
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(III)-catalyzed regioselective C–H activation dialkenylation/annulation cascade for rapid access to 6H-isoindolo[2,1-a]indole†
Fangming Zhang,
Xin Zhu,
Bo Luo and
Chengming Wang *
*
Department of Chemistry, College of Chemistry and Materials Science, Jinan University, Guangzhou, 511443, China. E-mail: cmwang2019@jnu.edu.cn
First published on 20th July 2021
Abstract
6H-isoindolo[2,1-a]indoles were accessed via a Rh(III)-catalyzed N–H free indole directed C–H activation dialkenylation/annulation cascade in moderate to excellent yields. This protocol also features: reaction procedures that are insensitive to air and moisture, excellent regioselectivity and good functional group tolerance.
The indole motif, which widely occurs in many natural products, pharmaceuticals and other functional molecules (Fig. 1), is evidently one of the most important skeletons.1 Over the past few decades, people have developed numerous synthetic routes towards indole.2 Among them, direct modification of indoles by transition-metal catalysis through a C–H activation strategy is quite interesting and is undoubtedly of great significance in consideration of atom economy and step simplicity, thus attracting much attention from both academia and industry.2b,3,4
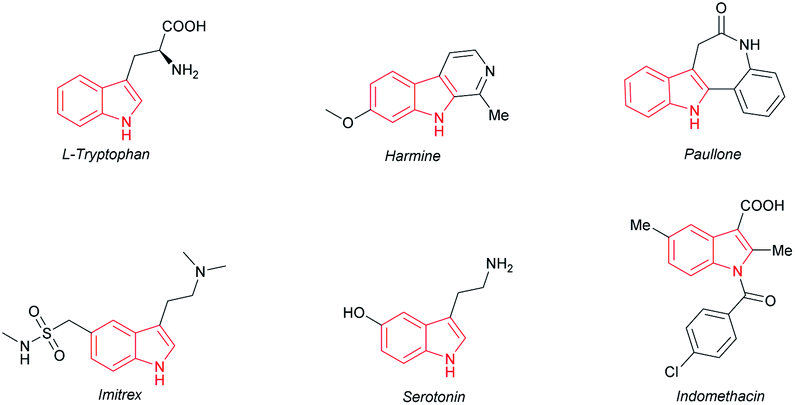 | ||
| Fig. 1 Compounds containing indole motif. | ||
Transition-metal catalyzed oxidative cross-coupling via a C–H activation pathway eliminating the need for preactivated reaction partners, has become one of the most powerful tools for molecule manipulation.2c,5 Since the pioneering work of Murai,6a Fujiwara and Moritani,6b,c this research area has undergone rapid developments. Generally, in order to achieve good reactivity and controlled selectivity, a directing group is usually needed.7 In this regard, various directing groups have been gradually designed and reported, including amides,8a amines,8b alcohols,8c carboxylic acids,8d esters,8e ketones,8f aldehydes,8g triazenes8h and N–H free indoles.9
Meanwhile, in contrast to the well-reported C–H arylation of indoles,10 direct selective alkenylation of 2-position of N–H free indole is still limited and challenging due to the electrophilic nature of the C-2 position.11 In 2005, Gaunt et al. reported Pd-catalyzed selective C-2 alkenylation of indoles,11b but the reaction efficiency is low and high catalyst loading is required (Scheme 1a). Another most effective strategy is pre-installing a directing group into the indole structure to control the selectivity (Scheme 1b).12 For example, Miura et al. disclosed Pd-catalyzed C-2 alkenylation reaction using carboxylic acid as a blocking and directing group.12a Carretero and co-workers explored N-pyridylsulfonyl as a directing group to functionalize indole at the C-2 position employing excess of alkenes in the presence of Pd(II) catalyst.12b
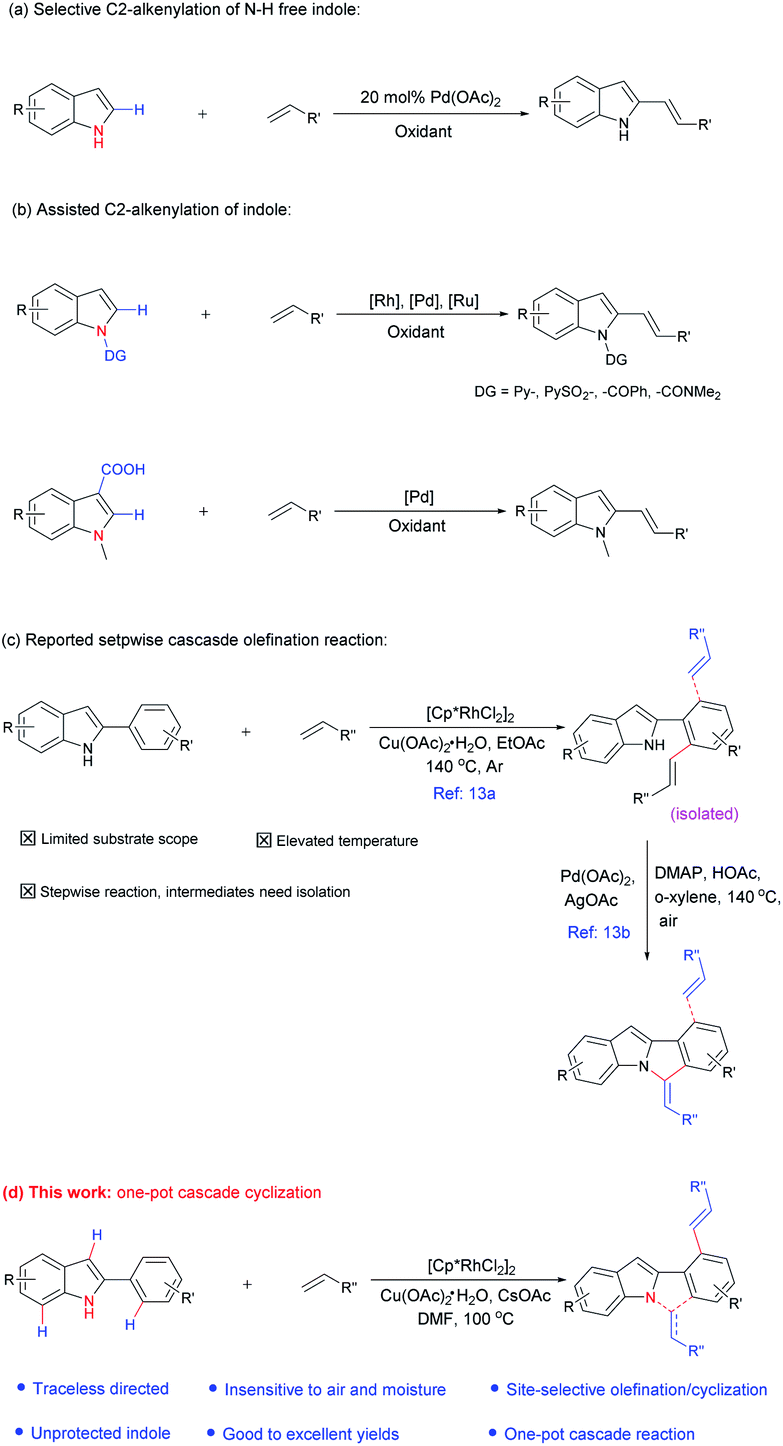 | ||
| Scheme 1 Transition-metal catalyzed C-2 alkenylation of indole. | ||
In the above-mentioned examples, high catalyst loading11b (as much as 20 mol%, Scheme 2a) or pre-installed directing groups were generally required.12 These directing groups, possessing functional groups containing a metal-binding heteroatom, remain part of the products after reaction. Such groups can rarely be conveniently removed under ambient conditions or undergo versatile cyclization reactions,12c-f which have greatly limited the structural diversity of the products and subsequent applications to complex molecule synthesis. Therefore, the need for exploration of traceless or easily removable directing groups that can address these drawbacks remains urgent. Recently, Huang et al. reported a N–H indole directed sequential cascade olefination/cyclization reaction (Scheme 1c).13 However, the substrate scope of this transformation is limited, stepwise operation procedure is required, thus make this process tedious. Herein, we reported a Rh-catalyzed N–H free indole directed dialkenylation followed by an intramolecular cascade cyclization reaction leading to the efficient synthesis of 6H-isoindolo[2,1-a]indole.
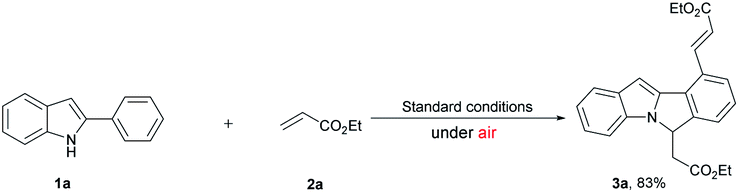 | ||
| Scheme 2 Reaction under air. | ||
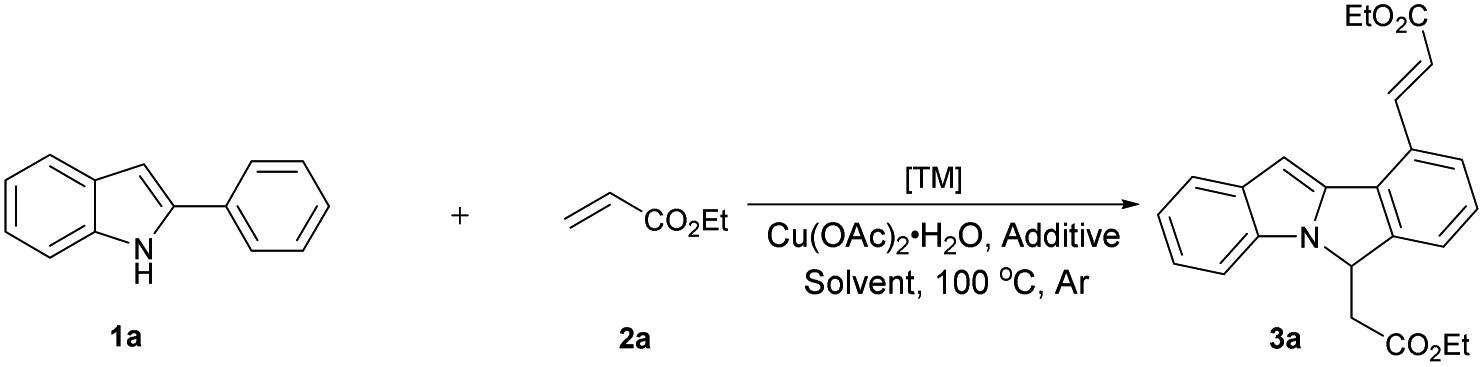
Our initial study was carried out by examining 2-phenyl indole 1a and ethyl acrylate 2a in the presence of [RhCp*Cl2]2 and Cu(OAc)2·H2O in DMF under argon atmosphere (Table 1). To our delight, the dialkenylation product 3a′ was isolated in 54% yield (entry 2). When base was added, fortunately, the desired cyclization product 3a was obtained in 55% yield (entry 4). In addition, the yield could be further improved to 86% when 2.5 equiv. ethyl acrylate 2a was employed (entry 13). Other solvents and bases failed to improve this result (entries 1, 3, 5 and 6). Catalysts proved to be critical to this transformation. Among the catalysts optimized, [RhCp*Cl2]2 appeared to be the best (entries 4, 10 and 11). The reaction was shut down in the absence of Rh catalyst or stoichiometric amounts of copper oxidant (entries 7–9). Finally, the optimized conditions were eventually identified as: 2-phenyl indole 1a (1.0 equiv.), ethyl acrylate 2a (2.5 equiv.), [RhCp*Cl2]2 (5 mol%), CsOAc (2.0 equiv.), and Cu(OAc)2·H2O (2.0 equiv.) in DMF under argon at 100 °C.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst | Additive | Yield |
| a Reaction on a 0.2 mmol scale, using 1a (1.0 equiv.), 2a (1.5 equiv.), additive (2.0 equiv.), Cu(OAc)2·H2O (2.0 equiv.), [TM] (3 mol%), solvent (1.5 mL), under N2, 21 h, isolated yield.b Additive (0.15 equiv.).c Without oxidant.d 10 mol% [Cu] under O2.e Without [Rh].f Using 2a (2.5 equiv.), [Rh] (5 mol%), 41 h. | ||||
| 1 | o-xylene | [Cp*RhCl2]2 | Na2CO3 | 10% |
| 2 | DMF | [Cp*RhCl2]2 | — | 54% |
| 3b | t-AmylOH | [Cp*RhCl2]2 | AgSbF6 | Trace |
| 4 | DMF | [Cp*RhCl2]2 | CsOAc | 55% |
| 5 | DMF | [Cp*RhCl2]2 | K3PO4·3H2O | 23% |
| 6 | DMF | [Cp*RhCl2]2 | t-BuOK | — |
| 7c | DMF | [Cp*RhCl2]2 | CsOAc | Trace |
| 8d | DMF | [Cp*RhCl2]2 | CsOAc | Trace |
| 9e | DMF | — | CsOAc | — |
| 10 | DMF | [RuCl2(p-cymene)]2 | CsOAc | Trace |
| 11 | DMF | RhCl(PPh3)3 | CsOAc | Trace |
| 12 | DMF | [Cp*RhCl2]2 | Cu(acac)2 | Trace |
| 13f | DMF | [Cp*RhCl2]2 | CsOAc | 86% |
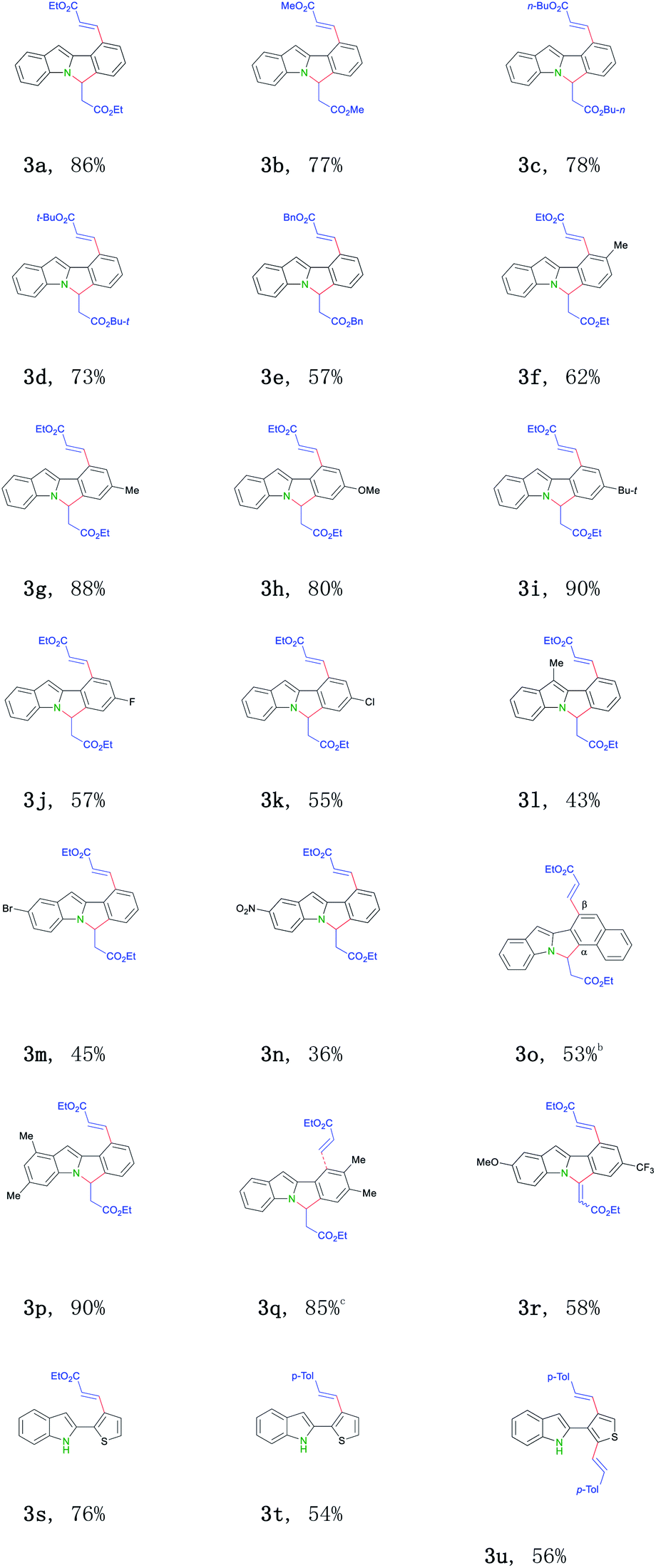
With the optimized conditions in hand, we next tend to investigate the scope of this transformation (Table 2). First, various activated olefins were tested. Good to excellent yields were obtained for different substituted acrylates (3a–e). Interestingly, when 2-thienyl substituted indole was employed, only mono-alkenylation product was got (3s). Significantly, unactivated 4-methyl styrene 2f also proceeded regularly in this transformation (3t–3u), further broadening the practical scope of this conversion. Next, different indole substrates were explored. The reaction proceeded smoothly for electron-rich substituted indoles with good yields (3f–i, 3l, 3p and 3q). Various 2-aryl indoles 1b–m with a broad substitution pattern and of different electronic nature at the ortho-, meta-, and para-positions on the phenyl ring can be well applied, thus smoothly affording the related 6H-isoindolo[2,1-a]indole products (3f–3q) in middle to good yields. Halogens did not interfere with this transition-metal catalyzed process and were well tolerated (3j–3k, 3m), thus provided possibilities for further modifications. Surprisingly, for CF3-substituted substrate, related cyclization olefination product was got (3r). Substrates bearing two methyl groups (3p and 3q), a substitution pattern widely found in indole motifs,14 also smoothly participated in this reaction. Currently, no electron-biased alkenes (for example: 1-octene) failed to afford the desired products for some unknown reasons.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:
The robustness of this Rh-catalyzed indole derivatization method was further examined under air instead of argon. Similar synthetic efficiency was got, thus further proving the practicality of this transformation (Scheme 2). It is worth dating that this dual C–H activation/annulation process is also insensitive to moisture. Commercially available solvent and reagents were directly used as received and were well-compatible in this reaction without any further purifications, which additionally expands the practical application of this conversion.
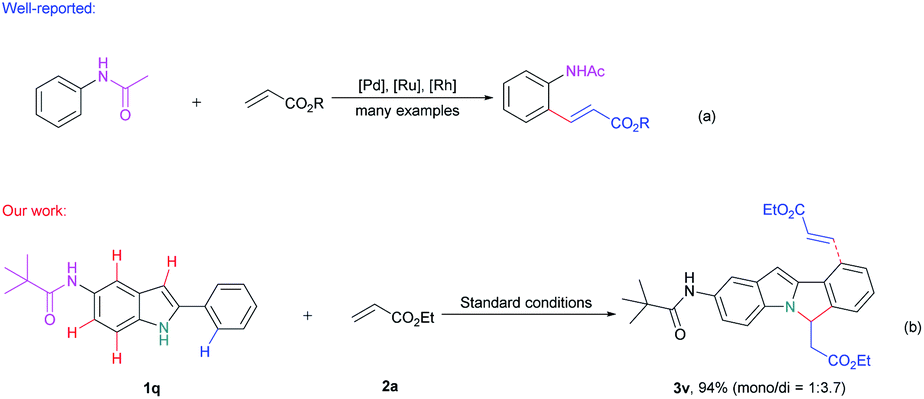
Next, we try to investigate the regioselectivity of this reaction. As it is showed in Scheme 3, N-(2-phenyl-1H-indol-5-yl)pivalamide 1q can smoothly undergo this conversion and only afforded the desired 6H-isoindolo[2,1-a]indole product 3v and no amide group directed C–H alkenylation product was detected,15 thus indicating the excellent selectivity of the C–H activation/annulation process.
 | ||
| Scheme 3 Regioselectivity of different directing groups. | ||
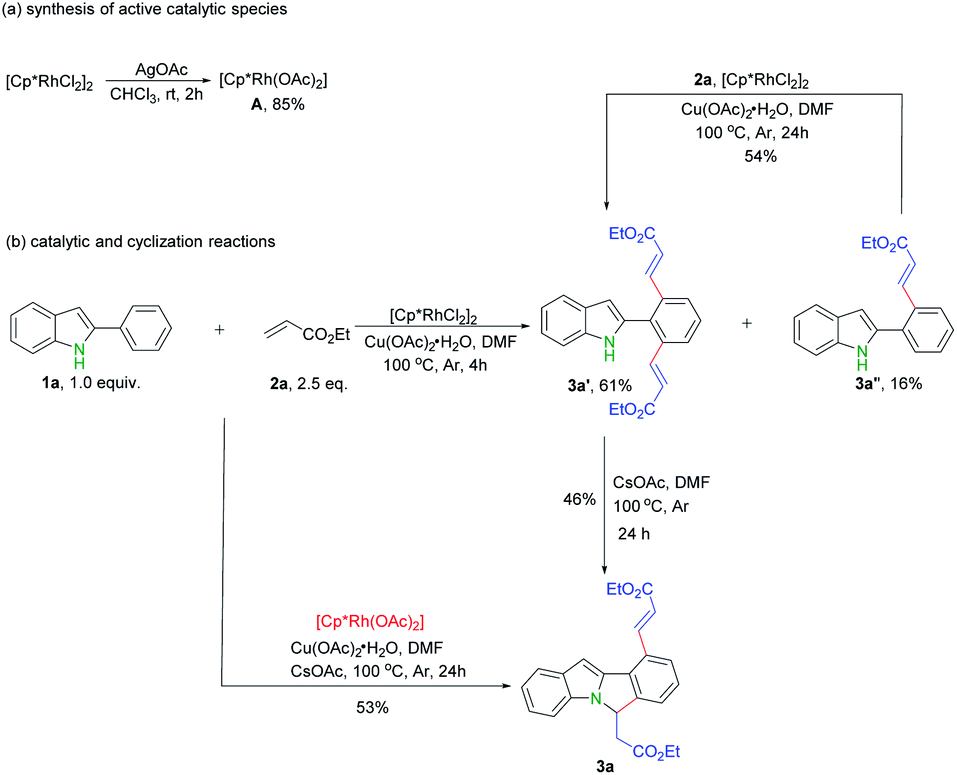
In order to shed lights on the mechanism of this transformation, a series of experiments were carried out. First, possible active rhodium catalytic species A was synthesized according to previous reports (Scheme 4a).13 Next, catalytic reaction of rhodium complex A with ethyl acrylate 2a under standard reaction conditions gave rise to desired product 3a in 53% yield (Scheme 4b), which indicate complex A possibly involves in this reaction. Thirdly, only mono- and di- alkenylation products were obtained in the absence of base while the cyclization product 3a produced by further extra addition of CsOAc (Scheme 4b). Moreover, the mono-alkenylation product 3a′′ can also be smoothly converted into the dialkenylation product 3a′ upon further treatments (Scheme 4b).
 | ||
| Scheme 4 Mechanism study. | ||
Finally, we proposed a mechanism for this transformation based on above experiments and reported literatures.9,13,16,17 First, [{Cp*RhCl2}2] dissociates and generates the active catalyst species [Cp*Rh(OAc)2] in the presence of cesium acetate.9d,16 C–H activation of 2-phenyl indole 1a by Rh(III) A produces rhodacyclic complex B,9 followed by olefin insertion and reductive elimination to afford 3a′′. One more this process gave rise to 3a′ and generated Rh(I), which can be further oxidized into Rh(III) by copper acetate and fulfilled the catalytic cycle.9,17 A base promoted intramolecular aza-Michael addition of 3a′ finally produced the cascade cyclization product 3a (Scheme 5)
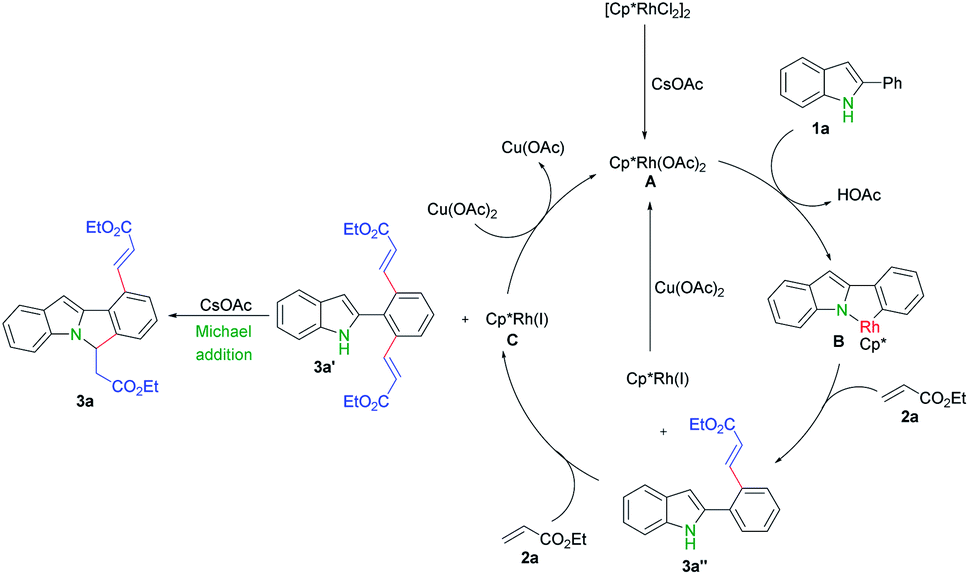 | ||
| Scheme 5 Proposed mechanism. | ||
In summary, we have reported a Rh-catalyzed N–H free indole directed C–H activation dialkenylation and cascade cyclization reaction. This strategy provides a general functionalization route of simple 2-aryl indole leading to the efficient synthesis of 6H-isoindolo[2,1-a]indoles. Excellent regioselectivity was obtained with substrates containing amide directing group. Further exploration of the synthetic utility of this chemistry is currently underway in our laboratory and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Fundamental Research Funds for the Central Universities” (21620318, 2019QNGG22). C. Wang also thanks the Jinan University (start-up fund) for additional support.Notes and references
- (a) R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed; (b) M. Somei and F. Yamada, Nat. Prod. Rep., 2005, 22, 73 RSC; (c) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (d) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29 RSC.
- (a) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS PubMed; (b) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (c) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (d) L. Ackermann, Synlett, 2007, 507 CrossRef CAS; (e) K. Krüger (née Alex), A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef; (f) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (g) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85 CrossRef CAS PubMed; (c) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed.
- (a) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed; (b) X. Gong, G. Song, H. Zhang and X. Li, Org. Lett., 2011, 13, 1766 CrossRef CAS PubMed; (c) S. R. Kandukuri and M. Oestreich, J. Org. Chem., 2012, 77, 8750 CrossRef CAS PubMed; (d) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed; (e) Y. Wu, Y. Yang, B. Zhou and Y. Li, J. Org. Chem., 2015, 80, 1946 CrossRef CAS PubMed.
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (f) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (g) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 1119 CrossRef CAS; (c) Y. Fujiwara, I. Moritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166 CrossRef CAS PubMed.
- (a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- (a) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed; (b) G. X. Cai, Y. Fu, Y. Z. Li, X. B. Wan and Z.-J. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed; (c) Y. Lu, D. H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (d) K. M. Engle, D. H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed; (e) S. H. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372 CrossRef CAS PubMed; (f) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef CAS PubMed; (g) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134 CrossRef CAS PubMed; (h) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242 CrossRef CAS PubMed.
- (a) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2068 CrossRef CAS PubMed; (b) L. Ackermann, L. Wang and A. V. Lygin, Chem. Sci., 2012, 3, 177 RSC; (c) Q. Han, X. Guo, Z. Tang, L. Su, Z. Yao, X. Zhang, S. Lin, S. Xiang and Q. Huang, Adv. Synth. Catal., 2018, 360, 972 CrossRef CAS; (d) Q. Zhang, Q. Li and C. Wang, RSC Adv., 2021, 11, 13030 RSC.
- For selected review on transition metal-catalyzed direct arylation of indole, see: (a) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673 CrossRef CAS ; For selected examples, see:; (b) L. Ackermann and S. Barfüsser, Synlett, 2009, 808 CrossRef CAS; (c) Z. Liang, B. Yao and Y. Zhang, Org. Lett., 2010, 12, 3185 CrossRef CAS PubMed; (d) S. Potavathri, K. C. Pereira, S. I. Gorelsy, A. Pike, A. P. LeBris and B. DeBoef, J. Am. Chem. Soc., 2010, 132, 14676 CrossRef CAS PubMed; (e) L. Wang, W.-B. Yi and C. Cai, Chem. Commun., 2011, 47, 806 RSC.
- (a) C. Jia, W. Lu, T. Kitamura and Y. Fujiwara, Org. Lett., 1999, 1, 2097 CrossRef CAS; (b) N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125 CrossRef CAS PubMed.
- For examples of C–H activation C2 alkenylation of indole assisted by a directing group, see: (a) A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159 CrossRef CAS PubMed; (b) A. García-Rubia, R. G. Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511 CrossRef PubMed; (c) V. Lanke and K. R. Prabhu, Org. Lett., 2013, 15, 2818 CrossRef CAS PubMed; (d) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem.–Eur. J., 2013, 19, 11863 CrossRef CAS PubMed; (e) L.-Q. Zhang, S. Yang, X. Huang, J. You and F. Song, Chem. Commun., 2013, 49, 8330 Search PubMed; (f) L. Yang, G. Zhang and H. Huang, Adv. Synth. Catal., 2014, 356, 1509 CrossRef CAS.
- A. Liu, Q. Han, X. Zhang, B. Li and Q. Huang, Org. Lett., 2019, 21, 6839 CrossRef CAS PubMed.
- B. D. Andrews, A. J. Poynton and I. D. Rae, Aust. J. Chem., 1972, 25, 639 CrossRef CAS.
- For selected examples using amide as directing group for C–H activation alkenylation, see: (a) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed; (b) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS PubMed; (c) J. Zhou, B. Li, F. Hu and B.-F. Shi, Org. Lett., 2013, 15, 3460 CrossRef CAS PubMed.
- (a) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294 CrossRef CAS PubMed; (b) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed.
- (a) B. Li, B. Zhang, X. Zhang and X. Fan, Chem. Commun., 2017, 53, 1297 RSC; (b) Y.-Q. Xia and L. Dong, Org. Lett., 2017, 19, 2258 CrossRef CAS PubMed; (c) X. Yang, Y. Li, L. Kong and X. Li, Org. Lett., 2018, 20, 1957 CrossRef CAS PubMed; (d) S. Guo, Y. Liu, Y. Wang, X. Zhang and X. Fan, J. Org. Chem., 2020, 85, 8910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02998c |
| This journal is © The Royal Society of Chemistry 2021 |