
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAppraisal of the greenness profile of a chromatographic method for the simultaneous estimation of carbamazepine and oxcarbazepine, along with two potential impurities and three formulation excipients
Nada S. Abdelwahab ab and
Maha M. Abdelrahman*a
ab and
Maha M. Abdelrahman*a
aPharmaceutical Analytical Chemistry, Faculty of Pharmacy, Beni-Suef University, Alshahed Shehata Ahmed Hegazy St., Beni-Suef, 62514, Egypt. E-mail: maha.abdelrahman@pharm.bsu.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University in Beni-Suef (NUB), Beni-Suef, Egypt
First published on 17th February 2021
Abstract
Structurally related carbamazepine (CBZ) and oxcarbazepine (OX) are two of the most commonly used antipsychotic drugs. The main impurities of CBZ, as described in both the USP and the BP, are iminodibenzyl (IMD) and iminostilbene (IST). Meanwhile, for non-pharmacopeial OX, the declared impurities include CBZ and IST. Prescribed oral suspensions of CBZ and OX contain additives including methyl paraben (MP), propyl paraben (PP) and sorbic acid (SA) as preservatives. An HPTLC method was introduced and developed for resolving the interference between CBZ, OX, their impurities, and the suspension additives in a single run, in addition to their quantitation with a high sensitivity that satisfies the USP requirements for the detection and quantitation of drug impurities. In the developed HPTLC method, CBZ and OX were measured in the range of 40–4000 ng per band, while IMD, IST, MP, PP and SA were in the range of 20–2000 ng per band, using a mixture of hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate:formic acid:acetic acid (8:2:0.5:0.3, by volume) and UV scanning at 254 nm. The greenness profile of the method was evaluated by two different tools, the analytical Eco-Scale and the Green Analytical Procedure Index (GAPI), then a comparison between their results was conducted. This is the first time that the studied drugs, along with their impurities and suspension additives, were analyzed by a HPTLC method in a single run and within the limits required by the USP guidelines.
:ethylacetate:formic acid:acetic acid (8:2:0.5:0.3, by volume) and UV scanning at 254 nm. The greenness profile of the method was evaluated by two different tools, the analytical Eco-Scale and the Green Analytical Procedure Index (GAPI), then a comparison between their results was conducted. This is the first time that the studied drugs, along with their impurities and suspension additives, were analyzed by a HPTLC method in a single run and within the limits required by the USP guidelines.
1. Introduction
Antipsychotics are a class of drugs known as neuroleptics and are used mainly for the management of psychotic disorders like paranoia, hallucinations or delusions, primarily in epilepsy, schizophrenia, and bipolar disorders (along with mood stabilizers).1 Some of the most commonly used antipsychotic drugs are carbamazepine (CBZ) and oxcarbazepine (OX), that are structurally close to tricyclic antidepressants.2Carbamazepine (CBZ) is officially a dibenzazepine derivative with psychotropic properties.2 It is listed in both the British Pharmacopeia (BP)3 and the United States Pharmacopeia (USP).4 Additionally, CBZ has different impurities and related substances3,4 including iminodibenzyl (IMD) and iminostilbene (IST). OX is a CBZ derivative with similar actions.2 OX is a non-official drug that has been found to have different impurities such as CBZ and IST.5 Moreover, CBZ and OX are individually formulated in tablet and suspension dosage forms, marketed as Tegretol® and Trileptal®, respectively. The marketed oral suspensions of CBZ and OX, as labeled by the manufacturer, contain methyl paraben (MP), propyl paraben (PP) and sorbic acid (SA) as preservatives. The chemical structures of all of the studied compounds are given in Fig. 1.
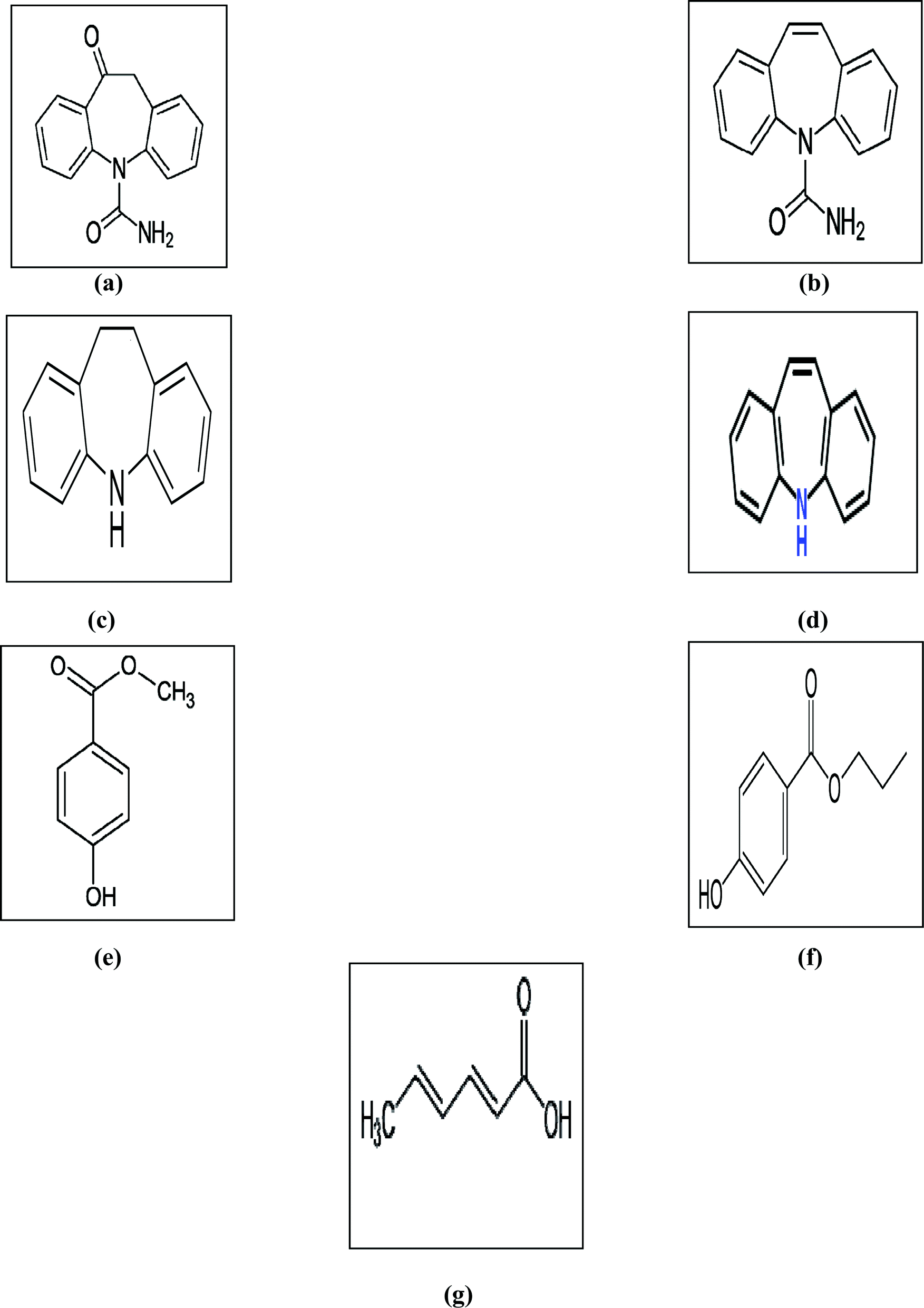 | ||
| Fig. 1 The chemical structures of (a) OX, (b) CBZ, (c) IMD, (d) IST, (e) MP, (f) PP and (g) SA. | ||
Upon reviewing the literature, it was observed that CBZ can be measured in different biological fluids and in tablet dosage form by different methods that were summarized in the review article published by Datar6 in 2015. Recently, it was analyzed in its pharmaceutical formulation by HPLC7,8 and LC-MS9–12 methods. Likewise, CBZ was analyzed by different HPLC methods in the presence of its impurities.13,14 OX was estimated in tablets by TLC-densitometric15 and different HPLC16,17 methods. It was also determined in the presence of its degradation products or related substances by TLC-densitometric,18 HPLC5,19–21 and LC-MS/MS22 methods. Additionally, it was analyzed along with its metabolites by HPLC23,24 and LC-MS/MS25–27 methods. Similarly, different methods including HPLC,28 LC-MS/MS,29,30 and supercritical fluid31 chromatographic methods were used for the determination of both CBZ and OX simultaneously in the presence of their metabolites.
Pharmaceutical safety and efficacy are important matters in drug therapy. It has been reported that the negative effects caused by drug impurities significantly affect drug safety. Therefore, the issues concerning the analytical estimation of impurities in pharmaceuticals are some of the most important subjects in modern pharmaceutical analysis.32 In the USP,4 it is specified that the total impurities should not exceed a maximum limit of 0.5% relative to the main drug. HPTLC methods are widely used planar chromatographic methods, which are applied to analyze several samples in parallel. In these chromatographic methods, disposable stationary phases are used which do not need a lot of sample clean up.33 Furthermore, the addition of MP, PP, and SA to oral suspensions was found to be a big problem during the analysis of active ingredients in pharmaceutical formulations.
Only one method has been previously developed in our laboratory for the determination of the studied components using a LC-MS-MS method.34 Although it attains high sensitivity and selectivity, it needs expensive instruments and chemicals. Similarly, all the published methods analyzed either CBZ or OX in tablets only, even though interference due to the suspension excipients is expected. All of this, in addition to the pharmaceutical importance of the studied drugs, persuaded us to develop a selective and sensitive method that is capable of simultaneously resolving and quantifying the two drugs (CBZ and OX), along with IMD and IST (as impurities), and the formulation excipients (MP, PP, and SA).
Nowadays, the analytical community is interested in protecting human health and the environment through reducing the harmful impacts of hazardous chemicals by using more clean methodologies. Chemists have paid attention to the application of the concepts of green analytical chemistry (GAC) in different analytical methods. Furthermore, the compromise between increasing the quality of the results and decreasing the environmental impact of a method is considered to be a great challenge. Recently, different tools have been applied to assess the greenness profile of any developed method, of which the analytical Eco-Scale and Green Analytical Procedure Index (GAPI) tools are widely used.
The proposed HPTLC method is the first established stability indicating chromatographic method for the quantitation of the seven components with minimal analysis costs and time. Besides, the method has the advantage of its high sensitivity, especially for the analyzed impurities, which met the requirements stated in the USP4 recommendations.
2. Experimental
2.1. Instruments
– The TLC Scanner (CAMAG) was controlled by WINCATS software. Scanning was performed in the absorbance mode with a 20 mm s−1 speed. A Linomat IV applicator was used for the band application with a 100μL syringe (CAMAG, Muttenz, Switzerland). And the source of radiation used was a deuterium lamp. It was operated with a band width of 5 mm, and 3 × 0.45 mm were selected as the slit dimensions. The chromatogram output data were obtained as integrated peak areas. HPTLC plates with a 0.25 mm coat of silica gel 60F254 (Merck, Germany) with dimensions of 15 × 20 cm were utilized.– A Sonix TV ss-series ultra-sonicator (USA) was used.
2.2. Samples
– Trileptal® tablets (Batch No. T1887) and Trileptal® suspension (Batch No. (W/V) H6725) were manufactured by Delpharm Huningue S.A.S., Huningue, France and licensed for Novartis Pharma AG, Basle, Switzerland. Their labels stated that they contained 300 mg of OX per tablet and 60 mg of OX per 1 mL suspension.
2.3. Solvents and chemicals
Analytical grade methanol, hexane, glacial acetic acid, formic acid, and ethyl acetate were obtained from El Nasr Pharmaceutical Chemicals Co., Abo-Zabaal, Giza, Egypt.2.4. Stock standard solutions
2.5. Procedure
:ethyl acetate:formic acid:acetic acid (8:2:0.5:0.3, by volume) to a distance of about 8 cm. Subsequently, the developed plates were dried and scanned at 254 nm. The radiation source applied was a deuterium lamp and the software was WINCATS. The peak areas of the studied components were recorded and the calibration graphs were established using the calculated integrated peak areas and the corresponding concentrations of each component.– For the tablet dosage forms (Tegretol® and Trileptal® tablets). Five tablets of each were accurately and separately weighed and ground. 50 mg of each of the powdered tablets was accurately transferred into two separate 100 mL volumetric flasks. After that, 75 mL methanol was added and the solutions were sonicated for about 15 min, then filtered, and the residue was washed with 5 mL methanol twice (2 × 5 mL) and the washing solution was then added to the filtrate. Finally, the volume of the prepared solutions was made up with methanol to obtain solutions of 0.5 mg mL−1 of either CBZ or OX.
– For the suspension dosage forms. 1.25 mL of the Tegretol® suspension and 0.5 mL of the Trileptal® suspension were accurately and separately transferred into two 50 mL volumetric flasks. After that, methanol was used to make up the volume to the mark then these were shaken well to prepare stock solutions of 0.5 and 0.6 mg mL−1 of CBZ and OX, respectively.
– Suitable dilutions within the linearity ranges of both drugs were made after proper filtration of the prepared solutions. Then, the procedure was followed to construct the calibration curves. The previously computed regression equations for CBZ and OX were employed to obtain the recovered concentrations in the analyzed dosage forms.
– The technique of standard addition was also used by producing mixtures of standard CBZ or OX solutions with their respective tablet and suspension solutions. Subsequently, the proposed HPTLC method was adopted and the concentrations of CBZ and OX were calculated using their relative regression equations.
3. Results and discussion
Carbamazepine and OX are widely prescribed tricyclic antipsychotics that are available in several dosage forms such as tablets and suspensions.Undoubtedly, pharmaceuticals intended for human consumption should be as fully characterized as possible. Assurance of a drug’s quality and safety is attained by monitoring and controlling its impurities. The presence of such impurities prohibits the accurate determination of the drug. Therefore, there is a demand for developing and validating a highly sensitive and selective analytical method for the analysis of these drugs without interference from their impurities. Synthesis-related impurities are expected to be found in the bulk forms and pharmaceutical formulations of CBZ and OX. Pharmacopoeias set strict limits for the purity of most drugs to ensure their efficacy and safety. The USP4 declared that the maximum concentration of any individual impurity should not be more than 0.2%, while the concentration of all impurities should not be more than 0.5% of the active pharmaceutical ingredient. Routine analysis of drug purity necessitates a simple and robust analytical method that provides sufficient resolution, accuracy and sensitivity.
In this manuscript, efforts were made to develop a sensitive, specific and accurate HPTLC method for the resolution of the active drugs, their impurities, and the labeled suspension additives. In addition, the environmental and health hazards of the developed method were reduced. The developed HPTLC method is more specific and economical than the official HPLC3,4 methods and any previously published chromatographic methods. In addition, it is the first developed method for resolving and quantifying the seven components using a single developing system and scanning wavelength. Moreover, it is the first developed method that has been applied to the determination of both CBZ and OX in their available suspension dosage forms, along with their suspension additives, MP, PP and SA.
One of the challenges in developing a specific method for resolving the cited mixture is the structural similarity between several pairs of the chosen components, like (CBZ and OX), (IMD and IST) and (MP and PP), leading to similar polarities for the members of each pair, and thus the same affinity with the stationary phase. In order to optimize the developed HPTLC method, different separation conditions were studied, such as the developing system composition, saturation time, different instrumental parameters, and the height of the used HPTLC plates.
The initial optimization steps started with the choice of the most appropriate developing system that affected the partition of the separated components between the stationary and mobile phases. Different systems were tested, like hexane:methanol (8:2, v/v), hexane:ethyl acetate (8:2, v/v), hexane:ethanol (8:2, v/v), and hexane:acetone (8:2, v/v). Unresolved spots between (CBZ and OX), (MP and PP) and (SA, IMD and IST) were obtained in all trials except when using hexane:ethyl acetate, which resulted in a slight separation between the studied components. The composition of the chosen developing system was then optimized by trying different ratios of hexane and ethyl acetate (from 7:3 to 9:1, v/v) where the best ratio was 8.5:1.5, v/v. The effect of the developing system pH was then studied using different volumes of acetic acid, formic acid, and ammonium hydroxide (0.2–0.5 mL each). No significant effect on the separation was found when using ammonium hydroxide (33%) solution. On the contrary, it was found that acetic acid was necessary to differentiate between the adjacent bands of OX and CBZ, while formic acid was essential for the good separation of MP, PP, SA, IMD and IST. Different combined ratios of formic acid and acetic acid were tried, and a developing system of hexane:ethyl acetate:formic acid:acetic acid (8.5:1.5:0.5:0.3, by volume) gave the greatest possible separation between the seven components. For the further improvement of the chromatographic resolution, HPTLC plates with different lengths were examined (10, 12, and 15 cm). The plate height was observed to significantly affect the separation efficiency and the chromatographic resolution between the seven components. The HPTLC plate of 15 cm length was the most suitable one. The effect of the saturation time of the stationary phase with the developing system was tested (15 and 30 min) and no significant effect of saturation time on the resolution was seen. Finally, different scanning wavelengths (215, 225, and 254 nm) were used in order to meet the detection and quantitation limits stated by the USP.4 Upon using 254 nm as a scanning wavelength, the highest sensitivity for all the studied components was attained.
At the end, the optimum chromatographic conditions were: the developing system: hexane:ethyl acetate:formic acid:acetic acid (8.5:1.5:0.5:0.3, by volume), saturation time: 15 min, HPTLC plate height: 15 cm and scanning wavelength: 254 nm. The obtained Rf values were 0.08, 0.16, 0.43, 0.55, 0.62, 0.72, and 0.83 for OX, CBZ, MP, PP, SA, IST, and IMD, respectively. The HPTLC densitogram is shown in Fig. 2.
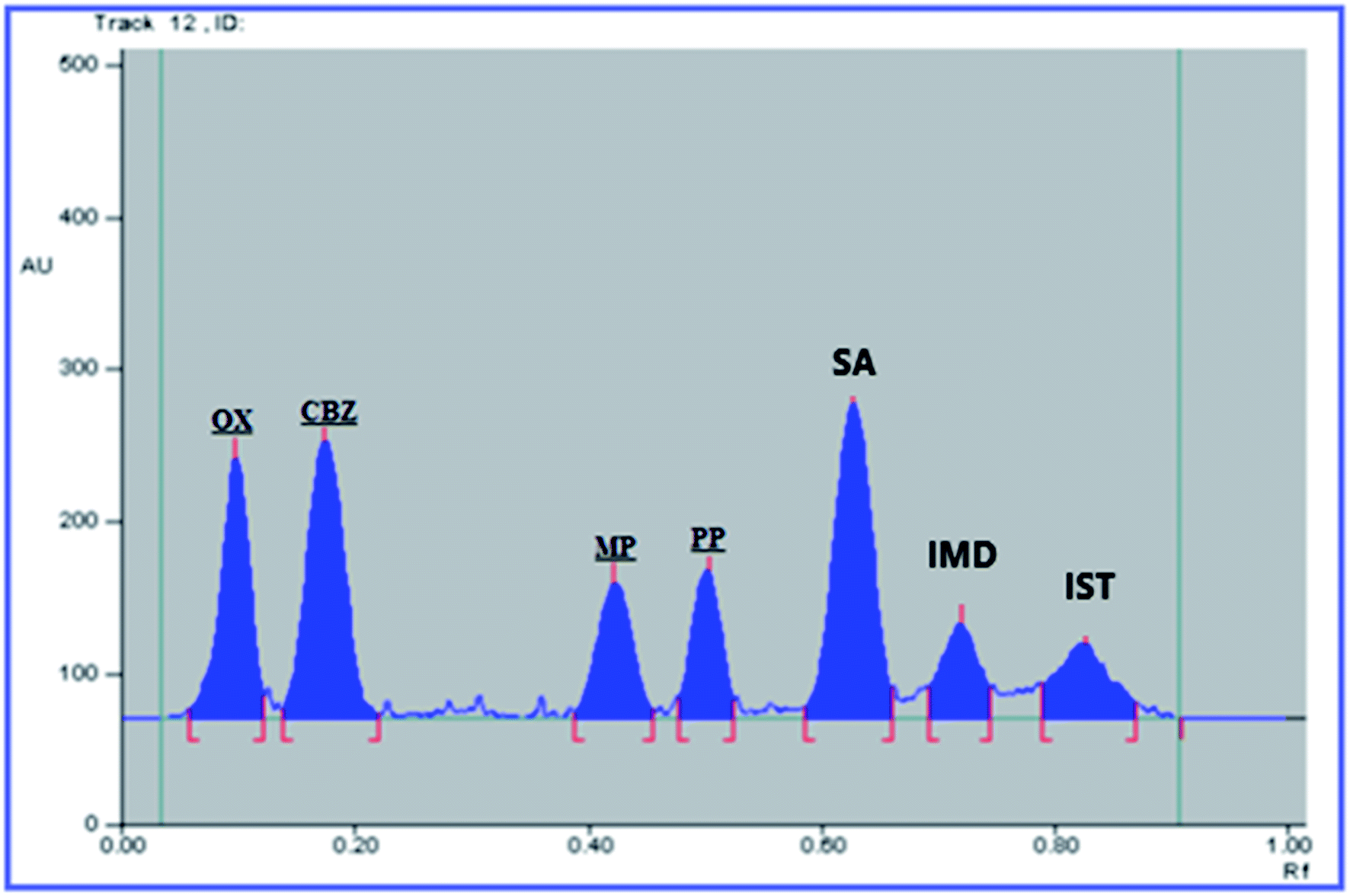 | ||
| Fig. 2 HPTLC densitogram of a mixture of OX (500 ng per band), CBZ (500 ng per band), IMD (200 ng per band), IST (200 ng per band), MP (200 ng per band), PP (200 ng per band) and SA (200 ng per band). | ||
3.1. Application of the method
After the proposed method was optimized, it was applied to analyze different samples of the pure components at different concentration ranges to check the method’s linearity. Linearity was achieved at different ranges: 40–4000 ng per band (for OX and CBZ) and 20–2000 ng per band (for IMD, IST, MM, PP, and SA). The regression equations were constructed using polynomial regression which gave a wider linear range for all the studied components compared to linear regression, as shown in Table 1.| Parameters | OX | CBZ | IMD | IST | MP | PP | SA |
|---|---|---|---|---|---|---|---|
| a Coefficient 1.b Coefficient 2.c Accuracy: mean of 9 concentrations for each component.d Average of 3 experiments.e Standard deviation of 3 concentrations of each component: (100, 1000 and 3000 ng per band) for OX and CBZ, and (100, 500 and 1500 ng per band) for IMD, IST, MP, PP, and SA, on the same day.f Standard deviation of 3 concentrations of each component: (100, 1000 and 3000 ng per band) for OX and CBZ, and (100, 500 and 1500 ng per band) for IMD, IST, MP, PP, and SA, on three successive days.g Where LOD = 3.3 × SD/S and LOQ = 10 × SD/S, where SD is the standard deviation of the intercept and S is the slope of the calibration curve. | |||||||
| Linearity range (ng per band) | 40–4000 | 20–2000 | 20–2000 | ||||
| Slope | −237.84 a | −615.48 a | −1769 a | −1512.10 a | −2265.70 a | −2631.60 a | −4209.80 a |
| 3807.90 b | 6069.10 b | 6937.4 b | 11537 b |
13314 b |
12313 b |
18288 b |
|
| Intercept | 226.12 | 205.20 | 571.47 | 293.40 | 118.14 | 323.50 | 1050.50 |
| Correlation coefficient (r) | 0.9999 | 0.9996 | 0.9997 | 0.9998 | 0.9998 | 0.9997 | 0.9997 |
| Accuracy (mean ± % RSD)c | 99.84 ± 1.38 | 100.10 ± 1.82 | 99.81 ± 1.11 | 99.94 ± 2.04 | 99.49 ± 1.47 | 98.96 ± 1.52 | 97.66 ± 0.38 |
|
|||||||
| Precision (% RSD) | |||||||
| Repeatabilityd,e | 0.98 | 1.08 | 1.39 | 0.64 | 1.46 | 1.64 | 2.07 |
| Intermediate precision d,f | 1.81 | 1.16 | 2.09 | 2.04 | 2.40 | 2.26 | 2.27 |
| LOD (ng per band)g | 12.50 | 13.00 | 6.00 | 6.40 | 6.60 | 6.60 | 6.65 |
| LOQ (ng per band)g | 38.00 | 39.00 | 18.50 | 19.25 | 19.75 | 19.90 | 20.00 |
After the evaluation of the method linearity, the validity of the method was checked for the available tablet and suspension dosage forms. The obtained results were found to be 99.27 ± 1.93, 95.57 ± 2.13 (for Tegretol® suspension and tablets, respectively) and 103.12 ± 1.92, 102.05 ± 1.29 (for Trileptal® suspension and tablets, respectively). As indicated in the results shown in Table 2, all were within the acceptable limits (90–110%). Additionally, to confirm the accuracy of the method, the standard addition technique was carried out on three different levels by spiking the dosage form sample solutions with different concentrations of pure CBZ and OX, separately. Then, the samples were analyzed using the developed method and the concentrations of pure drugs added were calculated. The calculated % recoveries are presented in Table 2 and all of these results confirmed that interference from the formulations’ excipients was absent, confirming the accuracy of the developed method.
| Pharmaceutical formulation | Tegretol® syrup | Tegretol® tablets | ||||
|---|---|---|---|---|---|---|
| Taken (ng per band) | Founda % ± % RSD | Standard addition recoveryb % ± SD | Taken (ng per band) | Founda % ± % RSD | Standard addition recoveryb % ± SD | |
| a Average of 6 determinations.b Average of 3 determinations. | ||||||
| CBZ | 150.00 | 99.27 ± 1.93 | 97.91 ± 0.66 | 150.00 | 95.57 ± 2.13 | 98.20 ± 1.20 |
3.2. Statistical comparison with the official method
Statistical comparisons between the suggested HPTLC method and the reference methods,4,5 using Student’s-t and F tests, were carried out and the values of the results were smaller than the theoretical ones, denoting that the proposed method differs non-significantly from the reference ones regarding accuracy and precision, as shown in Table 3.| Component | CBZ | OX | ||
|---|---|---|---|---|
| Method | HPTLC | Referencea | HPTLC | Referenceb |
| a HPLC method using a (4.6 mm × 250 mm) cyano column with 5–10 μm silica particles, a mobile phase of water/methanol/tetrahydrofuran (85:12:3, by volume) containing 0.22 mL formic acid and 0.5 mL triethylamine, flow rate = 1.5 mL min−1, and UV detection at 230 nm.b UHPLC method using a C18 column (100 × 2.1 mm, 1.9 μm particle size) using a gradient program of water (mobile phase A) and acetonitrile (mobile phase B), flow rate = 0.5 mL min−1, UV detection at 254 nm, and the column temperature was set to 30 °C.c The values between parentheses correspond to the theoretical values of t and F (P = 0.05). |
||||
| Mean ± SD | 100.10 ± 1.83 | 99.82 ± 2.03 | 99.84 ± 1.38 | 99.51 ± 1.75 |
| Variance | 3.33 | 4.13 | 1.91 | 3.07 |
| n | 7 | 6 | 9 | 6 |
| Student’s t-test | 0.26 (2.16)c | 0.40 (2.16)c | ||
| F-Test | 1.24 (4.39)c | 1.61 (3.69)c | ||
3.3. Method validation
The USP recommendations4 have been followed so as to assess and validate the performance of the method. During the validation step, different validation parameters were calculated like linearity and range, accuracy, precision, limit of detection (LOD) and limit of quantitation (LOQ). The results and details of the method validation are given in Table 1.To check the method specificity, variable synthetic mixtures containing different concentrations of the studied components were prepared and analyzed as described in the linearity section. Complete separation of all seven compounds was obtained, as proved by the densitogram in Fig. 2. Additionally, the percentage recoveries obtained from the analysis of the available suspension and tablet dosage forms, shown in Table 2, demonstrated the high specificity of the method and confirmed that the dosage forms’ additives did not interfere with the separated peaks of the active drugs. The densitograms in Fig. 3 and 4 show that MP, PP and SA (as the labeled suspension additives) did not interfere with the parent drugs.
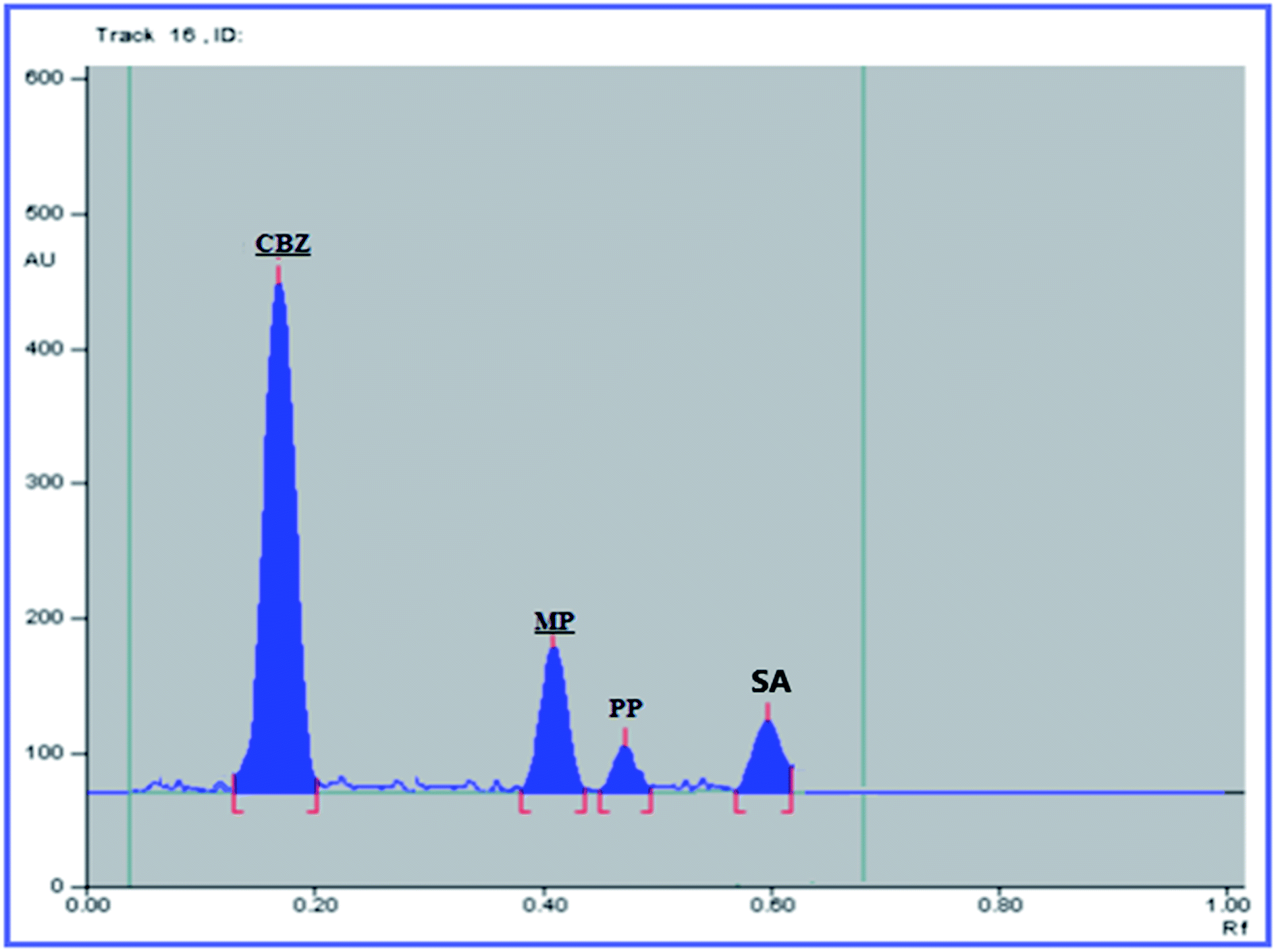 | ||
| Fig. 3 HPTLC densitogram of the mixture of the Tegretol® suspension (1000 ng per band CBZ). | ||
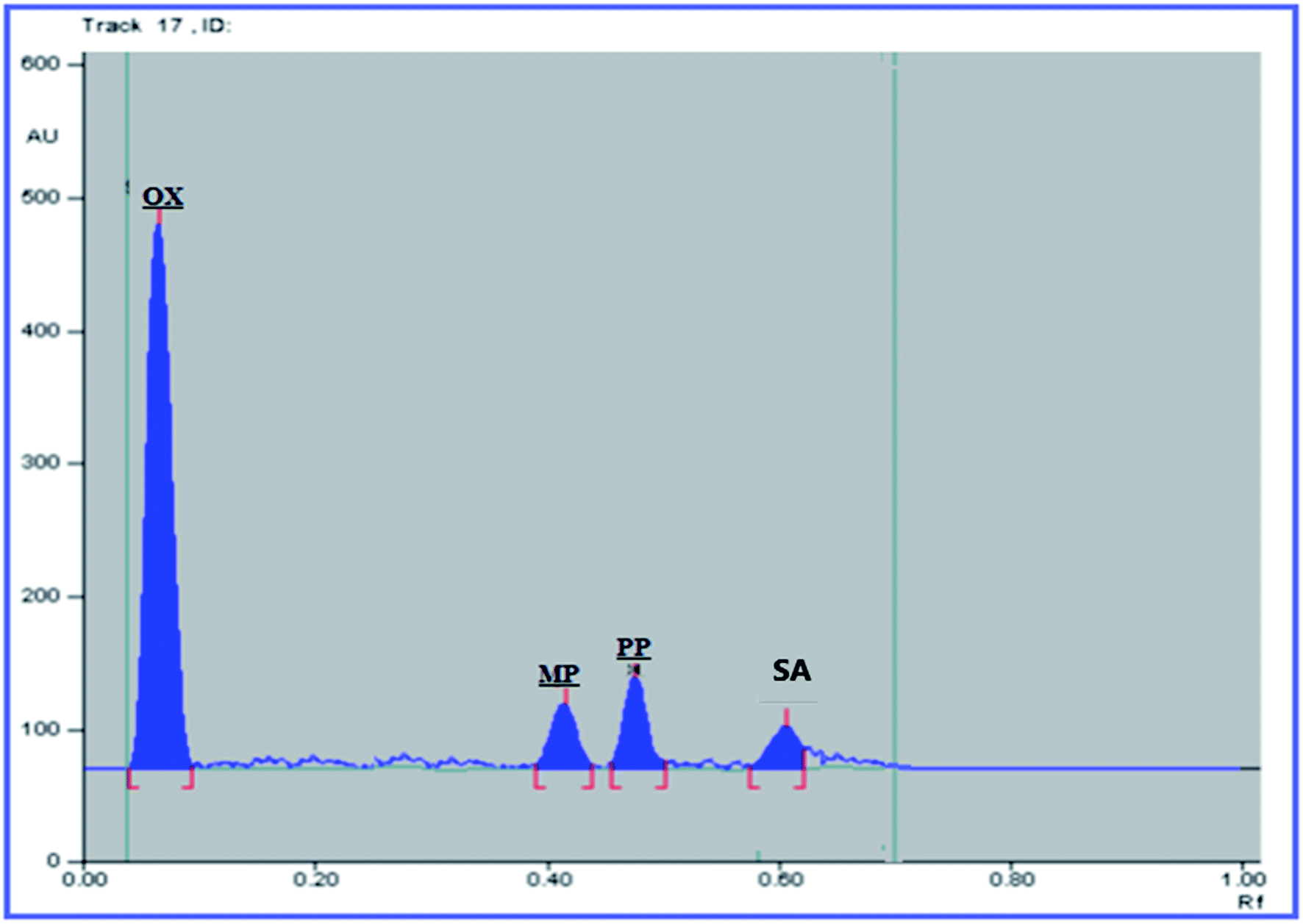 | ||
| Fig. 4 HPTLC densitogram of the Trileptal® suspension (1000 ng per band OX). | ||
In addition, the robustness of the suggested method was checked in order to evaluate its ability to remain constant even when small changes in some parameters were made. The studied parameters were the amount of formic acid (±0.05 mL) and acetic acid (±0.05 mL), and the saturation time (±5 minutes); the measured response was the Rf value for each of the studied components, and was represented as % RSD. Changes in both the formic acid amount and the saturation time had no significant effect on the studied response, while the amount of acetic acid significantly affected the resolution for both CBZ and OX. So, the amount of acetic acid should be exactly adjusted to 0.3 mL (in each 10 mL developing system) in order to obtain reproducible results.
3.4. System suitability testing parameters
Different parameters like the symmetry of peaks, the resolution, and selectivity factors were computed in order to test the system’s performance and the separation efficiency. The obtained results were within the acceptable limits, as shown in Table 4, which demonstrated the high efficiency of the chromatographic separation.| Parameters | OX | CBZ | MP | PP | SA | IST | IMD | Reference values33 |
|---|---|---|---|---|---|---|---|---|
| Rf | 0.08 ± 0.02 | 0.16 ± 0.02 | 0.42 ± 0.03 | 0.55 ± 0.02 | 0.62 ± 0.03 | 0.72 ± 0.02 | 0.83 ± 0.01 | — |
| Capacity factor (K) | 11.5 | 5.25 | 1.33 | 0.82 | 0.61 | 0.39 | 0.18 | — |
| Symmetry factor (T) | 0.92 | 1.07 | 1.04 | 1.00 | 0.97 | 1.00 | 1.05 | ∼1 |
| Resolution (Rs) | 1.13 | 3.51 | 1.52 | 2.15 | 1.50 | 1.70 | 1.70 | >1.5 |
| Selectivity (α) | 2.19 | 3.95 | 1.62 | 1.34 | 1.56 | 2.17 | 2.17 | >1 |
3.5. Greenness profile assessment
In this work, the environmental impact of the newly developed chromatographic method was considered for all steps, including sample preparation, method, development, analysis solvents, and the waste produced.35–39 In the present study, two approaches were applied to assess the greenness of the method. It is ideal to apply different tools to obtain a deeper view of the greenness of the method.| Parameters | Developed method | Penalty points | Reported HPLC4 | Penalty points | Reported HPLC5 | Penalty points | |
|---|---|---|---|---|---|---|---|
| Reagents | Consumed volume = volume of the developing system per run/number of samples on the HPTLC plate | Hexane | 8 | Water (Green nonhazardous solvent) | 0 | Water (Green nonhazardous solvent) | 0 |
| Consumed volume = 4 mL | |||||||
| Subtotal PP = 1 [solvent < 10 mL] | |||||||
| Signal word = 2 danger [more severe hazard = 2] | |||||||
| No. of pictograms = 4 | |||||||
| PP of solvent = subtotal PP × number of pictograms × signal word | Ethylacetate | 4 | Methanol | 6 | Methanol | 6 | |
| Consumed volume = 1 mL | Consumed volume = 1.8 mL | Consumed volume = 2.4 mL | |||||
| Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | |||||
| Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | |||||
| No. of pictograms = 2 | No. of pictograms = 3 | No. of pictograms = 3 | |||||
| Formic acid | 6 | Tetrahydrofuran | 6 | Tetrahydrofuran | 6 | ||
| Consumed volume = 0.25 mL | Consumed volume = 0.45 mL | Consumed volume = 0.6 mL | |||||
| Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | |||||
| Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | |||||
| No. of pictograms = 3 | No. of pictograms = 3 | No. of pictograms = 3 | |||||
| Acetic acid | 4 | Formic acid | 6 | Formic acid | 6 | ||
| Consumed volume = 0.15 mL | Consumed volume = 0.033 mL | Consumed volume = 0.004 mL | |||||
| Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | |||||
| Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | |||||
| No. of pictograms = 2 | No. of pictograms = 3 | No. of pictograms = 3 | |||||
| — | — | Triethylamine | 8 | Triethylamine | 8 | ||
| Consumed volume = 0.075 mL | Consumed volume = 0.01 mL | ||||||
| Subtotal PP = 1 [solvent < 10 mL] | Subtotal PP = 1 [solvent < 10 mL] | ||||||
| Signal word = 2 danger [more severe hazard = 2] | Signal word = 2 danger [more severe hazard = 2] | ||||||
| No. of pictograms =4 | No. of pictograms =4 | ||||||
| Instruments | Energy | ≤1.5 kWh per sample | 1 | ≤1.5 kW h per sample | 1 | ≤1.5 kW h per sample | 1 |
| Occupational hazard | Not a closed system | 3 | Analytical process hermetization | 0 | Analytical process hermetization | 0 | |
| Centrifuge | 1 | — | — | — | — | ||
| Wastes | <10 mL | 3 | >10 mL | 5 | >10 mL | 5 | |
| Total penalty points | 30 | 32 | 32 | ||||
| Analytical Eco-Scale total score | 70 | 68 | 68 | ||||
| Comment | Acceptable green analytical method | Acceptable green analytical method | Acceptable green analytical method | ||||
| GAPI assessment | 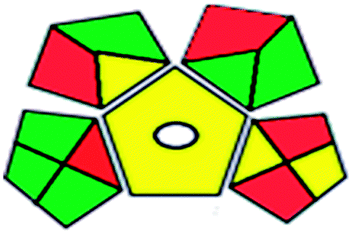 |
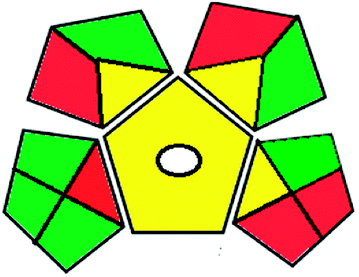 |
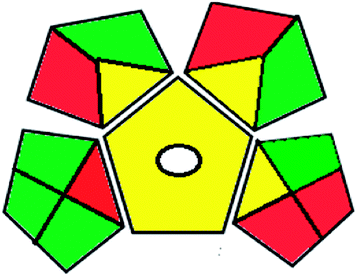 |
||||
The results of the two approaches used agreed with each other, proving the small environmental impact of the proposed HPTLC method.
3.6. Comparison with previously published chromatographic methods
The developed HPTLC method has a higher selectivity than the formerly published TLC-densitometric methods15,18 since it is applied for the simultaneous separation and quantitation of the studied drugs, their impurities and the suspension additives. The compared official HPLC methods3,4 for the analysis of CBZ and its related substances have similar greenness profiles, as proved by the results in Table 5. Conversely, the developed HPTLC method is greener than the published HPLC methods7,19–21 since acetonitrile is the organic modifier used in these methods and it has a high environmental impact. Compared with the other published HPLC methods7,8,13,14,16,17,19,20,21 and LC-MS-MS methods9–12,22 for the analysis of either CBZ or OX in their tablet formulations, the suggested HPTLC method has the advantage of saving time and money. Additionally, simple sample preparation steps are needed as well as low cost solvents, chemicals and instruments. However, the published LC-MS-MS methods have higher sensitivities for CBZ and OX but were not applied for the quantitation of either their impurities or excipients.4. Conclusion
For the first time, a selective, specific and accurate HPTLC method with little environmental impact was introduced for the simultaneous determination of the commonly used antipsychotic drugs CBZ and OX in the presence of their reported impurities and formulation excipients, in a single run. The suggested method is superior to any other formerly established chromatographic methods and is able to measure CBZ, OX, IMD, IST, MP, PP and SA with high sensitivity, which follows the USP regulations for drug impurity profiling. The HPTLC method has the advantages of lower solvent consumption, a short analysis time, and several samples can be determined simultaneously in the same run, which are important factors for quality control laboratories. The developed method can be used as an alternative to LC methods in laboratories lacking the facilities for LC instrumentation.Conflicts of interest
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.References
- S. Miyamoto, N. Miyake, L. F. Jarskog, W. W. Fleischhacker and J. A. Lieberman, Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents, Mol. Psychiatry, 2012, 17, 1206–1227, DOI:10.1038/mp.2012.47.
- Martindale, The Extra Pharmacopoeia: The Complete Drug Reference, ed. K. Profitt, Royal Pharmaceutical Society, London, UK, 35th edn, 2006 Search PubMed.
- The British Pharmacopoeia, Her Majesty’s, The Stationary Office, London, 2013 Search PubMed.
- The United States Pharmacopeia, National Formulary 35, United States Pharmacopeia Convention Inc., 30th edn, 2012 Search PubMed.
- J. Žirojević, K. Drljevic-Đurić and K. Đurđević, Determination of oxcarbazepine and its related substances using UHPLC method with UV detection, Arh. Farm., 2014, 64, 83–94 CrossRef.
- P. A. Datar, Quantitative bioanalytical and analytical method development of dibenzazepine derivative, carbamazepine: A review, J. Pharm. Anal., 2015, 5(4), 213–222 CrossRef.
- Z. Aydoğmuş, E. . M. Yılmaz, S. S. Aslan, Z. E. Taş and M. Üner, RP-HPLC method for the simultaneous determination of carbamazepine and nilotinib: application to solubility studies, Pharm. Chem. J., 2016, 3, 1–10 Search PubMed , http://www.tpcj.org.
- B. Kandilli, A. B. Uğur, M. Cetin and F. D. Miloğlu, A Simple HPLC-UV Method For Simultaneous Determination of Levetiracetam and Carbamazepine, Hacettepe Univ. J. Fac. Pharm., 2018, 38, 58–64 Search PubMed.
- T. A. Rodina, et al., Rapid HPLC/MS/MS for determination of carbamazepine and carbamazepine-10,11-epoxide, Pharm. Chem. J., 2016, 50, 419–423 CrossRef CAS.
- M. Abo El Hamd, M. Wada, R. Ikeda, S. Kawakami and K. Nakashima, Validation of an LC-MS/MS Method for the Determination of Propofol, Midazolam, and Carbamazepine in Rat Plasma: Application to Monitor Their Concentrations Following Co-administration, Biol. Pharm. Bull., 2015, 38, 1250–1253 CrossRef CAS.
- D. Andonie, Z. Gáll, P. Bosa, M. T. Dogaru and S. Vancea, Simultaneous Determination of Carbamazepine and Carbamazepine-10,11-epoxide in Different Biological Matrices by LC-MS/MS, Journal of Interdisciplinary Medicine, 2017, 2, 211–218 Search PubMed.
- J. Taibon, et al., An LC-MS/MS based candidate reference method for the quantification of carbamazepine in human serum, Clin. Chim. Acta, 2017, 472, 35–40 CrossRef CAS.
- P. Dodi, L. Ivanovi, A. Proti, M. L. Zeevi and B. M. Joci, Determination of carbamazepine and its impurities iminostilbene and iminodibenzyl in solid dosage form by column high-performance liquid chromatography, J. AOAC Int., 2010, 93, 1059–1068 CrossRef , PMID 20922935.
- P. Džodić, L. Živanović, A. Protić, M. Zečević and B. Jocić, Chemometrically assisted development and validation of LC for simultaneous determination of carbamazepine and its impurities iminostilbene and iminodibenzyl in solid dosage form, Chromatographia, 2009, 70, 1343–1351 CrossRef.
- T. S. Reddy and P. S. Devi, Validation of a high performance thin layer chromatographic method with densitometric detection for quantitative analysis of two anticonvulsants in tablets, J. Planar Chromatogr.–Mod. TLC, 2007, 20, 451–456 CrossRef.
- K. Raghavi, M. Sindhura, R. Prashanthi and B. N. Nalluri, RP-HPLC-PDA method development and validation for the estimation of oxcarbazepine in bulk and formulations, Acta Chromatogr., 2013, 25(3), 519–529 CrossRef CAS.
- A. Nosal-Wierciñska, et al., Electroanalytical and HPLC methods for the determination of oxcarbazepine in spiked human urine and tablet dosage form, Croat. Chem. Acta, 2014, 87(3), 213–219 CrossRef.
- D. S. Bhoite, S. N. Dhole, S. Bhoir, P. Sangole and S. Thorat, Development and validation of stability indicating HPTLC method for determination of oxcarbazepine in bulk and pharmaceutical formulation, Int. J. Pharm. Pharm. Sci., 2013, 5, 127–132 CAS.
- U. Bhaumik, et al., Stability-Indicating HPLC Method for the Determination of Oxcarbazepine in Pharmaceutical Formulation, Asian J. Chem., 2010, 22(3), 2051 CAS.
- P. S. Reddy, K. S. Babu and N. Kumar, Development and validation of a stability-indicating RP-LC method for the estimation of process-related impurities and degradation products of oxcarbazepine in pharmaceutical formulation, Acta Chromatogr., 2014, 26(2), 267–282 CrossRef CAS.
- P. Dwivedi, S. Yadav and J. Rao, Validated RP HPLC method for the determination of related substance of oxcarbazepine an antiepileptic drugs, Int. J. PharmTech Res., 2016, 9, 444–451 CAS.
- K. Raghavi, M. Sindhura, R. Prashanthi and B. N. Nalluri, Studies on forced degradation of oxcarbazepine using LC-MS compatible stability indicating RP-HPLC method, Int. J. PharmTech Res., 2012, 4, 3885–3893 CAS.
- M. A. Saracino, K. Tallarico and M. A. Raggi, Liquid chromatographic analysis of oxcarbazepine and its metabolites in plasma and saliva after a novel micro extraction by packed sorbent procedure, Anal. Chim. Acta, 2010, 661, 222–228 CrossRef CAS.
- S. Jin, Q. Zhao, D. Zhang, Z. Zhao and S. Mei, Development and validation of an improved HPLC-UV method for simultaneous determination of lamotrigine and oxcarbazepine and its active metabolite 10,11-dihydro-10-hydroxycarbazepine in human blood plasma and comparison with an UHPLC-MS/MS method, J. Anal. Sci. Technol., 2019, 10(1), 36 CrossRef CAS.
- U. H. Kashif and N. Kumar, Development and validation of LC/MS/MS method for the simultaneous quantitative analysis of oxcarbazepine and its metabolite 10-hydroxycarbazepine in K2EDTA plasma, Int. J. Pharm. Pharm. Sci., 2014, 6, 422–429 Search PubMed.
- Y. Mano, LC–MS-MS Determination of Oxcarbazepine and an Active Metabolite in Human Plasma for Clinical Application, J. Chromatogr. Sci., 2018, 56(8), 687–694 CAS.
- N. de Jesus Antunes, et al., Analysis of Oxcarbazepine and the 10-Hydroxycarbazepine Enantiomers in Plasma by LC-MS/MS: Application in a Pharmacokinetic Study, Chirality, 2013, 25(12), 897–903 CrossRef.
- A. Fortuna, J. Sousa, G. Alves, A. Falcão and P. Soares-da-Silva, Development and validation of an HPLC-UV method for the simultaneous quantification of carbamazepine, oxcarbazepine, eslicarbazepine acetate and their main metabolites in human plasma, Anal. Bioanal. Chem., 2010, 397, 1605–1615 CrossRef CAS.
- S. Deeb, D. A. McKeown, H. J. Torrance, F. M. Wylie, B. K. Logan and K. S. Scott, Simultaneous analysis of 22 antiepileptic drugs in postmortem blood, serum and plasma using LC/MS/MS with a focus on their role in forensic cases, J. Anal. Toxicol., 2014, 38, 485–494 CrossRef CAS.
- L. Yin, et al., Simultaneous determination of ten antiepileptic drugs in human plasma by liquid chromatography and tandem mass spectrometry with positive/negative ion-switching electrospray ionization and its application in therapeutic drug monitoring, J. Sep. Sci., 2016, 39, 964–972 CrossRef CAS.
- L. Wang, et al., Simultaneous determination of topiramate, carbamazepine, oxcarbazepine and its major metabolite in human plasma by SFC/ESI/MS/MS with polarity switching: application to therapeutic drug monitoring, Arabian J. Chem., 2019, 12, 4775–4783 CrossRef CAS.
- D. Jain and P. K. Basniwal, Forced degradation and impurity profiling: Recent trends in analytical perspectives, J. Pharm. Biomed. Anal., 2013, 86, 11–35 CrossRef CAS.
- P. S. Variyar, S. Chatterjee and A. Sharma, High-Performance Thin Layer Chromatography (HPTLC), M. M. Srivastava, Springer-Verlag Berlin Heidelberg, 2011 Search PubMed.
- M. M. Abdelrahman and N. S. Abdelwahab, Analysis of Carbamazepine, Oxcarbazepine, Their Impurities, and Non-Labeled Interfering Substances by Stability-indicating UPLC/MS/MS Method: Studying the Method’s Greenness Profile, Chromatographia, 2018, 81, 1503–1517 CrossRef CAS.
- H. Shaaban, New insights into liquid chromatography for more eco-friendly analysis of pharmaceuticals, Anal. Bioanal. Chem., 2016, 408, 6929–6944, DOI:10.1007/s00216-016-9726-2.
- E. S. Elzanfaly, M. A. Hegazy, S. S. Saad, M. Y. Salem and L. E. Abd El Fattah, Validated green high-performance liquid chromatographic methods for the determination of coformulated pharmaceuticals: A comparison with reported conventional methods, J. Sep. Sci., 2015, 38, 757–763, DOI:10.1002/jssc.201401151.
- R. Ferretti, L. Zanitti, A. Casulli and R. Cirilli, Green high-performance liquid chromatography enantioseparation of lansoprazole using a cellulose-based chiral stationary phase under ethanol/water mode, J. Sep. Sci., 2016, 39, 1418–1424, DOI:10.1002/jssc.201501329.
- Y. Yang, H. Gao, S. Hou, R. Su, H. Liu and J. Sun, A sensitive, high-throughput, and ecofriendly method for the determination of lumefantrine, artemether, and its active metabolite dihydroartemisinin by supercritical fluid chromatography and tandem mass spectrometry, J. Sep. Sci., 2018, 41(12), 2688–2696, DOI:10.1002/jssc.201800025.
- J. Płotka-Wasylka, A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index, Talanta, 2018, 181, 204–209 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |