu, L. Zhang, Q. Yu, Q. Zhao, C. Zhao and Y. Liu, Adv. Opt. Mater., 2017, 5, 1700770 CrossRef; (c) G. Liu, J. Zhu, Y. Zhou, Z. Dong, X. Xu and P. Mao, Org. Lett., 2018, 20, 5626–5630 CrossRef CAS; (d) G. Liu, L. Wang, Y. Zhou, N. Li, H. Zhang, J. Zhu, P. Mao and X. Xu, Dyes Pigm., 2020, 172, 107800 CrossRef CAS.
- R. A. Lorenzo, A. M. Carro and A. Concheiro, Anal. Bioanal. Chem., 2015, 407, 4927–4948 CrossRef CAS.
- (a) V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS; (b) Y. Liu, J. Genzer and M. D. Dickey, Prog. Polym. Sci., 2016, 52, 79–106 CrossRef.
- (a) J. Park, Q. Jiang, D. Feng and H. C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7188–7193 CrossRef CAS; (b) J. Park, Q. Jiang, D. Feng, L. Mao and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS; (c) Y. Qin, L. J. Chen, F. Dong, S. T. Jiang, G. Q. Yin, X. Li, Y. Tian and H. B. Yang, J. Am. Chem. Soc., 2019, 141, 8943–8950 CrossRef CAS; (d) G. Liu, X. Xu, Y. Chen, X. Wu, H. Wu and Y. Liu, Chem. Commun., 2016, 52, 7966–7969 RSC; (e) Q. J. Hu, Y. C. Lu, C. X. Yang and X. P. Yan, Chem. Commun., 2016, 52, 5470–5473 RSC.
- (a) A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS; (b) M. U. de la Orden, J. M. Montes, J. M. Urreaga, A. Bento, M. R. Ribeir, E. Perez and M. L. Cerrada, Polym. Degrad. Stab., 2015, 111, 78–88 CrossRef CAS; (c) L. Sun, Y. Yang, C. M. Dong and Y. Wei, Small, 2011, 7, 401–406 CrossRef CAS; (d) C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC; (e) W. Zhao, W. H. Ma, C. C. Chen, J. C. Zhao and Z. G. Shuai, J. Am. Chem. Soc., 2004, 126, 4782–4783 CrossRef CAS.
- (a) S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 324–327 CrossRef CAS; (b) M. Vlatković, B. L. Feringa and S. J. Wezenberg, Angew. Chem., Int. Ed., 2016, 55, 1001–1004 CrossRef; (c) J. Leng, G. Liu, T. Cui, S. Mao, P. Dong, W. Liu, X. Q. Hao and M. P. Song, Dyes Pigm., 2021, 184, 108838 CrossRef CAS.
- (a) G. Liu, Y. M. Zhang, X. Xu, L. Zhang and Y. Liu, Adv. Opt. Mater., 2017, 5, 1700770 CrossRef; (b) G. Liu, Y. M. Zhang, L. Zhang, C. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 12135–12140 CrossRef CAS.
- (a) N. Kamatham, J. P. Da Silva, R. S. Givens and V. Ramamurthy, Org. Lett., 2017, 19, 3588–3591 CrossRef CAS; (b) N. Kamatham, D. C. Mendes, J. P. Da Silva, R. S. Givens and V. Ramamurthy, Org. Lett., 2016, 18, 5480–5483 CrossRef CAS; (c) S. E. Border, R. Z. Pavlović, L. Zhiquan and J. D. Badjić, J. Am. Chem. Soc., 2017, 139, 18496–18499 CrossRef CAS; (d) M. A. Romero, N. Bas&lio, A. J. Moro, M. Domingues, J. A. González-Delgado, J. F. Arteaga and U. Pischel, Chem.–Eur. J., 2017, 23, 13105–13111 CrossRef CAS.
- Y. X. Wang, Y. M. Zhang and Y. Liu, J. Am. Chem. Soc., 2015, 137, 4543–4549 CrossRef CAS.
- X. Guan, Y. Chen, P. Guo, P. Li and Y. Liu, Chem. Commun., 2020, 56, 7187–7190 RSC.
- X. Wu, Y. Chen and Y. Liu, Adv. Sustainable Syst., 2019, 3, 1800165 CrossRef.
- S. Guo, X. Liu, C. Yao, C. Lu, Q. Chen, X. Y. Hu and L. Wang, Chem. Commun., 2016, 52, 10751–10754 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- (a) J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS; (b) M. Vendrell, D. Zhai, J. Cheng Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS; (c) T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- (a) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC; (b) Q. J. Hu, Y. C. Lu, C. X. Yang and X. P. Yan, Chem. Commun., 2016, 52, 5470–5473 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- B. Yuan, H. Wu, H. Wang, B. Tang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 706–710 CrossRef CAS.
- (a) C. Li, K. Han, J. Li, Y. Zhang, W. Chen, Y. Yu and X. Jia, Chem.–Eur. J., 2013, 19, 11892–11897 CrossRef CAS; (b) X. Wang, H. Deng, J. Li, K. Zheng, X. Jia and C. Li, Macromol. Rapid Commun., 2013, 34, 1856–1862 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characteristics of new compounds and other data. See DOI: 10.1039/d0ra08611h |
| This journal is © The Royal Society of Chemistry 2021 |