
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh acetone sensing properties of In2O3–NiO one-dimensional heterogeneous nanofibers based on electrospinning
Xiangxiang Fan *ab,
Yajuan Xua and
Wuming Heab
*ab,
Yajuan Xua and
Wuming Heab
aSchool of Information Engineering, Huzhou University, Huzhou 313000, China. E-mail: fanxiangxiang@zjhu.edu.cn
bZhejiang Province Key Laboratory of Smart Management & Application of Modern Agricultural Resources, Huzhou University, Huzhou 313000, China
First published on 17th March 2021
Abstract
Pure NiO nanofibers and the In2O3–NiO one-dimensional heterogeneous nanofibers were prepared by electrospinning, and the gas sensing properties to acetone were also investigated. Material characterization proved that the heterogeneous nanofibers were composed of In2O3 and NiO, and the nanofibers exhibited an enhanced sensitivity to acetone. At the optimal working temperature, the response of In2O3–NiO nanofibers to 50 ppm acetone was more than 10 times higher than that of pure NiO nanofibers. The minimum detection limit of the heterogeneous nanofibers reached 10 ppb, while the pure NiO nanofibers only reached 100 ppb. Among acetone and the comparison gases (methanol, ethanol, triethylamine, ethyl acetate, and benzene), the heterogeneous nanofibers achieved the highest response to acetone. In addition, the heterogeneous nanofibers exhibited an improved response–recovery rate and good long-term stability. These results indicated that the In2O3–NiO one-dimensional heterogeneous nanofibers have great potential in low-concentration acetone detection. Combined with the material properties, the mechanism of the enhanced sensing properties was discussed in detail for the In2O3–NiO heterogeneous nanofibers.
1. Introduction
Acetone is a volatile organic compound (VOC) and is widely used in industry. However, when the human body is exposed to acetone over a long time or at high concentration, the nerves and respiratory system can be damaged. In addition, acetone is a metabolite of glucose and fat in the human body. Because the concentration of acetone in breath gas is related to the blood glucose concentration, acetone can be used as a marker for the diagnosis of diabetes.1 Many researchers have paid attention to the detection of acetone, especially trace level acetone. Due to their simple preparation process and low cost, metal-oxide-semiconductor sensors have been widely used in the detection of acetone gas.2,3NiO is a p-type semiconductor, which has been used as a sensing material for VOCs sensors, because of the high VOCs catalytic activity and oxygen adsorption characteristics.4–6 The previous report has shown that the gas-sensing performance of p-type metal oxides is much lower than that of the n-type metal oxides.7 Construction of p–n heterogeneous nanomaterial is an effective way to improve the gas sensitivity of p-type metal oxides.8–10 Several n-type metal-oxides have been reported to improve acetone sensitivity of NiO, including Fe2O3, MoO3, ZnO, WO3, TiO2 and so on.11–15 Among these reports, the enhancement of Fe2O3 and MoO3 were more prominent for acetone sensing. Compared with the pure NiO, the response of the α-Fe2O3/NiO heterostructures to 100 ppm acetone increased by 6.9 times in Haq's report.11 The response of the α-MoO3@NiO core–shell material prepared was 6.6 times higher than that of the pure NiO in Xu's report.12 The sensitivity of NiO to acetone was greatly enhanced with p–n heterojunction, but the detection limit was still high, especially for breath detection (0.3–0.9 ppm for the healthy person). Therefore, the further study is necessary about the NiO-based sensors with lower detection limit of acetone.
In2O3 is an n-type metal oxide. Because of high conductivity and stability, In2O3 has been used to detect varieties of gases, including inorganic and organic gases.16–19 The In2O3–NiO sensing material performed well in VOCs detection.20–22 The response of In-doped NiO nanotubes prepared by Feng and co-workers to methanol was 5.2 times higher than that of the pure NiO.22 However, the investigation about the In2O3–NiO heterogeneous nanomaterial to enhance the acetone sensitivity has not been reported.
Electrospinning is a simple method to prepare one-dimensional nanofibers, which excelled in the preparation of high-performance gas sensing materials.23–25 In this work, the In2O3–NiO one-dimensional heterogeneous nanofibers were fabricated by this method. The influences of heterojunction on the acetone sensing properties and the enhanced-sensing mechanism were investigated.
2. Experimental
2.1 Preparation and characterization of nanofibers
Firstly, 0.9 g nickel acetate tetrahydrate (Ni(CH3COO)2·4H2O, AR, Aladdin) and 0.1 g indium nitrate hydrate (In(NO3)3·xH2O, 99.99% metals basis, Aladdin) were added into 10 mL mixed solvent of ethanol and N,N-dimethylformamide (mixed volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The solution was fully stirred by a magnetic stirrer at 40 °C until the solutes were completely dissolved. Secondly, 0.8 g polyvinylpyrrolidone (PVP, Mw = 1300000, Aladdin) was added into the above solution, and the solution was stirred again until a homogeneous solution formed. Thirdly, the homogeneous solution was transferred into a syringe, and the electrospinning was performed. The nozzle was a flat needle (26#) of stainless steel, the solution supply rate was 0.3 mL h−1, and the distance and voltage between the nozzle and the collection plate were 12 cm and 11 kV, respectively. Finally, the collected nonwoven film was placed in a resistance furnace for high-temperature sintering, in which the sintering temperature was 500 °C, the heating rate was 2 °C min−1, and the annealing time was 3 hours. The In2O3–NiO nanofibers were obtained after the organic material and metal-salts in the nonwoven film were completely decomposed, and the sample was named as N1. The In2O3–NiO nanofibers prepared with 0.8 g nickel acetate tetrahydrate and 0.2 g indium nitrate hydrate was named as N2.
:1). The solution was fully stirred by a magnetic stirrer at 40 °C until the solutes were completely dissolved. Secondly, 0.8 g polyvinylpyrrolidone (PVP, Mw = 1300000, Aladdin) was added into the above solution, and the solution was stirred again until a homogeneous solution formed. Thirdly, the homogeneous solution was transferred into a syringe, and the electrospinning was performed. The nozzle was a flat needle (26#) of stainless steel, the solution supply rate was 0.3 mL h−1, and the distance and voltage between the nozzle and the collection plate were 12 cm and 11 kV, respectively. Finally, the collected nonwoven film was placed in a resistance furnace for high-temperature sintering, in which the sintering temperature was 500 °C, the heating rate was 2 °C min−1, and the annealing time was 3 hours. The In2O3–NiO nanofibers were obtained after the organic material and metal-salts in the nonwoven film were completely decomposed, and the sample was named as N1. The In2O3–NiO nanofibers prepared with 0.8 g nickel acetate tetrahydrate and 0.2 g indium nitrate hydrate was named as N2.
The pure NiO nanofibers named N0 was prepared following the above process, but the solute was 1 g nickel acetate tetrahydrate.
The morphology of the nanofibers was analyzed by scanning electron microscope (SEM, GeminiSEM 300, ZEISS). X-ray diffraction (XRD, D8 ADVANCE, Bruker) was used to exam the crystal structure and crystalline quality of the metal oxides. The morphology, lattice structure and composition were investigated by transmission electron microscope and energy dispersive spectroscopy (TEM and EDS, Talos F200X, FEI). The specific surface area and the pore sizes were tested by Brunauer–Emmett–Teller method (BET, ASAP 2460, Micromeritics). The chemical composition and chemical state of the nanofibers were investigated with X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Fisher Scientific).
2.2 Fabrication and gas sensing test of the sensor
A ceramic tube with Au electrode was used as the substrate of the sensor, and a Ni–Cr alloy wire as the heater was inserted into the ceramic tube, as shown in Fig. 1(a). The obtained metal-oxides nanofibers were ground sufficiently to form the powder, and 10 mg powder was dispersed in 100 μL ethanol to form the paste. The paste was coated on the electrode of the ceramic tube by a small brush. After drying, the ceramic tube with the paste was annealed in the resistance furnace with annealing temperature of 300 °C for 3 hours. After the annealing, the ceramic tube was welded on the base with the solder, and the final sensor was obtained, as shown in Fig. 1(b). | ||
| Fig. 1 The photograph of (a) ceramic tube and Ni–Cr alloy heater, (b) the sensor, and (c) gas sensing test system. | ||
The gas sensing properties was tested by a static test method. The sensor was placed in a test chamber with the volume of 18 L. A certain volume of acetone saturated vapor was injected into the chamber to realize acetone environment with a specific concentration,26 and the gas sensing performance of the sensor was recorded by a data acquisition system. The gas sensing test system is shown in Fig. 1(c). The response of the sensor was defined as R = (Ia − Ig)/Ig (Ia is the current of the sensor in air, Ig is the current of the sensor in acetone environment), and the response and recovery time were defined as the time of the current reached 90% of the maximum change.
3. Results and discussion
3.1 Materials characterization
The morphologies of N0, N1 and N2 samples were analyzed with SEM images, as shown in Fig. 2(a–c). The film obtained by electrospinning was composed of one-dimensional fibers with smooth surface. According to the scale, the diameters of the three samples both were less than 100 nm. This indicates that the In2O3–NiO and NiO nanofibers were prepared based on electrospinning.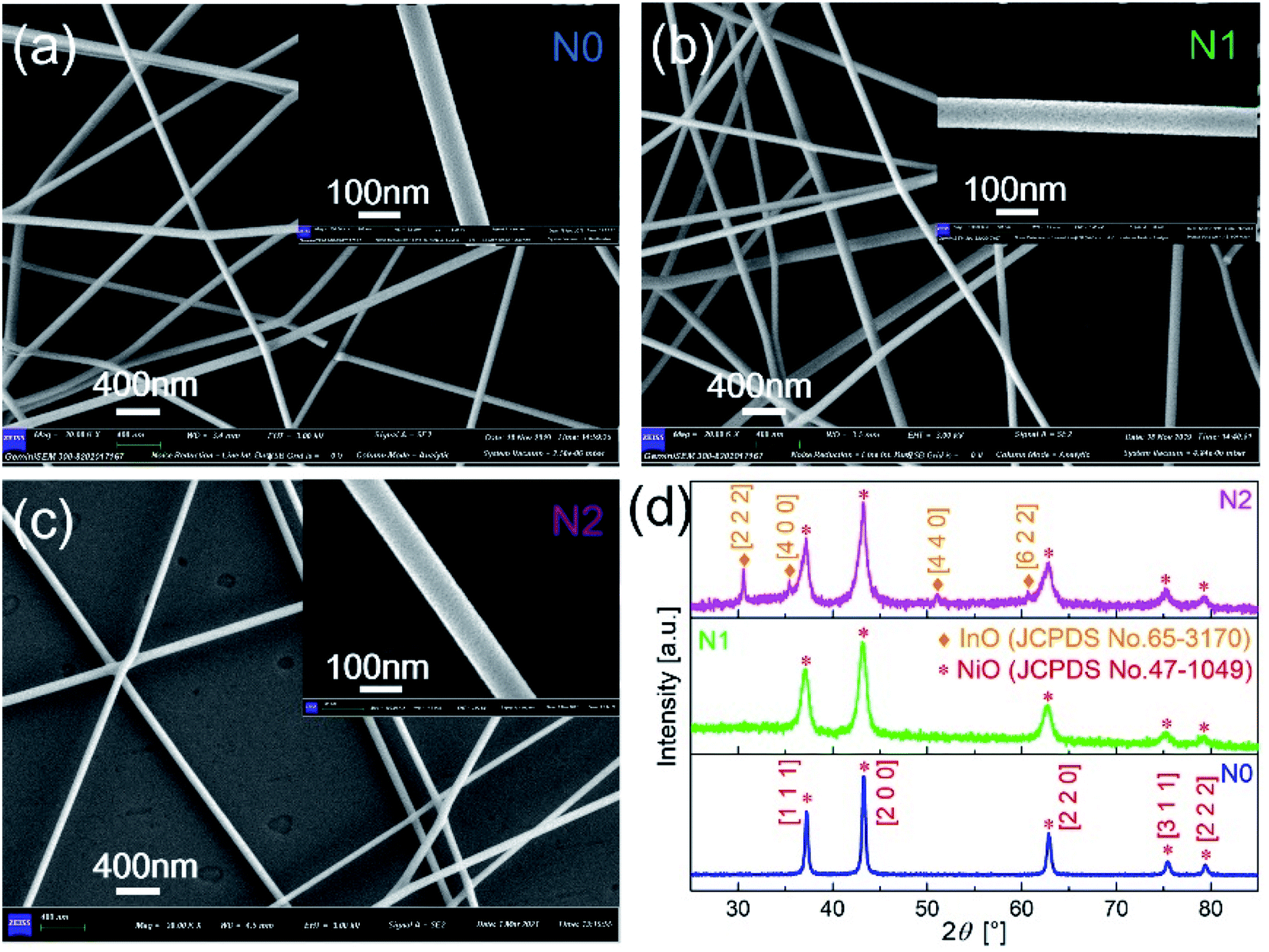 | ||
| Fig. 2 SEM images of (a) N0, (b) N1 and (c) N2 samples after sintering. (d) XRD patterns of N0, N1 and N2 samples. | ||
As shown in Fig. 2(d), the crystal structure and crystalline quality of the annealed fibers were tested by XRD. The strong and sharper diffraction peaks indicate that the crystal was formed. There were five diffraction peaks on the XRD patterns of N0 and N1 samples. The positions of the diffraction peaks corresponded to crystal direction of [1 1 1], [2 0 0], [2 2 0], [3 1 1] and [2 2 2] of NiO (JCPDS card number 47-1049). The XRD pattern of N1 didn't prove the presence of In2O3, because of the low content of In2O3 in the In2O3–NiO nanofibers. With the In2O3 content increases, four new peaks appeared on the XRD pattern of N2 sample. The positions of the four peaks corresponded to crystal direction of [2 2 2], [4 0 0], [4 4 0] and [6 2 2] of In2O3 (JCPDS card number 65-3170). This exhibits the existence of In2O3 grains and the formation of In2O3–NiO heterojunctions in N2 sample. Compared with the pure NiO nanofibers, the peak widths of the In2O3–NiO nanofibers were larger. According to peak of [2 0 0], the grain sizes in N1 and N2 samples were about 9.5 nm and 9.4 nm based on Scherrer equation, while the grain size of N0 was about 25.3 nm. This indicates that the grain sizes of the composite nanofiber were much smaller than that of pure one. The smaller grains were attributed to the presence of In2O3 as the second phase in the composite nanofibers, which hindered the aggregation of NiO nanocrystals.10,27
In order to clarify the existence of In2O3 in N1, the N1 sample was analyzed by TEM. The different interplanar distances were obtained in Fig. 3(b). The interplanar distance of 0.25 nm corresponded to the [400] lattice plane of In2O3, while the interplanar distance of d = 0.21 nm corresponded to the [200] lattice plane of NiO, which indicates that In2O3 formed a second phase in N1 sample. This exhibits that the In2O3–NiO heterojunctions also formed in N1 sample.
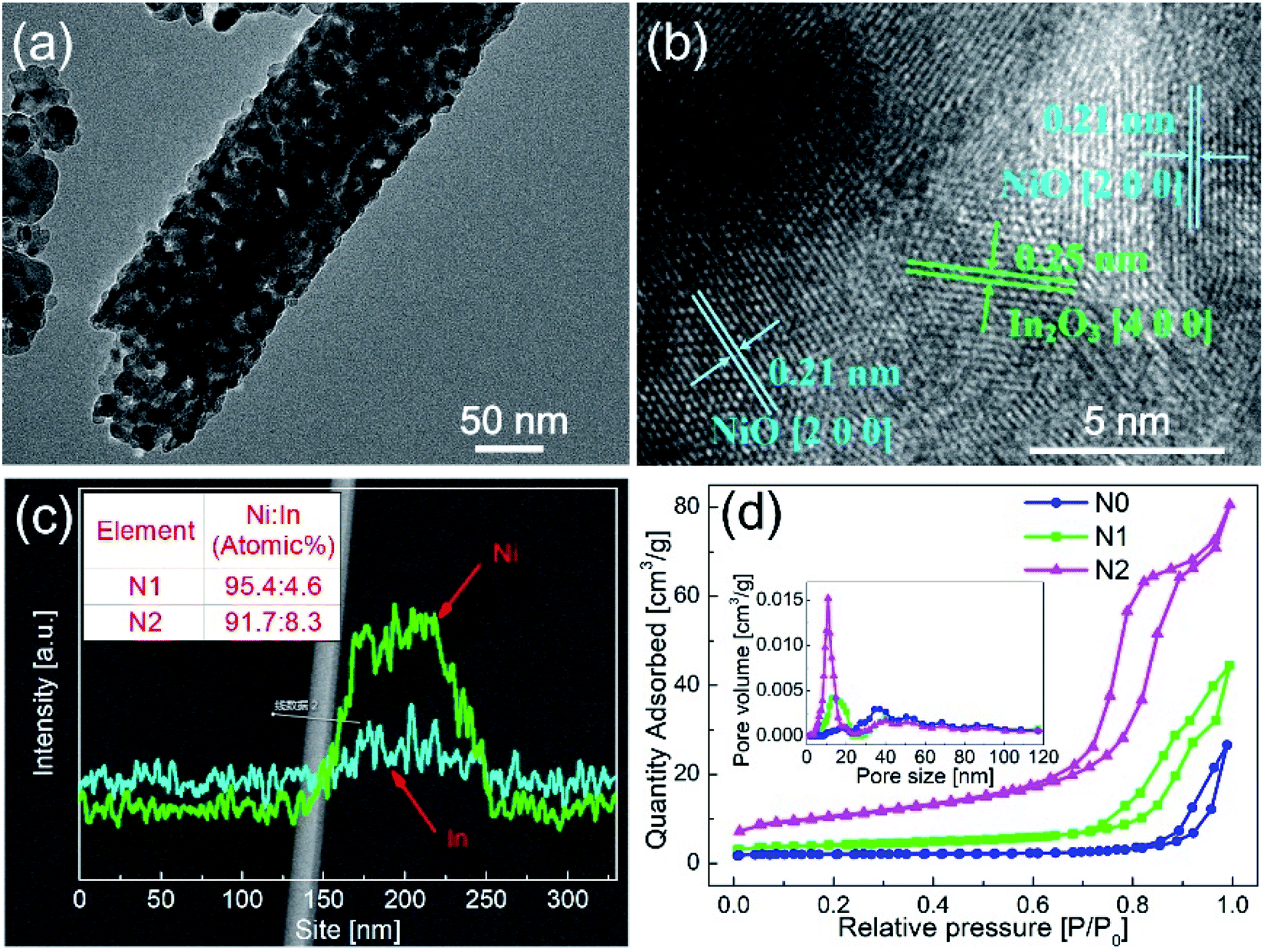 | ||
| Fig. 3 (a and b) TEM images of N1 sample after sintering. (c) EDS pattern of In2O3–NiO nanofibers. (d) N2 adsorption–desorption isotherms and BET pore size distribution curves of N0, N1 and N2 samples. | ||
In order to clarify the concentration and distribution of In and Ni in the heterogeneous nanofibers, EDS was used to scan the fiber along the radial direction, as shown in Fig. 3(c). The EDS pattern exhibits that Ni and In elements were evenly distributed in the In2O3–NiO nanofibers. After quantitative analysis, the atomic ratio of Ni and In in N1 and N2 samples were 95.4:4.6 and 91.7:8.3.
Fig. 3(d) shows the N2 adsorption–desorption isotherms and BET pore size distribution curves of the three samples. The BET surface areas of N1 and N2 samples were 13.7987 m2 g−1 and 36.274 m2 g−1, while that of N0 sample was 6.4691 m2 g−1. According to the BET pore size distribution curves, the pores of the two samples were mainly mesoporous. The pore sizes of N0 sample were mainly distributed around 50 nm. Besides the pores around 50 nm, N1 and N2 samples also had abundant pores distributed in the range of 10–20 nm. The BET test shows that the In2O3–NiO nanofibers exhibited larger specific surface area and richer porosity than that of the pure NiO nanofibers.
To further clarify the chemical composition and surface state of metal-oxides nanofibers, XPS was used to analyze all the three samples. Fig. 4(a) shows the full survey spectra of N0, N1 and N2 samples. N1 and N2 samples contained the elements of Ni, O, In and C, while In element didn't appear in N0 sample. Therefore, the existence of In element was further confirmed in N1 and N2 samples. Fig. 4(b and c) show the high-resolution spectra of In element. The peak positions of binding energy were located at 451.58 and 444.08 eV, corresponding to In 3d3/2 and In 3d5/2.
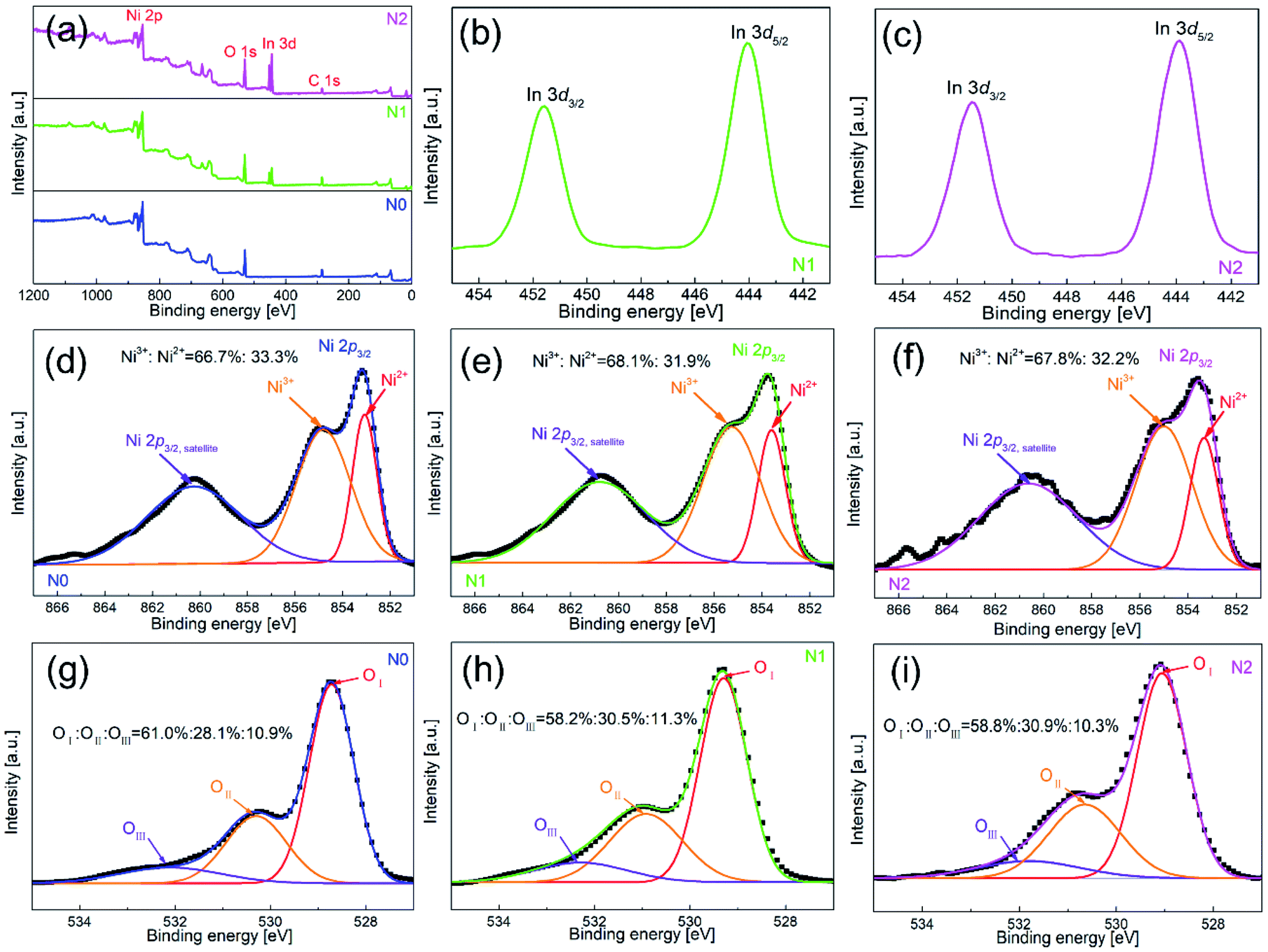 | ||
| Fig. 4 XPS spectra of (a) full survey for N0, N1 and N2 samples, and high resolution spectra for (d and g) N0, (b, e and h) N1 and (c, f and i) N2 samples. | ||
Fig. 4(d–f) show the high-resolution spectra of Ni element. There were two peaks of binding energy in the spectra, which were attributed to Ni 2p3/2,satellites and Ni 2p3/2, respectively. The peak of Ni 2p3/2 was divided into two peaks of 855.0 and 853.3 eV, and the two peaks corresponded to Ni3+ and Ni2+. By calculating the peak area of Ni 2p3/2, the contents of Ni3+ in N1 and N2 samples were 68.1% and 68.2%, which were slightly higher than that of N0 sample (66.7%). The increased content of Ni3+ was due to the oxidation of Ni2+ caused by the oxygen generated by In-doping in the lattice of NiO.21 However, the increase rate of Ni3+ was very small, which means that only a small part of In entered into the lattice of NiO.
Fig. 4(g–i) show the high-resolution spectra of O 1s for N0, N1 and N2 samples. The spectrum of O 1s was consisted of three peaks, which were located at 528.7 eV (OI), 530.3 eV (OII) and 532.1 eV (OIII). OI, OII and OIII were attributed to lattice oxygen, defective oxygen and chemically adsorbed oxygen, respectively. Among them, the amount of OII and OIII was closely related to gas sensitivity. OIII provided active sites for gas adsorption and reaction, and OIII promoted gas-sensing reaction.27 The OII + OIII proportions of N1 and N2 samples (41.8% and 40.9%) were slightly higher than that of N0 sample (39%). The OII + OIII proportion depended on the density of surface vacancies and the specific surface area. According to the XPS test, the amount of Ni3+ in the In2O3–NiO nanofibers was more than that of the pure NiO nanofibers due to the effect of In doping, which means that the vacancy density of the composite nanofibers increased.28 According to the BET test, the specific surface area of the In2O3–NiO nanofibers was also larger than that of the pure NiO nanofibers. Because of the two reasons, the In2O3–NiO nanofibers exhibited higher OII + OIII proportion.
3.2 Gas sensing properties
Fig. 5(a) shows the responses of N0, N1 and N2 samples to 50 ppm acetone at different operating temperatures. The responses of both samples increased first and then decreased with the temperature increasing. The maximum responses of N1 and N2 samples were obtained at 260 °C. Compared with the pure NiO nanofibers, the sensitivities of the In2O3–NiO nanofibers were greatly improved. The maximum responses of N1 and N2 samples to 50 ppm acetone were 4.5 and 11.8, which were more than 10 times higher than that of the pure NiO nanofibers.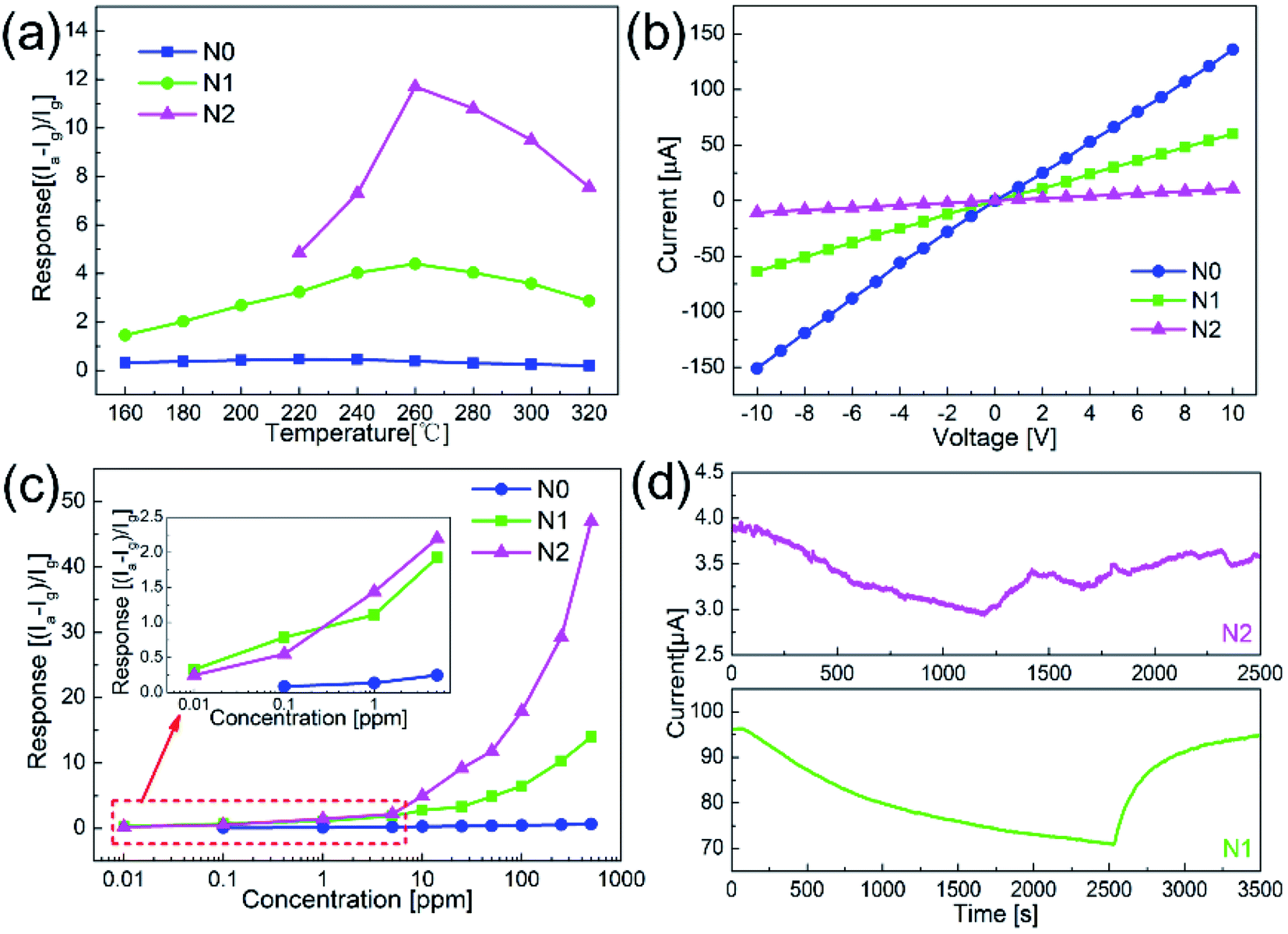 | ||
| Fig. 5 (a) Responses of N0, N1 and N2 samples to 50 ppm acetone at different temperature. (b) I–V curves of N0, N1 and N2 samples in atmospheric environment, (c) responses of N0, N1 and N2 samples to acetone with different concentrations, and (d) responses of N1 and N2 samples to 10 ppb acetone. (N2: 260 °C; N1: 260 °C; N0: 220 °C). | ||
Fig. 5(b) shows the I–V curves of N0, N1 and N2 samples in atmospheric environment. The currents of the three samples increased linearly with the increase of voltage. This indicates that a good ohmic contact was formed between the sensing material and the electrode. Because of the heterojunctions formed between In2O3 and NiO grains, the resistances of N1 and N2 samples became larger than that of N0 under the same voltage. With the increase of In concentration, the resistance of N2 was much greater than that of N1, which means that more heterojunctions were formed in N2.
Fig. 5(c) shows the responses of N0, N1 and N2 samples to acetone gas with different concentrations. As the concentration increased, the response values of the both samples became larger. Compared with the responses of N0 to acetone gas with different concentrations, the responses of N1 and N2 samples were larger. The detection limits of N1 and N2 samples both reached 10 ppb, while that of N0 was only 100 ppb. Fig. 5(d) shows the responses of N1 and N2 samples to 10 ppb acetone. The current values of the sensors both changed significantly. Compared with the smooth curve of N1 sample, the response curve of N2 has a larger noise signal because of the large resistance. For practical application, N1 with smaller resistance was more suitable as the acetone sensor.
A comparison of gas sensing properties between this work with the previous reported works is listed in Table 1. Compared with the listed metal-oxides (ZnO, WO3, TiO2),13–15,30 the In2O3–NiO heterogeneous nanofibers showed higher sensitivity and lower detection limit.
| Materials | Operating temperature | Response | Detecting limitation | Reference |
|---|---|---|---|---|
| a Response was defined as (Rg − Ra)/Ra. | ||||
| Fe2O3–NiO | 169 °C | 17.34a (100 ppm) | 5 ppm | 11 |
| MoO3–NiO | 350 °C | 19.3a (100 ppm) | 0.5 ppm | 12 |
| NiO–ZnO | 300 °C | — | 1 ppm | 13 |
| WO3–NiO | 300 °C | 3.4a (200 ppm) | 5 ppm | 14 |
| TiO2–NiO | 400 °C | 8.81a (200 ppm) | 5 ppm | 15 |
| NiO–ZnO | 260 °C | 6.5a (200 ppm) | 5 ppm | 30 |
| In2O3–NiO | 260 °C | 6.46 (100 ppm, N1), 17.9 (100 ppm, N2) | 10 ppb | This work |
Fig. 6(a) shows the response–recovery curves of N0 and N1 samples to 50 ppm acetone at the optimal working temperature. With the introduction of acetone gas, the current values of both samples decreased accordingly. When the acetone gas was exhausted, the current values returned to the original current values. The response time and recovery time of N1 sample were 20 and 558 s, which were shorter than those of N0 sample (24 and 610 s).
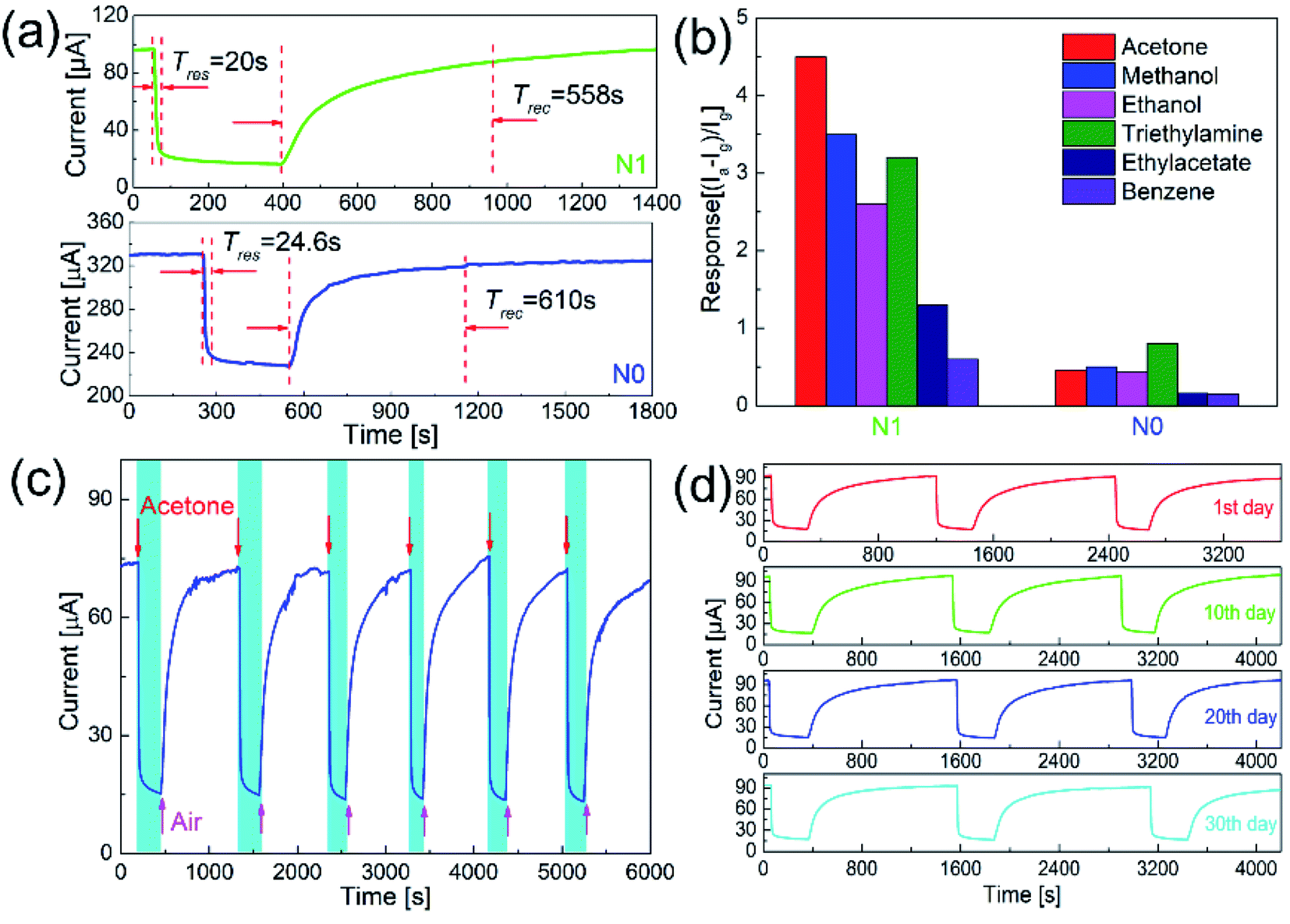 | ||
| Fig. 6 (a) Response curves of N0 and N1 samples to 50 ppm acetone, (b) responses of N0 and N1 samples to 50 ppm different gases, and (c and d) cycle gas sensing test of N1 sample to 50 ppm acetone (N1: 260 °C; N0: 220 °C). | ||
The response–recovery process depended on the adsorption and desorption of the gas. Since the operating temperature of N1 sample was slightly higher than that of N0 sample, the adsorption and desorption process on the surface of N1 sample was accelerated.29 The amount of oxygen defects and adsorbed oxygen on the surface also affected the response–recovery rate. The N1 sample with the larger amount of oxygen defects and adsorbed oxygen showed the faster response–recovery.21 In addition, the recovery time of the sensor was quite larger than the response time. This was due to the difference in the process of response and recovery. Response process involved the in-diffusion and reaction of acetone, while recovery process involved the in-diffusion, adsorption, dissociation, and ionization of oxygen on the surface.20 Therefore, recovery process took prolonged time.
Since p-type semiconductors exhibit good catalytic properties for VOCs, it's necessary to clarify the selectivity of N1 sample to acetone. Fig. 6(b) shows the responses of N1 and N0 samples to 50 ppm of different VOC gases (acetone, methanol, ethanol, triethylamine, ethyl acetate, and benzene). At the optimal working temperature, the responses of N0 sample to acetone, methanol, ethanol, triethylamine, ethyl acetate, and benzene were 0.46, 0.5, 0.43, 0.8, 0.16, and 0.15, respectively. For N0 sample, the responses to methanol and triethylamine were higher than acetone. The responses of N1 sample to acetone, methanol, ethanol, triethylamine, ethyl acetate, and benzene were 4.5, 3.5, 2.6, 3.2, 1.3, and 0.6, respectively. N1 sample showed the highest response to acetone among these VOC gases.
The gas sensitivity of the sensor depended on the gas reaction on the surface. The reaction energy and the number of gas molecules were most important factors in the reaction. With the temperature increased, sufficient reaction energy was obtained by the gas, but the desorption rate of gas was accelerated. Due to the combined effect of these two factors, the optimal reaction temperatures for various gas-sensing materials were different.26 Under the optimal operating temperature, the surface reaction intensity reached the maximum, and N1 sample obtained the maximum response.
Long-term stability is an important indicator to measure the performance of a sensor. Fig. 6(c) shows the cycle test to acetone of N1 sample. During the cycle test, the sensor achieved the repeated response–recovery, which shows a good stability. As shown in Fig. 6(d), the response curves and values of the sensor were almost unchanged over one month, which shows a good long-term stability.
3.3 Gas-sensing mechanism
Acetone sensitivity of the pure NiO and In2O3–NiO nanofibers mainly depended on the adsorption and reaction of gases on the surface. According to the above response curves, the NiO and In2O3–NiO nanofibers both showed the gas sensing properties of p-type semiconductor. When the samples were placed in air, oxygen gas molecules were adsorbed on the surface and form oxygen ions (O2−, O−, O2−) by capturing electrons from the materials. As exposed to acetone atmosphere, the adsorbed oxygen ions reacted with acetone. With the reaction, the electrons captured by oxygen were released back to the materials and recombined with the holes, which resulted the decrease of the holes. For p-type semiconductor, the majority carriers were holes. Therefore, the resistances of the samples increased and the current decreased. When acetone was exhausted, oxygen gas molecules were re-adsorbed on the surface of the materials and the currents returned to the original values. The reaction processes could be expressed as follows:31| O2(ads) + e− → O2−(ads) |
| O2−(ads) + e− → 2O−(ads) |
| CH3COCH3(gas) + 4O2−(ads) → 3H2O(gas) + 3CO2(gas) + 4e− |
| CH3COCH3(gas) + 8O−(ads) → 3H2O(gas) + 3CO2(gas) + 8e− |
The improved acetone sensitivity of the In2O3–NiO nanofibers was mainly attributed to the following reasons. First, inhibition of grains growth. The In2O3 inhibited the growth of NiO crystal leading to a reduction of the grains size. Small-sized particles possessed larger specific surface area which provided more adsorption sites.32 Second, improvement of surface characteristics. The amount of oxygen defects and adsorbed oxygen on the surface of the heterogeneous nanofibers increased, which promoted the gas adsorption and reaction.27
Moreover, adjustment of heterojunction was the most important reason. Fig. 7(a) shows the band structure of NiO and In2O3 in air. The hole accumulation layer formed near the surface of NiO, and its resistance depended on the thickness of the hole accumulation layer. As shown in Fig. 7(b), because of the difference in the Fermi energy level of NiO and In2O3, the electrons from conduction band of In2O3 were transferred to the valence band of NiO to maintain the Fermi energy level balance. The p–n heterojunctions formed near the contact interface of In2O3 and NiO crystal grains in the composite nanofibers, and a hole depletion layer formed on the side of NiO. As a conduction channel, the hole accumulation layer was narrowed down due to the appearance of the depletion layer, resulting in an increase of the resistance.33 When the acetone gas reacted on the surface of the In2O3–NiO nanofibers, more electrons were transferred from the side of In2O3 to the side of NiO, causing the hole depletion layer to further widen. In this way, the conduction channel was further narrowed down, resulting in a further increase of the resistance. With the adjustment effect of the heterojunction on the conduction channel, the resistance changes of the In2O3–NiO nanofibers became larger, and the composite nanofibers exhibited a higher acetone-sensing response.
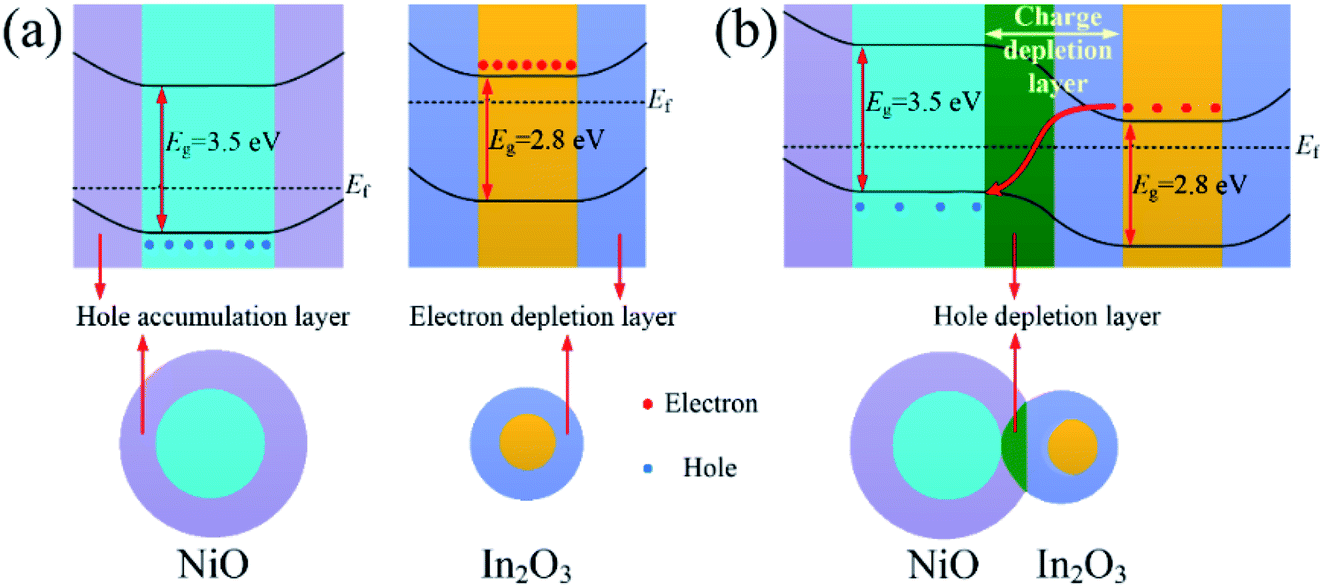 | ||
| Fig. 7 Band structure of (a) NiO and In2O3 and (b) In2O3–NiO in air. | ||
4. Conclusions
The In2O3–NiO heterogeneous nanofibers with different In concentration were prepared by electrospinning. In element mainly presented in the In2O3–NiO nanofibers as the second phase of In2O3, and a small amount of In entered into the lattice of NiO crystal. Because of the existence of In2O3, the growth of NiO grains was inhibited. Compared with the pure NiO nanofibers, the surface oxygen defects and the adsorbed oxygen increased on the surface of the heterogeneous nanofibers. Because of the adjustment of heterojunction, the inhibition of grain growth and the improvement of surface properties, the heterogeneous nanofibers exhibited acetone ultra-sensing properties. For the In2O3–NiO nanofibers, the response to 50 ppm acetone was more than 10 times higher than that of the pure NiO nanofibers, and the minimum detection limit reached 10 ppb. The heterogeneous nanofibers also exhibited the improved response–recovery rate. In addition, the composite fibers showed the better acetone selectivity and long-term stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Zhejiang Provincial Natural Science Foundation (LQ20F010001) and Scientific Research Project of Education Department of Zhejiang Province (Y201839153).References
- J. C. Andersom, W. J. Lamm and M. P. Hlastala, Measuring airway exchange of endogenous acetone using a single-exhalation breathing maneuver, J. Appl. Physiol., 2006, 100, 880–889 CrossRef PubMed.
- K. He, Z. Jin, X. F. Chu, W. J. Bi, W. Wang, C. Wang and S. T. Liu, Fast response–recovery time toward acetone by a sensor prepared with Pd doped WO3 nanosheets, RSC Adv., 2019, 9, 28439–28450 RSC.
- Y. Xiong, X. Chang, X. R. Qiao, K. Li, L. Zhu, F. J. Xia, X. F. Li, Q. B. Zheng, W. Xing and Q. Z. Xue, Co-MOF-74 derived Co3O4/graphene heterojunction nanoscrolls for ppb-level acetone detection, Sens. Actuators, B, 2019, 300, 127011 CrossRef CAS.
- S. X. Cao, L. L. Peng, B. T. Liu, T. Han, C. Zhao, D. C. Zhu and Y. Y. Tang, Hydrothermal synthesis of novel lotus-root slice NiO architectures with enhanced gas response properties, J. Alloys Compd., 2019, 798, 478–483 CrossRef CAS.
- Q. J. Wang, C. Y. Li, P. Sun, F. M. Liu, G. Y. Lu and X. Li, Hydrothermal and sintering synthesis of porous sheet-like NiO for xylene gas sensor, Mater. Res. Express, 2019, 6, 1150e6 CrossRef.
- C. Su, L. Zhang, Y. T. Han, C. Ren, X. W. Chen, J. Hu, M. Zeng, N. T. Hu, Y. J. Su, Z. H. Zhou and Z. Yang, Controllable synthesis of crescent-shaped porous NiO nanoplates for conductometric ethanol gas sensors, Sens. Actuators, B, 2019, 296, 126642 CrossRef CAS.
- H. J. Kim and J. H. Lee, Highly sensitive and selective gas sensors using p-type oxide semiconductors: overview, Sens. Actuators, B, 2014, 192, 607–627 CrossRef CAS.
- X. G. San, M. Li, D. Y. Liu, G. S. Wang, Y. B. Shen, D. Meng and F. L. Meng, A facile one-step hydrothermal synthesis of NiO/ZnO heterojunction microflowers for the enhanced formaldehyde sensing properties, J. Alloys Compd., 2018, 739, 260–269 CrossRef CAS.
- U. T. Nakate, R. Ahmad, P. Patil, Y. S. Wang, K. S. Bhat, T. Mahmoudi, Y. T. Yu, E. K. Suh and Y. B. Hahn, Improved selectivity and low concentration hydrogen gas sensor application of Pd sensitized heterojunction n-ZnO/p-NiO nanostructures, J. Alloys Compd., 2019, 797, 456–464 CrossRef CAS.
- J. N. Zhang, H. B. Lu, C. Liu, C. J. Chen and X. Xin, Porous NiO-WO3 heterojunction nanofibers fabricated by electrospinning with enhanced gas sensing properties, RSC Adv., 2017, 7, 40499 RSC.
- M. U. Haq, Z. Wen, Z. Y. Zhang, S. Khan, Z. R. Lou, Z. Z. Ye and L. P. Zhu, A two-step synthesis of nanosheet-covered fibers based on α-Fe2O3/NiO composites towards enhanced acetone sensing, Sci. Rep., 2018, 8, 1705 CrossRef PubMed.
- K. Xu, S. L. Duan, Q. Tang, Q. Zhu, W. Zhao, X. Yu, Y. Yang, T. Yu and C. L. Yuan, P-N heterointerface-determined acetone sensing characteristics in α-MoO3@NiO core@shell nanobelts, CrystEngComm, 2019, 21, 5834–5844 RSC.
- G. Kavitha, K. T. Arul and P. Babu, Enhanced acetone gas sensing behavior of n-ZnO/p-NiO nanostructures, J. Mater. Sci.: Mater. Electron., 2018, 29, 6666–6671 CrossRef CAS.
- S. Choi, J. K. Lee, W. S. Lee, C. Lee and W. I. Lee, Acetone sensing of multi-networked WO3-NiO core-shell nanorod sensors, J. Korean Phys. Soc., 2017, 71, 487–493 CrossRef CAS.
- G. J. Sun, H. Kheel, S. Choi, S. K. Hyun and C. Lee, Prominent gas sensing performance of TiO2-core/NiO-shell nanorod sensors, J. Nanosci. Nanotechnol., 2017, 17, 4099–4102 CrossRef CAS.
- J. W. Ma, H. Q. Fan, W. M. Zhang, J. N. Sui, C. Wang, M. C. Zhang, N. Zhao, A. K. Yadav, W. J. Wang, W. Q. Dong and S. R. Wang, High sensitivity and ultra-low detection limit of chlorine gas sensor based on In2O3 nanosheets by a simple template method, Sens. Actuators, B, 2020, 305, 127456 CrossRef CAS.
- P. F. Zhou, Y. B. Shen, W. Lu, S. K. Zhao, T. T. Li, X. X. Zhong, B. Y. Cui, D. Z. Wei and Y. H. Zhang, Highly selective NO2 chemiresistive gas sensor based on hierarchical In2O3 microflowers grown on clinoptilolite substrates, J. Alloys Compd., 2020, 828, 154395 CrossRef CAS.
- D. P. Xue, S. S. Zhang and Z. Y. Zhang, Hydrothermal synthesis of methane sensitive porous In2O3 nanosheets, Mater. Lett., 2019, 252, 169–172 CrossRef CAS.
- J. Cao, N. R. Zhang, S. M. Wang, C. Chen and H. M. Zhang, Researching the crystal phase effect on gas sensing performance in In2O3 nanofibers, Sens. Actuators, B, 2020, 305, 127475 CrossRef CAS.
- X. M. Hou, Y. K. Sun and B. Bai, Fabrication of cubic p–n heterojunction-like NiO/In2O3 composite microparticles and their enhanced gas sensing characteristics, J. Nanomater., 2016, 7589028 Search PubMed.
- H. J. Kim, H. M. Jeong, T. H. Kim, J. H. Chung, Y. C. Kang and J. H. Lee, Enhanced ethanol sensing characteristics of In2O3-decorated NiO hollow nanostructures via modulation of hole accumulation layers, ACS Appl. Mater. Interfaces, 2014, 6, 18197–18204 CrossRef CAS PubMed.
- C. H. Feng, X. Y. Kou, B. Chen, G. B. Qian, Y. F. Sun and G. Y. Lu, One-pot synthesis of In doped NiO nanofibers and their gas sensing properties, Sens. Actuators, B, 2017, 253, 584–591 CrossRef CAS.
- T. Han, S. Y. Ma, X. L. Xu, X. H. Xu, S. T. Pei, Y. Tie, P. F. Cao, W. W. Liu, B. J. Wang, R. Zhang and J. L. Zhang, Rough SmFeO3 nanofibers as an optimization ethylene glycol gas sensor prepared by electrospinning, Mater. Lett., 2020, 268, 127575 CrossRef CAS.
- X. Y. Huang, Z. T. Chi, J. Liu, D. H. Li, X. J. Sun, C. Yan, Y. C. Wang, H. Li, X. D. Wang and W. F. Xie, Enhanced gas sensing performance based on p-NiS/n-In2O3 heterojunction nanocomposites, Sens. Actuators, B, 2020, 304, 127305 CrossRef CAS.
- K. C. Hsu, T. H. Fang, I. T. Tang, Y. J. Hsiao and C. Y. Chen, Mechanism and characteristics of Au-functionalized SnO2/In2O3 nanofibers for highly sensitive CO detection, J. Alloys Compd., 2020, 822, 153475 CrossRef CAS.
- X.-X. Fan, X.-L. He, J.-P. Li, X.-G. Gao and J. Jia, Ethanol sensing properties of hierarchical SnO2 fibers fabricated with electrospun polyvinylpyrrolidone template, Vacuum, 2016, 128, 112–117 CrossRef CAS.
- C. Yan, H. B. Lu, J. Z. Gao, G. Q. Zhu, F. Yin, Z. B. Yang, Q. R. Liu and G. Li, Synthesis of porous NiO-In2O3 composite nanofibers by electrospinning and their highly enhanced gas sensing properties, J. Alloys Compd., 2017, 699, 567–574 CrossRef CAS.
- J. Zhang, D. W. Zeng, Q. Zhu, J. J. Wu, Q. W. Huang and C. S. Xie, Effect of nickel vacancies on the room-temperature NO2 sensing properties of mesoporous NiO nanosheets, J. Phys. Chem. C, 2016, 120, 3936–3945 CrossRef CAS.
- X. X. Fan, Y. J. Xu, C. Y. Ma and W. M. He, In situ growth of Co3O4 nanoparticles based on electrospray for an acetone gas sensor, J. Alloys Compd., 2021, 854, 157234 CrossRef CAS.
- R. Zhang, J. W. Shi, T. T. Zhou, J. C. Tu and T. Zhang, A yolk-double-shelled heterostructure-based sensor for acetone detecting application, J. Colloid Interface Sci., 2019, 539, 490–496 CrossRef CAS PubMed.
- Y. Lu, Y. H. Ma, S. Y. Ma, W. X. Jin, S. H. Yan, X. L. Xu, X. H. Jiang, T. T. Wang, H. M. Yang, H. Chen and Z. Qiang, Synthesis of cactus-like NiO nanostructure and their gas-sensing properties, Mater. Lett., 2016, 164, 48–51 CrossRef CAS.
- C. Wang, J. Y. Liu, Q. Y. Yang, P. Sun, Y. Gao, F. M. Liu, J. Zheng and G. Y. Lu, Ultrasensitive and low detection limit of acetone gas sensor based on W-doped NiO hierarchical nanostructure, Sens. Actuators, B, 2015, 220, 59–67 CrossRef CAS.
- H. Y. Gao, Q. Yu, K. Chen, P. Sun, F. M. Liu, X. Yan, F. M. Liu and G. Y. Lu, Ultrasensitive gas sensor based on hollow tungsten trioxide-nickel oxide (WO3-NiO) nanoflowers for fast and selective xylene detection, J. Colloid Interface Sci., 2019, 535, 458–468 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |