
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT calculations for single-atom confinement effects of noble metals on monolayer g-C3N4 for photocatalytic applications†
Cheng Yangab,
Zong-Yan Zhao*c,
Hai-Tang Weia,
Xi-Yu Denga and
Qing-Ju Liu *a
*a
aSchool of Materials and Energy, National Center for International Research on Photoelectric and Energy Materials, Yunnan Key Laboratory for Micro/Nano Materials & Technology, Yunnan University, Kunming 650091, P. R. China. E-mail: qjliu@ynu.edu.cn; Fax: +86-871-65032713; Tel: +86-871-65032713
bSchool of Optoelectronic and Communication Engineering, Yunnan Open University, Kunming 650223, P. R. China
cFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, P. R. China. E-mail: zzy@kmust.edu.cn; Fax: +86-871-65107922; Tel: +86-871-65109952
First published on 21st January 2021
Abstract
Graphitic carbon nitride, as a very promising two-dimensional structure host for single atom catalysts (SACs), has been studied extensively due to its significant confinement effects of single atoms for photocatalytic applications. In this work, a systematic investigation of g-C3N4 confining noble metal single atoms (NM1@g-C3N4) will be performed by using DFT calculations. The geometric structure calculations indicate that the most favorable anchored sites for the NM1 is located in the six-fold cavity, and the deformed wrinkle space of g-C3N4 helps the NM1 to be stabilized in the six-fold cavity. The electronic structure calculations show that the conduction band of NM1@g-C3N4 moved down and crossed through the Fermi level, resulting in narrowing the band gap of the NM1@g-C3N4. Moreover, the confined NM1 provide a new channel of charge transport between adjacent heptazine units, resulting in a longer lifetime of photo-generated carriers except Ru, Rh, Os and Ir atoms. Furthermore, the d-band centres of NM1 in NM1@g-C3N4 show that Rh1@, Pd1@, Ir1@ and Pt1@g-C3N4 SACs may have better photocatalytic performance than other NM1@g-C3N4 SACs. Finally, Pt1@g-C3N4 SACs are considered to have higher photocatalytic activity than other NM1@g-C3N4 SACs. These results demonstrate that the confinement effects of noble metals on monolayer g-C3N4 not only makes the single atom more stable to be anchored on g-C3N4, but also enhances the photocatalytic activity of the system through the synergistic effect between the confined NM1 and the monolayer g-C3N4. These detailed research may provide theoretical support for engineers to prepare photocatalysts with higher activity.
1. Introduction
Environmental pollution and the energy crisis caused by excessive use of fossil fuels are the two major problems that have seriously affected the sustainable development of human being in the world today. Catalytic technologies play a critical role in solving two above-mentioned problems, which have been extensively used in many fields, such as chemical industry, production,1,2 clean energy technologies3,4 and environmental treatment.5,6 Traditional supported metal catalysts, especially noble metals, including ruthenium (Ru), rhodium (Rh), palladium (Pd), silver (Ag), osmium (Os), iridium (Ir), platinum (Pt), gold (Au), have excellent catalytic performance. Therefore, they are applied in a wide range of heterogeneous catalysis fields.7–10 As is well-known, heterogeneous catalysts have the advantages of a stable structure and easy separation of products. Meanwhile, they also have some weakness, such as the low metal utilization rate and the low catalyst efficiency, which leads to the wasting of noble metal resources, the increase of production costs and the catalysts to be bulky and heavy. In contrast, homogeneous catalysts have a large number of homogeneous single active sites, which make the catalysts show high catalytic activity and selectivity. However, low stability and difficult separation of products is homogeneous catalysts' Achilles heel. Fortunately, single-atom catalysts (SACs) that were first put forward by Zhang's group in 2011 (ref. 11) are provided with both advantages of heterogeneous and homogeneous catalysts and become the bridge and tie to link both up.12–14 For the past few years, SACs with the unique electronic structure and unsaturated coordination environment have drawn extensive attention around the world and can be used effectively in many catalytic reactions due to their low noble metal loadings and high utilization rates of noble metal atoms.15–17 Unfortunately, the specific surface area of noble metal particles increases dramatically with the decrease of the size of noble metal particles to single atom, resulting in a sharp increase in the free energy of the noble metal surface.18 That is, it is very easy for agglomeration to form large particles during the preparation of the catalysts and the catalytic reaction, resulting in deactivation of the catalysts. Therefore, it is very important for SACs to search the suitable confinement supports to effectively confine single atom.19–21 As a matter of fact, confinement supports can effectively increase kinetic stabilization of a single atom to prevent the aggregation and create well-dispersed SACs by the strong metal–support interactions. Most importantly, confinement supports can also form new electronic structures and new coordination environment to affect the catalytic activity and selectivity.22 This phenomenon is known as the confinement effects, which have already aroused enormous interest in catalytic research field. In recent years, many confinement supports for SACs have been widely researched, such as FeOx,23 CeO2,24 WOx,25 Al2O3,26 MnO2,27 SiO2,28 ThO2,29 TiC,30 Mo2C,31 metal–organic frameworks (MOFs),32 and so on.Two-dimensional (2D) materials have been extensively studied and used in a variety of applications.33,34 Comparing with traditional three-dimensional (3D) materials, 2D materials are more suitable to confine noble metal single atoms to construct SACs due to their unique physical and chemical properties,35 including (1) 2D materials have excellent mechanical strength, high chemical and thermal stability; (2) 2D materials can confine more noble metal single atoms to provide more active sites because of their large surface area; (3) 2D materials can regulate the electronic structure of noble metal single atoms to improve the catalytic performance through synergistic effect;36,37 (4) the intrinsic catalytic activities of the pristine 2D materials can be triggered by the confined noble metal single atoms;38–40 (5) the vacant structure on both sides of 2D materials is helpful for the adsorption, diffusion and dissociation of reactants and reaction products.
Graphitic carbon nitride (g-C3N4), as an emerging kind of 2D material with layered structure, is a typical synthetic polymer semiconductor composed of sp2 carbon (C) and sp2 nitrogen (N) atoms forming π-conjugated system.41 In fact, g-C3N4 can be regarded as a special case of graphene via doping of N atoms. But, unlike N-doped graphene, g-C3N4 has more N species and a lot of huge N-coordinating cavities available for confining single atoms. More interestingly, pristine g-C3N4, as a very promising photocatalyst in itself,42 has been widely researched in the field of photocatalysis, including contaminant degradation,43 hydrogen production44,45 and carbon dioxide (CO2) reduction.46,47 However, the photocatalytic property of pristine g-C3N4 has been limited due to its serious recombination of photo-generated carriers and narrow light absorption range.42 Thus, to promote the photocatalytic performance of pristine g-C3N4, some SACs photocatalysts via g-C3N4 confining noble metal single atoms (NM1@g-C3N4, NM = Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) have been researched in recent years. For instance, Gao and co-workers demonstrated that the single atoms of Pd and Pt supported on g-C3N4 as an efficient photocatalyst significantly increase catalysts' ability to absorb visible-light and effectively reduce the reaction barriers during CO2 reduction.48 Density functional theory (DFT) calculations showed that the preferred product is HCOOH and CH4 on Pd@g-C3N4 SACs and Pt@g-C3N4 SACs from CO2 reduction, respectively. Moreover, Xiong et al. demonstrated that the charge transfer between confined Pt single atoms and ligand g-C3N4 has great effect on photocatalytic reactions. Compared with pristine g-C3N4, Pt2+ single atoms confined in g-C3N4 as catalyst showed higher photocatalytic activity due to its novel electronic structure.49 Further, Su and co-workers reported that high-valence PtII ingle atoms confined in g-C3N4 can modulate the valence band structure and lower the valence band maximum level of the semiconductor catalyst to tune photocatalytic water splitting performance of pristine g-C3N4.50 This work proposed a new way for preparing high efficient photocatalysts by modifying the valence band structure of that. In brief, noble metal single atoms confined in g-C3N4 are a very practical and effective way to improve the photocatalytic performance.
Although the above reports have greatly enhanced our understanding on monolayer g-C3N4 as the structure hosts for NM1@g-C3N4 SACs, a comprehensive understanding of confinement effects on NM1@g-C3N4 SACs for photocatalysis is still missing. In this work, based on DFT calculations, a systematic study on NM1@g-C3N4 SACs will be conducted for the first time to reveal the single-atom confinement effects of noble metals on g-C3N4 for photocatalysis. The results will help in obtaining a better understanding of confinement effects of NM1@g-C3N4 SACs and a new clue of designing many novel photocatalysts with high catalytic activity, stability and selectivity.
2. Computational methods
In this work, all calculations were conducted with Cambridge serial total energy package (CASTEP) module based on DFT.51 The interaction between valence electrons and the ionic core was described by using the ultra-soft pseudo-potential.52 The form of the Perdew–Burke–Ernzerhof (PBE) functional at the generalized gradient approximation (GGA) level was employed as the exchange–correlation functional.53 A plane wave basis set were used to describe electron wave functions with the cut-off energy of 500 eV. The empirical correction method (DFT-D) from the scheme of Tkatchenko and Scheffler (TS) was employed to describe the van der Waals interactions.54 The irreducible brillouin zone was sampled by Monkhorst–Pack k-point meshes of 3 × 3 × 1 and 5 × 5 × 1 during geometry optimization and electronic properties calculations in the reciprocal space, respectively. The lattice parameters and the positions of atoms were relaxed until the maximum force, the maximum stress, the maximum displacement, maximal energy change and the self-consistent field tolerance were less than 0.03 eV Å−1, 0.05 GPa, 1 × 10−3 Å, 1 × 10−5 eV per atom and 1 × 10−6 eV per atom, respectively. In this paper, the calculated band gap value of pristine monolayer g-C3N4 (1.16 eV) was far less than the experimental result reported previously (2.70 eV) by Wang and his co-worker,42 due to the well-known limitations of the GGA method in describing the electronic structures of the materials. Furthermore, since g-C3N4 is a covalent system, the band gap correction of g-C3N4 is failed using the GGA + U method. However, the hybrid approach which can give the correct band gap is not suitable to describe widely the electronic stricture of g-C3N4 system due to its huge time cost.55 In this work, although the band gap value of the system is underestimated, the calculated results can still be used to accurately describe the photocatalytic performance of the system.To study the stability of NM atoms confined in g-C3N4, the binding energy was computed by the following formula.
| Eb = ENM1@g-C3N4 − Eg-C3N4 − ENM1 | (1) |
3. Results and discussion
3.1. Optimized structure of pristine monolayer g-C3N4
The tri-s-triazine-based (heptazine) g-C3N4 with perfect layered structures was chosen due to its excellent thermal stability among all the allotropes of g-C3N4.56 The bulk crystal structure of g-C3N4 was optimized based on the calculation parameters provided in Section 2. The calculation results are as follows: a = b = 7.127 Å, c = 6.289 Å, α = β = 90° and γ = 120.001°, which are very consistent with previous calculation results,57,58 and indicates that the computational methods are effective and reliable in this paper.Further, the monolayer g-C3N4 was constructed by cutting the bulk crystal structure of g-C3N4 along the (001) plane. The 2 × 2 × 1 g-C3N4 supercell consisting of 32 N atoms and 24 C atoms were used in this study. Meanwhile, the vacuum space was built to be 15 Å along z direction, which was enough to avoid periodic interactions between repeated slabs. Based on the computational methods provided in Section 2, the slab model of the monolayer g-C3N4 was optimized. The optimized structure of 2 × 2 × 1 g-C3N4 supercell, whose calculated lattice constant was 7.13 Å, as shown in Fig. 1(a). The calculated results were in good agreement with the previous theoretical59,60 and experimental61 results. As can be seen in Fig. 1(a), the 2 × 2 × 1 g-C3N4 supercell consists of four heptazine structural motifs (C6N7) that are connected to each other by bridge N atoms labelled as N3. Obviously, a large N-coordinating cavity (six-fold cavity) was formed by six unsaturated N atoms of four heptazine structural motifs. In each of these heptazine structural motifs, there were two classes of non-equivalent C atoms and N atoms, respectively, which were labelled as C1, C2, N1 and N2, as shown in Fig. 1(a).
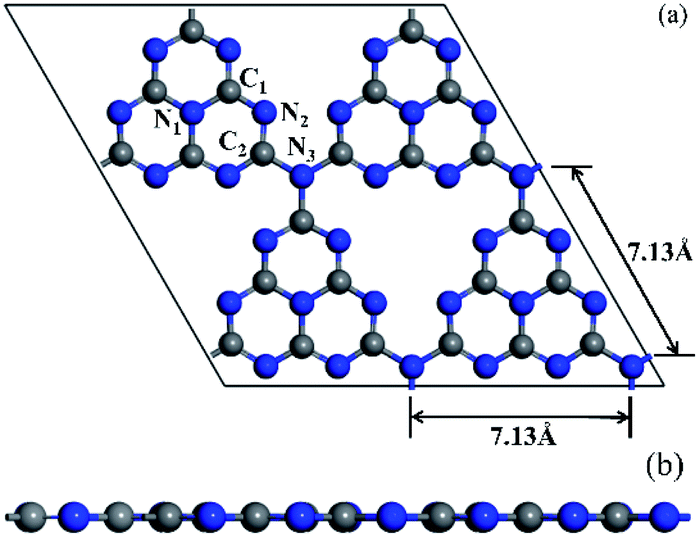 | ||
| Fig. 1 Top view (a) and side view (b) of optimized structure for 2 × 2 × 1 g-C3N4 supercell. The balls in gray and blue represent the C and N atoms, repectively. Roman numbers labeled on the C and N atoms represent different sites, repectively. | ||
The calculated bond lengths of C1–N1, C1–N2, C2–N2 and C2–N3 are 1.39 Å, 1.33 Å, 1.34 Å and 1.47 Å, respectively, are in good agreement with the results of the published literature.59,62 Furthermore, it is evident from Fig. 1(b) that the optimized structure of monolayer pristine g-C3N4 is a stable planar configuration.
3.2. Geometric structures of NM single atoms confined in monolayer g-C3N4
In order to find the most favourable adsorption configuration for NM single atoms confined in pristine g-C3N4, all kinds of adsorption sites around the six-fold cavity were considered on the 2 × 2 × 1 g-C3N4 supercell, as shown in Fig. 2. Each NM single atom species was placed at the S1–S7 sites, respectively, forming 56 kinds of NM1@g-C3N4 configurations in total. The binding energies were obtained when these configurations were fully relaxed, as shown in Table 1. As can be seen from this table, the maximum binding energies are −5.42 eV, −4.08 eV, −3.55 eV, −1.29 eV, −5.17 eV, −3.57 eV, −3.60 eV and −0.001 eV for Ru, Rh, Pd, Ag, Os, Ir, Pt and Au single atom at adsorption site S2, S2, S2, S1, S2, S2, S2, and S1, respectively. Here, the binding energies were negative, which represented exothermic processes. These results indicate that the site S2, S2, S2, S1, S2, S2, S2, and S1 in the six-fold cavity are energetically favourable for Ru, Rh, Pd, Ag, Os, Ir, Pt and Au single atoms confined in g-C3N4. However, the Au1@g-C3N4 with positive Eb values (Eb ≈ 0 eV for site S1, 0.15 eV < Eb < 0.44 eV for other sites) at all sites is thermodynamic instability. Therefore, it is difficult for Au single atom to be introduced into the monolayer g-C3N4 to prepare Au1@g-C3N4 catalysts. Further, as can be found, the binding energy decreased with increasing atomic number and met 4d > 5d trend, as shown in Fig. 3(a). Meanwhile, the average bond lengths between noble metal atoms and N atoms for eight kinds of noble metal atoms at the different adsorption sites were also obtained, as listed in Table 2. From this table, it had the shortest NM–N bond lengths for eight kinds of noble metal atoms at the site S1 or S2 in the six-fold cavity. These sites where the shortest bond lengths occurred correspond exactly to the sites where the maximum binding energies occurred. The results suggest that the shorter the bond length is, the greater the binding energy is. For instance, the Ru–N bond length of 1.89 Å is shortest with a maximum binding energy of −5.42 eV, while the Au–N bond length of 2.42 Å is longest with a minimum binding energy of −0.001 eV. As opposed to the behaviour of the bond lengths and the binding energies, the bond length increased with increasing atomic number and meets 4d < 5d trend, as shown in Fig. 3(b).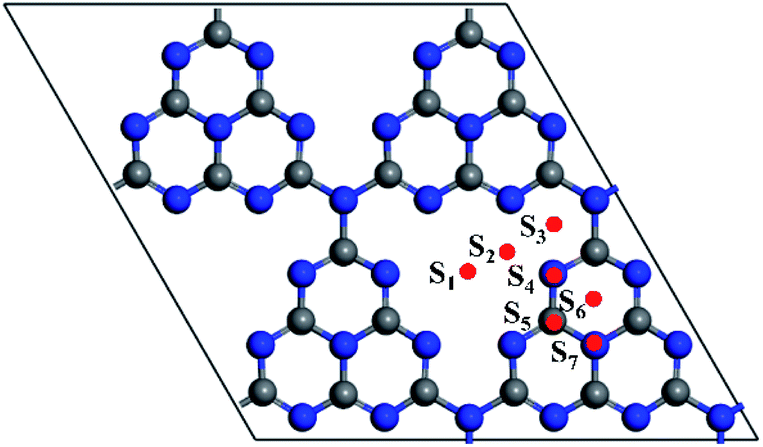 | ||
| Fig. 2 The possible deposition sites for NM atoms. Position: S1, centre of the six-fold cavity; S2, corner of the six-fold cavity; S3, top of the five-membered ring; S4, top of N atom; S5, top of C atom; S6, top of the six-membered ring; S7, top of three six-membered rings. The balls in gray and blue represent the C and N atoms, repectively. | ||
| Sites | Eb for NM1@g-C3N4/eV | |||||||
|---|---|---|---|---|---|---|---|---|
| Ru | Rh | Pd | Ag | Os | Ir | Pt | Au | |
| a Moved to S1.b Moved to S2.c Moved to S6. | ||||||||
| S1 | b | b | b | −1.29 | −3.28 | b | b | −0.001 |
| S2 | −5.42 | −4.08 | −3.55 | a | −5.17 | −3.57 | −3.60 | a |
| S3 | −4.19 | −2.32 | b | 0.12 | −3.71 | 0.65 | b | 0.43 |
| S4 | −4.47 | −1.89 | −0.81 | a | 0.11 | 1.17 | −0.61 | 0.44 |
| S5 | b | −0.68 | −2.49 | 0.27 | −1.52 | 0.86 | −0.66 | 0.39 |
| S6 | −2.97 | 0.22 | −0.12 | 0.30 | −0.19 | 1.31 | −0.13 | 0.15 |
| S7 | c | 1.06 | −0.45 | 0.32 | 1.41 | 2.55 | −0.01 | 0.43 |
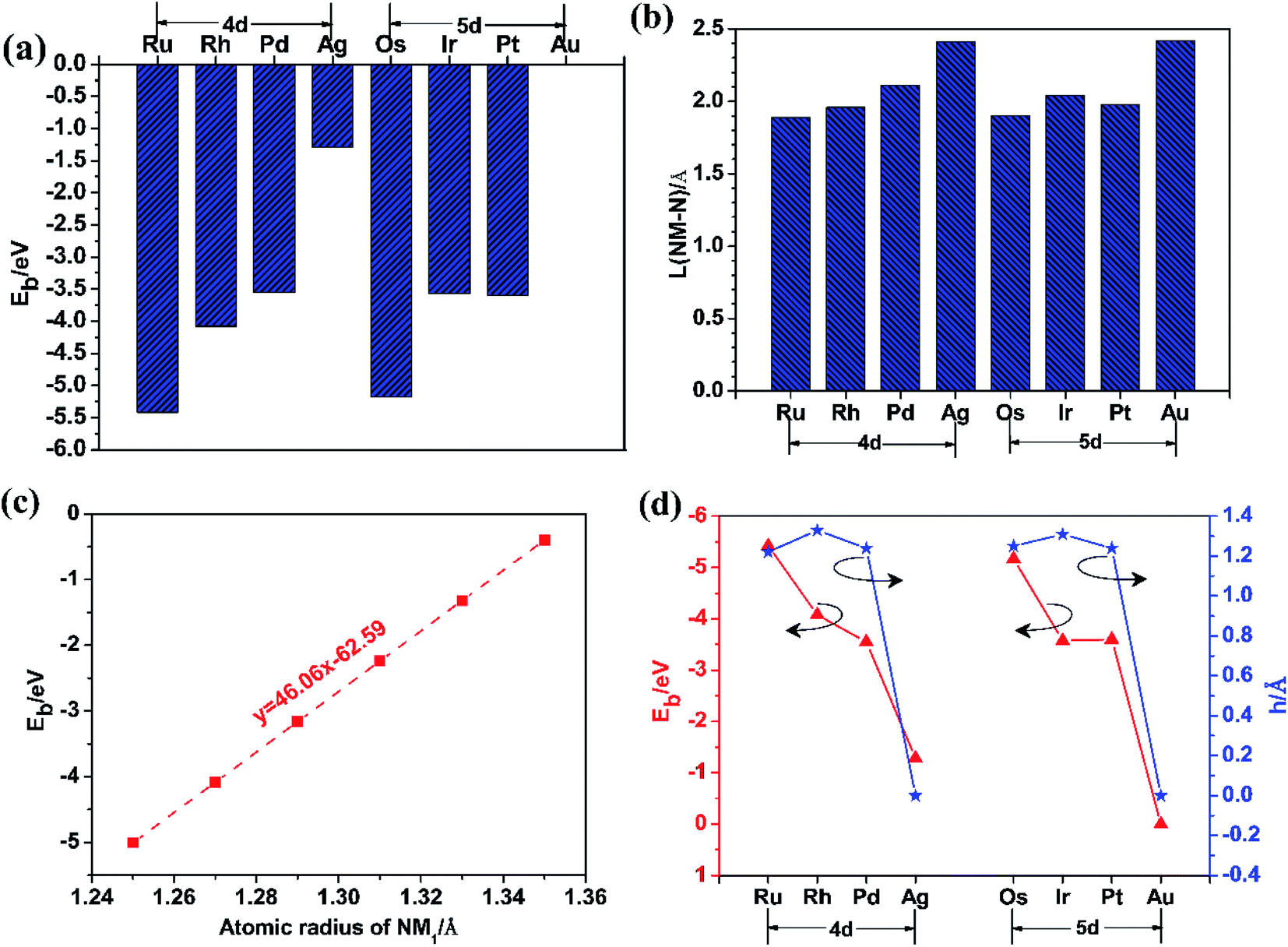 | ||
| Fig. 3 (a) The binding energy (Eb) of NM1; (b) the average bond length (L) between NM1 and N atoms (NM–N); (c) the binding energy (Eb) of NM1 versus the atomic radius of NM1; (d) the binding energy (Eb) of NM1 and the geometric deformation (h). | ||
| Sites | L(NM–N) for NM@g-C3N4/Å | |||||||
|---|---|---|---|---|---|---|---|---|
| Ru | Rh | Pd | Ag | Os | Ir | Pt | Au | |
| a Moved to S1.b Moved to S2.c Moved to S6. | ||||||||
| S1 | b | b | b | 2.41 | 2.28 | b | b | 2.42 |
| S2 | 1.89 | 1.96 | 2.05 | a | 1.90 | 2.01 | 1.98 | a |
| S3 | 1.97 | 2.04 | b | — | 1.97 | 1.99 | b | — |
| S4 | 1.92 | 2.05 | 2.10 | a | 1.91 | 2.04 | 2.04 | — |
| S5 | b | 2.12 | 2.28 | — | 1.95 | 2.09 | 2.25 | — |
| S6 | 2.63 | 2.23 | — | — | 2.03 | 2.19 | 2.37 | — |
| S7 | c | 2.01 | 2.10 | — | 1.99 | 2.04 | 2.00 | — |
More interestingly, the binding energy for eight kinds of noble metal atoms at the site S1 or S2 in the six-fold cavity linearly decreased with increasing atomic radius of NM1, as shown in Fig. 3(c). This indicates that the larger the atomic radius of NM1 is, the weaker the confinement effect of the six-fold cavity for NM1 is. This is because as the radius of NM1 increase, the confinement configurations become more and more unstable. Moreover, just as shown in Fig. S2 in the ESI,† the introduction of NM1 resulted in the deformation of pristine g-C3N4 from a flat shape to a wrinkled shape. This deformation can be characterized on the basis of the thickness (h) of the slab NM1@g-C3N4 SACs, as Fig. 3(d) and S2 in the ESI.† Even more importantly, as shown in Fig. 3(d), the deformation quantity (h) and the binding energy decreased with increasing atomic number, and followed the same variation trend on 4d and 5d. The results indicate that the larger the deformation is in each period, the larger the binding energy is; on the contrary, the smaller the deformation is, the smaller the binding strength is. This may be due to the fact that the single atoms are more easily anchored in the deformed wrinkle space than in the flat structure of g-C3N4. The deformed wrinkle space provided an extremely important confined environment for the single atoms. In other words, these results confirm the importance of wrinkle for NM1@g-C3N4 SACs.
In brief, it can be suggested from the above analysis of energy and spatial configuration that the most favourable adsorption sites are S2, S2, S2, S1, S2, S2, S2, and S1 for single Ru, Rh, Pd, Ag, Os, Ir, Pt and Au atom confined in pristine g-C3N4 catalysts, respectively. In other words, the most favourable confined sites for NM single atoms are located in the six-fold cavity, while Au single atom was hard to confine in g-C3N4 due to its small binding energy in the monolayer g-C3N4. Moreover, in order to clearly show the most favourable confined configurations of NM1@g-C3N4, the corresponding top view and side view of that are displayed in Fig. S1 and S2 in the ESI,† respectively.
3.3. Electronic structures of NM single atoms confined in monolayer g-C3N4
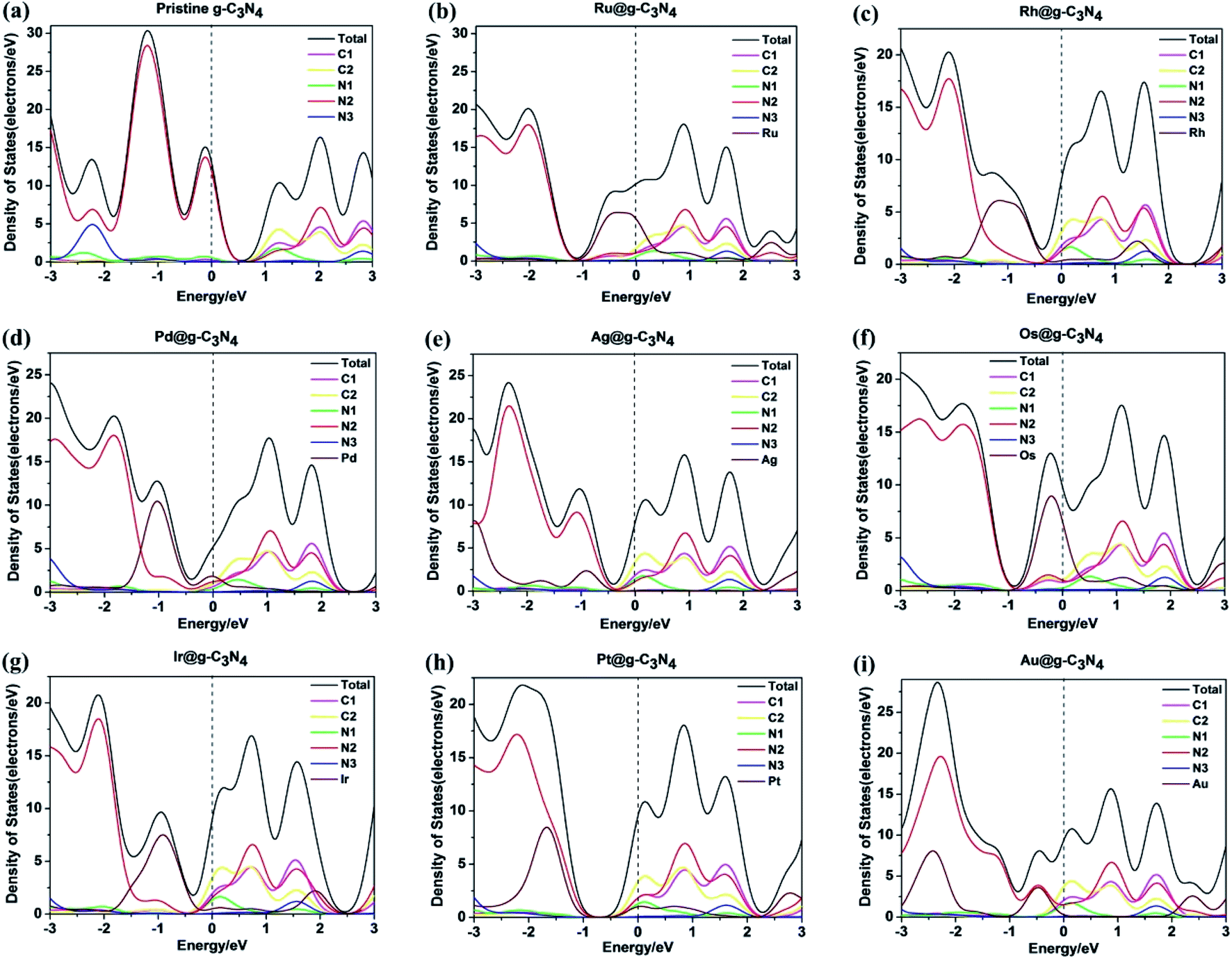 | ||
| Fig. 4 Density of states for (a) pristine g-C3N4 and NM1@g-C3N4, NM1 = (b) Ru1, (c) Rh1, (d) Pd1, (e) Ag1, (f) Os1, (g) Ir1, (h) Pt1, (i) Au1. The Fermi energy is set to zero. | ||
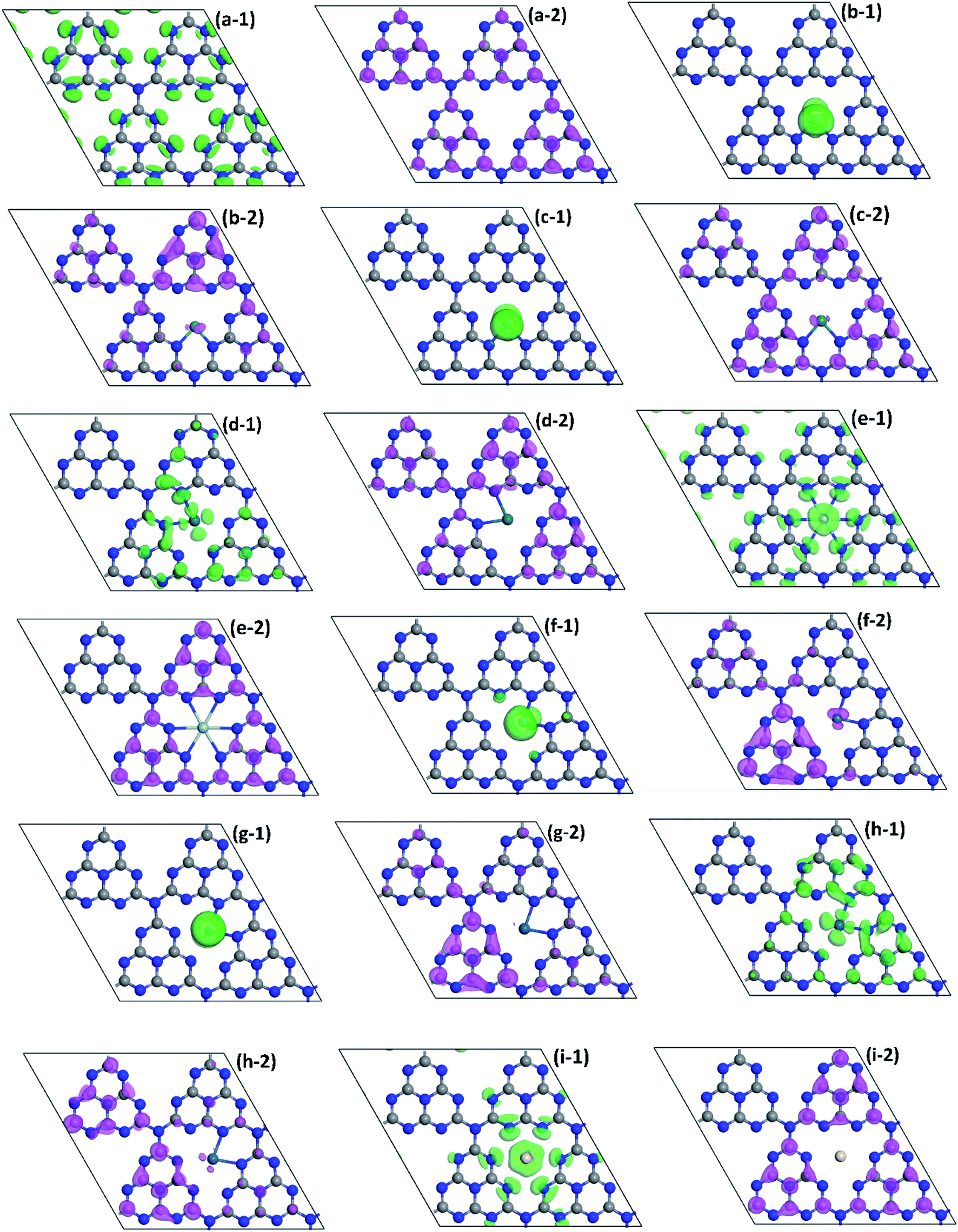 | ||
| Fig. 5 HOMO (green part) of (a-1) pristine g-C3N4 and NM1@g-C3N4, NM1 = (b-1) Ru1, (c-1) Rh1, (d-1) Pd1, (e-1) Ag1, (f-1) Os1, (g-1) Ir1, (h-1) Pt1, (i-1) Au1; LUMO (purple part) of (a-2) pristine g-C3N4 and NM1@g-C3N4, NM1 = (b-2) Ru1, (c-2) Rh1, (d-2) Pd1, (e-2) Ag1, (f-2) Os1, (g-2) Ir1, (h-2) Pt1, (i-2) Au1. The isovalue is set as 0.05 e Å−3. The balls in gray and blue represent the C and N atoms, repectively. | ||
As shown in Fig. 4(b)–(i), after introducing noble metal single atoms into the monolayer g-C3N4, the DOS of NM1@g-C3N4 had been changed greatly compared with that of the pristine g-C3N4 in Fig. 4(a). For all NM1@g-C3N4 systems, noble metal single atoms participated mainly in the constitution of VB, while the compositions of CB were almost unchanged compared with the pristine g-C3N4. Significantly, only a handful of noble metal single atoms participated in the constitution of CB. In particular, as for Ru1@, Rh1@, Pd1@, Os1@ and Ir1@g-C3N4, the VB is contributed by noble metal single atoms and a few of N2 atoms. As regards Ag1@, Pt1@ and Au1@g-C3N4, the VB is contributed by the N2 atoms and a part of noble metal single atoms. Most remarkably, the CB moved down and passed through the Fermi level due to the introduction of noble metal single atoms, resulting in narrowing the band gap of the NM1@g-C3N4 systems. This will effectively improve the visible light response of the systems.
Similarly, to better understand the structure of band edges, the corresponding HOMO and LUMO of VBM and CBM of monolayer NM1@g-C3N4 are shown in Fig. 5(b)–(i). As can be seen from these figures, the HOMO and LUMO of all the monolayer NM1@g-C3N4 had been redistributed compared to that of pristine monolayer g-C3N4 in Fig. 5(a). The redistribution of HOMO and LUMO resulted in the variation of the active sites. Meanwhile, the separation of the HOMO and LUMO can effectively promote the photocatalytic performance of 2D materials. More specifically, the distributions of the HOMO and LUMO can be roughly divided into four categories. For monolayer Ru1@, Rh1@, Os1@ and Ir1@g-C3N4, the HOMO mainly covered the noble metal single atoms, while the LUMO covered almost all triazine units except the N2 and N3 atoms. In this case, the non-localized distribution of the HOMO and LUMO can effectively promote the separation of photo-generated electron–hole pairs. Photo-generated carriers, however, cannot move freely in adjacent heptazine units because there is neither the HOMO nor the LUMO on the N3 atoms and the N2 atoms next to the noble metal single atoms, resulting in short carrier lifetime and poor photocatalytic activity. For monolayer Ag1@ and Au1@g-C3N4, the HOMO is distributed on the noble metal single atoms and the N2 atoms, while the LUMO is located on three adjacent heptazine units around Ag and Au single atoms except the N2 atoms. In this instance, the distribution of the HOMO and LUMO are localized mainly in Ag and Au atoms and three adjacent heptazine units around them. From Fig. 5(e) and (i), after introducing Ag and Au single atoms, photo-generated carriers can be transported through Ag and Au single atom channels among three adjacent heptazine units under visible-light irradiation. Thus, the introduction of Ag and Au single atoms will enhance the carrier mobility on g-C3N4 to some extent. For Pd1@g-C3N4, the HOMO mainly covered on the noble metal single atom and two adjacent heptazine units including it, while the LUMO covered almost all triazine units except Au single atoms, the N2 and N3 atoms. For Pt1@g-C3N4, the distributions of the HOMO are similar to that of Pd1@g-C3N4, while the LUMO is distributed on two heptazine units that is not adjacent to Au single atoms. Obviously, for Pd1@g-C3N4 and Pt1@g-C3N4, because of the remarkable delocalized distribution of the HOMO and LUMO, photo-generated electron–hole pairs can be effectively separated. And more notably, as can been found clearly from Fig. 5(d-1) and (h-1), the HOMO is distributed on the N2 atoms next to Pd and Pt atoms. Therefore, it is reasonable to believe that photo-generated carriers can be transported freely between two adjacent heptazine units through the channels formed by C–N–Pd–N–C or C–N–Pt–N–C chains. This will effectively prolong the carrier lifetime and reduce the recombination rate of photo-generated electron–hole pairs, resulting in high photocatalytic activity. These results are in good agreement with the DOS analysis. In a word, based on the above discussions, it can be inferred that Pd1@, Pt1@, Ag1@ and Au1@g-C3N4 may have better photocatalytic activity than Ru1@, Rh1@, Os1@ and Ir1@g-C3N4.
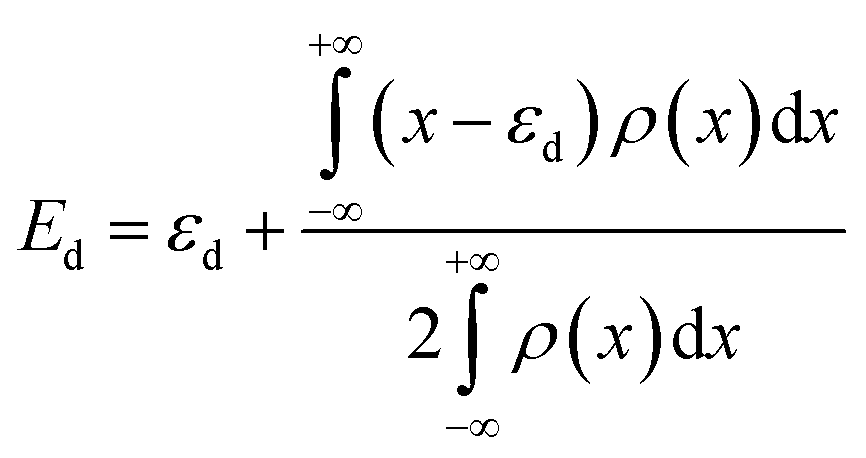 | (2) |
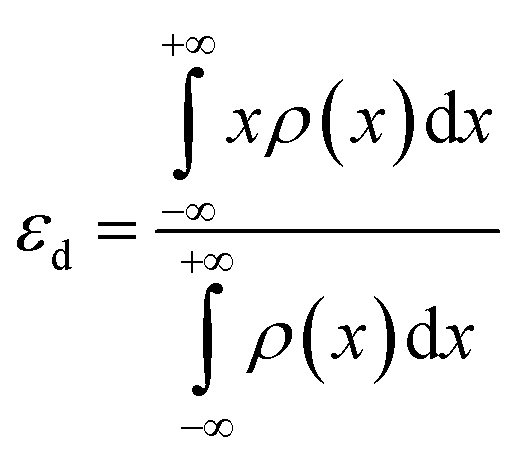 | (3) |
The d-band centres of noble metal single atoms in NM1@g-C3N4 SACs are shown in Fig. 6. As we know, the d-band centres cannot be too large or too small, otherwise it will not match well with the energy levels of the adsorbates. As can be seen from Fig. 6, the d-band centres show similar change tendency for 4d and 5d noble metals. And, the energy curves for 4d and 5d noble metals cross near the d-band centres of Rh1@, Pd1@, Ir1@ and Pt1@g-C3N4 SACs. Meanwhile, the d-band centres of Rh1@, Pd1@, Ir1@ and Pt1@g-C3N4 SACs are located in the middle, while the other d-band centres are either too large or too small. In other words, the energy levels matching between Rh1@, Pd1@, Ir1@ and Pt1@g-C3N4 SACs and the adsorbates are better than that between Ru1@, Ag1@, Os1@ and Au1@g-C3N4 SACs and the adsorbates. Therefore, it can be predicted that Rh1@, Pd1@, Ir1@ and Pt1@g-C3N4 SACs may have better photocatalytic performance than Ru1@, Ag1@, Os1@ and Au1@g-C3N4 SACs.
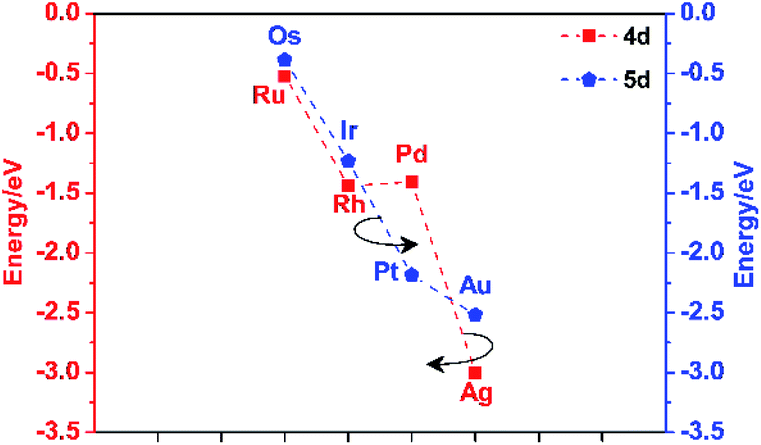 | ||
| Fig. 6 The d-band centres of noble metal single atoms in NM1@g-C3N4 SACs. | ||
Combined with the above discussions, it is reasonable to believe that Pd1@g-C3N4 and Pt1@g-C3N4 SACs may have more excellent photocatalytic activity than the other NM1@g-C3N4 (NM = Ru, Rh, Ag, Os, Ir, Au) SACs.
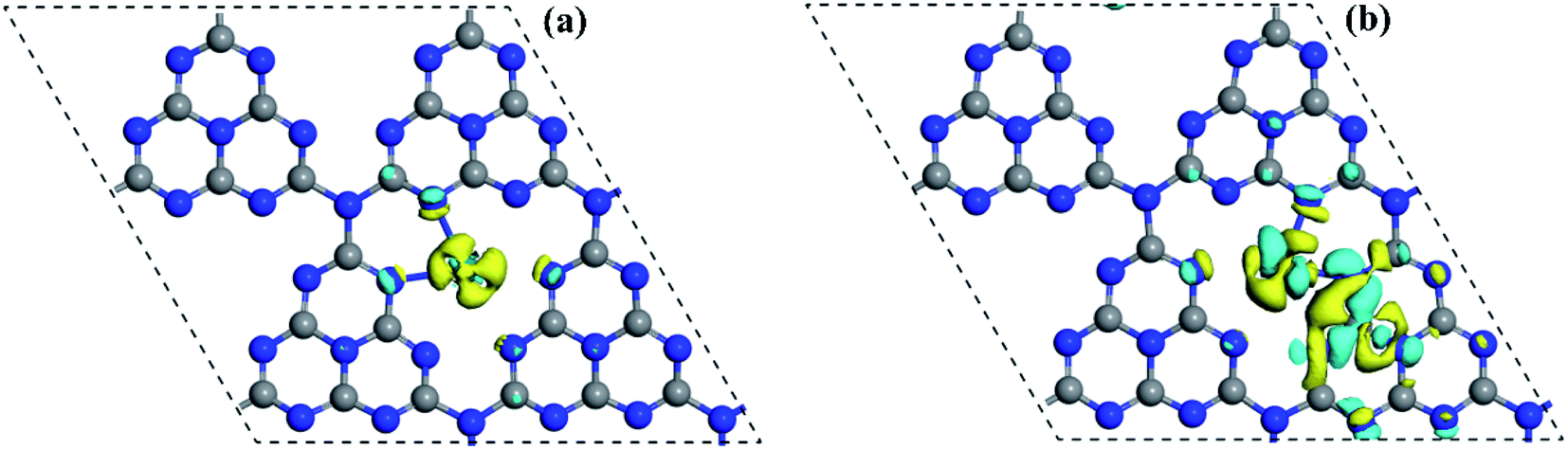 | ||
| Fig. 7 Plots of 3D charge density difference of (a) Pd1@g-C3N4 and (b) Pt1@g-C3N4 at isosurfaces of ±0.05 e Å−3. The blue and yellow colours represent charge accumulation and depletion, respectively. The balls in gray and blue represent the C and N atoms, repectively. | ||
| Species | Mulliken charges | Bond population |
|---|---|---|
| Ru | +0.71 | 0.23 |
| Rh | +0.69 | 0.25 |
| Pd | +0.44 | 0.18 |
| Ag | +0.31 | 0 |
| Os | +0.60 | 0.34 |
| Ir | +0.52 | 0.33 |
| Pt | +0.31 | 0.25 |
| Au | +0.25 | 0.01 |
In brief, based on all the above discussion, it is reasonable to believe that Pt1@g-C3N4 SACs may have higher photocatalytic activity than the other NM1@g-C3N4 SACs under the same conditions, and can be considered as one of the most promising NM1@g-C3N4 SACs.
4. Conclusions
In summary, the geometric structures and electronic structures of pristine monolayer g-C3N4 and eight types SACs of the noble metal single atom confined in g-C3N4 were studied systematically by using DFT calculations. The calculation results of the geometric structures show that the most favourable confined sites for NM single atoms are located in the six-fold cavity. Remarkably, Au single atom is difficult to be confine in g-C3N4 due to its small binding energy. At the same time, the deformed wrinkle space of g-C3N4 contributed to the stability of the noble metal single atom in the six-fold cavity of g-C3N4. The “confining” not only means that the noble metal single atom is constrained by the coordination environment of g-C3N4, but also means that new electronic state are formed due to the strong electron interaction between the noble metal single atom and g-C3N4, so as to realize the modulation of the photocatalytic activity. After introducing the confined noble metal single atoms into the monolayer g-C3N4, the CB of the systems moved down and crossed through the Fermi level, causing the band gap of the NM1@g-C3N4 systems to be narrowed, thus increasing its response to visible light. Meanwhile, photo-generated electron–hole pairs can be effectively separated for NM1@g-C3N4 due to the remarkable delocalized distribution of the HOMO and LUMO. Moreover, after introducing the confined Ag and Au single atoms, photo-generated carriers can be transported through Ag and Au single atom channels among three adjacent heptazine units under visible-light irradiation. And, for Pd1@g-C3N4 and Pt1@g-C3N4 SACs, photo-generated carriers can be transported freely between two adjacent heptazine units through the channels formed by C–N–Pd–N–C or C–N–Pt–N–C chains, thus prolonging the lifetime of photo-generated carriers. Especially, Pt1@g-C3N4 single-atom catalysts are considered to have higher photocatalytic activity than the other NM1@g-C3N4 single-atom catalysts. These results provide a new insight into understand single-atom confinement effects of noble metals on monolayer g-C3N4 for photocatalytic applications, and a new idea for preparing high-efficiency photocatalysts.Author contributions
Cheng Yang: investigation, data curation, writing-original draft; Zong-Yan Zhao: methodology, software; Qing-Ju Liu: supervision, conceptualization, methodology, software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 51562038), Yunnan Yunling Scholars Project and the Key Project of Natural Science Foundation of Yunnan (2018FY001(-011)), and Scientific Research Foundation of Yunnan Provincial Education Department (2020J0489).Notes and references
- P. M. Yeletsky, O. O. Zaikina, G. A. Sosnin, R. G. Kukushkin and V. A. Yakovlev, Fuel Process. Technol., 2020, 199, 106239 CrossRef CAS.
- J. Ren, J.-P. Cao, F.-L. Yang, X.-Y. Zhao, W. Tang, X. Cui, Q. Chen and X.-Y. Wei, Energy Convers. Manage., 2019, 183, 182–192 CrossRef CAS.
- Y. Zhao, M. Que, J. Chen and C. Yang, J. Mater. Chem. C, 2020, 8, 16258–16281 RSC.
- J. Yu, S. Chang, X. Xu, X. He and C. Zhang, J. Mater. Chem. C, 2020, 8, 8887–8895 RSC.
- X. Liu, L. Zhu, X. Wang and X. Meng, Environ. Sci. Pollut. Res., 2020, 27, 13590–13598 CrossRef CAS.
- H. Çağlar Yılmaz, E. Akgeyik, S. Bougarrani, M. El Azzouzi and S. Erdemoğlu, J. Dispersion Sci. Technol., 2020, 41, 414–425 CrossRef.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189 CrossRef CAS.
- R. Lu, L. He, Y. Wang, X.-Q. Gao and W.-C. Li, Chin. J. Catal., 2020, 41, 350–356 CrossRef CAS.
- J.-H. Wu, F.-Q. Shao, X.-Q. Luo, H.-J. Xu and A.-J. Wang, Appl. Surf. Sci., 2019, 471, 935–942 CrossRef CAS.
- K. Negi, A. Umar, M. S. Chauhan and M. S. Akhtar, Ceram. Int., 2019, 45, 20509–20517 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS.
- F. Chen, X. Jiang, L. Zhang, R. Lang and B. Qiao, Chin. J. Catal., 2018, 39, 893–898 CrossRef CAS.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., 2016, 128 Search PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544 CAS.
- J. Zhang, X. Wu, W.-C. M. Cheong, W. Chen, R. Lin, J. Li, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 1002 CrossRef.
- G. Liu, A. Robertson, M. Li, W. Kuo, M. Darby, M. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. Warner and S. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, L. Junjie, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- M. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS.
- X. Sun, J. Lin, Y. Zhou, L. Li, Y. Su, X. Wang and T. Zhang, AIChE J., 2017, 63, 4022–4031 CrossRef CAS.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS.
- M. Zhou, M. Yang, X. Yang, X. Zhao, L. Sun, W. Deng, A. Wang, J. Li and T. Zhang, Chin. J. Catal., 2020, 41, 524–532 CrossRef CAS.
- T. Len, C. Dessal, F. Morfin, J. L. Rousset, P. Afanasiev and L. Piccolo, Presented in Part at the 25ièmes journées du Groupe Français de Spectroscopie Vibrationnelle (GFSV), Ecully, France, 2019-05-13, 2019 Search PubMed.
- H. Zhang, S. Sui, X. Zheng, R. Cao and P. Zhang, Appl. Catal., B, 2019, 257, 117878 CrossRef CAS.
- R. Huang, Y. Cheng, Y. Ji and R. J. Gorte, Nanomaterials, 2020, 10 Search PubMed.
- B. Long, Y. Tang and J. Li, Nano Res., 2016, 9, 3868–3880 CrossRef CAS.
- Y. Wang, X. Zhang, C. Cheng and Z. Yang, Appl. Surf. Sci., 2018, 453, 159–165 CrossRef CAS.
- Q. Li, Z. Ma, R. Sa, H. Adidharma, K. A. M. Gasem, A. G. Russell, M. Fan and K. Wu, J. Mater. Chem. A, 2017, 5, 14658–14672 RSC.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS.
- H. Xiong and F. Yang, Opt. Express, 2020, 28, 5306–5316 CrossRef.
- V. Dutta, P. Singh, P. Shandilya, S. Sharma, P. Raizada, A. K. Saini, V. K. Gupta, A. Hosseini-Bandegharaei, S. Agarwal and A. Rahmani-Sani, J. Environ. Chem. Eng., 2019, 7, 103132 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS.
- Y. Wang, Z. Rong, Y. Wang, T. Wang, Q. Du, Y. Wang and J. Qu, ACS Sustainable Chem. Eng., 2017, 5, 1538–1548 CrossRef CAS.
- Y. Wang, Z. Rong, Y. Wang and J. Qu, J. Catal., 2016, 333, 8–16 CrossRef CAS.
- Y. Tu, P. Ren, D. Deng and X. Bao, Nano Energy, 2018, 52, 494–500 CrossRef CAS.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- T. Xu, D. Wang, L. Dong, H. Shen, W. Lu and W. Chen, Appl. Catal., B, 2019, 244, 96–106 CrossRef CAS.
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS.
- J. Pan, Z. Dong, B. Wang, Z. Jiang, C. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, Appl. Catal., B, 2019, 242, 92–99 CrossRef CAS.
- Y. Huo, J. Zhang, K. Dai, Q. Li, J. Lv, G. Zhu and C. Liang, Appl. Catal., B, 2019, 241, 528–538 CrossRef CAS.
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song and J. Zhu, Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS.
- H. Su, W. Che, F. Tang, W. Cheng, X. Zhao, H. Zhang and Q. Liu, J. Phys. Chem. C, 2018, 122, 21108–21114 CrossRef CAS.
- M. Segall, P. J. Lindan, M. Probert, C. Pickard, P. Hasnip, S. Clark and M. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B, 1990, 41, 7892–7895 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- X. G. Ma, Y. H. Lv, J. Xu, Y. F. Liu and Y. F. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
- H. W. Zuo, C. H. Lu, Y. R. Ren, Y. Li, Y. F. Zhang and W. K. Chen, Acta Phys.-Chim. Sin., 2016, 32, 1183–1190 CAS.
- B. Zhu, J. Zhang, C. Jiang, B. Cheng and J. Yu, Appl. Catal., B, 2017, 207, 27–34 CrossRef CAS.
- J. Cui, S. Liang, X. Wang and J. Zhang, Mater. Chem. Phys., 2015, 161, 194–200 CrossRef CAS.
- J. B. Michael, J. O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177–8182 CrossRef.
- S. M. Aspera, M. David and H. Kasai, Jpn. J. Appl. Phys., 2010, 49, 115703 CrossRef.
- X. Jin, R. Wang, L. Zhang, R. Si, M. Shen, M. Wang, J. Tian and J. Shi, Angew. Chem., 2020, 132, 6894–6898 CrossRef.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- J. Liang, Q. Yu, X. Yang, T. Zhang and J. Li, Nano Res., 2018, 11, 1599–1611 CrossRef CAS.
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09815a |
| This journal is © The Royal Society of Chemistry 2021 |