
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis and characterization of the octahedral CoIII complex of a pyrrolopyrrolizine derivative formed with dicyanovinylene-bis-(meso-aryl)dipyrrin†
Ji-Young Shin *
*
Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: jyshin@chembio.nagoya-u.ac.jp; Fax: +81-52-747-6771
First published on 8th January 2021
Abstract
A low spin state and diamagnetic CoIII complex 1 possessing pyrrolopyrrolizine ligands formed with dicyanovinylene-bis-(meso-aryl)dipyrrin was synthesized via the thermally activated metalation with CoCl2 and isolated via column chromatography. The nuclear magnetic resonance of complex 1 revealed diamagnetism, thereby confirming the structure of the octahedral CoIII-complex of strong-field ligands. The resulting molecular structure of 1 was elucidated by the X-ray diffraction analysis. An arrangement of two pyrrolizine-ligands for the metal chelation was found in the AB–BA order, which was distinct from the case observed during the formation of bis-NiII-expanded porphyrinoids.
Introduction
Dipyrrin molecules have garnered considerable attention from synthetic and applied chemists due to their proficiency in various wide-ranged applications from medicinal/medical sciences to photo-electronic devices.1 Especially, dipyrrin derivative-generated neutral metal complexes, whose separation, purification, and further handling are significantly beneficial.2 Thus, dipyrrin segments, have been used in manufacturing either singly expanded molecules or largely assembled arrays/polymers that act as proficient building units.32,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) has been a suitable oxidant to completely obtain delocalized π-electron conjugations.4 It has been established that DDQ-adducts were formed during the oxidation of dipyrromethane possessing electron-withdrawing meso-aryl. The subsequent transformations of DDQ-adducts were also investigated by treating with Lewis acid in various alcohols and with Lewis base, respectively. Dichlorofuranone-derivatives were formed in a concerted reaction involving the nucleophilic attack of alcohol, the partial elimination of the molecule, followed by the rearrangement of the π-conjugation pathway when treated with Lewis acid (B in Fig. 1).5 Dicyanovinylene-derivatives were formed in a concerted reaction involving the initiation by the nucleophilic attack of the Lewis base, the partial elimination of the DDQ-segment, followed by the regeneration of a π-conjugation network when treated with the Lewis base (A in Fig. 1).5a
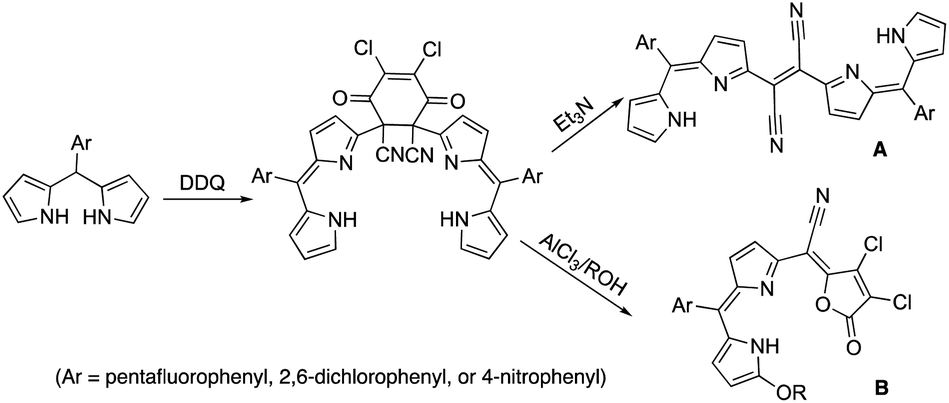 | ||
| Fig. 1 The formation of DDQ adduct and further transformation towards compounds A and B in the presence of Lewis base and Lewis acid. | ||
Furthermore, the nickel metalation of pentafluoro-phenyl-substituted dicyanovinylene compound A-C6F5 afforded enantiomeric pairs of regioisomeric pairs of bis-nickel complexes of expanded porphyrinoids (Fig. 2) when treated with a transition-metal Lewis acid such as Ni(OAc)2.6 It was comprehended that the bicyclic pyrrolizine moiety was functionalized during this transformation. It was reported that the use of a semi-metallic Lewis-acid, BF3·OEt2 or a basic-metal Lewis-acid, InBr3, resulted in the formation of pyrrolodiazepino-pyrrolizine derivatives (Fig. 3) due to a setback in creating sizeable macrocyclic structures.7
 | ||
| Fig. 2 The formation of bis-NiII-expanded porphyrinoids during the metalation of A-C6F5 with Ni(OAc)2. | ||
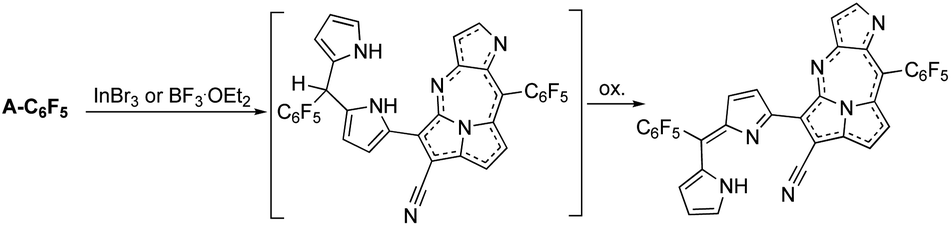 | ||
| Fig. 3 The formation of pyrrolodiazepino-pyrrolizines in the presence of a non-transition metal Lewis acid, either BF3OEt2 or InBr3. | ||
Other transition-metal sources were then stimulated to foster the investigation of different metal-complexes of cyclic/chain ligands. It is noteworthy to mention that the use of CoCl2 afforded a different alignment (AB–BA order) of ligands as compared with the sequence obtained in the presence of Ni(OAc)2 (AB–AB order). The resulting cobalt complex indicated a low spin, diamagnetic, and octahedral CoIII complex formed due to facing two strong-field ligands in the same direction. The formation of bis-nickel(II)-expanded porphyrins, involving cross-condensations and further oxidations were successful within the AB–AB alignment of ligands. Herein, the synthetic and structural details are reported.
Results and discussion
Dicyanovinylene-bis(meso-aryl)dipyrrin (DCVAD) was prepared following a previously method and used in the formation of cobalt complexes with CoCl2 (Scheme 1).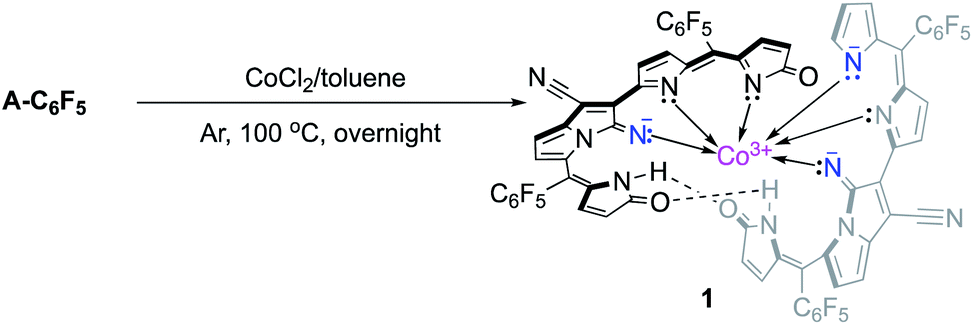 | ||
| Scheme 1 The synthesis of CoIII complex 1. | ||
A toluene solution of DCVAD with CoCl2 was heated to 100 °C overnight, from which CoIII complex 1 was isolated as a yellow fraction via silica gel column chromatography. The low solubility of complex 1 made it difficult to isolate on the silica gel, and the isolation yield was about 10%. A by-product exhibited a larger molecular mass value (m/z = 2130.82) than that of complex 1, whose structure was predicted to have two cobalt cations that coordinated with three ligand units. The mass per charge ratio (m/z) in the MALDI-TOF mass spectrometry of 1 was 1491.89, which was consistent with [M + H]+ for the molecular formula C68H20CoF20N12O3. The growth of single crystals for the X-ray diffraction analysis was achieved with a solvent combination of chloroform and methanol by the inter-conversion vaporization and slow diffusion of two solvents, which resulted in fine-fit single-crystallization and appropriate diffractions to refine the molecular structure (Fig. 4 and S1–S3†).
 | ||
| Fig. 4 The crystal structure of 1: (a) a complex unit, (b) and (c) the first ligand moiety positioning horizontally, and (d) and (e) the second ligand moiety positioning vertically. Figures (c) and (e) show the dihedral angles between the terminal pyrrolones and the coordinating parts in the horizontal and vertical ligand moieties, respectively. | ||
As shown in Fig. 4, each molecular origin of A-C6F5 first formed bicyclo-pyrrolizine, whose initiation inhibited the catalytic reaction of the transition-metal (CoCl2) Lewis-acid, which occurred at vinylene-nitrile. Consequently, the neighbouring pyrrole attached the nitrile carbon nucleophilically, resulting in the formation of a bicycle-pyrrolizine segment. Simultaneously, the conjugation pathway of π-electrons was reorganized for the proficient ligation of the metal-chelation. In the terminal pyrrole rings, the nucleophilic addition of water occurred, and the π-conjugation pathways were fulfilled by the deprotonation of hydroxyl groups, resulting in 2-pyrrolones. Herein, the cobalt-centre was associated with six coordination sites to complete the ligation, in which one terminal pyrrole out of the four still retained α-free confirmation from the nucleophilic addition of water due to the restricted stabilization of the enol-form compared with that of the keto-form. It is noteworthy to mention that two distinct terminal pyrroles took a relative orientation to each other and formed intramolecular hydrogen bonds (H-bonds), which did not contribute to metalation, but eventually stabilized the overall structure of compound 1 (dashed lines representing H-bonds in Fig. 4a).
Nevertheless, almost the same ligand was held between two ligands towards the cobalt centre; one possessing a α-free pyrrole-terminal and the other that possessed all pyrrolone-terminals disordered the position of ligands in the crystalline state. The octahedral structure compromising the CoIII centre holds two ligand units, associating with the planarity of two ligands, as shown in Fig. 4b and d. Mean deviations within 20 atoms involved in ligands main skeleton except for the terminal pyrrole/pyrrolone realized hydrogen-bonds and adjacent meso-methin carbons were calculated as 0.174 Å for (b) and 0.137 Å for (d). A ligand unit that places vertically in Fig. 4a is presented in (b) and (c), while the other unit that places horizontally in the figure is presented in (d) and (e). Furthermore, it was implied that terminal pyrroles, which were not involved in the metalation, were standing up from the mean planes of chelating parts due to the following factors. Firstly, the steric interference was induced in individual ligand units; the terminal pyrrolone that was not involved in the metal coordination was sterically hindered with the CoIII-coordinating part. Secondly, hydrogen-bonding interactions between terminal pyrrolones, led to the relative orientation. As a result, dihedral angles were measured as 51.23° for the ligand unit (b) and 51.53° for the ligand unit (d) (Fig. 4c and e). Hydrogen-bond lengths for N–H⋯O were 2.871 and 2.909 Å.
The metal-chelating imino-/amino-Ns of fully conjugated multi-pyrrolyl ligands play as strong field ligands, in which a low spin state of CoIII was compromised. The low spin, diamagnetic hexa-coordination of CoIII was exhibited with 1H NMR spectroscopy (Fig. 5). Two terminal pyrroles possessing H-bond interactions, where the amino-Ns of pyrroles acted as the proton donors of H-bonds, resonated two singlet-peaks of NH at a downfield shifted area, 10.48 and 10.21 ppm due to the deshielding effect of H-bond, whose peaks were assigned with the addition of D2O as disappearing peaks (spectra a and c).
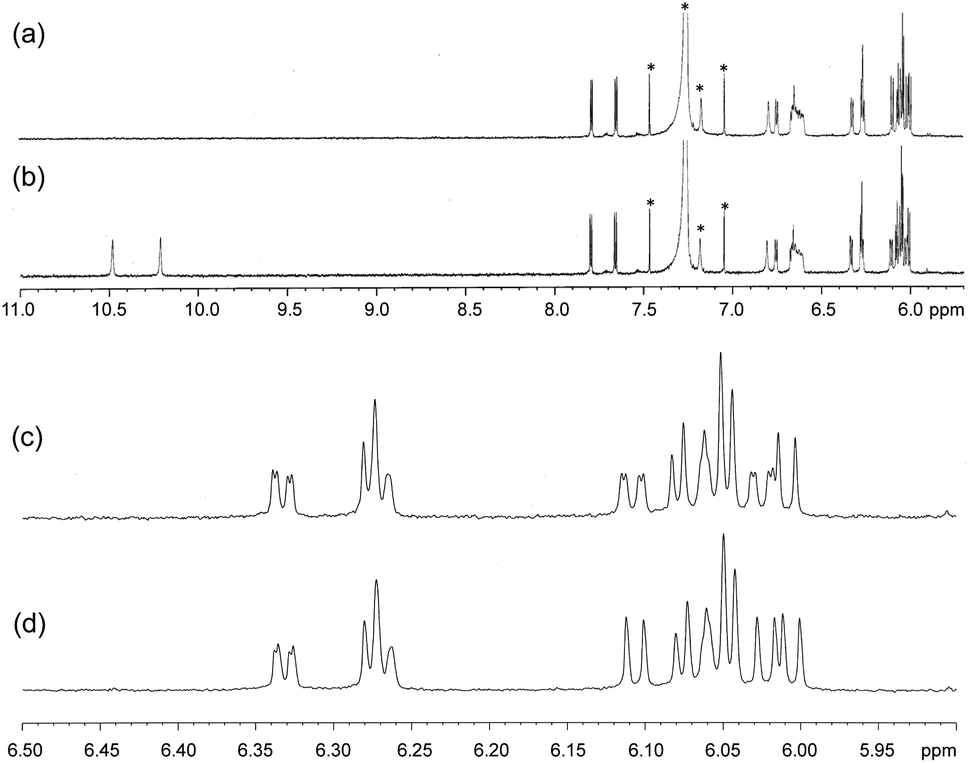 | ||
| Fig. 5 The 1H NMR spectra of complex 1 in CDCl3 after (a and c) and before (b and d) the addition of D2O. | ||
Furthermore, the peaks of pyrrole-βHs were assigned to 6.00–7.81 ppm. Because of the distinct structures of the metal-chelating ligands with the CoIII centre, twenty protons and twenty fluorines (four p-Fs and eight each for o- and m-Fs of four pentafluorophenyl groups) were assigned in the 1H and 19F NMR spectra, respectively (Fig. 5 and 6). A few peaks overlapped as random cases. The homonuclear HH-COSY NMR exhibits eight correlation pairs of the β-protons of pyrrole rings and one correlation peak for the α- and β-protons of the α-free terminal pyrrole with nine sets of proton–proton correlations, as shown in Fig. 7. Furthermore, 19F NMR reveals individually located, twenty fluorine peaks (Fig. 6). The fluorine–fluorine correlations are demonstrated with the FF-COSY NMR analysis (Fig. S8†), whose spectrum shows fluorine sets for four meso-pentafluorophenyls as marked with four different symbols. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of 2-pyrrolones of 1 expose broad bands overlapping around 1694 cm−1 in CH2Cl2, which is significant for the 2-pyrrolone carbonyl group inducing a weakened energy vibration frequency as compared to common carbonyl groups (Fig. S9†).
O stretching vibration of 2-pyrrolones of 1 expose broad bands overlapping around 1694 cm−1 in CH2Cl2, which is significant for the 2-pyrrolone carbonyl group inducing a weakened energy vibration frequency as compared to common carbonyl groups (Fig. S9†).
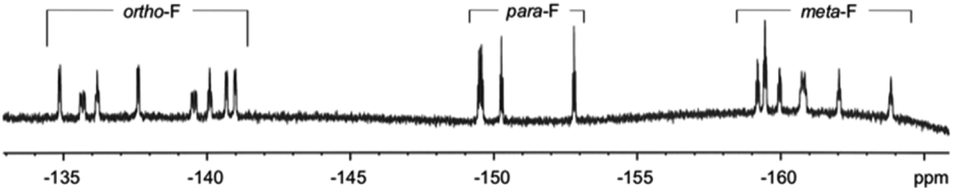 | ||
| Fig. 6 The 19F NMR spectrum of 1 in CDCl3. | ||
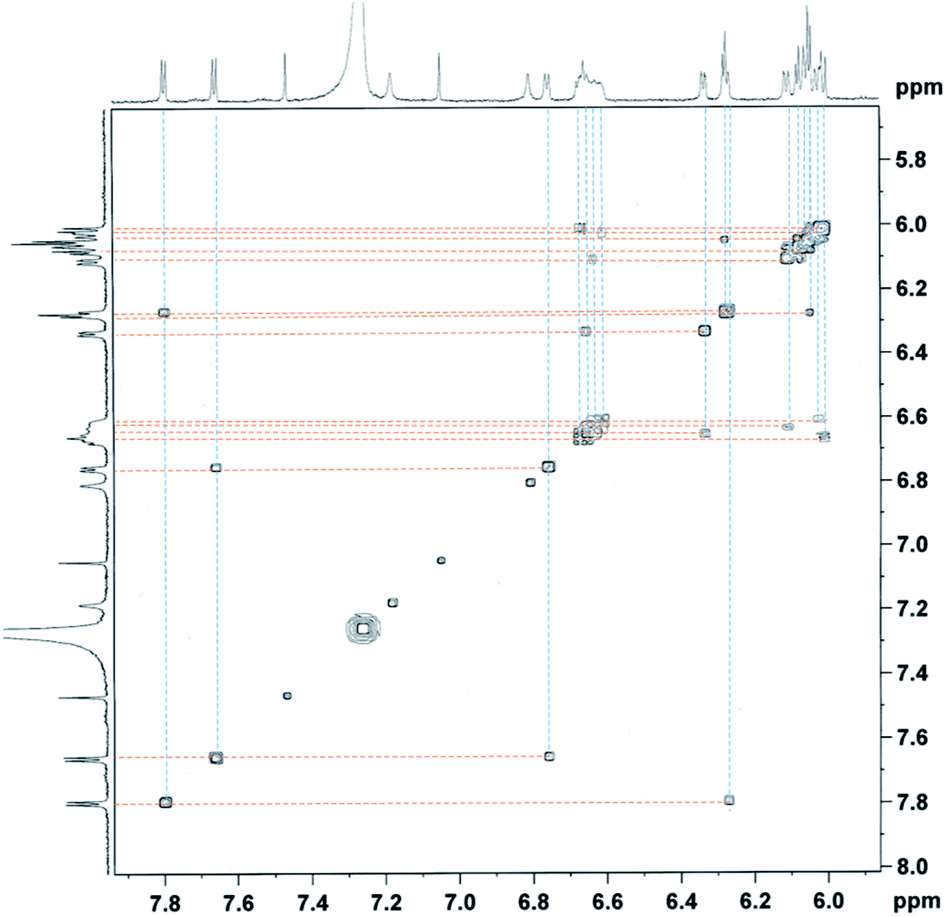 | ||
| Fig. 7 The HH COSY NMR (F1 = F2 = 500 MHz) spectrum of 1 in CDCl3. | ||
CH2Cl2 solution of 1 exhibits a vastly elongated absorption band of about 1100 nm and demonstrates strong absorbance nearly (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε = 4.01) at λmax = 795 nm (Fig. 8).
ε = 4.01) at λmax = 795 nm (Fig. 8).
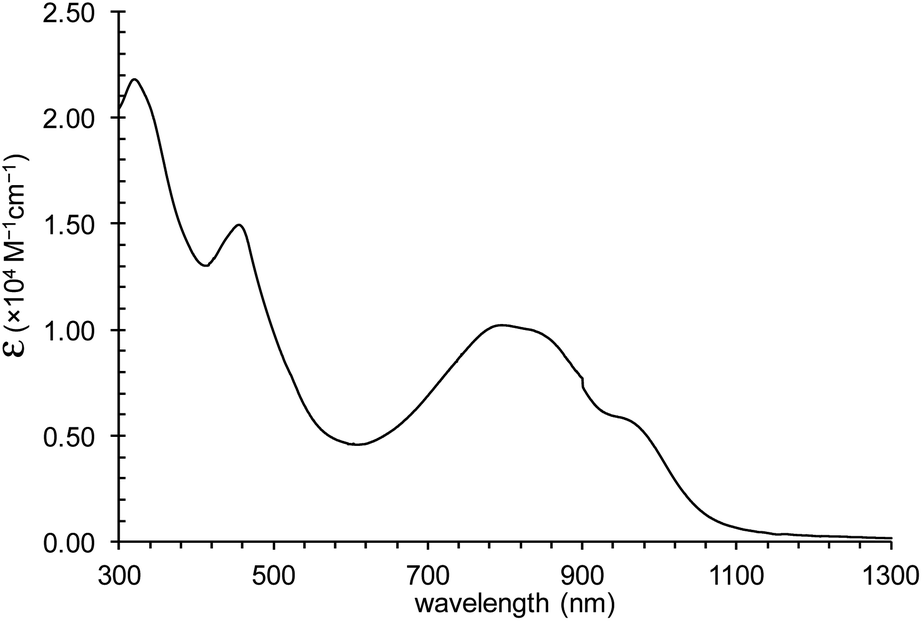 | ||
| Fig. 8 The UV-vis-NIR absorption spectrum of complex 1 in CH2Cl2. | ||
The difference in potentials for 1 between the 1st oxidation (0.722 V) and 1st reduction (0.194 V) is measured as 0.528 V with cyclic voltammetry (Fig. 9). The small HOMO–LUMO gap of 0.528 V much less than 1.0 V is in good agreement with the absorption band, largely bathochromic shifted, and tailed until 1300 nm. The first reduction process was reversible distinct from the CoIII octahedral complex containing macrocyclic, fewer liberty ligands that exposed irreversible waves for the first reduction process due to the increased liberty on the axial ligands of CoII state via the transformation from CoIII to CoII.8 Furthermore, unlikely broad waves are observed between reduction potentials, which probably streamed from the liberty and propositioned to the broaden band at the absorption spectrum. The second reduction at −0.763 V for Co(II)/Co(I) was reversible, and no decomposition was reflected in any window of the potential range, 1.0–1.6 V.
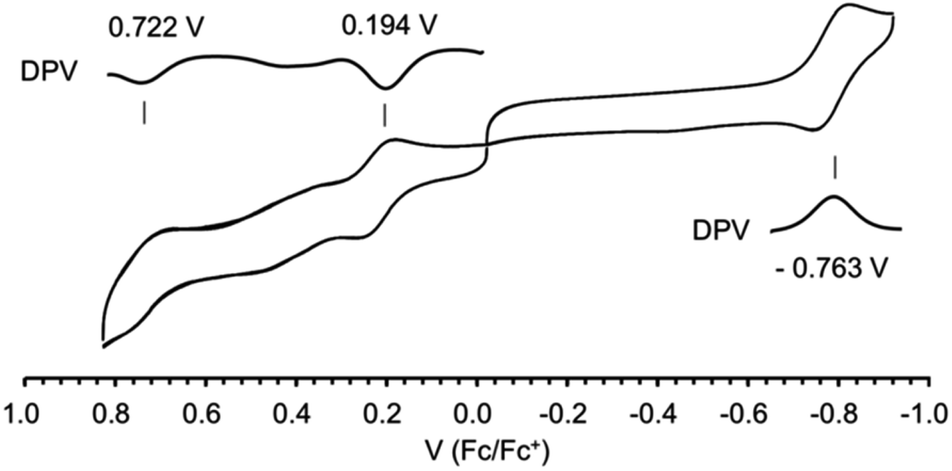 | ||
| Fig. 9 The cyclic voltammograms of complex 1 in CH2Cl2 (0.1 M nBu4NPF6). Working electrode: glassy carbon, counter electrode: Pt, reference cell: Ag/AgCl, and Fc/Fc+: ferrocene/ferrocenium couple. | ||
Experimental
Materials and methods
All reagents obtained from commercial suppliers were used without further purification. Products were column chromatographed on a silica gel (silica gel 300 or 400). The nuclear magnetic resonance (NMR) spectra of synthetic compounds were recorded using a Bruker Avance 300 MHz NMR spectrometer using the CDCl3 solvent and trimethylsilane (TMS) as an internal standard. The X-ray diffraction patterns of the single crystal of CoIII complex 1 were collected at 93 K with a Varimax Saturn N instrument operated with the Rigaku operation software package. The molecular structure was analyzed using direct methods with the aid of SHELXS-97 software, and the refinement was accomplished using the SHELXL-97 program. All hydrogen atoms were placed in calculated positions.Preparation of CoIII complex 1
The precursor molecule A-C6F5 was prepared following a previously method.5a A toluene solution (90 mL) of A-C6F5 (60 mg, 86.4 μmol) and CoCl2·6H2O (120 mg, 0.924 mmol) was heated to 100 °C for 9 h and then cooled down. The resulting solution was uploaded on aluminium oxide in a column to separate two fractions using eluents having gradually increased polarities, from neat CH2Cl2 to 1% MeOH in CH2Cl2. The first green fraction was separated to obtain the diazepino-pyrrolizine compound (last structure in Fig. 3) as a failed/retained ligand in cobalt metalation: spectral data elucidated the compound.7 The second fraction with a yellowish-green colour was then collected and washed with water to remove aluminium oxide dissolved in the increased polar eluent. Using brine and CH2Cl2, the organic layer was washed, separated, and dried over anhydrous Na2SO4. The residue was then precisely purified on a silica gel using neat CH2Cl2. A brownish yellow coloured fraction, fluorescent under the projection of a 365 nm UV lamp was collected to obtain compound 1 (10% yield). The 1H NMR spectrum of the compound exposes the two significant singlets (10.48 and 10.21 ppm) of amino-NHs, supporting the molecular structure that preserves pyrroles, but not the metalation. Furthermore, the growth of single crystals was achieved with a CHCl3 solution and MeOH via vapor diffusion.Spectral data of 1‡
λmax (nm (log ε), CH2Cl2): 320 (4.34), 456 (4.17), 795 (4.01); δH (500 MHz, 298 K, CDCl3): 10.48 (bs, 1H, NH), 10.21 (bs, 1H, NH), 7.80 (d, J = 4.5 Hz, 1H), 7.66 (d, J = 4.5 Hz, 1H), 6.81 (s, 1H), 6.68 (d, J = 5.4 Hz, 1H), 6.67–6.58 (m, 3H), 6.34 (d, J = 4.8 Hz, 1H), 6.28 (d, J = 3.6 Hz, 1H), 6.27 (d, J = 3.7 Hz, 1H), 6.11 (d, J = 5.6 Hz, 1H), 6.08 (d, J = 3.7 Hz, 1H), 6.06 (d, J = 5.3 Hz, 2H), 6.05 (d, J = 3.7 Hz, 1H), 6.03 (d, J = 5.7 Hz, 1H), 6.01 (d, J = 5.4 Hz, 1H); δF (470.57 MHz, 298 K, calibrated with external CF3COOH): −134.9 (d, J = 23.5 Hz, 1F, o-F), −135.7 (dd, ddJ = 51.8 Hz, dJ = 23.5 Hz, 1F, o-F), −136.2 (dd, ddJ = dJ = 23.5 Hz, 1F, o-F), −137.6 (dd, ddJ = 7.1 Hz, dJ = 23.5 Hz, 1F, o-F), −139.5 (dd, ddJ = 51.8 Hz, dJ = 23.5 Hz, 1F, o-F), −140.1 (dd, ddJ = dJ = 23.5 Hz, 1F, o-F), −140.7 (d, J = 23.5 Hz, 1F, o-F), −140.9 (d, J = 23.5 Hz, 1F, o-F), −149.5 (t, J = 23.5 Hz, 1F, p-F), −149.6 (t, J = 23.5 Hz, 1F, p-F), −150.3 (t, J = 23.5 Hz, 1F, p-F), −152.8 (t, J = 23.5 Hz, 1F, p-F), −159.2 (td, tdJ = 4.7 Hz, tJ = 23.5 Hz, 1F, m-F), −159.5 (td, tdJ = 4.7 Hz, tJ = 23.5 Hz, 2F, m-F), −159.9 (td, tdJ = 4.7 Hz, tJ = 23.5 Hz, 1F, m-F), −160.7 (t, J = 23.5 Hz, 1F, m-F), −160.9 (t, J = 23.5 Hz, 1F, m-F), −162.1 (t, J = 23.5 Hz, 1F, m-F), −163.9 (t, J = 23.5 Hz, 1F, m-F); MALDI TOF MS: calcd for [M + nH]+, M = C68H19CoF20N12O3: 1492.087 (calcd), 1492.858 (obs.); MALDI TOF MS: calcd for C68H19CoF20N12O3 ([M + nH]+): 1492.087; found: 1492.858.Conclusions
In conclusion, the CoIII complex 1 was synthesized and isolated from a toluene solution of dicyanovinylene-bis(meso-aryl)dipyrrin via thermal metalation of the pyrrolo-pyrrolizine derivative. The molecular structure of 1 was elucidated by the X-ray diffraction analysis, in which two pyrrolizine-ligands chelating to the CoIII atom oriented in an AB–BA alignment with the octahedral CoIII complex. Furthermore, NMR spectroscopic and X-ray crystallographic details disclosed the complex 1 to be diamagnetic, low spin octahedral Co(III)-complex of strong-field ligands.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the JSPS KAKENHI grant number JP18K04925. J.-Y. Shin acknowledges the Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University for its great support for the provision of experimental and analytical instruments as well as indispensable advice.Notes and references
- (a) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC; (b) C. S. Kue, S. Y. Ng, S. H. Voon, A. Kamkaew, L. Y. Chung, L. V. Kiew and H. B. Lee, Photochem. Photobiol. Sci., 2018, 17, 1691 RSC; (c) C.-L. Hou, Y. Yao, D. Wang, J. Zhang and J.-L. Zhang, Org. Chem. Front., 2019, 6, 2266 RSC; (d) J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 106 Search PubMed; (e) L. Lecarme, A. Kochem, L. Chiang, J. Moutet, F. Berthiol, C. Philouze, N. Leconte, T. Storr and F. Thomas, Inorg. Chem., 2018, 57, 9708 CrossRef CAS; (f) J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108 RSC; (g) D. Perl, S. W. Bisset and S. G. Telfer, Dalton Trans., 2016, 45, 2440 RSC.
- (a) H. Fischer and H. Orth in Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1940, vol. 2, pp.1–151 Search PubMed; (b) H. Falk in The Chemistry of Linear Oligopyrroles and Bile Pigments, Springer, Vienna, 1989 CrossRef; (c) C. Brückner, Y. Zhang, S. J. Rettig and D. Dolphin, Inorg. Chim. Acta, 1997, 263, 279 CrossRef; (d) R. S. Singh, R. P. Paitandi, R. K. Gupta and D. S. Pandey, Coord. Chem. Rev., 2020, 414, 213269 CrossRef CAS; (e) S. A. Baudron, Dalton Trans., 2020, 49, 6161 RSC.
- (a) C. V. K. Sharma, G. A. Broker, J. G. Huddleston, J. W. Baldwin, R. M. Metzger and R. D. Rogers, J. Am. Chem. Soc., 1999, 121, 1137 CrossRef CAS; (b) H. Yanagi, H. Mukai, K. Ikuta, T. Shibutani, T. Kamikado, S. Yokoyama and S. Mashiko, Nano Lett., 2002, 2, 601 CrossRef CAS; (c) Q. Chen and D. Dolphin, Can. J. Chem., 2002, 80, 1668 CrossRef CAS; (d) A. Thompson, S. J. Rettig and D. Dolphin, Chem. Commun., 1999, 631 RSC; (e) Y. Zhang, A. Thompson, S. J. Rettig and D. Dolphin, J. Am. Chem. Soc., 1998, 120, 13537 CrossRef CAS; (f) A. Thompson and D. Dolphin, Org. Lett., 2000, 2, 1315 CrossRef CAS; (g) R. Taniguchi, S. Shimizu, J.-Y. Shin, H. Furuta and A. Osuka, Tetrahedron Lett., 2003, 44, 2505 CrossRef CAS; (h) J.-Y. Shin, D. Dolphin and B. O. Patrick, Cryst. Growth Des., 2004, 4, 659 CrossRef CAS; (i) J.-Y. Shin, B. O. Patrick and D. Dolphin, CrystEngComm, 2008, 10, 960 RSC; (j) Q. Miao, J.-Y. Shin, B. O. Patrick and D. Dolphin, Chem. Commun., 2009, 2541 RSC; (k) Z. Zhang and D. Dolphin, Chem. Commun., 2009, 6931 RSC; (l) L. Ma, J.-Y. Shin, B. O. Patrick and D. Dolphin, CrystEngComm, 2008, 10, 1539 RSC; (m) Y. Chiba, T. Nakamura, R. Matsuoka and T. Nabeshima, Synlett, 2020, 31, 1663 CrossRef CAS.
- (a) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153 CrossRef CAS; (b) J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS; (c) L. Yu, K. Muthukumaran, J. V. Sazanovich, C. Kirmaier, E. Hindin, J. R. Diers, P. D. Boyle, D. f. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2003, 42, 6629 CrossRef CAS; (d) G. R. Geier III, J. F. B. Chick, J. B. Callinan, C. G. Reid and W. P. Auguscinski, J. Org. Chem., 2004, 69, 4159 CrossRef; (e) L. Capella, P. C. Montevecchi and D. Nanni, J. Org. Chem., 1994, 59, 7379 CrossRef CAS; (f) C. Brücker, V. Karunaratne, S. J. Rettig and D. Dolphin, Can. J. Chem., 1996, 74, 2182 CrossRef; (g) J. S. Lindsey, in The porphyrin Handbook ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 1 Search PubMed; (h) Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427 CrossRef CAS; (i) J.-Y. Shin, H. Furuta, K. Yoza, S. Igarashi and A. Osuka, J. Am. Chem. Soc., 2001, 123, 7190 CrossRef CAS; (j) T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831 CrossRef CAS.
- (a) J.-Y. Shin, B. O. Patrick and D. Dolphin, Org. Biomol. Chem., 2009, 7, 2032 RSC; (b) J.-Y. Shin and D. Dolphin, New J. Chem., 2011, 35, 2483 RSC.
- N. H. Faialaga, S. Ito, H. Shinokubo, Y. Kim, K. Kim and J.-Y. Shin, Dalton Trans., 2017, 46, 10802 RSC.
- J.-Y. Shin, RSC Adv., 2019, 9, 40031 RSC.
- A. Mahammed, B. Mondal, A. Rana, A. Dey and Z. Gross, Chem. Commun., 2014, 50, 2725 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, Mass, and absorption spectra as well as X-ray packing diagrams. CCDC 1870416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09452h |
‡ Crystallographic data of 1 (CCDC 1870416): (C68H20CoF20N12O3). (CHCl3).0.38(CH3OH) Mr = 1623.19, T = 93(2) K, crystal size = 0.09 × 0.09 × 0.01 mm3, Green prism, Mo radiation, triclinic, space group ![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 1 (#2), a = 11.204(5) Å, b = 16.844(5) Å, c = 18.858(5) Å, α = 63.767(5)°, β = 83.998(5)°, γ = 77.380(5)°, V = 3115.1(19) Å3, Z = 2, Pcalcd = 1.728 g cm−1, R1(F) = 0.0972 (I > 2(l)), wR2(F2) = 0.2944 (all), GoF = 1.072. Disordered solvent molecules were refined in one chloroform and around 0.38 methanol molecules per one molecule of 1. Furthermore, one of the terminal pyrrolones, positioning the closet to the terminal pyrrole of the partner ligand in the cobalt complex was also disordered: two terminal parts were disordered and placed close to each other by forming hydrogen bonds, N–H⋯O. 1 (#2), a = 11.204(5) Å, b = 16.844(5) Å, c = 18.858(5) Å, α = 63.767(5)°, β = 83.998(5)°, γ = 77.380(5)°, V = 3115.1(19) Å3, Z = 2, Pcalcd = 1.728 g cm−1, R1(F) = 0.0972 (I > 2(l)), wR2(F2) = 0.2944 (all), GoF = 1.072. Disordered solvent molecules were refined in one chloroform and around 0.38 methanol molecules per one molecule of 1. Furthermore, one of the terminal pyrrolones, positioning the closet to the terminal pyrrole of the partner ligand in the cobalt complex was also disordered: two terminal parts were disordered and placed close to each other by forming hydrogen bonds, N–H⋯O. |
| This journal is © The Royal Society of Chemistry 2021 |