
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo methods for the preparation of sitagliptin phosphate via chemical resolution and asymmetric hydrogenation†
Fei Ye *,
Zhifeng Zhang,
Wenxia Zhao,
Jianhai Ding,
Yali Wang and
Xueyan Dang
*,
Zhifeng Zhang,
Wenxia Zhao,
Jianhai Ding,
Yali Wang and
Xueyan Dang
School of Chemistry and Chemical Engineering, Ningxia Normal University, Guyuan, 756000, China. E-mail: yefei0310@outlook.com; yefeiqqq@163.com
First published on 25th January 2021
Abstract
Two effective processes have been developed for the preparation of sitagliptin phosphate. The approach of chemical resolution obtained R-sitagliptin in five steps from commercially available starting materials using the inexpensive NaBH4 to reduce the enamine and then using (−)-di-p-toluoyl-L-tartaric acid to resolve racemates in 11% yield overall. The route successfully avoids the use of expensive noble metal as catalysts compared with traditional synthesis methods, resulting in greatly reduced costs and simplified synthetic routes. Other alternative asymmetric hydrogenation of β-ketomide routes for the synthesis of sitagliptin were found, two of the intermediates were synthesized for the first time.
Introduction
Type 2 diabetes1–4 mellitus is a commonly and rapidly growing disease, with millions of new cases reported annually worldwide. Diabetes has become a serious threat to human health and brings enormously high economic costs, not only to individuals and families, but also to the global health system.5 It is estimated that approximately 5 million people died from diabetes and its complications in 2015, representing one of the leading causes of death globally.6–8 Dipeptidyl peptidase-4 (DPP-4) inhibitors, enzymes that act to degrade and inactivate glucagon-like peptide-1 (GLP-1), are a novel class of anti-hyperglycemic agents for the treatment of Type 2 diabetes mellitus. GLP-1 is an incretin hormone that stimulates insulin secretion and biosynthesis, inhibits glucagon release in a glucose-dependent manner, and also aids gastric emptying and reducing appetite.9–11 Sitagliptin,12–15 which was originally known as MK-0431 under the trade name Januvia, is a chiral beta-amino acid derivative that contains both a trifluorophenyl moiety and a trifluoromethylated triazole,16 and also is the first agent developed in the class of DPP-4 inhibitors approved by the FDA in 2006. Because of its safety, efficiency, and being well-tolerated in various treatment regimens, it has tremendous commercial value (more than 11.4 billion dollars annually since 2018)17 and thus has captured the interest of many pharmaceutical companies and research groups.Although researchers from Merck have reported three generations of processes for the preparation of sitagliptin on a large scale, it does not diminish the interest of other research groups in sitagliptin. The key problem with the synthesis of sitagliptin is how to install optically pure chiral central, β-amino acid. In Merck's first-generation synthesis process (Scheme 1),18 starting from achiral β-keto ester 2, chirality was introduced in form of a hydroxyl group in β-hydroxy acid 3 through a ruthenium-catalyzed asymmetric hydrogenation.19–21 The hydroxyl group of 3 was transformed into protected amino acid 5 through several steps, followed by direct coupling to the triazolopiperazine 6 afforded sitagliptin 1 after cleavage of the N-benzyloxy group and salt formation. Despite the overall yield being quite high at 52%, multiple steps and isolations resulted in large amounts of waste and poor atom-economy.
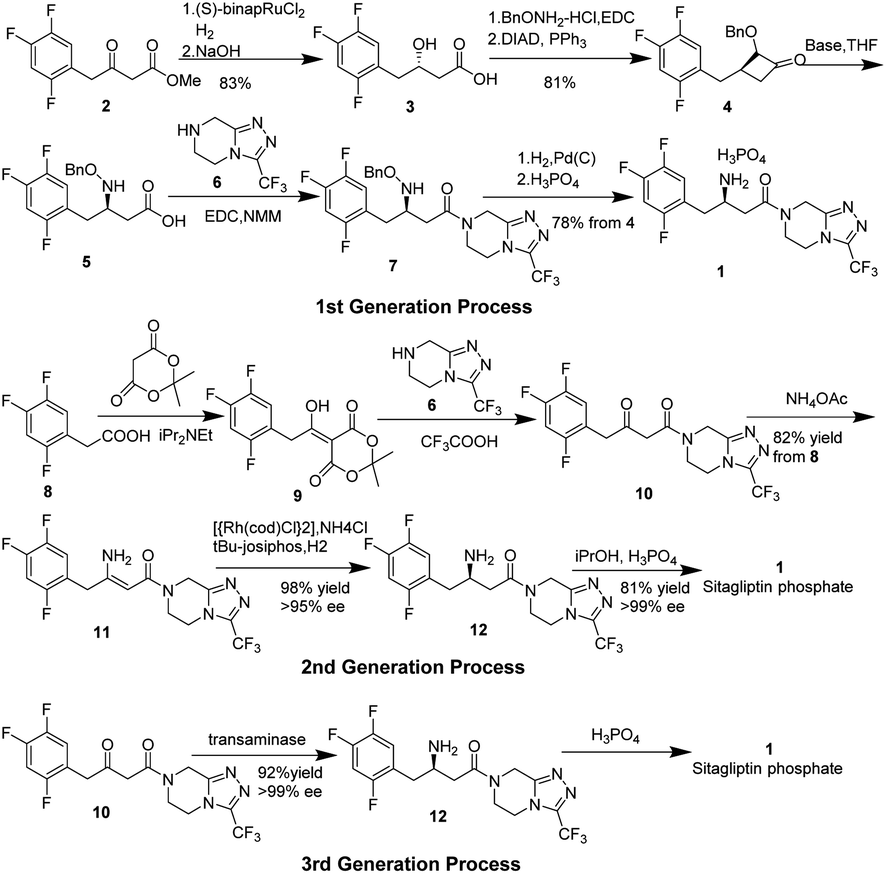 | ||
| Scheme 1 Merck's three generations synthesis processes. | ||
In their second-generation process,22 dehydrositagliptin 1123 was firstly synthesized by a three-step one-pot process starting from trifluorophenyl acetic acid 8, which contained within its structure the entire carbon skeleton of 1. Crystallization and a simple filtration could give 11 in 82% overall yield with 99 wt% purity. The researchers then used the rhodium complex of t-Bu JOSIPHOS to asymmetrically hydrogenate the unprotected enamine amide 11 to install the chiral amine center into 12 in 98% conversion along with up to 95% ee.23,24 Finally, sitagliptin 1 was recrystallized and isolated as its phosphate monohydrate salt in 84% yield with an upgrade in enantiomeric excess up to 99% ee.25 Although this 2nd generation process for the sitagliptin manufacture has led to a significant reduction in waste as compared to the 1st generation process, its endgame leftover room for improvement because of the inherent drawbacks of utilizing an expensive transition metal mediated hydrogenation step. This process needs the specialized high-pressure equipment and a process for the complete removal of the precious metal from the product stream, both of which were significant cost drivers.
To circumvent these drawbacks, Merk and Codexis researchers have recently developed a highly efficient biocatalytic process (the 3rd generation process)26 to prepare sitagliptin 1. In the presence of the R-selective transaminase enzyme, direct amination of prositagliptin ketone to enantiomeric pure sitagliptin 1 (up to 99% ee) was carried out in multipurpose under mild conditions, and thus eliminated all the drawbacks of the 2nd generation process. Not only does the 3rd generation process lead to a reduction of the total waste produced (19%) as compared to the 2nd generation process, but also increase the overall yield (13%) and productivity (53%). This biocatalytic route should become an important auxiliary asymmetric synthesis in a pilot-scale production.
Besides, recently, several synthesis methods of sitagliptin have been reported on laboratory scale, including biocatalytic asymmetric reductive amination of β-ketomide,26,27 enzymatic resolution of racemates,28 chemical kinetic resolution,16,29 and stereoselective reduction of an enamine derivative using NaBH4.30,31 Although some of these synthetic routes are very efficient, given the enormous industrial interest, an inexpensive, convenient, industrially viable, and greener synthetic route is still in urgent need.
In this work, we report for the first time the process of chemical resolution of racemates 13 to sitagliptin 1 followed by reduction of enamine 11 using NaBH4 (Scheme 2) and the process of directly asymmetric hydrogenation of β-ketomide 10 to 14, a chiral intermediate of sitagliptin (Scheme 3).
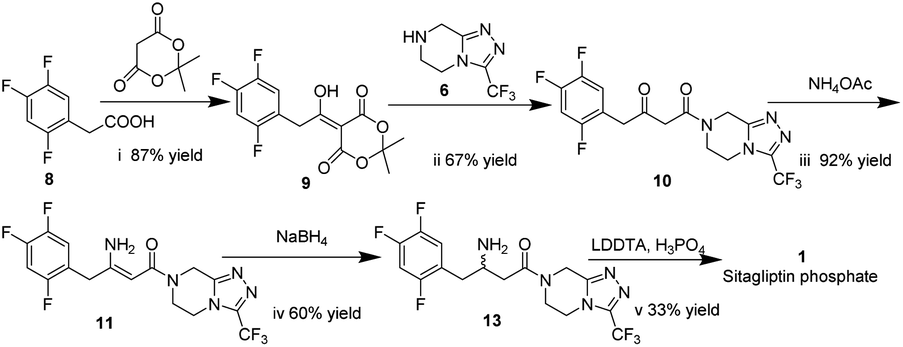 | ||
| Scheme 2 The process for the preparation of sitagliptin phosphate via chemical resolution. | ||
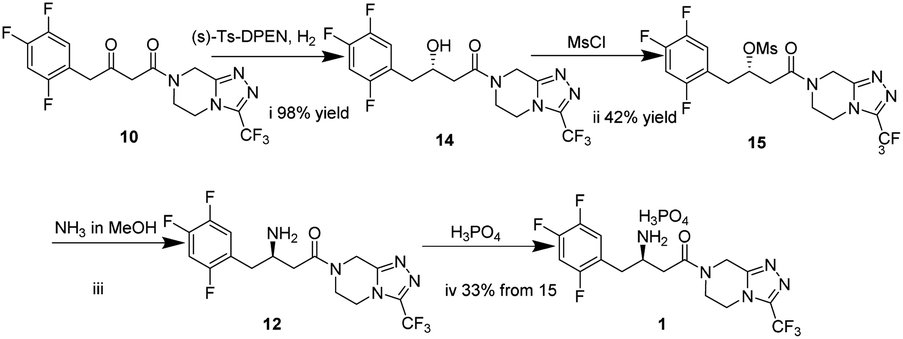 | ||
| Scheme 3 The process for the preparation of sitagliptin phosphate via asymmetric hydrogenation. | ||
Results and discussion
Chemical resolution preparation of sitagliptin 1
Meldrum's adduct 9 was firstly prepared from 2,4,5-trifluorophenyl acetic acid 8 utilizing the ability of Meldrum's acid to act as an acyl anion equivalent at 50 °C for 5 h in 87% yield. This process involves the activation of 2,4,5-trifluorophenyl acetic acid 8, and we chose N,N-carbonyl di-imidazole (CDI)32,33 as active agent instead of pivaloyl chloride34 to simplify the experiment. 9 was then coupled to the 3-(trifluoromethyl)-1,2,4-triazolo-[4,3-a]piperazine hydrochloride 6 using i-Pr2NEt, to provide β-ketomide 10 in 67% yield. Treatment of 10 with AcONH4 and ammonia water results in formation of enamine 11. As the reaction temperature was raised to 58 °C, the desired product 11 began to crystallize from the reaction mixture. Upon to the mild temperature, 11 was isolated as a white crystalline solid through simple filtration in 92% yield with 99 wt% purity. Next the reduction of enamine 11 was performed with NaBH4 at −15 °C in 4.5 h, monitored by HPLC until enamine 11 disappeared. It was found that the reductive reaction of 11 and NaBH4 could not be performed very well. Therefore, several additional chemicals were screened to improve the conversion of the reduction reaction.As indicated in Table 1, these additives play a catalyst role in reaction mixtures, and significantly improve the conversion of the reductive reaction. BF3 diethyl etherate yields the highest conversion (entry 2). However, because of the extreme toxicity of BF3 diethyl etherate, we chose MsOH as the additive of the reductive reaction.
|
|||
|---|---|---|---|
| Entry | Reduction | Additive | Conversion (%) |
| a All reactions were carried out as follows: THF (50 mL) was firstly cooled to −10 °C. NaBH4 (2.33 g, 61.68 mmol) and an additive such as MsOH (5.9 g, 61.68 mmol) were added dropwise at −10 to −5 °C, and then enamine 11 (5.0 g, 12.34 mmol) and isopropanol (30 mL) were added. The reaction mixture was aged at −15 °C for 4.5 h, monitored by HPLC. Isolated yield after ethyl acetate extraction. | |||
| 1 | NaBH4 | None | 0 |
| 2 | NaBH4 | BF3 diethyl etherate | 95 |
| 3 | NaBH4 | MsOH (methanesulfonic acid) | 93 |
| 4 | NaBH4 | Acetic acid | 10 |
| 5 | NaBH4 | TFA (trifluoroacetic acid) | 2 |
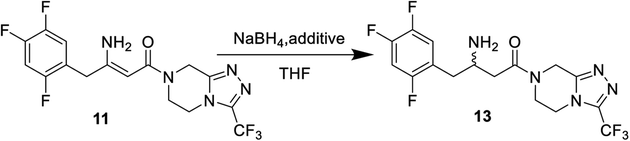
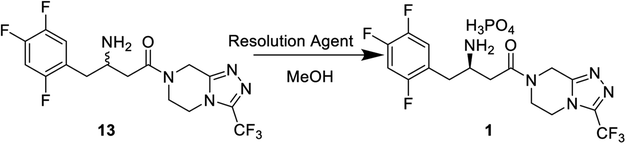
After the reductive reaction, the resolution of racemates 13 was carried out by treatment with di-p-toluoyl-L-tartaric acid in MeOH to form R-sitagliptin 1 in 33% yield with 99 wt% purity and 96% ee. At the same time, we compared the effects of the two L-tartaric acids on the resolution, as shown in Table 2. Lastly, the separated filtrate was treated to obtain S-sitagliptin phosphate with 90% ee.
|
|||
|---|---|---|---|
| Entry | Resolution agent | Solvent | Configuration (ee%) |
| a All reactions were carried out using racemates 13 (0.5 g, 1.23 mmol) and resolution agents such as (−)-di-p-toluoyl-L-tartaric acid (0.24 g, 0.62 mmol) in solvent (15 mL) at 65 °C for 1 h. White sitagliptin tartrate was obtained after filtration. And tartrate was hydrolyzed by using ammonia water (10 mL), and extracted twice by ethyl acetate (30 mL × 2). The organic layer was concentrated and isopropanol (15 mL) was added. Then 85 wt% H3PO4 was added dropwise and the reaction mixture was aged at 78 °C for 1 h to afford sitagliptin 1. | |||
| 1 | (−)-L-Tartaric acid | MeOH | 4% |
| 2 | (−)-Di-p-toluoyl-L-tartaric acid | MeOH | 96% |
Asymmetric hydrogenation of β-ketoester 10 to sitagliptin 1 via three steps
The difficulty of this route (Scheme 3) lies in the asymmetric reduction of 10. The carbonyl could be reduced by the solution of formic acid and Et3N in the presence of a chiral catalyst at room temperature in the literature.35,36 Although the reaction mixture of 10 and formic acid were heated to 80 °C, the reaction did not proceed. The β-ketoester 10 was hydrogenated by hydrogen under 435 psi at 65 °C for 12 h to afford (S)-3-hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-1-(2,4,5-trifluorophenyl)butan-1-one 14. Treatment of 14 with MsCl resulted in (S)-4-oxo-4-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-1-(2,4,5-trifluorophenyl)butan-2-yl methanesulfonate 15 in 42% yield with 98 wt% purity and 7% ee. And then methanesulfonate 15 reacted with NH3 in MeOH under 145 psi to form R-sitagliptin 1 in 33% yield with 99 wt% purity and 7% ee. Although this approach failed to afford adequate results, an alternative route of the synthesis of sitagliptin was developed, 14 and 15 were firstly synthesized as new substrates.Conclusion
In this study, we established two new efficient routes of the synthesis of sitagliptin phosphate. The approach of chemical resolution of racemates 13 to sitagliptin 1 not only successfully obtained R-sitagliptin with up to 96% ee but also afforded S-sitagliptin with 90% ee by using chiral resolution agent, (−)-di-p-toluoyl-L-tartaric acid. Reduction of enamine 11 and chemical resolution of racemates 13 as two key steps, makes the synthetic route simple and practical and greatly reduce the cost.The approach of asymmetric hydrogenation of β-ketoester 10 to 1 using [Ru(cymene)Cl]2 and (s)-Ts-DPEN as a chiral catalyst to directly hydrogenate 10 and reacts with MsCl to form the methanesulfonate 15, a key intermediate, and finally successfully obtained R-sitagliptin 1. Although this approach failed to afford idealized optical purity results, an alternative route of the synthesis of sitagliptin was found, compound 14 and 15 were firstly synthesized as new substrates.
Experimental section
General
All of the reagents and solvents were purchased from commercial sources and used without purification. Melting points were determined on a Reichert micro-hot-stage apparatus. The IR spectra were recorded on an FSM-2201 spectrometer with Fourier transform from samples dispersed in mineral oil. High resolution mass spectra were recorded in ESI mode using magnetic sector mass analyzer and TOF mass spectrometer. 1H and 13C spectra were recorded with a 400 MHz NMR instrument at 295 K. Chemical shifts are reported relative to the residual signals of tetramethylsilane in CDCl3 or deuterated solvent CDCl3/DMSO-d6 for 1H and 13C NMR spectroscopy, respectively. Multiplicities are reported as follows: singlet (s), doublet (d), broad singlet (br s), doublet of doublets (dd), doublet of triplets (dt), triplet (t), quartet (q), and multiplet (m). All of the reactions were monitored by HPLC or TLC. Tetrahydrofuran was distilled over sodium before use.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 heptane/IPAc. The mixture was cooled in an ice-bath and filtered, and then the solid is rinsed with 2:1 heptane/IPAc. After drying, the Meldrum's acid adduct 9 (14.6 g, 99 wt%) was obtained as a white solid in 87% yield. Mp 121–123 °C; ESI-MS (m/z): (317, M+ + H); IR (KBr), v, cm−1: 3448, 3001, 1734, 1653, 1581, 1525, 1428, 1338, 1307, 1203, 1153, 1024, 922, 789. 1H NMR (400 MHz, CDCl3) δ 15.49 (s, 1H), 7.15 (m, 1H), 6.95 (m, 1H), 4.45 (s, 2H), 1.76 (s, 6H).
:1 heptane/IPAc. The mixture was cooled in an ice-bath and filtered, and then the solid is rinsed with 2:1 heptane/IPAc. After drying, the Meldrum's acid adduct 9 (14.6 g, 99 wt%) was obtained as a white solid in 87% yield. Mp 121–123 °C; ESI-MS (m/z): (317, M+ + H); IR (KBr), v, cm−1: 3448, 3001, 1734, 1653, 1581, 1525, 1428, 1338, 1307, 1203, 1153, 1024, 922, 789. 1H NMR (400 MHz, CDCl3) δ 15.49 (s, 1H), 7.15 (m, 1H), 6.95 (m, 1H), 4.45 (s, 2H), 1.76 (s, 6H).Racemates 13
THF (50 mL) was charged into a 250 mL three-neck flask and cooled to −10 °C. NaBH4 (2.33 g, 61.68 mmol) and MsOH (5.9 g, 61.68 mmol) were added dropwise at −10 to −5 °C, and then enamine 11 (5.0 g, 12.34 mmol) and isopropanol (30 mL) were added. The reaction mixture was aged at −15 °C for 4.5 h, monitored by HPLC. When the reaction was finished, water was added and ethyl acetate (60 mL × 2) was extracted twice. The organic layer was dried by MgSO4 and concentrated under diminished pressure. Oil racemates 13 (3.0 g, 78 wt%) was obtained in 60% yield.Sitagliptin 1
Racemates 13 (0.5 g, 1.23 mmol) was dissolved in methanol (15 mL) and solution of (−)-di-p-toluoyl-L-tartaric acid (0.24 g, 0.62 mmol) in isopropanol (15 mL) was added. Then the mixture was heated to 65 °C and stirred for 1 h, and white solids began to precipitate. The reaction mixture was cooled to ambient temperature and aged for an additional 2 h before filtration. The cake was recrystallized by methanol solution in water (1:1). Sitagliptin tartrate was obtained in 96% ee.
Sitagliptin tartrate was directly dissolved in water (30 mL) and the pH of the mixture was adjusted to 10–12 with 28 wt% ammonia water (10 mL), and extracted twice by ethyl acetate (30 mL × 2). The organic layer was concentrated and isopropanol (15 mL) was added. Then 85 wt% H3PO4 was added dropwise and the reaction mixture was aged at 78 °C for 1 h. The slurry wan then cooled to room temperature and aged for an additional 0.5 h before filtration. The wet cake was washed with 15 mL cold ethanol and dried at 40 °C in a vacuum oven to afford sitagliptin 1 (0.21 g, 99 wt%) in 33% yield and 96% ee. Mp 218–220 °C; ESI-MS (m/z): (408, M+ + H); IR (KBr), v, cm−1: 3196, 3027, 2861, 1649, 1521, 1453, 1167, 1093, 850. 1H NMR (400 MHz, DMSO-d6) δ 7.29 (m, 1H), 7.16 (m, 1H), 4.94 (s, 2H), 4.30 (d, J = 17.6 Hz, 1H), 4.27 (d, J = 17.6 Hz, 1H), 4.00 (m, 2H), 3.12 (m, 2H), 2.99–3.08 (m, 1H), 2.92 (dd, J = 17.1 Hz, 1H), 2.87 (m, 1H); 13C NMR (CDCl3) δ 36.2, 38.0, 39.1, 40.0, 40.1, 41.6, 42.4, 43.2, 43.6, 48.5, 105.6, 118.2, 1119, 121.7, 143.7, 146.6, 148.8, 149.7, 150.4, 156.1, 170.2, 170.58.
:1) (150 mL). The eluant was cooled to −10 °C and stirred for 2 h, and white solids precipitated. After an additional 2 h age, the mixture was filtered. The wet cake was washed with ethyl acetate (50 mL) and dried at ambient temperature in a vacuum oven to obtain white crystalline 15 (5.1 g, 98 wt%) in 42% yield and 7% ee. Mp 157–159 °C; ESI-MS (m/z): (487, M+); IR (KBr), v, cm−1: 3044, 2934, 1658, 1511, 1443, 1338, 1172, 1015, 528. 1H NMR (400 MHz, DMSO-d6) δ 7.54–7.44 (m, 2H), 5.19 (m, 1H), 4.98 (t, 1H), 4.92–4.84 (m, 1H), 4.24 (m, 1H), 4.12–4.08 (m, 1H), 3.96–3.94 (m, 2H), 3.09–3.04 (m, 3H), 3.02 (s, 3H), 2.94–2.83 (m, 1H).Sitagliptin 1
(S)-4-Oxo-4-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-1-(2,4,5-trifluorophenyl)butan-2-yl methanesulfonate 15 (3.0 g, 6.17 mmol) and the prepared solution of NH3 in MeOH (13 wt% 60 mL) were charged into the flask. The reaction mixture was heated to 65 °C and stirred for 8 h under 145 psi. MTBE (methyl tert-butyl ether) (50 mL) and 0.5 M H3PO4 (50 mL) were added after concentrated. The pH of the mixture was adjusted to 8–9 with the solution of NaOH. The aqueous layer was separated, and the organic layer was concentrated and isopropanol (40 mL) and water (40 mL) were added. Then 0.5 M H3PO4 (1 mL) was added dropwise and the mixture was heated to 70 °C and stirred for 2 h. The slurry was filtered after cooled to the room temperature. The wet cake was washed with 15 mL isopropanol and dried at 40 °C in a vacuum oven to obtain sitagliptin 1 (1.2 g, 99 wt%) in 33% yield and 7% ee. ESI-MS (m/z): (408, M+ + H); 1H NMR (400 MHz, CDCl3) δ 7.00–7.25 (m, 5H), 4.61–4.91 (m, 2H), 4.31–4.36 (m, 1H), 3.84–3.99 (s, 2H), 2.78–3.82 (m, 2H), 2.66–2.72 (m, 1H), 2.59–2.64 (m, 1H), 2.51 (s, 2H); 13C NMR (D2O) δ: 30.9, 33.7, 38.0, 41.8, 43.1, 48.2, 105.8, 115.4, 118.5, 120.9, 145.5, 148.7, 157.2, 170.2.Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this article.Acknowledgements
This work was partially supported by the Natural Science Foundation for Ningxia Province (Grants no. 2018AAC03235), Liupanshan Resources Engineering Technology Central (Grants no. HGZD19-10), the Research Award Fund for First-class Discipline Construction (Education Discipline) in Higher Education Institutions of Ningxia (Grants no. NXYLXK2017B11), Development of Science and Technology of Ningxia (Grants no. 2019BEB04019).References
- For leading references, see: N. A. Thornberry and A. E. Weber, Curr. Top. Med. Chem., 2007, 7, 557–568 CrossRef CAS.
- S. L. Gwaltney II and J. A. Stafford, Annu. Rep. Med. Chem., 2005, 40, 149–165 Search PubMed.
- B. Ahren, M. Landin-Olsson, P. A. Jansson, M. Svenson, D. Holmes and A. Schweizer, J. Clin. Endocrinol. Metab., 2004, 89, 2078–2084 CrossRef CAS.
- International Diabetes Federation (IDF), Diabetes Atlas, 2019, 9th edn, http://www.idf.org/diabetesatlas Search PubMed.
- See: (a) www.diabetes.org; (b) http://www.who.int.
- E. Adeghate, H. Kalasz, G. Veress and K. Tekes, Curr. Med. Chem., 2010, 17, 517–551 CrossRef CAS.
- E. Adeghate, E. Feher and H. Kalasz, Expert Opin. Invest. Drugs, 2015, 24, 1–15 CrossRef CAS.
- F. Peng, Y. Chen and C. Chen, J. Org. Chem., 2017, 82(17), 9023–9029 CrossRef CAS.
- A. Weber, J. Med. Chem., 2004, 48, 4135–4141 CrossRef.
- D. Drucker, Expert Opin. Invest. Drugs, 2003, 12, 87–100 CrossRef CAS.
- S. A. Miller and E. L. St. Onge, Ann. Pharmacother., 2006, 40, 1336 CrossRef CAS.
- D. Kim, L. Wang, M. Beconi and G. J. Eiermann, J. Med. Chem., 2005, 48, 141–151 CrossRef CAS.
- C. S. Shultz and S. W. Krska, Acc. Chem. Res., 2007, 40, 1320–1326 CrossRef CAS.
- L. H. Xia, Chin. J. New Drugs, 2007, 16, 979–981 CAS.
- R. Pathak and M. B. Bridgeman, Pharmacol. Ther., 2010, 35, 509–513 Search PubMed.
- H. Mei, J. Han, K. D. Klika, K. Izawa, T. Sato, N. A. Meanwell and V. A. Soloshonok, Eur. J. Med. Chem., 2020, 186, 111826 CrossRef CAS.
- S. Matej, F. Rok and G. Stanislav, ACS Omega, 2020, 5(10), 5356–5364 CrossRef.
- K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Palucki, N. R. Rivera, D. Steinhuebel, J. D. Armstrong III, D. Askin and E. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634–639 CrossRef CAS.
- J. Xu, H. O. Ok and E. J. Gonzalez, Bioorg. Med. Chem. Lett., 2004, 14, 4759–4762 CrossRef CAS.
- U. Schollkopf, U. Groth and C. Deng, Angew. Chem., Int. Ed. Engl., 1981, 20, 798–799 CrossRef.
- D. Kim, L. Wang, M. Beconi, G. J. Eiermann and M. H. Fisher, J. Med. Chem., 2005, 48, 141–151 CrossRef CAS.
- K. B. Hansen, H. Yi, F. Xu, N. Rivera, A. Clausen and M. Kubryk, J. Am. Chem. Soc., 2009, 131, 8798–8804 CrossRef CAS.
- F. Xu, J. D. Armstrong, G. X. Zhou and B. Simmons, J. Am. Chem. Soc., 2004, 126, 13002–13009 CrossRef CAS.
- Y. Hsiao, N. R. Rivera, T. Rosner and S. W. Krska, J. Am. Chem. Soc., 2004, 126, 9918–9919 CrossRef CAS.
- A. M. Clausen, B. Dziadul, K. L. Cappuccio and M. Kaba, Org. Process Res. Dev., 2006, 10, 723–726 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz and J. Brands, Science, 2010, 329, 305–309 CrossRef CAS.
- Y. Wei, S. Xia and C. He, Biotechnol. Lett., 2016, 38, 841–846 CrossRef CAS.
- L. L. Zeng, Y. J. Ding, G. C. Zhang, H. R. Song and W. H. Hu, Chin. Chem. Lett., 2009, 20, 1397 CrossRef CAS.
- S. Zhou, J. Wang, X. Chen, J. L. Aceña, V. A. Soloshonok and H. Liu, Angew. Chem., Int. Ed., 2014, 53, 7883–7886 CrossRef CAS.
- K. Lin, Z. Cai and W. Zhou, Synth. Commun., 2013, 43, 3281 CrossRef CAS.
- O. Gutierrz, D. Metil and N. Dwivedi, Org. Lett., 2015, 17, 1742–1745 CrossRef.
- A. J. Blacker, Process for the Preparation of aromatic Amines, WO2004110976, 2004, 09, 06 Search PubMed.
- D. Spencer, I. Norihiro and N. Eugenia, Process to Chiral Beta-amino acid Derivatives, WO2004085661 , 2004, 03, 19.
- Y. Xiao, D. Joseph and W. Shane, Process for the Preparation of Chiral Beta amino Acid Derivatives by Asymmetric Hydrogenation, WO2004085378, 2004, 03, 15 Search PubMed.
- B. H. Lipshutz, K. Noson and W. Chrisman, J. Am. Chem. Soc., 2001, 123, 12917–12918 CrossRef CAS.
- H. Shimizu, I. Nagasaki and K. Matsumura, Acc. Chem. Res., 2007, 40, 1385–1393 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10273c |
| This journal is © The Royal Society of Chemistry 2021 |