
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening hydrolysis of spiro-epoxyoxindoles using a reusable sulfonic acid functionalized nitrogen rich carbon catalyst†
Parth Patel ab,
Raj Kumar Takac,
Bhavesh Parmarcd,
Shilpa Dabasac,
Brijesh Patelac,
Eringathodi Sureshcd,
Noor-ul H. Khan*abc and
Saravanan Subramanian*ac
ab,
Raj Kumar Takac,
Bhavesh Parmarcd,
Shilpa Dabasac,
Brijesh Patelac,
Eringathodi Sureshcd,
Noor-ul H. Khan*abc and
Saravanan Subramanian*ac
aInorganic Materials and Catalysis Division, CSIR-Central Salt & Marine Chemicals Research Institute, G. B. Marg, Bhavnagar-364002, Gujarat, India. E-mail: khan251293@yahoo.co.in; saravanans@csmcri.res.in
bCharotar University of Science and Technology, Changa, Anand-388421, Gujarat, India
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
dAnalytical and Environmental Science Division and Centralized Instrument Facility CSIR-Central Salt & Marine Chemicals Research Institute, G. B. Marg, Bhavnagar-364 002, Gujarat, India
First published on 1st April 2021
Abstract
Controlling the product selectivity of a ring-opening hydrolysis reaction remains a great challenge with mineral acids and to an extent with homogeneous catalysts. In addition, even trace amounts of metal impurities in a bioactive product hinder the reaction progress. This has necessitated the development of robust and metal-free catalysts to offer an alternative sustainable route. We report a nitrogen-rich sulfonated carbon as a catalyst derived from an inexpensive precursor for the synthesis of bioactive vicinal diols of spiro-oxindole derivatives. The well-characterized catalyst shows wide generality with different electronic and steric substituents in the substrates under mild reaction conditions. Hot filtration test confirms no leaching of the acid moiety and the catalyst could be reused for four cycles with retention of activities.
Introduction
The development of an effective strategy for the construction of small molecules with biologically relevant molecular architectures has been the subject of intense research in recent years.1–7 3,3-Substituted oxindoles are fascinating molecules with unique structural skeleton pertaining to several complex natural products such as XEN 402, chitosenine, spirotryprostatin A, (−)-horsfiline (Fig. 1a).8–10 In an ideal scenario, the transformation of isatin derivatives to spiro-epoxyindoles followed by a ring-opening reaction and/or installation of functionalities leads to an oxindole core, with varying degree of substitutions for broad spectrum of applications.11–26 The densely functionalized spiro-fused heterocyclic motif has drawn much attention of biologists and synthetic chemists. In this class of molecules, 3-hydroxy, 3-substituted-2-oxindole derivatives are powerful building blocks and exhibit significant biological activities such as proteasome inhibitory activity27 and other therapeutic activities28 (Fig. 1b). Given the ubiquity of the hydroxy substituted oxindole core, the ring-opening of spiro-epoxyoxindoles avails a simple and practical approach to achieve vicinal diols and alkoxyalcohols with a spiro-quaternary center. In fact, the hydrolysis of epoxide is an important reaction in living organisms for the detoxification of exogeneous substances catalyzed by epoxide hydrolases.29,30 Vicinal diols, in turn, are versatile intermediates for antifreezes, polyester resins, pharmaceuticals, and cosmetics.31 Ring-opening of epoxides by various nucleophiles has been reported using Lewis acids, Lewis bases, one-electron transfer reagents, metal complexes and supported metal oxides.32–37 However, there remain challenges such as low regioselectivity in the case of Lewis acid catalyzed ring-opening of epoxides with alcohols, and in case of metal complexes, deactivation occurs through the loss of counterions during reactions, which are yet to overcome.35 On the other hand, the increasing environmental concerns have triggered the exploration of reusable solid-acid catalysts, and becoming more important in academic and industrial research.38–46 While gratifyingly acknowledging the literature reports and their proven value, still there has been a constant pursuit for a cost-effective catalytic system that could work under mild reaction conditions resulting the product with high yield and selectivity. Recently, carbon-based metal-free catalysts have been studied extensively due to the availability of abundant sources and low cost nature. Among the carbon materials, heteroatom doped carbon has received special attention in diverse catalytic applications. Particularly, nitrogen doped carbon materials are quite well-known for tuning the electronic distribution of the carbon-based network due to its size similarity with carbon and higher electronegativity. On the other hand, sulfonic acid functionalized carbon materials, a class of solid-protonic acids characterized by its high Brønsted acidity, low-production cost and eco-friendly alternative to liquid H2SO4, are promising catalysts for industrial applications.47 Ironically, SO3H-functionalized carbon materials have not been explored for their catalytic activity on the hydrolysis of epoxides. Encouraged by this observation and our ongoing interest in spiro-epoxyoxindole derivatives48 and the development of a cost-effective bifunctional catalytic system,30,49 herein we report the synthesis of nitrogen-rich sulfonated carbon as a cost-effective and reusable heterogeneous catalyst for the first-time and its application in the ring-opening reaction of bioactive spiro-epoxyoxindole derivatives. The developed heterogeneous catalytic system features (a) an inexpensive, reusable and metal-free catalyst, (b) mild reaction conditions, and (c) high yields of desired vicinal diol products. | ||
| Fig. 1 (a) Examples of biological important spirocyclic oxindoles, (b) examples of 3-hydroxy, 3-substituted-2-oxindole derivatives. | ||
Results and discussion
Developing a catalyst for transformations involving bioactive starting materials is not a trivial task. The construction of tertiary alcohols bearing different carbon substituents is challenging and has a potential role in biological applications.50,51 With this in mind, we are interested in studying the hydrolytic ring-opening of spiro-epoxyoxindoles that lead to the formation of differently substituted alcohols. We commenced our catalytic study using p-toluenesulfonic acid (p-TSA) as a homogeneous catalyst for the hydrolysis of N-benzyl spiro-epoxyoxindole as a model substrate using dry acetonitrile at 50 °C (Table 1, Entry 1). As expected, the formation of a diol product was observed, but not as a sole-product, along with a noticeable amount of N-benzyl isatin, the precursor of the starting epoxide. This is possibly due to the strong acidity of p-TSA and being in the same phase of the starting material. This finding shed light to develop a heterogeneous solid-acid catalyst for liquid phase hydrolytic ring-opening reaction. Thus, to retain the catalytic performance of sulfonic acid, we envisaged to introduce the same functionality on the surface of the nitrogen-doped carbon support and make the catalytic process an alternative to conventional acids. On the outset based on our previous experience, we envisaged that a catalyst developed from a cucurbit[6]uril (CB6) based molecule will meet the demands since it (1) is easy to synthesize and scale up, (2) has rich carbon and heteroatom (nitrogen and oxygen) ratio, and (3) is tunable for further functionalization. Consequently, we initiated the synthesis from commercially available and inexpensive reagents following the previously reported procedure52 with slight modifications. The obtained white CB6 powder was used further to prepare Cal-CB6 by pyrolysis and subsequently, upon sulfonation resulted in the black SO3H@N–C/CB6 (Fig. 2). The prepared catalyst was characterized using a series of analytical techniques.
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
| a Reaction conditions: N-benzyl spiro-epoxyoxindole (0.2 mmol), H2O (300 μL), 20 mg catalyst, 0.5 mL dry CH3CN, at 50 °C for 24 h.b Reaction time – 3 h. Conversion was determined by using gas chromatography. | |||
| 1 | PTSAb | 96 | 32 |
| 2 | SO3H@N–C/CB6 | 97 | 99 |
| 3 | CB6 | 18 | 82 |
| 4 | Cal-CB6 | 9 | 91 |
| 5 | SO3H@CB6 | 58 | 52 |
| 6 | Amberlyst-15 | 32 | 46 |
| 7 | SO3H@C | 43 | 87 |
| 8 | No catalyst | n.r. | — |
| 9 | SO3H@CHT | 43 | 99 |

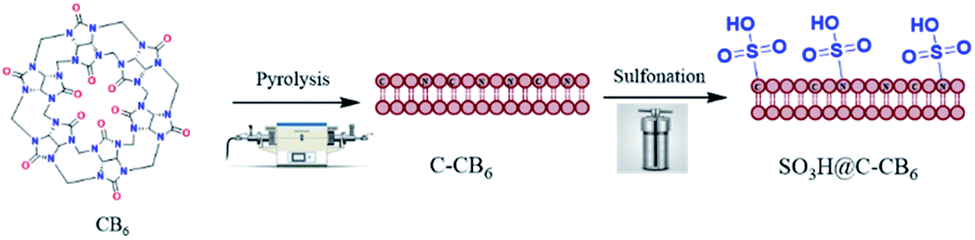 | ||
| Fig. 2 Preparation of sulfonic acid functionalized nitrogen-rich carbon SO3H@N–C/CB6. | ||
The functional groups attached to Cal-CB6 and SO3H@N–C/CB6 were evaluated by FT-IR spectroscopy (Fig. 3a). Cal-CB6 showed a peak at 1631 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations in carboxylic acid and 3200–3500 cm−1 due to O–H stretching vibration in the hydroxyl group. Upon sulfonation, SO3H@N–C/CB6 shows distinct peaks at 1064 and 1224 cm−1, which are assigned to symmetric OS stretching vibrations of sulfonic groups. This confirms that the SO3H groups were successfully anchored to the Cal-CB6 framework.
O stretching vibrations in carboxylic acid and 3200–3500 cm−1 due to O–H stretching vibration in the hydroxyl group. Upon sulfonation, SO3H@N–C/CB6 shows distinct peaks at 1064 and 1224 cm−1, which are assigned to symmetric OS stretching vibrations of sulfonic groups. This confirms that the SO3H groups were successfully anchored to the Cal-CB6 framework.
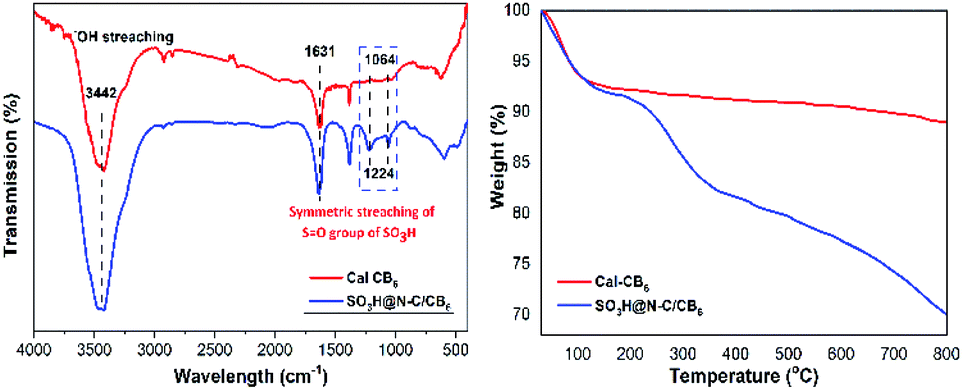 | ||
| Fig. 3 (a) FTIR analysis (b) TGA analysis of Cal-CB6 and SO3H@N–C/CB6. | ||
The TGA curves of SO3H@N–C/CB6 and Cal-CB6 are depicted in Fig. 3b. SO3H@N–C/CB6 shows a two-step weight loss; an initial 8% weight loss was observed from 30 to 140 °C due to the evaporation of moisture and volatile material. However, Cal-CB6 shows lower weight loss than SO3H@N–C/CB6 due to the presence of a sulfonic group along with captured moisture in the latter. Furthermore, a second weight loss in Cal-CB6 (1%) and SO3H@N–C/CB6 (10%) occurred in the 140−350 °C temperature region.
The XRD pattern exhibits (Fig. S2†) two broad, weak diffraction peaks (2θ = 15–30°, 40–50°) attributed to amorphous carbon composed of aromatic carbon sheets oriented in a considerably random fashion.
The XPS survey spectrum of the sulfonated material SO3H@N–C/CB6 is provided in Fig. 4. It demonstrates five peaks centered at approximately 168, 233, 284.5, 401 and 531.5 eV. These can be assigned to S 2p, S 2s, C 1s, N 1s and O 1s, respectively. The C1s spectrum can be deconvoluted into three peaks including CC at 284.8 eV, C–S, C–N at 286.5 eV and OC–O at 289.1 eV. There are two peaks associated with the O 1s spectrum, centered at 531.6 eV (C–O) and 532.7 eV (S–O). The S2p core level of SO3H@N–C/CB6 could be deconvoluted into two peaks at 168.5 eV (S–OH) and 169.6 eV (SO) and thus this result confirms the presence of the SO3H groups in SO3H@N–C/CB6.53 The surface morphology of the synthesized sulfonated material was characterized by scanning electron microscopy (Fig. S3†) and the analyzed images showed irregular hollow network with amorphous carbon nature.
 | ||
| Fig. 4 XPS profile (a) survey, (b) C 1s, (c) O 1s, (d) S 2p of SO3H@N–C/CB6. | ||
With the characterized catalyst, we then focused on the catalytic study of the synthesized material. Indeed, the reaction catalyzed by SO3H@N–C/CB6 afforded the desired product in 97% conversion (Table 1, Entry 2). In order to understand the catalyst activity, we screened different materials such as CB6, C-CB6, SO3H@CB6, Amberlyst-15, and SO3H@C (sulfonated carbon) under the same reaction conditions.
The results are summarized in Table 1; the study using CB6 and Cal-CB6 depicts the role of sulfonation in the catalytic activity (Entries 3 and 4), whereas, the materials such as SO3H@CB6, Amberlyst-15, and SO3H@C also resulted in slightly improved conversion of 58, 32, and 43%, respectively. The lower activity of Amberlyst-15 indicates the strong influence of its inherent acidity on the epoxide, resulting in the reaction to be unselective. Furthermore, under our reaction conditions, the hydrolysis of the model substrate does not proceed without the catalyst (Entry 8).
In order to investigate further the effect of nitrogen doped carbon, we tested the sulphonic acid functionalized chitosan54 and observed a conversion of 43% under the optimized reaction conditions. This might be due to the change in physical and chemical features with the varied amount of nitrogen and sulphonic acid functionalization in the material.
Encouraged with the catalytic activity, we then screened other reaction parameters such as temperature, solvent and catalyst loading. With temperature variation studies, we identified that the reaction becomes sluggish by decreasing the temperature from 50 °C (Chart 1a). Solvents play a key role in the ring-opening reaction of epoxides. Thus, we then varied the solvent and a significant solvent effect was observed for the ring-opening hydrolysis of the model substrate and the results are displayed in Chart 1b. Other than acetonitrile, all the other polar aprotic solvents resulted in the hydrolysed product in less conversion.
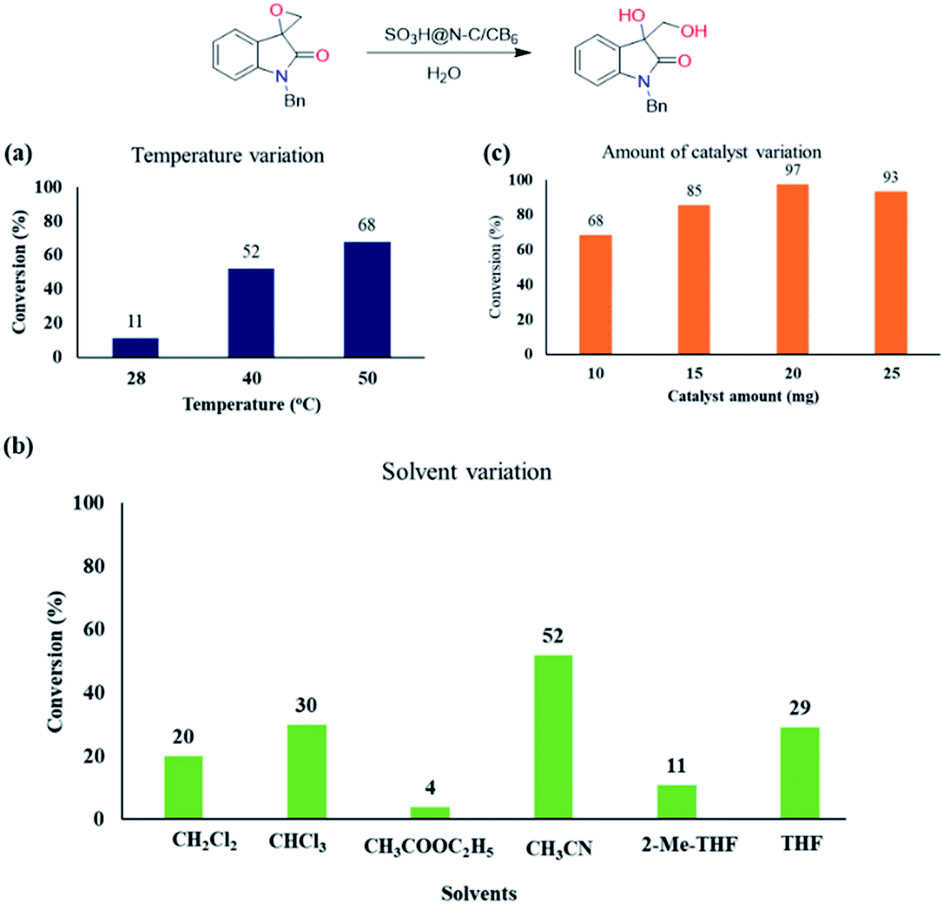 | ||
| Chart 1 Optimization of reaction conditionsa. Reaction conditions (a): Catalyst (20 mg), N-benzyl spiro-epoxyoxindole (0.2 mmol), H2O (300 μL), dry CH3CN (0.5 mL), time 24 h. (b): Catalyst (20 mg), N-benzyl spiro-epoxyoxindole (0.2 mmol), H2O (300 μL), solvents (0.5 mL), time 24 h. (c): Catalyst (mg), N-benzyl spiro-epoxyoxindole (0.2 mmol), H2O (300 μL), dry CH3CN (0.5 mL), Time 24 h. aConversion was determined by gas chromatography. | ||
Next, we optimized the catalyst loading by varying it in the series of 10 to 25 mg (Chart 1c). On decreasing the catalyst loading from 20 to 10 and 15 mg, the yield of the diol product also decreased from 97% to 85% and 70%, respectively, maintaining the same reaction time of 24 h. Increasing the catalyst loading to 25 mg showed detrimental effect in the product formation. Thus, we optimized the catalyst loading as 20 mg for the ring-opening hydrolysis of spiro-epoxyindoles.
Substrate scope
With the optimal reaction condition in hand, the scope and limitation of the catalyst was examined for the hydrolysis of various substrates (Fig. 5). The effect of protecting groups on the oxindole derivative was also investigated and observed excellent yields of the desired product (2a–2c). In general, most of the studied substrates underwent a smooth and clean reaction to afford the vicinal diol product with good to excellent yields, but the substrates having withdrawing groups gave slightly less yield (2f–2i). Furthermore, we extended the protocol for hydrochlorination and alcoholysis with excellent yield (2k and 2j, Fig. 6) and the characterization of the product confirms the regioselectivity of the process.55 | ||
| Fig. 5 Substrate (0.2 mmol), H2O (300 μL), catalyst (20 mg), CH3CN (0.5 mL), 50 °C. Conversion was determined by gas chromatography. | ||
 | ||
| Fig. 6 (a) Alcoholysis and (b) hydrochlorination of spiro-epoxyoxindoles using SO3H@N–C/CB6. Substrate (0.2 mmol), nucleophile (300 μL in case of 2j and 2.2 equiv. in 2k), catalyst (20 mg), CH3CN (0.5 mL), 50 °C. Conversion was determined by gas chromatography. | ||
It is essential to confirm the molecular structure of a new molecule with single-crystal XRD analysis; thereby, we tried recrystallizing the vicinal diol products and the quality of the obtained crystals was good enough to confirm the structures. Apart from this, all the products were characterized by 1H and 13C NMR spectroscopy, and the crystal structures of five new substituted 3,3-dihydroxy oxindole products have been established (Fig. 7).
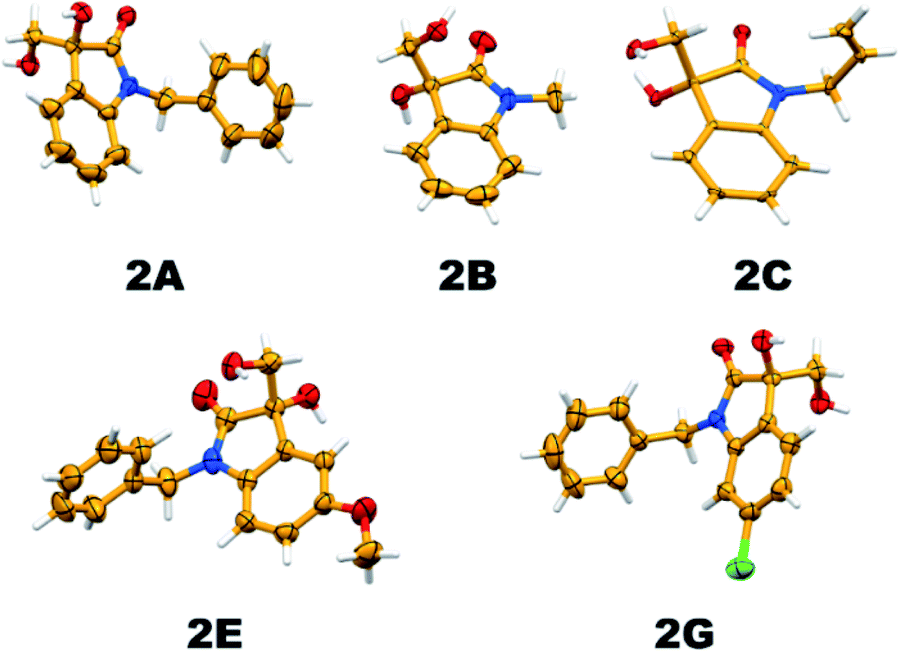 | ||
| Fig. 7 Thermal ellipsoid plot depicting the crystal structure of hydrolysis products (2A, 2B, 2C, 2E and 2G; 40% probability factor for the thermal ellipsoids). | ||
Based on the catalytic study, a plausible mechanism for the ring-opening hydrolysis of spiro-epoxyindoles is proposed (Scheme 1). Initially, the spiro-epoxide gets protonated (I) by the sulfonic acid moiety in the catalyst. The protonated epoxide then opened up the ring via SN1 fashion to form the benzylic carbocationic intermediate (II).56 Successively, the oxygen from the water molecule attacks the generated carbocation intermediate, followed by the abstraction of a proton by the nitrogen-rich center of the catalyst, resulting in the smoother formation of the diol product.
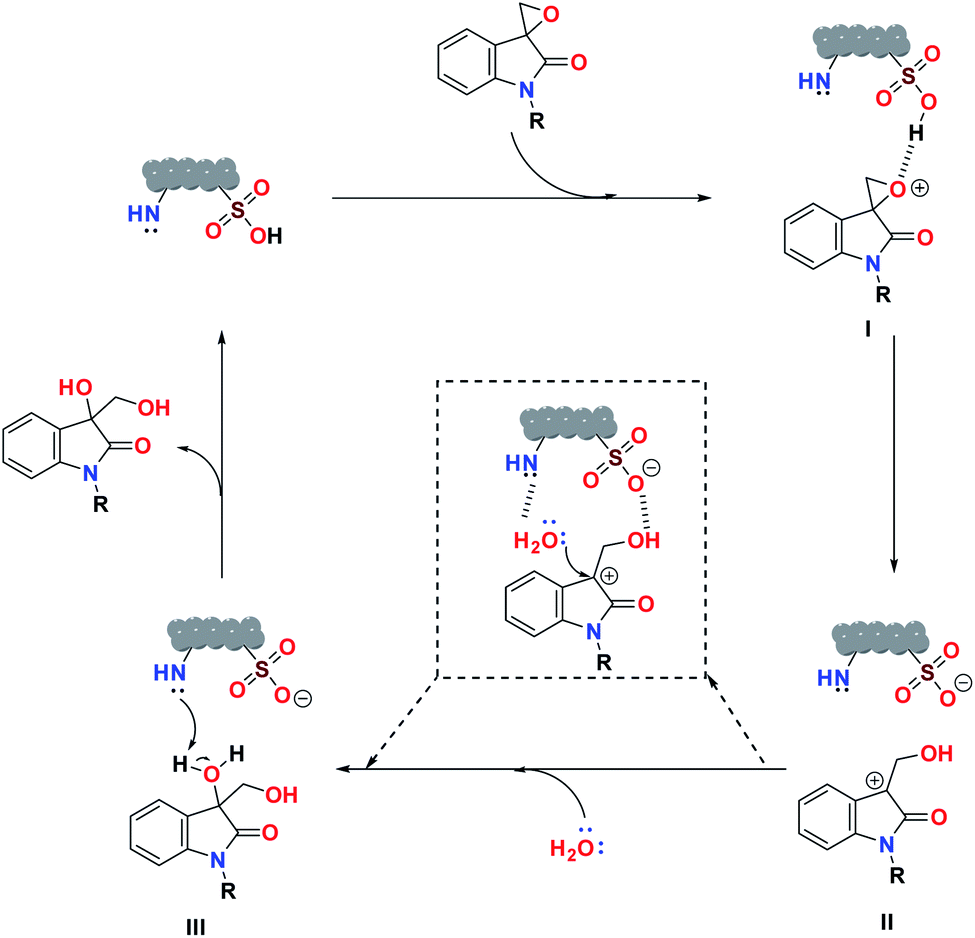 | ||
| Scheme 1 A plausible mechanism for the ring-opening hydrolysis of spiro-epoxyindoles catalyzed by SO3H@N–C/CB6 | ||
Catalyst reusability
The reusability reactions were carried out as described in the experimental procedure. The reusability of SO3H@N–C/CB6 was tested by carrying out four runs (Fig. 8). After each run, the catalyst was recovered by filtration and was washed with acetone and dried at 80 °C for 6 h in a vacuum oven. The dried catalyst was then used for the next cycle. The catalyst showed retention of activity for four cycles with good yield. | ||
| Fig. 8 Reusability study of SO3H@N–C/CB6 catalyst for the hydrolytic ring-opening reaction of spiro-epoxyoxindoles. | ||
Hot filtration test
To examine the leaching of the sulfonic acid group at reaction temperature, a hot filtration test was performed. Then, ring-opening of N-benzylspiroepoxide with water using the catalyst SO3H@N–C/CB6 and acetonitrile as solvent at 50 °C occurred. After 4 h of reaction time, the solid SO3H@N–C/CB6 catalyst was filtered off and the reaction mass was allowed to react further. We found that no further reaction occurred after the hot filtration test. This experimental finding suggests that there is no leaching from the SO3H@N–C/CB6 catalyst during the progress of a reaction.Conclusions
A highly active SO3H@N–C/CB6 catalyst was synthesized by a calcination process followed by its functionalization using sulfuric acid. The catalytic system has the following salient features: (a) the catalyst was thermally stable, (b) the catalyst eliminates intraparticle diffusion resistance, and (c) the catalyst does not show any leaching behavior. SO3H@N–C/CB6 proves to be efficient for the synthesis of versatile vicinal diol products, which is difficult to achieve with homogeneous catalysts. The catalyst was well-characterized using XRD, FT-IR spectroscopy, TGA, XPS and elemental analysis. The catalyst can be reused four times with minor changes in the catalytic activity.Experimental section
All reagents and solvents were purchased from commercial sources and were used without further purification. NMR spectra were obtained on a Bruker spectrometer at (600 MHz and 151 MHz for 1H and 13C NMR, respectively) and referenced internally with TMS; splitting patterns were reported as s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet, br = broad. IR spectra were recorded using the KBr pellet method on a Perkin-Elmer GX FTIR spectrometer. For each IR spectra, 10 scans were recorded at 4 cm−1 resolution. TGA analysis was carried out using Mettler Toledo Star SW 8.10. TG analysis was performed in nitrogen environment while the heating rate was ramped from room temperature to 800 °C at 10 °C min−1. Powder X-ray diffraction (PXRD) data were collected using a PANalytical Empyrean (PIXcel 3D detector) system with CuKα radiation. Single crystal structures were determined using a BRUKER D8 QUEST diffractometer.Synthesis of glycoluril and CB6
Glycoluril was synthesized according to the previously published procedure.52 Typically, in a single necked 500 mL RB flask, 39–40% glyoxal (40 mL; 0.35 mol) and urea (53.4 g, 0.89 mol) were taken, and the resulting solution was stirred for 30 min. Subsequently, water (80 mL) was added to the reaction mixture and further stirred for 20 min followed by the addition of 8.5 mL conc. HCl in 20 mL of water very slowly. After the complete addition of acid (pH = 1.5–2), the reaction mixture was refluxed on an oil bath that generated a white solid precipitate. The heating and stirring were continued for about 60 min, then it was cooled to room temperature, poured into ice water, filtered, and the white solid was washed with cold water (3 × 50 mL) and with acetone (2 × 40 mL). The resulting white solid was allowed to dry for 6 h at 85 °C (yield: 32.70 g, 66%).CB6 was synthesized according to the previous procedure.57 In a 250 mL RB flask, which was equipped with a condenser and dean-stark apparatus, glycoluril (15 g, 106 mmol) and 37% aq. formaldehyde were taken to which conc. H2SO4 (15 mL) and water (100 mL) were added, and the resulting mixture was heated for 12 h. During that time water was removed from the reaction mixture, and after that the reaction mixture was stirred at 160 °C for the next 24 h. Then the reaction mixture was cooled to room temperature and poured into water (250 mL). A yellowish precipitate was filtered off, and the solid mass was dissolved in conc. HCl. Next, the clear brown solution was diluted with water to get a white precipitate that was washed with water and dried at 140 °C (yield: 81%).
Procedure for pyrolysis of CB6
The dried CB6 was first ground to fine powder and transferred to a calcination boat. Then, the calcination boat was placed in a calcination tubular furnace for pyrolysis at 800 °C with a temperature gradient of 5 °C min−1 for 2 h under argon atmosphere. The entire pyrolysis process was carried out in an inert atmosphere until the calcined material was cooled down to <40 °C. After cooling the furnace temperature (<40 °C), its closed chamber was opened, and the black colored Cal-CB6 was obtained (40–42% yield).N. B. The temperature of the furnace should be cooled down to <40 °C under the inert atmosphere. Otherwise, the carbon powder burns instantly with air contact.
Procedure for sulfonation of Cal-CB6
Sulfonation of Cal-CB6 was performed by adding 2 g of Cal-CB6 to a Teflon autoclave. 20 mL of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentrated H2SO4:HNO3 was added slowly to the Teflon vessel, and the slurry was mixed well for 10 min and allowed to attain room temperature. The Teflon autoclave was kept in a heating oven for 12 h at 150 °C. After heating, the Teflon vessel was allowed to cool down to room temperature. The catalyst slurry was thoroughly washed with ultrapure water until it reaches pH 7, and then dried at 70 °C for 12 h. A total amount of 1.95 g of product was obtained after the purification process.
:1 concentrated H2SO4:HNO3 was added slowly to the Teflon vessel, and the slurry was mixed well for 10 min and allowed to attain room temperature. The Teflon autoclave was kept in a heating oven for 12 h at 150 °C. After heating, the Teflon vessel was allowed to cool down to room temperature. The catalyst slurry was thoroughly washed with ultrapure water until it reaches pH 7, and then dried at 70 °C for 12 h. A total amount of 1.95 g of product was obtained after the purification process.
Typical procedure for the synthesis of spiro-epoxyoxindole
Spiroepoxide was synthesized by the previously reported procedure.58,59 A mixture of trimethylsulfoxonium iodide (2.0 mmol) and cesium carbonate (4.0 mmol) in dry CH3CN was stirred at 50 °C for 1 h under a nitrogen atmosphere to generate the sulfur ylide. An appropriate solution of spiro-epoxyoxindole (2.0 mmol) in dry CH3CN (10 mL) was then added dropwise over 10 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was filtered through a pad of Celite and the filtrate was evaporated to dryness. The crude product was purified by column chromatography using silica gel 100–200 mesh as the stationary phase and n-hexane/ethyl acetate 90:10 as the mobile phase.
Typical procedure for the hydrolysis of spiro-epoxyoxindole
The catalyst (20 mg) was weighed out in an oven dried sample vial. Spiro-epoxyoxindole (0.2 mmol) and dry CH3CN (0.5 mL) was added and stirred. To this solution, the nucleophile (300 μL) was added and subsequently the reaction mixture was heated to 50 °C and allowed to run for 24 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was filtered and the filtrate was evaporated to dryness. The crude product was purified by column chromatography using silica gel 100–200 mesh as the stationary phase and n-hexane/ethyl acetate 90:10 as the mobile phase.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The registration number of this publication is CSIR-CSMCRI – 216/2020. Financial support from DST and CSIR for SRF (PP), RA (BP) and analytical support by AESD&CIF of CSIR-CSMCRI are gratefully acknowledged. SS thanks DST for the INSPIRE Faculty award (DST/INSPIRE/04-I/2017/000003).References
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2003, 2209–2219 CrossRef.
- N. Ye, H. Chen, E. A. Wold, P.-Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS PubMed.
- M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS PubMed.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- S. Subramanian, H. A. Patel, Y. Song and C. T. Yavuz, Adv. Sustainable Syst., 2017, 1, 1700089 CrossRef.
- S. B. Annes, R. Saritha, S. Subramanian, B. Shankar and S. Ramesh, Green Chem., 2020, 22, 2388–2393 RSC.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933 CrossRef CAS PubMed.
- Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein, V. Ferey, J.-C. Carry, J. R. Deschamps, D. Sun and S. Wang, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed.
- G. Zhu, G. Bao, Y. Li, W. Sun, J. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2017, 56, 5332–5335 CrossRef CAS PubMed.
- S. Hajra, S. Maity and S. Roy, Adv. Synth. Catal., 2016, 358, 2300–2306 CrossRef CAS.
- B. M. Trost and M. K. Brennan, Synthesis, 2009, 2009, 3003–3025 CrossRef.
- F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165–5181 RSC.
- L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef CAS.
- Y. Liu, H. Wang and J. Wan, Asian J. Org. Chem., 2013, 2, 374–386 CrossRef CAS.
- M. M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef CAS.
- H. Wu, Y.-P. He and F. Shi, Synthesis, 2015, 47, 1990–2016 CrossRef CAS.
- M. M. C. Lo, C. S. Neumann, S. Nagayama, E. O. Perlstein and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 16077–16086 CrossRef CAS PubMed.
- X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819–13825 CrossRef CAS PubMed.
- A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2010, 12, 5254–5257 CrossRef CAS PubMed.
- D. Xie, L. Yang, Y. Lin, Z. Zhang, D. Chen, X. Zeng and G. Zhong, Org. Lett., 2015, 17, 2318–2321 CrossRef CAS PubMed.
- T. Arai, H. Ogawa, A. Awata, M. Sato, M. Watabe and M. Yamanaka, Angew. Chem., Int. Ed., 2015, 54, 1595–1599 CrossRef CAS PubMed.
- P.-F. Zheng, Q. Ouyang, S.-L. Niu, L. Shuai, Y. Yuan, K. Jiang, T.-Y. Liu and Y.-C. Chen, J. Am. Chem. Soc., 2015, 137, 9390–9399 CrossRef CAS PubMed.
- B. K. Albrecht and R. M. Williams, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11949–11954 CrossRef CAS PubMed.
- P. Sharma, K. R. Senwar, M. K. Jeengar, T. S. Reddy, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Eur. J. Med. Chem., 2015, 104, 11–24 CrossRef CAS PubMed.
- M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed., 2014, 53, 8142–8145 CrossRef CAS PubMed.
- S. Nandi, P. Patel, N.-u. H. Khan, A. V. Biradar and R. I. Kureshy, Inorg. Chem. Front., 2018, 5, 806–813 RSC.
- A. Lundin, I. Panas and E. Ahlberg, Phys. Chem. Chem. Phys., 2007, 9, 5997–6003 RSC.
- B. Tang, W. Dai, G. Wu, N. Guan, L. Li and M. Hunger, ACS Catal., 2014, 4, 2801–2810 CrossRef CAS.
- A. Parulkar, R. Joshi, N. Deshpande and N. A. Brunelli, Appl. Catal., A, 2018, 566, 25–32 CrossRef CAS.
- A. Parulkar, A. P. Spanos, N. Deshpande and N. A. Brunelli, Appl. Catal., A, 2019, 577, 28–34 CrossRef CAS.
- N. Deshpande, A. Parulkar, R. Joshi, B. Diep, A. Kulkarni and N. A. Brunelli, J. Catal., 2019, 370, 46–54 CrossRef CAS.
- B. Tang, W.-C. Song, S.-Y. Li, E.-C. Yang and X.-J. Zhao, New J. Chem., 2018, 42, 13503–13511 RSC.
- R. Tak, M. Kumar, T. Menapara, N. Gupta, R. I. Kureshy, N.-u. H. Khan and E. Suresh, Adv. Synth. Catal., 2017, 359, 3990–4001 CrossRef CAS.
- W. M. Van Rhijn, D. E. De Vos, B. F. Sels and W. D. Bossaert, Chem. Commun., 1998, 317–318, 10.1039/A707462J.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448–2459 CrossRef CAS.
- J. A. Melero, G. D. Stucky, R. van Grieken and G. Morales, J. Mater. Chem., 2002, 12, 1664–1670 RSC.
- D. Das, J.-F. Lee and S. Cheng, J. Catal., 2004, 223, 152–160 CrossRef CAS.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403–9413 CrossRef CAS PubMed.
- Q. Yang, J. Liu, J. Yang, M. P. Kapoor, S. Inagaki and C. Li, J. Catal., 2004, 228, 265–272 CrossRef CAS.
- Q. Yang, M. P. Kapoor, S. Inagaki, N. Shirokura, J. N. Kondo and K. Domen, J. Mol. Catal. A: Chem., 2005, 230, 85–89 CrossRef CAS.
- D. J. Macquarrie, S. J. Tavener and M. A. Harmer, Chem. Commun., 2005, 2363–2365, 10.1039/B501431J.
- B. Rác, Á. Molnár, P. Forgo, M. Mohai and I. Bertóti, J. Mol. Catal. A: Chem., 2006, 244, 46–57 CrossRef.
- L. J. Konwar, P. M. Arvela and J. P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS PubMed.
- R. K. Tak, P. Patel, S. Subramanian, R. I. Kureshy and N. H. Khan, ACS Sustainable Chem. Eng., 2018, 6, 11200–11205 CrossRef CAS.
- P. Patel, S. Nandi, M. S. Maru, R. I. Kureshy and N. H. Khan, J. CO2 Util., 2018, 25, 310–314 CrossRef CAS.
- F. Kühn, S. Katsuragi, Y. Oki, C. Scholz, S. Akai and H. Gröger, Chem. Commun., 2020, 56, 2885–2888 RSC.
- Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS.
- S. Nandi, P. Patel, A. Jakhar, N. H. Khan, A. V. Biradar, R. I. Kureshy and H. C. Bajaj, ChemistrySelect, 2017, 2, 9911–9919 CrossRef CAS.
- M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi and A. Rossi, RSC Adv., 2015, 5, 75953–75963 RSC.
- J. H. Advani, A. S. Singh, N. H. Khan, H. C. Bajaj and A. V. Biradar, Appl. Catal., B, 2020, 268, 118456 CrossRef CAS.
- R. K. Tak, N. Gupta, M. Kumar, R. I. Kureshy, N. H. Khan and E. Suresh, Eur. J. Org. Chem., 2018, 2018, 5678–5687 CrossRef CAS.
- S. Hajra, S. Roy, S. Maity and S. Chatterjee, J. Org. Chem., 2019, 84(4), 2252–2260 CrossRef CAS PubMed.
- P. Patel, S. Nandi, T. Menapara, A. V. Biradar, R. K. Nagarale, N. H. Khan and R. I. Kureshy, Appl. Catal., A, 2018, 565, 127–134 CrossRef CAS.
- B. Parmar, P. Patel, R. S. Pillai, R. K. Tak, R. I. Kureshy, N. H. Khan and E. Suresh, Inorg. Chem., 2019, 58, 10084–10096 CrossRef CAS PubMed.
- R. K. Tak, P. Patel, S. Subramanian, R. I. Kureshy and N. H. Khan, ACS Sustainable Chem. Eng., 2018, 6, 11200–11205 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1961234–1961238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01161h |
| This journal is © The Royal Society of Chemistry 2021 |