
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of biogenic amines in alcoholic beverages using a novel fluorogenic compound as derivatizing reagent†
Fangfang Zhu‡
,
Honglong Liu‡,
Wenqing Zhang,
Chuanyuan Du,
Hao Zhu,
Xiaodi Du,
Xiaosong Hu * and
Yawei Lin*
* and
Yawei Lin*
Department of Chemistry, School of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, 430070 Wuhan, China. E-mail: xhu@whut.edu.cn; linyawei2012@whut.edu.cn
First published on 1st June 2021
Abstract
Biogenic amines (BAs) are organic nitrogenous compounds that are responsible for several biological events. If their concentration reaches the threshold level, it can cause mild to serious health problems in human. A novel bis-styrylphenyl Meldrum's acid derivative (BSMAD) was synthesized and served as a fluorescent turn-on pre-column derivatizing reagent for the quantitative analysis of BAs. A method for the determination of BA by high-performance liquid chromatography (HPLC) was established, which has a low detection limit (0.4 nmol L−1), excellent linearity (R2 ≥ 0.9946) and repeatability (RSD ≤ 3.7% intra-day, RSD ≤ 5.8% inter-day). The proposed method was successfully applied for the determination of BAs in several alcoholic beverages, including yellow wine, red wine, cooking wine, and beer. Satisfactory recoveries were obtained in the range of 94.6–100.5%. Compared with other methods, this pre-column derivatization method using BSMAD is simple, reliable, highly sensitive, and of low interference, providing an effective method for future studies of BAs in different matrices.
1 Introduction
Biogenic amines (BAs) are a class of low molecular weight basic nitrogenous compounds with biological activity, which are usually formed from amino acids by decarboxylation, and by amination and transamination of aldehydes or ketones.1–5 According to the structure of BAs, they can be classified into three different categories: putrescine (PUT), cadaverine (CAD), spermine (SPN), spermidine (SPD), and other aliphatic amines; tyramine (TYM), phenylethylamine (PEA), and other aromatic amines; histamine (HIM), tryptamine (TRM), and other heterocyclic amines. BAs are typical active components in organisms, and an appropriate amount of them can promote normal physiological activities. However, excessive intake of BAs will produce adverse reactions. HIM is the most toxic BA, and excessive HIM can lead to headache, dyspepsia, abnormal blood pressure, and even neurotoxicity. TYM is next most toxic, and excessive TYM can also cause headaches and hypertension. CAD and PUT are less toxic, but they can inhibit HIM and TYM-related metabolic enzyme activities, increasing the amount of them and enhancing bodily discomfort symptoms. TRM and PEA can also lead to hypertension when excessive amounts exist in the human body. SPN and SPD are less toxic and can promote cell proliferation.1–5BAs exist not only in living organisms but also in a variety of foods, especially in fermented foods such as cheese, sausage, beer, wine, rice wine, and condiments. They present in many aquatic and meat products as well. HIM and other BAs have been used as the primary chemical markers of food bacterial spoilage, and their content can serve as an index of food freshness.1–5 The toxicological level of these BAs is very difficult to establish, because it depends on the presence of other amines and each BA has different characteristics. Different concentration thresholds for BAs have been reported in the literature and legal regulations. A concentration threshold of 100 mg HIM/kg food and 2 mg L−1 alcoholic beverage have been suggested. Values of 100–800 mg kg−1 for TYM and PEA have been suggested as the legal upper limit for the existence of them in food.1 The quantitative analysis of BAs in food is of great significance to promote food safety and protect human health, though different concentration threshold of them might exist. More and more studies have been carried out on BAs' quantitative analysis in various kinds of meat products, aquatic products, and other foods.4,6–29
BAs determination in food is usually complicated because their concentrations are low, and the food samples usually need some pre-processing steps. Common methods for BAs determination are chromatographic ones, such as thin-layer chromatography (TLC),6,7 capillary electrophoresis (CE),8–11 gas chromatography (GC),12–16 and high-performance liquid chromatography (HPLC).17–29 HPLC is the most widely used because of its high resolution, sensitivity, efficiency, and reliability. Nevertheless, a single chromatographic approach to simultaneously determine mixture of BAs without derivatization is not feasible due to the lack of adequate chromatographic or absorption/fluorescence properties. A derivatization step is usually necessary before chromatographic separation and detection.
Fluorescent reagents are widely used in the pre-column or post-column derivatization in high-performance liquid chromatography coupled with fluorescence detection (HPLC-FLD), which dramatically enhances the sensitivity of the detection of BAs. 6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC),17,18 dansyl chloride (DNS-Cl),19–23 9-fluorenylmethyl chloroformate (FMOC-Cl),24,25 and O-phthalaldehyde (OPA)26–29 are some popular derivatizing reagents used for the determination of BAs. AQC, DNS-Cl, and FMOC-Cl all have intrinsic fluorescence and may generate fluorescent by-products during the derivatization process, which complicates BAs' quantitative analysis. OPA is a fluorogenic derivatizing reagent, which may avoid the shortcomings of AQC, DNS-Cl, and FMOC-Cl. However, BAs derivatives generated from OPA are usually unstable, and OPA itself is sensitive to air and light. Thus, there is still an urgent need for novel fluorogenic derivatizing reagents for BAs.
In the present work, a novel fluorogenic bis-styrylphenyl Meldrum's acid derivative (BSMAD, Fig. 1) was designed and synthesized for BAs' quantitative analysis. BSMAD has a bis-styrylphenyl structure, which can serve as a fluorogenic moiety30 and a Meldrum's acid derivative form, which can react with amines due to its α,β-unsaturated nature, according to Anslyn group's work.31,32 BSMAD is nonfluorescent itself; however, a fluorescent derivative is formed upon the reaction with nucleophilic amines. This work aimed to achieve a new method using BSMAD as the derivatizing reagent allowing the qualitative and quantitative analysis of BAs in foods with HPLC-FLD. Reaction parameters were optimized to achieve high derivatization yields under mild conditions. The method was validated by analyzing BAs in alcoholic beverages. The study focused on HIM, TYM, TRM, PUT, CAD, and PEA, since they represent BAs with higher toxicity. The established method has a low detection limit (0.4 nmol L−1), excellent linearity (R2 ≥ 0.9946), and high repeatability (RSD ≤ 3.7% intra-day, RSD ≤ 5.8% inter-day), which suggests that it has a great potential in the sensitive analysis of BAs in various sources.
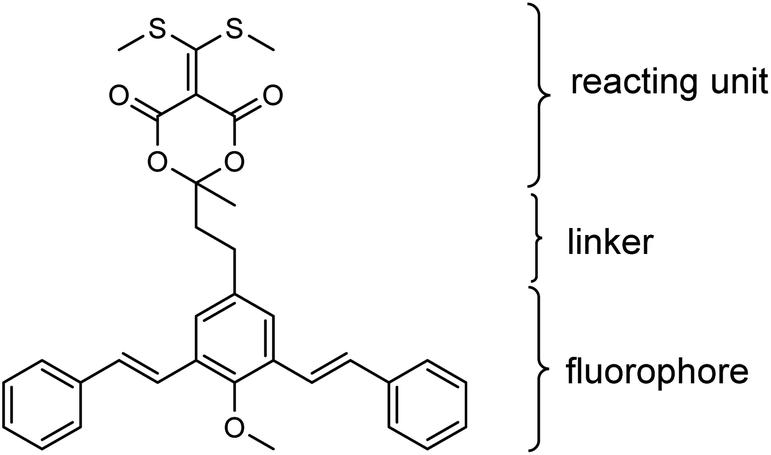 | ||
| Fig. 1 The chemical structure of fluorogenic bis-styrylphenyl Meldrum's acid derivative (BSMAD), the bis-styrylphenyl structure serves as a fluorophore and the Meldrum's acid derivative part serves as a reacting unit able to react with amines. | ||
2 Experimental
2.1 Reagents and chemicals
Unless stated otherwise, all reagents were of analytical grade. BSMAD was synthesized in our laboratory (Section 2.9 and ESI†). 4-(4-Hydroxyphenyl)-2-butanone (99%+) were purchased from Aladdin. Chloroform-d (99.8%+), dimethyl sulfoxide (DMSO, 99.9%+), DMSO-d6 (99.8%+), N,N-dimethylformamide (DMF, 99.9%+), styrene (99.5%+), palladium(II) acetate (99%+), histamine dihydrochloride (HIM, 99+%), tyramine (TYM, 98%+), putrescine dihydrochloride (PUT, 99+%), cadaverine dihydrochloride (CAD, 99+%), tryptamine (TRM, 98%), and 2-phenylethylamine (PEA, 99%) were purchased from Innochem (Beijing, China). Ammonium hydroxide (25–28%), potassium iodide (99.5%+), iodine (99.0%+), sodium thiosulfate pentahydrate (99.5%+), sodium chloride (99.5%+), sodium sulfate (99.5%+), potassium carbonate (99.5%+), concentrated sulfuric acid (99.5%+), acetic anhydride (98.5%+), triethylamine (99.9%), triphenylphosphine (99.5%+), petroleum ether, ethyl acetate (99.5%+), tetrahydrofuran (THF, 99.9%+), potassium hydrogen phthalate, sodium tetraborate, sodium dihydrogen phosphate (99+%), and disodium hydrogen phosphate dodecahydrate (99+%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). N,N-Diisopropylethylamine (99.5%+), methyl iodide (99.5%+), propanedioic acid (99.0%+), carbon disulphide (99.0%), propanone (99.5%+), dichloromethane (99.0%+), triphenylphosphine (99.5%+) were purchased from Shanghai Qinba Chemical Co., Ltd. HPLC grade of acetonitrile (ACN, 99.9%) was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Water was Milli-Q grade. Filter (N66, 13 mm × 0.22 μm) was purchased from Jinteng (Tianjin, China). All HPLC solvents were filtered through a 0.22 μm membrane and degasified in an ultrasonic bath for 30 min.PBS buffer (pH 7.4, 0.2 mol L−1) was prepared by mixing 81.0 mL of disodium hydrogen phosphate (0.2 mol L−1) and 19.0 mL of sodium dihydrogen phosphate (0.2 mol L−1). Unless noted otherwise, the concentration of all PBS reagents in the study was 10 mmol L−1. A specific amount of BSMAD was weighted and dissolved in ACN to prepare a stock solution with a concentration of 10 mmol L−1 as the fluorescent derivatization reagent. Specific amounts of BAs (HIM, TYM, PUT, CAD, TRM, and PEA) were dissolved in ACN-PBS (50/50, v/v) to prepare a stock solution with a concentration of 10 mmol L−1 as the standard solution of BAs. These standard stock solutions were stored in the refrigerating chamber of a refrigerator at −20 °C.
2.2 Apparatus
Analytical TLC was performed with silica gel GF254 plates. The 1H and 13C NMR spectra were recorded at 25 °C using a 500 MHz NMR spectrometer (Bruker Avance III). Chemical shifts (δ) are reported in PPM using tetramethylsilane (TMS) as an internal standard (δ = 0 ppm), or using the deuterated solvent signal as reference. Spin–spin coupling constants (J) are given in Hz. Infrared spectra were registered on a Nicolet iS5 (Thermo Fisher Scientific) in potassium bromide tablets. Chromatographic analyses were performed on an LC-20AT system (Shimadzu, Japan) with a fluorescence detector (RF-20A, Shimadzu, Japan) and a UV detector (SPD-20A, Shimadzu, Japan). Separations were performed on a reversed-phase HPLC column (Agilent Eclipse XDB-C18, 5 μm, 4.6 mm × 150 mm). UV-Vis spectra were recorded with a UV757CRT UV-Vis spectrophotometer (Shanghai spectrum instrument Co., Ltd). Fluorescence spectra were obtained by an LS55 (PerkinElmer, UK) fluorescence spectrometer. A PB-10 pH meter (Sartorius, USA) was used to determine the pH values.2.3 Fluorescent analysis
2 mL of BSMAD (2 μmol L−1) and 2 mL of amine (2 μmol L−1) solution reacted at 25 °C for 2 h to obtain the derivative. The maximum absorption wavelength was obtained by scanning on a UV757CRT UV-Vis spectrophotometer. The derivative's maximum absorption wavelength was then set as the excitation wavelength on an LS-55 fluorescence spectrometer, and the emission spectrum was scanned in the range of 350 to 600 nm. Then the emission wavelength was set at the maximum value, and the excitation wavelength was scanned in the range of 200 to 400 nm to obtain the maximum of it. A similar fluorescent analysis was also carried out on BSMAD itself.2.4 Derivatisation procedure
In the standard derivatization procedure, 60 μL mixture of standard BA sample (HIM, TYM, PUT, CAD, TRM, and PEA, 0.01 mmol L−1), 120 μL BSMAD solution (1 mmol L−1), and 60 μL ACN-PBS (50/50, v/v, pH 7.4) solution were mixed in an Eppendorf tube. It was vortexed and then kept in a thermostatic water-bath (25 °C) for 2 h and then subjected to HPLC-FLD analysis.2.5 Chromatographic separation
Chromatographic analyses were carried out on an HPLC system using a C18 column (5 μm, 4.6 × 150 mm). The excitation and emission wavelengths were set at 310 and 405 nm, respectively. 20 μL of the prepared test solution was injected into the HPLC. Mobile phase A was 97% water/3% tetrahydrofuran, and mobile-phase B was 97% acetonitrile/3% tetrahydrofuran. The elution condition was: 0–60 min, 65% solvent B. The flow rate was kept at 1 mL min−1, and the LC column temperature was set at 25 °C, which was pre-balanced with the mobile phase for 30 min before analysis.2.6 LC-MS analysis
The target analyte was characterized by Q-Exactive plus (Thermo Fisher Scientific). Easy NLC 1000 combined with InertSustain-C18 column (5 μm, 4.6 mm × 150 mm, Agilent eclipse) was used to separate the analytes. The positive ion mode was used for detection, and the scanning range was set at 50–2000 m/z. The primary mass spectrometry scanning mode was SIM (selective ion detection) mode. The nano upgraded electric spray ion (ESI) source has a voltage of 2.25 kV and a column temperature of 25 °C. Mobile phase A was 97% water/3% tetrahydrofuran, and mobile phase B was 97% acetonitrile/3% tetrahydrofuran. The elution condition was as follows: 0–60 min, 65% solvent B the flow rate was 300 nL min−1. Xcalibur 3.1 software was used for data processing.2.7 Method validation
The proposed method was validated according to the ref. 33. The linearity was determined by analyzing standard amine mixtures (HIM, TYM, PUT, CAD, TRM, and PEA) with different concentrations, starting from the limit of quantification (LOQ, S/N = 10) values to 5 μmol L−1. Intra-day precision was determined by six replicates analyses of standard amines solution on the same day. Inter-day precision was measured using the same method on three different days under the same operational parameters. Recoveries of the six amines were calculated by analyzing the samples spiked with amine standards at medium and high concentrations (0.4 μmol L−1 and 1.0 μmol L−1) before the sample preparation procedure. The concentration of amines recovered (Crecovered) and amines spiked (Cspiked) were compared to calculate the recovery percentage using the following equation: recovery = [Crecovered/Cspiked] × 100%.2.8 Sample processing
60 μL wine sample was filtered by 0.22 μm filter membrane and then transferred into a 1 mL sample tube. 1 mL ACN-PBS (50/50, v/v) was added to adjust the wine sample's pH to 7.4, which was then used for the following quantitative analysis based on HPLC-FLD.2.9 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as eluent to give compound 1.
:1 as eluent to give compound 1.Compound 1 was a light brown solid, yield: 31.0%; 1H NMR (500 MHz, CDCl3) δ 7.51 (s, 2H), 5.64 (s, 1H), 2.75–2.78 (m, 2H), 2.70–2.73 (m, 2H), 2.15 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 207.1, 151.9, 139.0, 137.2, 82.1, 44.8, 30.1, 27.5; HRMS: m/z calculated for C10H10I2O2 [M + H]+ 416.8843, found 416.8803.
:1 as eluent to give compound 2.Compound 2 was colorless oil, yield: 98.0%; 1H NMR (500 MHz, CDCl3): δ 7.59 (s, 2H), 3.83 (s, 3H), 2.76–2.79 (m, 2H), 2.71–2.74 (m, 2H), 2.16 (s, 3H); 13C NMR (126 MHz, CDCl3): δ 206.8, 157.1, 141.0, 139.6, 90.3, 60.6, 44.5, 30.0, 27.7; HRMS: m/z Calculated for C11H12I2O2 [M + H]+ 430.8999, found 430.9007.
:1 as eluent to give compound 3.Compound 3 was a white solid, yield: 73.0%; 1H NMR (500 MHz, DMSO-d6): δ 7.63 (d, J = 7.5 Hz, 4H), 7.56 (s, 2H), 7.37–7.42 (m, 6H), 7.29–7.32 (m, 4H), 3.74 (s, 3H), 2.86–2.89 (m, 2H), 2.80–2.83 (m, 2H), 2.14 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 208.1, 154.2, 137.7, 130.7, 130.2, 129.2, 128.2, 127.0, 125.9, 122.8, 62.4, 44.7, 30.2, 29.3. HRMS: m/z Calculated for C27H26O2 [M + H]+ 383.2006, found 383.1980.
:1 as eluent to give compound 4.Compound 4 was a white solid, yield: 24.0%; 1H NMR (500 MHz, DMSO-d6): δ 7.62–7.64 (m, 6H), 7.36–7.42 (m, 8H), 7.28–7.33 (m, 2H), 4.24 (d, J = 20.5 Hz, 1H), 3.99 (d, J = 20.5 Hz, 1H), 3.75 (s, 3H), 2.79 (t, J = 8.0 Hz, 2H), 2.36 (t, J = 8.0 Hz, 2H), 1.81 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 164.0, 153.8, 137.2, 136.5, 130.3, 129.8, 128.8, 127.8, 126.5, 125.5, 122.2, 106.8, 62.0, 40.9, 36.7, 28.5, 25.2. HRMS: m/z Calculated for C30H28O5 [M + H]+ 469.2010, found 469.2020.
:1 as eluent to give product BSMAD.BSMAD was a yellow solid, yield: 10.0%; 1H NMR (500 MHz, DMSO-d6): δ 7.63 (d, J = 7.5 Hz, 4H), 7.60 (s, 2H), 7.35–7.42 (m, 8H), 7.28–7.32 (m, 2H), 3.74 (s, 3H), 2.78–2.81 (m, 2H), 2.66 (s, 6H), 2.32–2.35 (m, 2H), 1.79 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 192.1, 159.2, 153.8, 137.2, 136.7, 130.4, 129.8, 128.8, 127.8, 126.5, 125.5, 122.2, 104.1, 103.0, 62.1, 40.6, 28.5, 24.4, 20.9. HRMS: m/z Calculated for C33H32O5S2 [M + H]+ 573.1764, found 573.1764.
3 Results and discussion
3.1 Fluorogenic reagent and its derivatization reaction with TRM
Meta-phenylene vinylene (m-PV) unit has tuneable physical, optical, and electronic properties, making it a popular fluorophore in various research areas.30 We incorporated m-PV moiety into a Meldrum's acid derivative and synthesized compound BSMAD, expecting it to serve as a conjugate acceptor for amines with the fluorescent property. Anslyn group's study on the Meldrum's acid derivative has proved that the α,β-unsaturated nature of it can undergo 1,4-conjugate addition reactions with amine nucleophiles.31,32 The synthetic route of derivative reagent BSMAD is shown in Scheme 1. Firstly, bis-styrylphenyl fluorophore was assembled by Heck reaction, and the keto carbonyl group was then converted to a Meldrum's acid derivative. The characterizations of the intermediates and final product are shown in Fig. S1–S15.†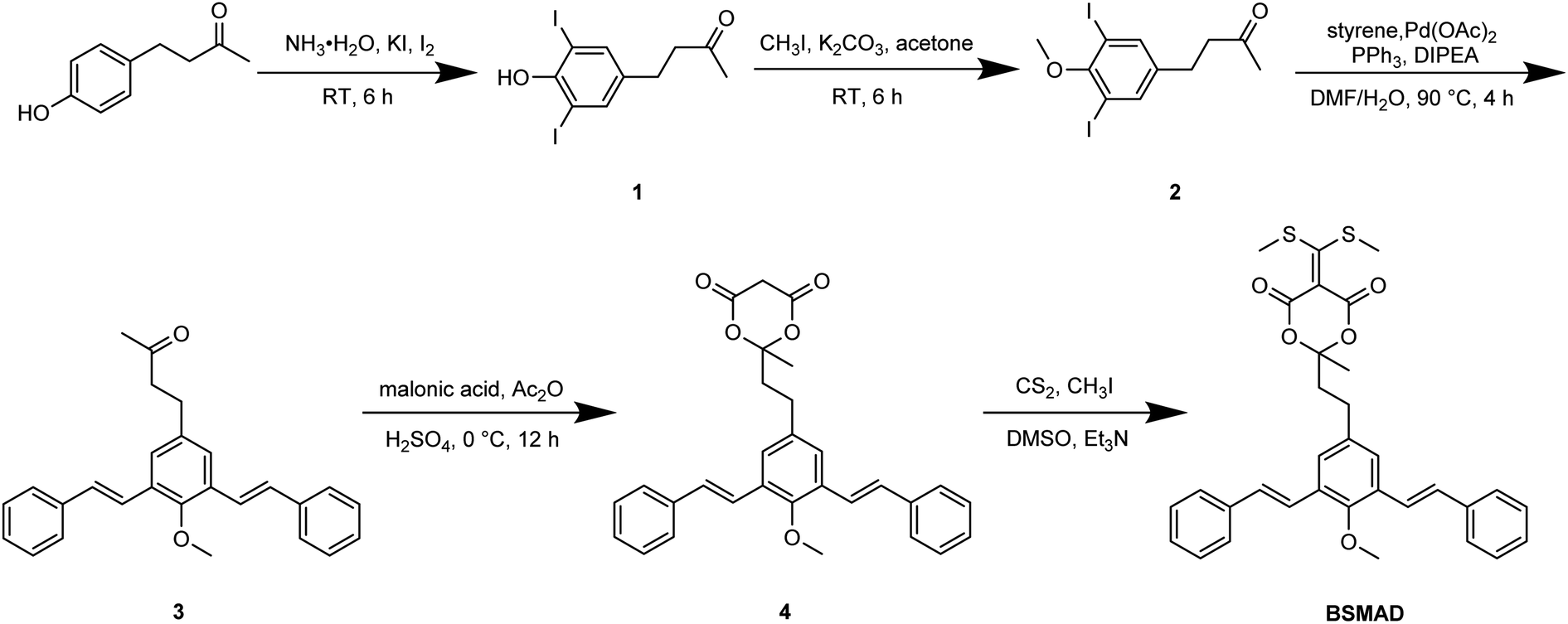 | ||
| Scheme 1 The synthetic route of derivating reagent BSMAD. | ||
Fluorescent analysis on BSMAD showed that it was nonfluorescent. However, when TRM was added to the BSMAD solution, blue fluorescence was turned on almost instantly (Fig. 2). Fluorescent analysis of the resulting solution on a fluorescence spectrometer indicated that a product with a fluorescent excitation wavelength of 310 nm and an emission wavelength of 405 nm formed. Similar fluorescent excitation and emission spectra were aquired for BSMAD derivatives of HIM, TYM, PUT, CAD, and PEA (Fig. S16†).
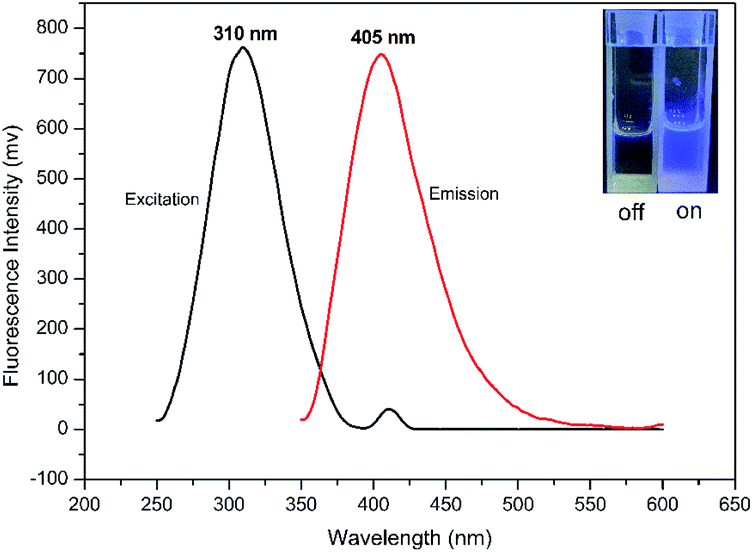 | ||
| Fig. 2 The fluorescent excitation and emission spectra of the derivatization product (TRM-BSMAD), λex = 310 nm, λem = 405 nm. Inset: Images of the solution of BSMAD before and after the reaction with TRM under 365 nm UV light; the BSMAD solution was nonfluorescent and derivatization product (TRM-BSMAD) was fluorescent, indicating the fluorogenic nature of BSMAD. | ||
A possible explanation for the fluorogenic nature of the derivatization reaction between BSMAD and the amine is illustrated in Fig. 3. The sulfur atoms in BSMAD might quench the m-PV units' fluorescence through the process of photoinduced electron transfer (PET). When one of the methylthio group leaves upon the addition of an amine, the PET process is significantly suppressed, and the fluorescence of bis-styrylphenyl fluorophore is thus recovered.
 | ||
| Fig. 3 The fluorogenic reaction of BSMAD with an amine to afford a fluorescent derivative. The addition of an amine to the conjugate acceptor may suppress the PET process, and thus turn on the fluorescence of bis-styrylphenyl fluorophore. | ||
A more detailed derivatization study was carried out using TRM as the model BA to react with BSMAD for 2 h at 25 °C followed by HPLC-FLD analysis. Compared with the blank (see Fig. 4A), one new peak (tR: 35.6 min) is observed in the HPLC chromatogram (Fig. 4B) and identified as the TRM-BSMAD derivative (Fig. 4C). The eluate corresponding to that peak was then collected and subjected to ESI-MS detection. In the ESI-MS spectrum, the [M + H]+, [M + NH4]+ peaks of the TRM-BSMAD derivative (m/z 685.2731, 702.2995) with high intensities are observed, which agree very well with the theoretical value (m/z 685.2731, 702.2995). These results prove that TRM was successfully derivatized with BSMAD to form the TRM-BSMAD.
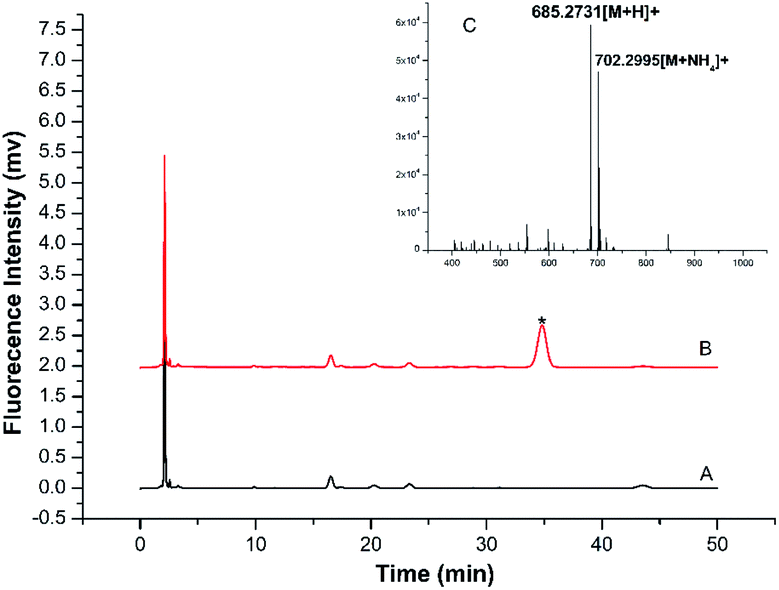 | ||
| Fig. 4 (A) Chromatogram of blank sample (B) representative chromatogram obtained from standard TRM solution (20 μmol L−1) after derivatization with BSMAD. The derivative TRM-BSMAD has a retention time of 35.6 min. The elution followed an isocratic mode and the mobile phase was acetonitrile-water (7.0/3.0, v/v). The flow rate was 1.0 mL min−1. (C) ESI-MS spectrum of TRM-BSMAD, [M + H]+, [M + NH4]+ Calculated: m/z = 685.2731, 702.2995, found: m/z = 685.2731, 702.2995. | ||
3.2 Optimization of derivatization reaction
Some important derivatization reaction conditions were optimized by a single factor optimization method to obtain higher fluorescence intensity. The BSMAD concentration, reaction temperature, reaction time, and pH were optimized using the peak areas of the derivatives of six BAs (0.05 mmol L−1 each) as the evaluation standard. All the reactions were conducted three times in parallel to reduce the error. The results are shown in Fig. 5.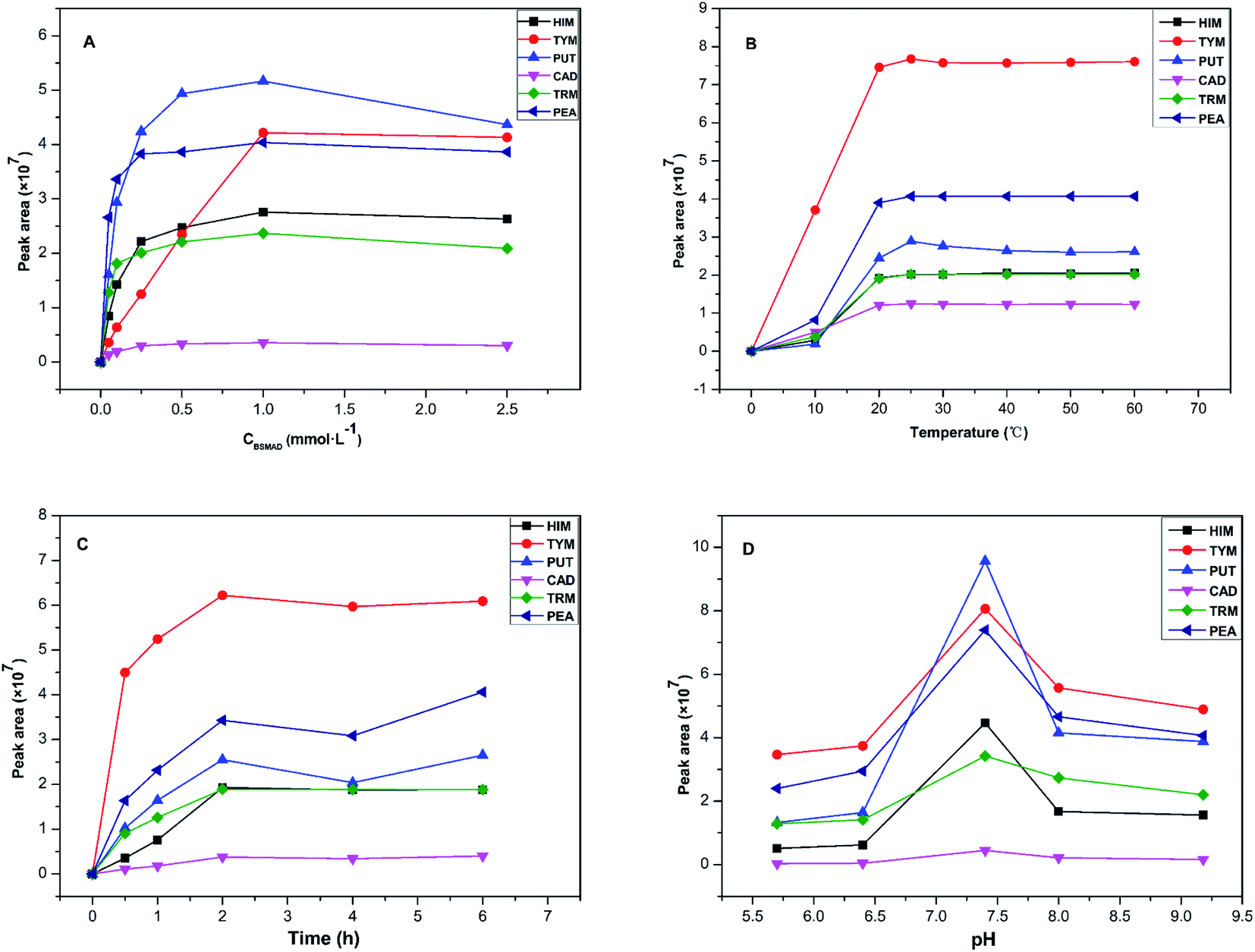 | ||
Fig. 5 Effect of derivatization conditions on peak areas of the derivatives, including (A) derivatization reagent concentration, (B) reaction temperature, (C) reaction time, (D) pH. The data represents the average of the three measurements. ■ = HIM derivative,  = TYM derivative, = TYM derivative,  = PUT derivative, = PUT derivative,  = CAD derivative, = CAD derivative,  = TRM derivative, and = TRM derivative, and  = PEA derivative. = PEA derivative. | ||
In summary, the best pre-column derivatization method was confirmed as follows: a probe concentration of 0.1 mmol L−1, a reaction temperature of 25 °C, a pH value of 7.4, and a reaction time of 2 h.
3.3 Analysis of amine mixtures
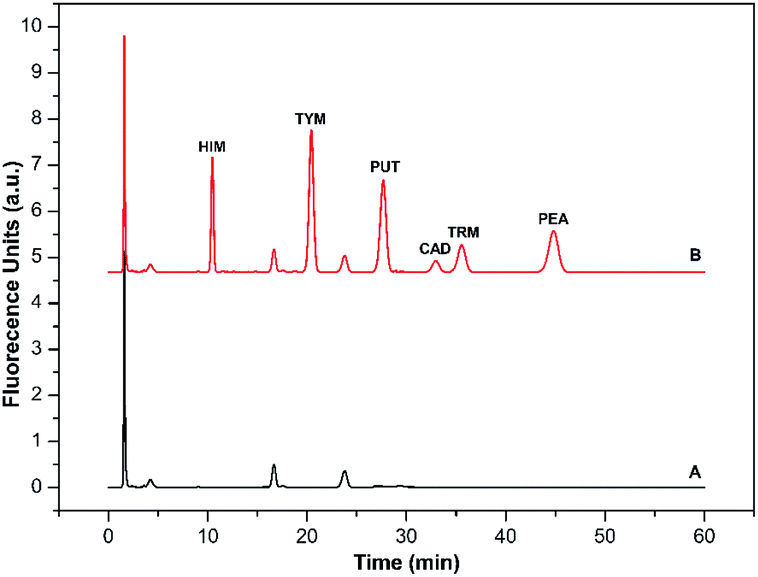 | ||
| Fig. 6 (A) Typical chromatogram of reagent blank carried through the same derivatization procedure except for the addition of BAs; (B) typical chromatogram of a standard mixture of six BAs (5.0 μmol L−1 each). Shown are the derivatives of HIM (tR: 10.2 min), TYM (tR: 20.5 min), PUT (tR: 28.3 min), CAD (tR: 33.7 min), TRM (tR: 35.7 min), and PEA (tR: 45.4 min). Mobile phase: A was 97% water/3% tetrahydrofuran, and B was 97% acetonitrile/3% tetrahydrofuran. The elution condition was 0–60 min, 65% solvent B, flow rate: 1.0 mL min−1. | ||
| Amine | Range (nmol L−1) | Regression equationsa | R2 | LODb (nmol L−1) | LOQb (nmol L−1) | RSDc (%) | |
|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | ||||||
| a X = sample concentration (nmol L−1), Y = peak area (mV min).b Signal-to-noise ratio = 3.c n = 6. | |||||||
| HIM | 2–5000 | Y = 320.61X − 1.34 × 104 | 0.9989 | 0.4 | 1.3 | 2.4 | 3.6 |
| TYM | 2–5000 | Y = 2055.95X + 3.92 × 106 | 0.9946 | 0.4 | 1.3 | 1.4 | 3.3 |
| PUT | 2–5000 | Y = 865.18X + 1.39 × 105 | 0.9961 | 0.8 | 2.7 | 3.7 | 5.8 |
| CAD | 2–5000 | Y = 31.42X + 2.17 × 105 | 0.9957 | 0.8 | 2.7 | 3.3 | 4.2 |
| TRM | 2–5000 | Y = 365.56X − 8.75 × 103 | 0.9986 | 0.8 | 2.7 | 2.9 | 3.9 |
| PEA | 2–5000 | Y = 1040.29X + 1.2 × 105 | 0.9981 | 0.8 | 2.7 | 1.8 | 2.3 |
These results prove that the new derivatization method can provide sensitive and selective BAs analysis with high precision.
3.4 Real sample analysis
BAs exist in living organisms and various foods, especially in fermented ones. To evaluate the method's applicability, six BAs in four alcoholic beverages, including yellow wine, red wine, cooking wine, and beer samples, were analyzed qualitatively and quantitatively. BSMAD was used as the derivatizing reagent, and the whole process was completed under optimal derivatization and chromatographic conditions. After simple sample preparation procedures (described in Section 2.8), the samples were derivatized with BSMAD, and sent directly for HPLC-FLD analysis. The whole real sample analysis took several hours for each one and cost of it is reasonable compared with other approaches. The HPLC results of these alcoholic beverages unspiked and spiked with standard BAs mixture solutions are shown in Fig. 7. All six BAs were well separated and detected without any interference. The detailed data are listed in Tables 2 and 3. It can be seen from the tables that the recoveries of the actual samples are between 94.6% to 100.5%, and the reproducibility of the method is high (RSD is 1.37% to 3.68%). These results show that the established method has good accuracy and reproducibility for the real sample analysis.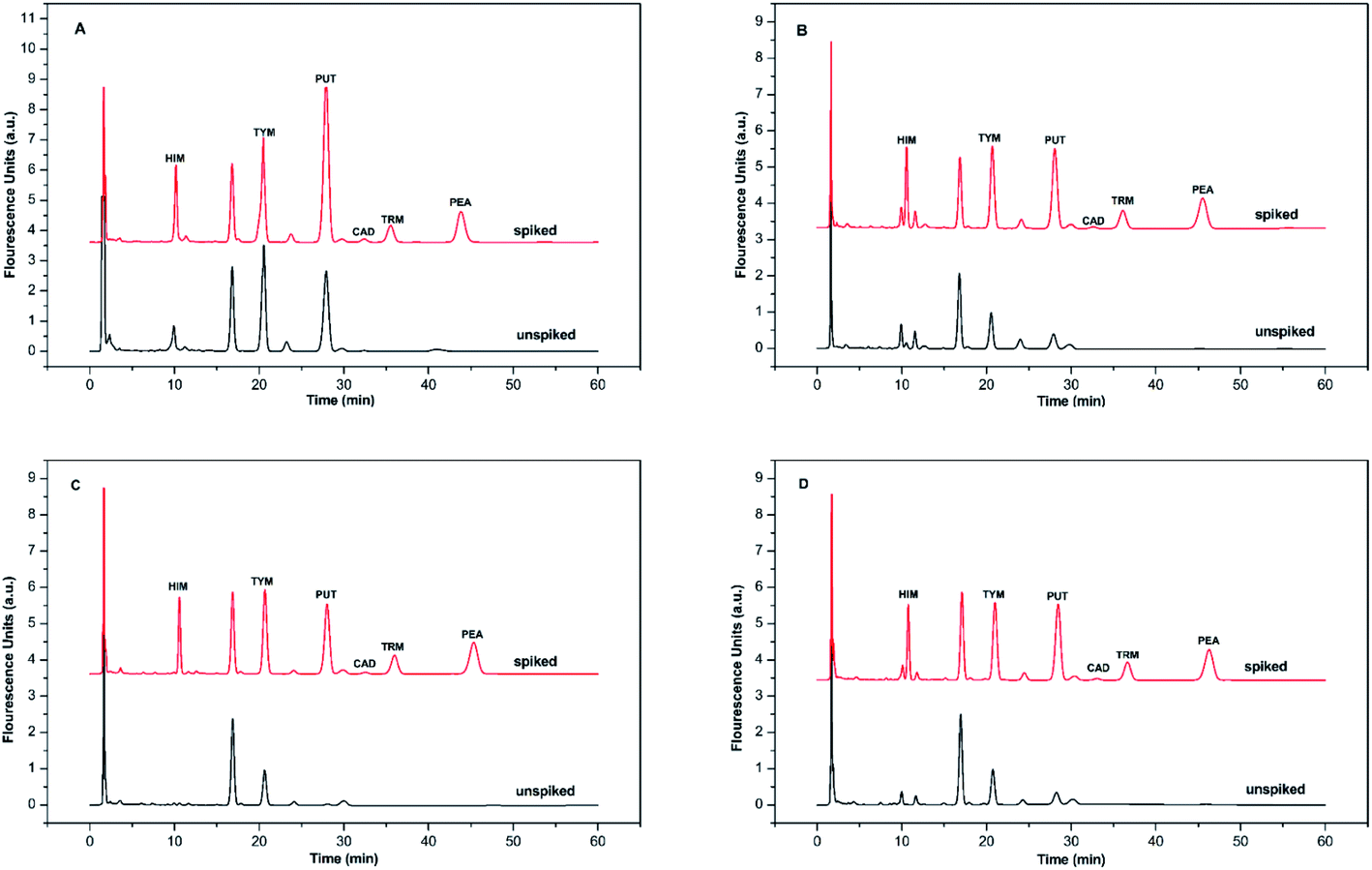 | ||
| Fig. 7 The chromatograms obtained from alcoholic beverage sample and the same sample spiked with HIM, TYM, PUT, CAD, TRM, and PEA, 1.0 mmol L−1 each. Mobile phase: A was 97% water/3% tetrahydrofuran, and B was 97% acetonitrile/3% tetrahydrofuran. The elution condition was 0–60 min, 65% solvent B, flow rate: 1.0 mL min−1. Chromatographic conditions are the same as described in Fig. 6. (A) yellow wine simple, (B) red wine simple, (C) cooking wine simple, (D) beer simple. | ||
| Amines | Content ± SD (μg L−1) | |||
|---|---|---|---|---|
| Yellow wine | Red wine | Cooking wine | Beer | |
| HIM | 13.10 ± 0.31 | 7.84 ± 0.31 | 2.59 ± 0.18 | 1.72 ± 0.31 |
| TYM | 29.39 ± 0.03 | 18.07 ± 0.03 | 15.92 ± 0.12 | 15.23 ± 0.04 |
| PUT | 12.39 ± 0.46 | 8.22 ± 0.46 | 3.36 ± 0.46 | 12.40 ± 0.46 |
| CAD | 5.23 ± 1.32 | 3.33 ± 0.38 | 4.02 ± 0.19 | 3.68 ± 0.22 |
| TRM | — | 2.15 ± 0.47 | — | — |
| PEA | 6.81 ± 0.22 | 5.57 ± 0.52 | — | — |
| Total | 66.92 ± 2.35 | 45.18 ± 2.18 | 25.89 ± 0.95 | 33.03 ± 1.03 |
| Amines | Added (μmol L−1) | Yellow wine | Red wine | Cooking wine | Beer | ||||
|---|---|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| HIM | 0.4 | 97.9 | 1.80 | 98.5 | 3.50 | 98.1 | 4.12 | 100.0 | 2.13 |
| 1.0 | 97.5 | 2.00 | 97.7 | 2.02 | 95.8 | 3.70 | 98.4 | 3.19 | |
| TYM | 0.4 | 102.5 | 2.41 | 98.9 | 1.44 | 95.2 | 3.60 | 96.5 | 2.02 |
| 1.0 | 100.1 | 3.12 | 97.0 | 1.19 | 96.9 | 2.98 | 97.9 | 3.53 | |
| PUT | 0.4 | 100.3 | 3.34 | 99.5 | 2.99 | 94.6 | 2.86 | 100.3 | 1.91 |
| 1.0 | 99.3 | 2.28 | 99.8 | 2.37 | 96.0 | 1.39 | 99.3 | 2.18 | |
| CAD | 0.4 | 98.2 | 1.37 | 96.5 | 2.30 | — | — | — | — |
| 1.0 | 98.8 | 2.00 | 97.0 | 1.58 | — | — | — | — | |
| TRM | 0.4 | 94.7 | 2.75 | 95.5 | 1.23 | — | — | — | — |
| 1.0 | 96.8 | 3.68 | 95.3 | 1.61 | — | — | — | — | |
| PEA | 0.4 | 96.6 | 1.70 | 100.2 | 2.84 | 100.0 | 1.56 | 96.6 | 1.78 |
| 1.0 | 98.0 | 2.03 | 99.7 | 1.95 | 100.5 | 2.79 | 98.0 | 3.42 | |
These results show that HIM, TYM, PUT, and PEA are present in all the alcoholic beverages studied. The contents of HIM, TYM, and PUT are relatively high, indicating that they need to be carefully monitored in these products due to their toxicities. Additionally, no TRM was observed in the yellow wine sample. Neither TRM nor PEA was detected in cooking wine and beer. It is possibly the consequence of different treatment processes in various alcoholic beverages.
3.5 Comparison with other methods
As shown in Table 4, all other methods using pre-column derivatization have LODs higher than 6.98 nmol L−1, while our method has a much lower LOD range, which is between 0.4 and 0.8 nmol L−1. Most of other derivatization reagents need to be pretreated before derivatization. However, our method does not require any prior purification or extraction, and the derivatized sample is directly subjected to HPLC-FLD analysis. Moreover, our derivatization method with BSMAD can be conducted at room temperature. Therefore, our approach has the advantages of high detection sensitivity and good reproducibility. We believe BSMAD is more suitable for qualitative and quantitative BAs analysis according to all these features it owns.| Matrix | Method | Labeling reagent | Extraction methods | Derivatize temperature | LOD (nmol L−1) | Reference |
|---|---|---|---|---|---|---|
| a A solid-phase extraction (SPE) with mixed-mode resins method before HPLC-FLD analysis.b A modified liquid–liquid extraction(LLE) with high performance liquid chromatography-fluorescence detector (HPLC-FLD).c High performance liquid chromatography-fluorescence detector (HPLC-FLD) with ionexchange solid-phase extraction cartridge (PCX-SPE). | ||||||
| Wine | HPLC-FLD | 6-Aminoquinolyl-N-hydroxysuccinimidyl (AQC) | SPE | 65 °C | 68.63–188.15 | 18a |
| Wine | HPLC-FLD | Dansyl chloride (DNS-Cl) | LLE | 60 °C | 6.98–450.45 | 23b |
| Intestine | HPLC-FLD | 9-Fluorenylmethyl chloroformate (FMOC-Cl) | — | 40 °C | 80–400 | 24 |
| Fish | HPLC-FLD | O-Phthalaldehyde (OPA) | PCX-SPE | 25 °C | 16.20 | 28c |
| Alcoholic beverages | HPLC-FLD | Bis-styrylphenyl Meldrum's acid (BSMAD) | — | 25 °C | 0.4–0.8 | This work |
4 Conclusions
In summary, we have detailed a new pre-column derivatization method based on a new fluorogenic probe of BSMAD, which has been applied to determine HIM, TYM, PUT, CAD, TRM, and PEA for the first time. BAs can be labeled with BSMAD with high derivatization yields under mild conditions, producing stable derivatives that have significant signal responses for both fluorescence and MS detection. Additionally, combined with LC separation, the method is selective and of low interference, allowing simultaneous analysis of six BAs in alcoholic beverage samples without complicated purification or extraction procedures. We obtained sub-nmol L−1 level detection of BAs, which is much more sensitive than most other methods. And is good enough for the determination of any BAs in alcoholic beverages or other foods in regard to their concentration thresholds. The results show good reproducibility and high accuracy. We expect that this new probe can find wide applications in the labeling of various BAs and other vital biomolecules bearing amino groups.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21472144), the Fundamental Research Funds for the Central Universities (WUT: 2020IB026 and WUT: 2021IA003), and the National Innovation and Entrepreneurship Training Program for College Students (202010497026) for financial support.References
- M. H. S. Santos, Int. J. Food Microbiol., 1996, 29, 213–231 CrossRef CAS.
- S. Bodmer, C. Imark and M. M. Kneubühl, Inflammation Res., 1999, 48, 296–300 CrossRef CAS PubMed.
- K. J. Broadley, Pharmacol. Ther., 2010, 125, 363–375 CrossRef CAS PubMed.
- M. Papageorgiou, D. Lambropoulou, C. Morrison, E. Kłodzińska, J. Namieśnik and J. Płotka-Wasylka, TrAC, Trends Anal. Chem., 2018, 98, 128–142 CrossRef CAS.
- D. Fayzrakhmanov, K. Ekici, A. K. Omer, B. Ziganshin, F. Nezhmetdinova and R. Shaydullin, BIO Web of Conferences, 2020, vol. 17, p. 00232 Search PubMed.
- E. Garcia-Moruño, A. V. Carrascosa and R. Muñoz, J. Food Prot., 2005, 68, 625–629 CrossRef PubMed.
- A. Costantini, M. Cersosimo, V. D. Prete and E. Garcia-Moruno, J. Food Prot., 2006, 69, 391–396 CrossRef CAS PubMed.
- W. Li, Y. Pan, Y. Liu, X. Zhang, J. Ye and Q. Chu, Chromatographia, 2014, 77, 287–292 CrossRef CAS.
- V. Adımcılar, N. Öztekin and F. Bedia Erim, Food Analytical Methods, 2018, 11, 1374–1379 CrossRef.
- E. A. Gubartallah, A. Makahleh, J. P. Quirino and B. Saad, Molecules, 2018, 23, 1112 CrossRef PubMed.
- X. Sun, X. Yang and E. Wang, J. Chromatogr. A, 2003, 1005, 189–195 CrossRef CAS PubMed.
- C. Almeida, J. O. Fernandes and S. C. Cunha, Food Control, 2012, 25, 380–388 CrossRef CAS.
- M. Papageorgiou, D. Lambropoulou, C. Morrison, J. Namiesnik and J. Plotka-Wasylka, Talanta, 2018, 183, 276–282 CrossRef CAS PubMed.
- J. Plotka-Wasylka, V. Simeonov and J. Namiesnik, J. Chromatogr. A, 2016, 1453, 10–18 CrossRef CAS PubMed.
- J. Huang, N. Gan, F. Lv, Y. Cao, C. Ou and H. Tang, J. Sep. Sci., 2016, 39, 4384–4390 CrossRef CAS PubMed.
- J. Y. Hong, N. H. Park, M. S. Oh, H. S. Lee, H. Pyo and J. Hong, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 940, 94–103 CrossRef CAS PubMed.
- P. Hernandez-Orte, A. Pena-Gallego, M. J. Ibarz, J. Cacho and V. Ferreira, J. Chromatogr. A, 2006, 1129, 160–164 CrossRef CAS PubMed.
- J. L. Ordonez, R. M. Callejon, M. L. Morales and M. C. Garcia-Parrilla, Food Chem., 2013, 141, 2713–2719 CrossRef CAS PubMed.
- J. J. Learey, S. Crawford-Clark, B. J. Bowen, C. J. Barrow and J. L. Adcock, J. Sep. Sci., 2018, 41, 4430–4436 CrossRef CAS PubMed.
- S. J. Liu, J. J. Xu, C. L. Ma and C. F. Guo, Food Chem., 2018, 266, 275–283 CrossRef CAS PubMed.
- C. Molins-Legua, P. Caxmpíns-Falcó, A. Sevillano-Cabeza and M. Pedrón-Pons, Analyst, 1999, 124, 477–482 RSC.
- H. Zhu, S. Yang, Y. Zhang, G. Fang and S. Wang, Anal. Methods, 2016, 8, 3747–3755 RSC.
- Y. Liu, F. Han, Y. Liu and W. Wang, Food Analytical Methods, 2020, 13, 911–922 CrossRef.
- W. Engelmann, A. Zeyner, K. D. Markuske and J. R. Aschenbach, Chromatographia, 2009, 70, 1207–1213 CrossRef CAS.
- V. Lozanov, B. Benkova, L. Mateva, S. Petrov, E. Popov, C. Slavov and V. Mitev, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 860, 92–97 CrossRef CAS PubMed.
- S. Iijima, Y. Sato, M. Bounoshita, T. Miyaji, D. J. Tognarelli and M. Saito, Anal. Sci., 2013, 29, 539–545 CrossRef CAS PubMed.
- A. Weremfo, M. K. Eduafo, H. A. Gyimah and S. Abassah-Oppong, J. Food Qual., 2020, 2020, 1–6 CrossRef.
- A. Kounnoun, M. El Maadoudi, F. Cacciola, L. Mondello, H. Bougtaib, N. Alahlah, N. Amajoud, A. El Baaboua and A. Louajri, Chromatographia, 2020, 83, 893–901 CrossRef CAS.
- G. C. L. Reis, F. B. Custódio, B. G. Botelho, L. R. Guidi and M. B. A. Gloria, J. Food Compos. Anal., 2020, 86, 103375–103382 CrossRef CAS.
- P. Cheruku, J. H. Huang, H. J. Yen, R. S. Iyer, K. D. Rector, J. S. Martinez and H. L. Wang, Chem. Sci., 2015, 6, 1150–1158 RSC.
- K. L. Diehl, I. V. Kolesnichenko, S. A. Robotham, J. L. Bachman, Y. Zhong, J. S. Brodbelt and E. V. Anslyn, Nat. Chem., 2016, 8, 968–973 CrossRef CAS.
- D. A. Fulton, Nat. Chem., 2016, 8, 899–900 CrossRef CAS PubMed.
- FDA, Guidance for Industry, Bioanalytical Method Validation, FDA, CDER, CVM, rev 1, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01436f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |