
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA model of modified meta-iodobenzylguanidine conjugated gold nanoparticles for neuroblastoma treatment†
Kween Saimuanga,
Khomson Suttisintongb,
Narongpol Kaewchangwatb,
Eknarin Thanayupongb,
Yodsathorn Wongngam b,
Putthiporn Charoenphunc,
Rujira Wanotayand,
Abdelhamid Elaissarie,
Suradej Hongengf,
Duangporn Polpanich*b and
Kulachart Jangpatarapongsa*a
b,
Putthiporn Charoenphunc,
Rujira Wanotayand,
Abdelhamid Elaissarie,
Suradej Hongengf,
Duangporn Polpanich*b and
Kulachart Jangpatarapongsa*a
aCenter for Research and Innovation, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand. E-mail: kulachart.jan@mahidol.edu
bNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand. E-mail: duangporn@nanotec.or.th
cDepartment of Diagnostic and Therapeutic Radiology, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand
dDepartment of Radiological Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
eUniv. Lyon, University Claude Bernard Lyon-1, CNRS, ISA-UMR 5280, 69622 Villeurbanne, France
fDepartment of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand
First published on 20th July 2021
Abstract
Iodine-131 meta-iodobenzylguanidine (131I-mIBG) has been utilized as a standard treatment to minimize adverse side effects by targeting therapies to bind to the norepinephrine transporter (NET) expressed on 90% of neuroblastoma cells. However, only a minority of patients who receive 131I-mIBG radiotherapy have clinical responses, and these are usually not curative. In this study, novel ligand-conjugated gold nanoparticles (GNPs) based on mIBG were synthesized and evaluated biologically with neuroblastoma cells in vitro. To induce specific internalization to the tumor cells and utilize it as a model for radioenhancement, 127I-modified mIBG was successfully synthesized and grafted covalently to the surface of carboxylated PEG-GNPs. 49.28% of the novel mIBG derivative was grafted on carboxylated PEG-GNPs. The particles were stable and not toxic to the normal fibroblast cell line, L929, even at the highest concentration tested (1013 NPs per mL) at 24, 48, and 72 h. Moreover, the cellular uptake of the model was decreased significantly in the presence of a NET inhibitor, suggesting that there was specific internalization into neuroblastoma cells line (SH-SY5Y) via the NET. Therefore, this model provides useful guidance toward the design of gold nanomaterials to enhance the efficiency of 131I-mIBG treatment in neuroblastoma patients. However, the investigation of radio-therapeutic efficiency after radioisotope 131I substitution will be further conducted in a radiation safety laboratory using an animal model.
Introduction
Neuroblastoma is an embryonal tumor arising from neural crest – derived progenitor cells. It is the most common extra-cranial solid tumor in infants and children between the ages of 1 and 5 years, and accounts for nearly 15% of all pediatric cancer mortality,1 yet it is rare in people older than 10.2 A large international neuroblastoma patient cohort reported that neuroblastoma occurs in adrenal (47%) and abdominal/retroperitoneal regions (24%), as well as being observed in the neck (2.7%), thoracic (15%), pelvic (3%) and other regions (7.9%).3 The International Neuroblastoma Risk Group (INRG) system classifies patients into four groups [very low-risk, low-risk, intermediate-risk (IR), and high-risk (HR)] based on stage at diagnosis, age, pathology, and genomic characterization.4–6 Previous studies reported that approximately 50% of patients have metastatic disease at the time of the initial diagnosis and distant recurrence is a major obstacle to the treatment of these patients.7 The pillars of treatment of neuroblastoma consist of chemotherapy, surgical resection, and radiotherapy. Modern treatment regimens incorporate (i) chemotherapy and surgery, (ii) consolidation therapy via myeloablative therapy (MAT) with autologous hematopoietic stem cell transplant (AHSCT) and/or irradiation, and (iii) post-consolidation/maintenance therapy.6,8 Yet nearly 50% of HR patients develop resistance to chemotherapeutic agents, resulting in a high likelihood of relapse.9,10 Thus, the need for novel and more effective treatment regimens for neuroblastoma is imperative.11–13One of the novel therapeutic approaches under active investigation to improve outcomes is iodine-131 meta-iodobenzylguanidine (131I-mIBG). mIBG is an analog of the catecholamine norepinephrine, which can be labeled with a radioactive isotope, 131I, to use clinically for treatment. The advantage of 131I-mIBG is a minimization of adverse side effects by targeting therapies to bind with the norepinephrine transporter (NET).14–17 Previous studies indicate that 90% of neuroblastomas and other cancers of sympathetic neuronal precursors express NET. It is reported that 131I-mIBG exhibits activity against refractory neuroblastoma, with response rates ranging from 10–50%. However, previous studies suggest that only 30% of children who receive 131I-mIBG radiotherapy for neuroblastoma have any clinical response, and these responses are usually not curative.15,18–20
The extensive nanotechnology research in recent decades has prompted its application as an alternative to overcome some of the limitations of conventional treatment. There are many potential metal nanoparticles owning anti-cancer activities, including hafnium oxide, superparamagnetic iron oxide, selenium and magnesium oxide nanoparticles.21–23 Hafnium oxide nanoparticles could improve the pathological response in controlled phase 3 clinical trial in patients with soft tissue sarcoma.21 Gholami et al. demonstrated that superparamagnetic iron oxide nanoparticles labelled with radiotherapeutic isotopes could enhance the radiation dose up to 20%24 while Singh group mentioned anti-cancer properties of selenium and magnesium oxide NPs in term of ROS production leading to cancer cell death.22,23 Among them, gold nanoparticles (GNPs)-based therapy has been developed as a novel potential strategy as delivery vehicles for drugs, contrast agents and radiation enhancers/radiosensitizers, and in diagnosis.25 Due to crucial physical and chemical properties mentioned in previous clinical studies which are (i) chemical inertness; (ii) surface properties; (iii) electronic structure; and (iv) optical properties.26–28 By owning the enhanced permeability and retention (EPR) effect, nanoparticles tend to accumulate in tumor sites more than the normal tissues. Together with the property of GNPs to enhance radiation due to their high atomic number (Z), conjugation of GNPs with specific agents can reduce the therapeutic radiation dose and further limit the damage to healthy tissues while eradicating the tumor.29–32 Not only external sources of radiation, GNPs could enhance the efficacy of radioisotope in term of radiotherapy. Recent studies demonstrated that polymer-grafted GNPs significantly enhanced the killing potential of a systemic radioiodine treatment in vitro and in vivo.33,34 Moreover, Yook et al. reported that panitumumab-modified GNPs complexed to the β-particle-emitter, 177Lu, enhanced cell death in triple negative breast cancer.35 To utilize the nanoparticles in human body, functionalization of the nanoparticle's surface has been employed. Polyethylene glycol (PEG) is the most widely used polymer due to its safety in human approved by the Food and Drug Administration (FDA). By owning unique hydrophilicity and electrical neutrality, PEG coating could improve biophysical and chemical properties of the nanoparticles. In nanomedicine, PEG could reduce RES uptake and prevent immune recognition of the nanoparticles, also called ‘stealth effect’, thus prolong the circulation time.36–41
In this study, we developed mIBG-conjugated carboxylated PEG-GNPs with the aim of enhancing the accumulation of the particles within the tumor while sparing the adjacent normal tissues. Modified mIBG (127I-MoM-Boc) was synthesized from 1,3-bis(tert-butoxycarbonyl) guanidine (N,N′-di-Boc-guanidine) and 1,4-bis(bromomethyl)-2-iodobenzene. The modification was made at C-4 of mIBG to maintain the affinity binding to NET,42–45 and characterized using 1H, 13C nuclear magnetic resonance (NMR) and mass spectrometry. Then the 127I-MoM-Boc was conjugated to carboxylated PEG-GNPs; deprotection was subsequently performed to obtain 127I-MoM-PEG-GNPs. Diagram describing three main steps of synthesis research is displayed in Scheme 1. Binding efficiency (%) was determined indirectly from supernatant using UV-vis spectrophotometry. The product was characterized and prepared for cytotoxicity testing in normal cells using an MTS assay. Internalization testing, with/without NET inhibitor, was performed using inductively coupled plasma mass spectrometry (ICP-MS) in a neuroblastoma cell line (SH-SY5Y) to observe the specific binding of 127I-MoM-PEG-GNPs to the cancer cells.
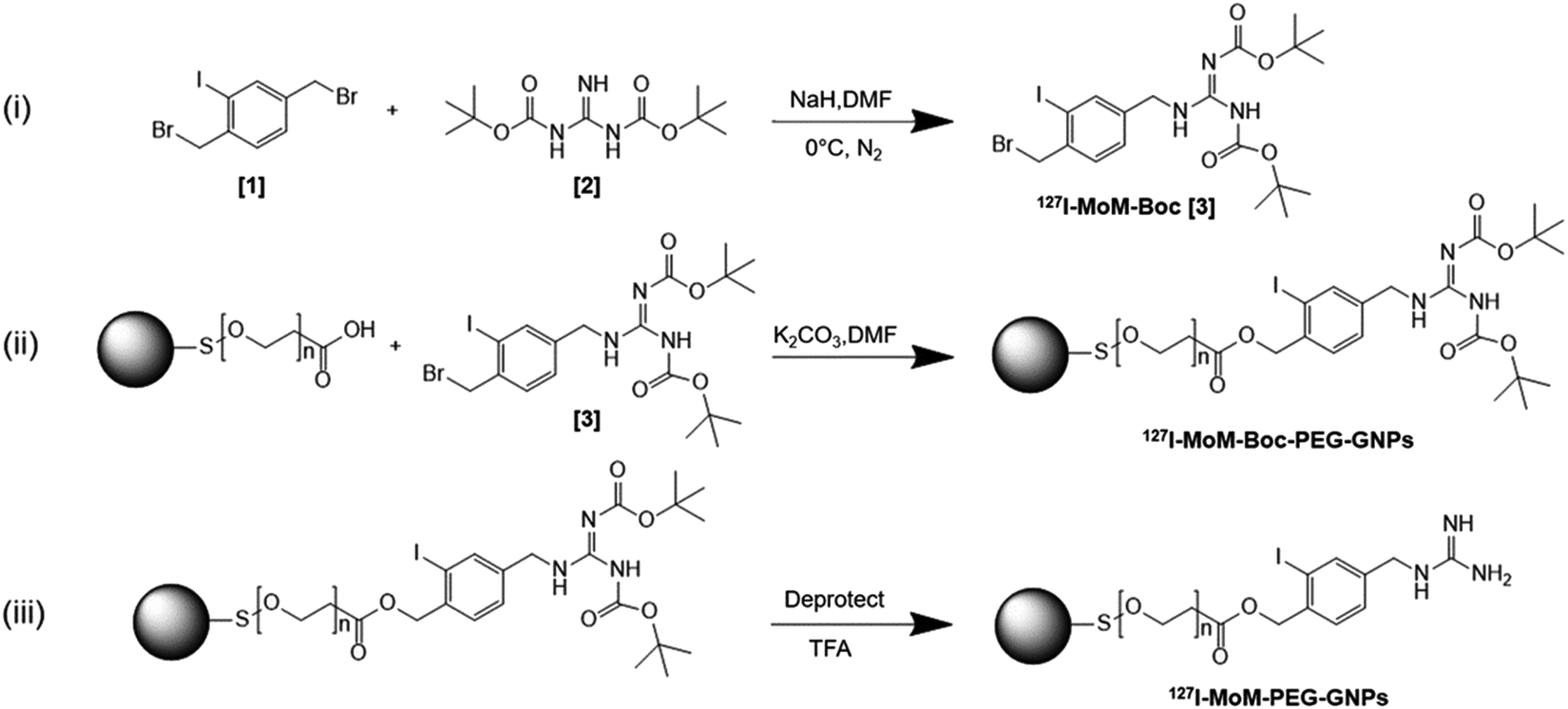 | ||
| Scheme 1 Synthesis of 127I-MoM-PEG-GNPs. | ||
Experimental
Materials
In this study, all chemicals used were of analytical grade. 1,3-Bis(tert-butoxycarbonyl) guanidine (C11H21N3O4) and 1,4-bis(bromomethyl)-2-iodobenzene (C8H7Br2I), used as precursors to synthesize 127I-MoM-Boc, were purchased from Angene Int. (Nanjing, China) and Chem-space (New Jersey, USA), respectively. Carboxylated PEG-GNPs (particle diameter 15 nm), ethyl acetate (C4H8O2), potassium carbonate (K2CO3), trifluoroacetic acid (C2HF3O2), dry dimethylformamide (C3H7NO), and dichloromethane (CH2Cl2) were purchased from Sigma-Aldrich (Missouri, USA). All aqueous solutions were prepared using deionized (DI) water (resistivity of 18.2 MΩ cm) obtained from PURELAB® water purification systems (ELGA LabWater, UK).Cell culture
Murine fibroblast (L929) and human neuroblastoma (SH-SY5Y) cell line were obtained from Faculty of Medical Technology and Faculty of Medicine at Ramathibodi Hospital, Mahidol University, Thailand. Those cell lines were cultured in Dulbecco modified eagle medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Gibco, USA). Cells were maintained in a tissue culture incubator at 37 °C and 5% CO2.Synthesis of 127I-modified meta-iodobenzylguanidine-Boc
127I-MoM-Boc was primarily synthesized. First, 22.57 mg of NaH (60% dispersion in paraffin oil; 13.54 mg, 0.56 mmol in final) was washed with hexane 3 times and dried. Then dry dimethylformamide (DMF) was added, and the solution was stirred on ice. 100 mg (0.26 mmol) of 1,4-bis (bromomethyl)-2-iodobenzene [1] and 66.51 mg (0.26 mmol) of 1,3-bis(tert-butoxycarbonyl) guanidine (N,N′-di-Boc-guanidine) [2] were dissolved with dry DMF (total 2.57 mL). Next, dissolved 1,3-bis(tert-butoxycarbonyl) guanidine was added in the solution in one portion, and the reaction mixture was continuously stirred at 0 °C for 5 min. Then, dissolved 1,4-bis(bromomethyl)-2-iodobenzene was added to the mixture and stirred for an additional 10 min. Ethyl acetate was used to quench the reaction. The mixture was partitioned between ethyl acetate and water, and the water layer was extracted twice with ethyl acetate and brine. Water was discarded from the ethyl acetate layer using sodium sulfate. Then the ethyl acetate layer was filtered, evaporated, and dried under reduced pressure. After drying 127I-MoM-Boc [3] under reduced pressure, the crude was 127 mg of a light-yellow oil (87.13%). 1H NMR (CDCl3) δ 9.42 (s, 1H), 9.24 (s, 1H), 7.78 (s, 1H), 7.37 (d, 1H), 7.25 (d, 2H), 5.08 (s, 2H), 4.55 (s, 2H), 1.47 (s, 9H), 1.36 (d, 9H); 13C NMR (126 MHz, CDCl3): δ 27.84, 28.24, 38.41, 46.28, 78.99, 84.55, 99.48, 127.87, 130.11, 138.84, 139.19, 141.07, 154.57, 160.41, 162.48; HRMS calcd for C19H28BrIN3O4 568.0300 (M + H)+, obsd 568.0307 (Fig. S1–S3†).Synthesis of 127I-modified meta-iodobenzylguanidine-Boc-PEGylated gold nanoparticles
To conjugate 127I-MoM-Boc with carboxylated PEG-GNPs, first the concentration of –COOH present on the surface of PEG-GNPs was titrated with NaOH (1 mM) using a pH meter (Mettler Toledo, USA). Next, 0.4 mg of 127I-MoM-Boc (0.7 μmol) and 0.2 mg of potassium carbonate (1.4 μmol) were dissolved in DMF (total 1.75 mL). Dissolved 127I-MoM-Boc was added to 1 mL of carboxylated PEG-GNPs (0.35 μmol) and stirred until the solution was homogenous. Dissolved potassium carbonate was then added in the solution and stirred overnight. Next, DI water was added to quench the reaction. The solution was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min twice to remove DMF and then resuspended with DI water. The supernatant was collected to further determine binding efficiency (%) of 127I-MoM-Boc-PEG-GNPs using UV-vis spectrophotometry at 260 nm. The morphology and particle size distribution were determined by transmission electron microscopy (TEM, JEM-2100, JEOL, Japan) while zeta potential and λmax were determined using Zetasizer (Ultra Pro, Malvern, UK) and UV-vis spectrophotometry (Shimadzu, Japan), respectively.
000 rpm for 30 min twice to remove DMF and then resuspended with DI water. The supernatant was collected to further determine binding efficiency (%) of 127I-MoM-Boc-PEG-GNPs using UV-vis spectrophotometry at 260 nm. The morphology and particle size distribution were determined by transmission electron microscopy (TEM, JEM-2100, JEOL, Japan) while zeta potential and λmax were determined using Zetasizer (Ultra Pro, Malvern, UK) and UV-vis spectrophotometry (Shimadzu, Japan), respectively.
Deprotection of Boc group
127I-MoM-PEG-GNPs were obtained as follows. Trifluoroacetic acid (TFA) (0.138 μmol) was diluted in dichloromethane (DCM) and added to 1 mL of 127I-MoM-Boc-PEG-GNPs (0.069 μmol). After stirring for 60 min, the reaction was quenched by DCM and DI water. The product, which was in the DI water layer, was separated and centrifuged at 12000 rpm for 30 min. The pellet was resuspended in DI water and stored at 4 °C. Particle morphology and size distribution of the 127I-MoM-PEG-GNPs were determined by TEM, while zeta potential and λmax were determined by Zetasizer and UV-vis spectrophotometry, respectively.
To leave only 127I-MoM which was covalently bound to –COOH of PEG located on GNPs, the physically attached 127I-MoM was removed from the solution. First, 127I-MoM-PEG-GNPs solution was centrifuged at 12000 rpm for 30 min and the supernatant discarded. The pellet of product was resuspended by phosphate buffer (PB) in pH 7.0. Then, the solution was shaken at 300 rpm for 120 min. After that, the solution was centrifuged and the supernatant was kept and dried at 80 °C overnight to remove physically attached 127I-MoM. The product was resuspended in DI water with adjustment of OD to 1, which represented 1.64 × 1012 nanoparticles (NPs) per mL (according to manufacturer's recommendation) for further experiments. Particle morphology and size distribution of the product after removing physically attached 127I-MoM were determined by TEM while zeta potential and λmax were determined by Zetasizer and UV-vis spectrophotometry, respectively. Energy-dispersive X-ray spectroscopy (EDX) of 127I-MoM-PEG-GNPs was also performed to analyse the element of the product.
Stability test of 127I-modified meta-iodobenzylguanidine PEGylated gold nanoparticles
To investigate the stability of the particles in DI water, 127I-MoM-PEG-GNPs was stored 4 weeks at 4 °C after synthesis. UV-vis spectrophotometry and TEM were utilized to determine the size and dispersion of the particles.Cytotoxicity of 127I-modified meta-iodobenzylguanidine PEGylated gold nanoparticles
To investigate the cytotoxicity of 127I-MoM-PEG-GNPs, the normal fibroblast cell line, L929, was employed according to ISO 10993-5 with modifications. First, L929 cells were suspended in 10% FBS supplemented DMEM were added to 96-well plates (2.5 × 103 cells per well) and incubated overnight at 37 °C in a humidified incubator with 5% CO2. Then the seeded cells were treated with 127I-MoM-PEG-GNPs at various concentrations (0, 108, 109, 1010, 1011, 1012, 2.5 × 1012, 5 × 1012, and 1013 NPs per mL). After 24, 48, and 72 h, MTS solution (Promega, USA) was added to each well (20 μL per well) and cells were incubated for 2 h at 37 °C in the dark. The solution was then measured for absorbance at 490 nm using a microplate reader (Biotek, USA).Inhibition assay
To investigate cellular uptake and specificity of 127I-MoM-PEG-GNPs for neuroblastoma cells, the uptake of 2 × 1011 NPs per mL of 127I-MoM-PEG-GNPs was quantitated in the presence of 100 nM desipramine, a NET inhibitor. SH-SY5Y cells were seeded to 6-well plates (5 × 105 cells per well) and incubated overnight at 37 °C in a humidified incubator with 5% CO2. Cells were treated with 100 nM desipramine 24 h prior to addition of 127I-MoM-PEG-GNPs, while untreated cells were used as controls. After an additional 24 h-incubation, cells were washed with 1× PBS, harvested, counted, and then trypsinized. After centrifuged at 1800 rpm for 5 min at room temperature, the pellet was digested using aqua regia for 10 min. The sample was then diluted to 2% aqua regia with DI water. The concentrations of internalized gold cocultured with the NET inhibitor were determined by ICP-MS and reported as the concentration of gold (fg) per cell.Results and discussion
Preparation and characterization of modified meta-iodobenzylguanidine PEGylated gold nanoparticles
The concentration of –COOH of PEG GNPs was 350 μM after titration with 1 mM NaOH (35 μL, pH 8.3). The synthesis of 127I-MoM-Boc-PEG-GNPs was then performed by conjugating 127I-MoM-Boc to carboxylated PEG-GNPs. Binding efficiency (%) was measured indirectly using UV-vis spectrophotometry at 260 nm, by assessment of supernatant collected after centrifugation to determine the residue 127I-MoM-Boc. The result showed that 49.28% of 127I-MoM-Boc was bound to carboxylated PEG-GNPs.The sizes of carboxylated PEG-GNPs and 127I-MoM-Boc-PEG-GNPs, as characterized by TEM, were 14.0 and 14.1 nm with polydispersity index (PDI) 1.005 and 1.005 (Fig. 1a–d), respectively, suggesting monodispersion.
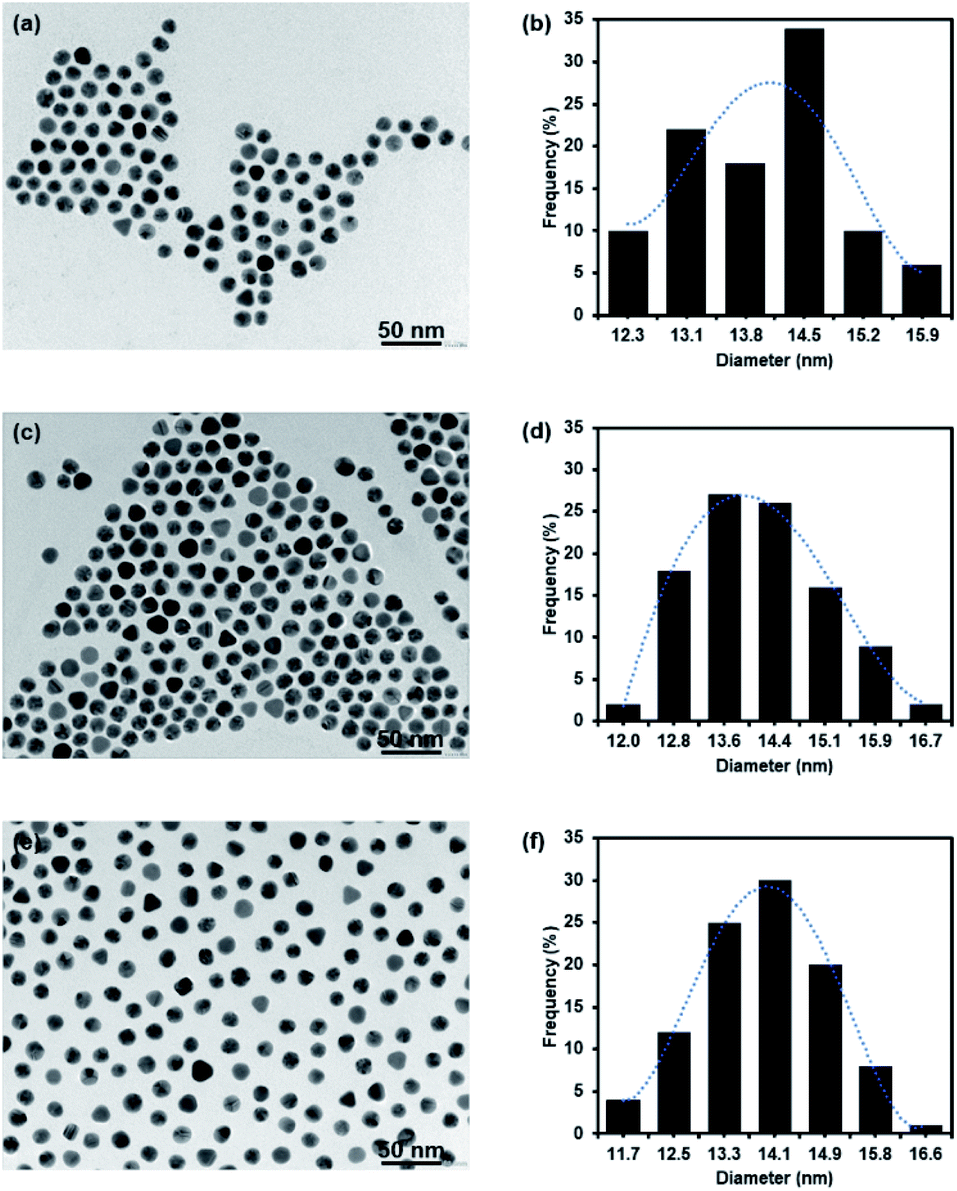 | ||
| Fig. 1 TEM micrograph and histogram of GNPs. (a and b) Carboxylated PEG-GNPs; (c and d) 127I-MoM-Boc-PEG-GNPs; (e and f) 127I-MoM-PEG-GNPs in DI water. | ||
The product was characterized further by UV-vis spectrophotometry. The graph (Fig. 2) shows that after coupling 127I-MoM-Boc with carboxylated PEG-GNPs, the maximum wavelength was shifted from 521 nm to 523 nm. The zeta potential of the particles after conjugation was changed from −27.40 mV to −22.38 mV in DI water. A red shift of GNPs is due to change in the local refractive index (RI).46 Oliveira and Zhang, et al. demonstrated that the absorption peak is red-shifted as the concentration of organic molecules anchored to the GNPs increases.47,48 However, the zeta potential of the particles was less negative due to the absence of carboxyl groups representing the completion of chemical coating.49,50
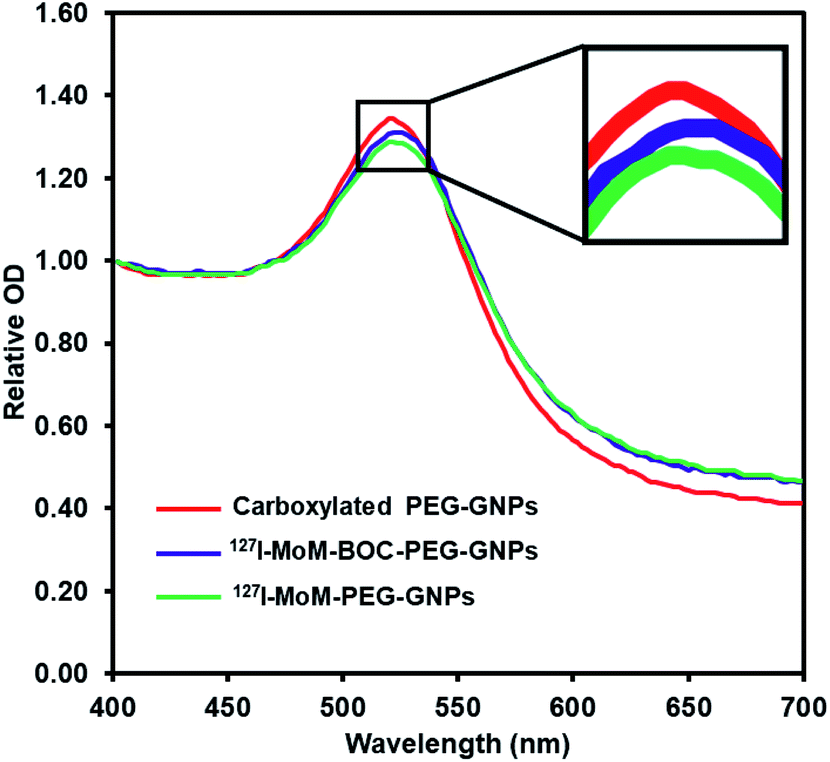 | ||
| Fig. 2 UV spectra of: carboxylated PEG-GNPs red line with maximum peak at 521 nm; 127I-MoM-Boc-PEG-GNPs blue line with maximum peak at 523 nm; 127I-MoM-PEG-GNPs green line with maximum peak at 523 nm. | ||
After deprotection, 127I-MoM-PEG-GNPs were washed and resuspended in DI water. The products were then characterized by TEM. Results presented in Fig. 1e and f showed that the size of the nanoparticles was 13.9 nm with PDI 1.006, suggesting monodispersion. In addition, the product was characterized by UV-vis spectrophotometry. Fig. 2 showed that after deprotection of the Boc group, there was no shift of the maximum wavelength (523 nm), which indicated that there was no aggregation of the particles. In addition, the zeta potential of the particles after deprotection became more positive (from −22.38 mV to −19.87 mV) in DI water due to the presence of free guanidine groups.45 Moreover, the element of 127I-MoM-PEG-GNPs was analysed using EDX analysis. The result indicated that % mass of iodine was 33.71 (Fig. 4S†) suggesting the conjugation of 127I-MoM to carboxylated PEG-GNPs after deprotection.
Stability test of 127I-modified meta-iodobenzylguanidine PEGylated gold nanoparticles
After 4 week storage at 4 °C, the stability test of 127I-MoM-PEG-GNPs was investigated using UV-vis spectrophotometry and TEM. UV-vis spectrophotometry demonstrated a single peak with the maximum wavelength at 523 nm after 4 week storage, which was equal to that of freshly prepared 127I-MoM-PEG-GNPs (Fig. 3a), suggesting no size changing and no particle aggregation. TEM image revealed the size and morphology of the particles which was 13.9 nm with PDI 1.006 in spherical shape (Fig. 3b and c), thus no size and morphology changing after 4 week storage. The results indicated that 127I-MoM-PEG-GNPs are stable for extended periods after the synthesis. Several studies mentioned that functionalizing GNPs with PEG increases stability, both in vivo and in vitro.51,52 Zhang et al. demonstrated that GNPs coated with high molecular weight, 5000 Da, were more stable than GNPs coated with low molecular weight PEG, 2000 Da. They mentioned that GNPs coated with PEG5000 exhibited the highest colloidal stability and did not aggregate in phosphate buffer saline (PBS) containing 10% fetal bovine serum (FBS).52 Therefore, carboxylated PEG5000 GNPs was utilized in this study to increase the stability of the particles. Not only providing high stability, PEG increases circulation time and reducing protein adsorption resulting in making the particles more available for uptake by target organs and owning higher chance to benefit from the EPR effect promoted by the tumor leaky vasculature.53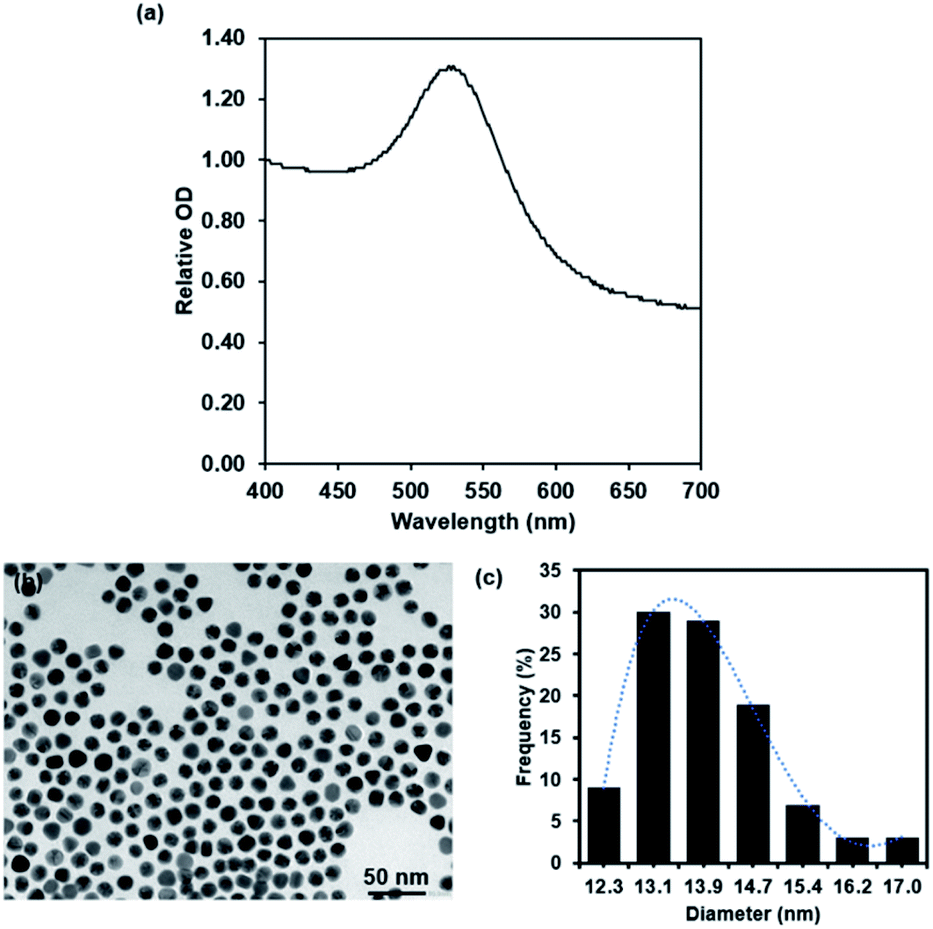 | ||
| Fig. 3 UV spectra (a) and TEM micrograph and histogram of 127I-MoM-PEG-GNPs (b and c) after 4 week storage at 4 °C. | ||
Cytotoxicity of 127I-modified meta-iodobenzylguanidine PEGylated gold nanoparticles to normal fibroblast cells
MTS assay was performed to investigate the cytotoxicity of carboxylated PEG-GNPs and 127I-MoM-PEG-GNPs to the normal fibroblast cell line, L929, in various concentrations [0, 108, 109, 1010, 1011, 1012, 2.5 × 1012, 5 × 1012, and 1013 NPs per mL] at 24, 48, and 72 h. The results showed that % cell survival was higher than 80% at every concentration and time point studied for both carboxylated PEG-GNPs and 127I-MoM-PEG-GNPs, suggesting that pre- and post-modification of carboxylated PEG-GNPs were not harmful to the normal cells (Fig. 4 and 5). To use as a model for radioenhancement, the size, charge, concentration, and surface modification of GNPs each play an important role. Review of the literature showed that 5–20 nm GNPs can be effectively utilized as sensitizers of radiation therapy.30–32,54 In addition, the particles could have ability to cross the blood–brain barrier (BBB) to the central nervous system (CNS) which is a potential sanctuary site for neuroblastoma as the previous studies have shown that the rate of CNS recurrence is >10%.7,55 Physiologically, end-feet of astrocytes locate on the surface area of the capillary basement membrane. The gap between astrocytic end-feet and the capillary endothelium is approximately 20 nm. Therefore, the penetration of the NPs through BBB is critical size-dependent with the upper penetration limit being approximately 20 nm.56–58 Besides, surface charge can directly affect the toxicity and the fate of the particles once entering in human body. Neutral or negatively charged NPs could decrease the plasma protein adsorption and lower nonspecific cellular uptake resulting in a longer blood circulation half-life than positively charged NPs.59 The positively charged NPs are also reported to cause the BBB destruction while neutral NPs, on the other hand, negatively charged NPs were found to have no effect on BBB integrity.60–62 Therefore, negatively charged 15 nm GNPs were employed in this study due to the advantages mentioned earlier. In the matter of concentration, our results suggested that the IC50 of 127I-MoM-PEG-GNPs could be higher than 1013 NPs per mL. Our maximum concentration (1013 NPs per mL) was greater than published upper limits (1012 NPs per mL)56,63–65 indicating that 127I-MoM-PEG-GNPs could have a low toxicity to the normal cells. In terms of surface modification, PEG, a hydrophilic coiled polymer, was employed to this study due to several advantages to improve the physical and chemical properties of NPs, including biocompatibility, processability, and biodegradability. Liu, et al. noted that PEG at low concentrations (≤5 mg mL−1) was not toxic to the normal cells and could be neglected.66 Likewise, in this study, PEG may have helped the model to become more biocompatible with the normal L929 cells, even at high concentrations.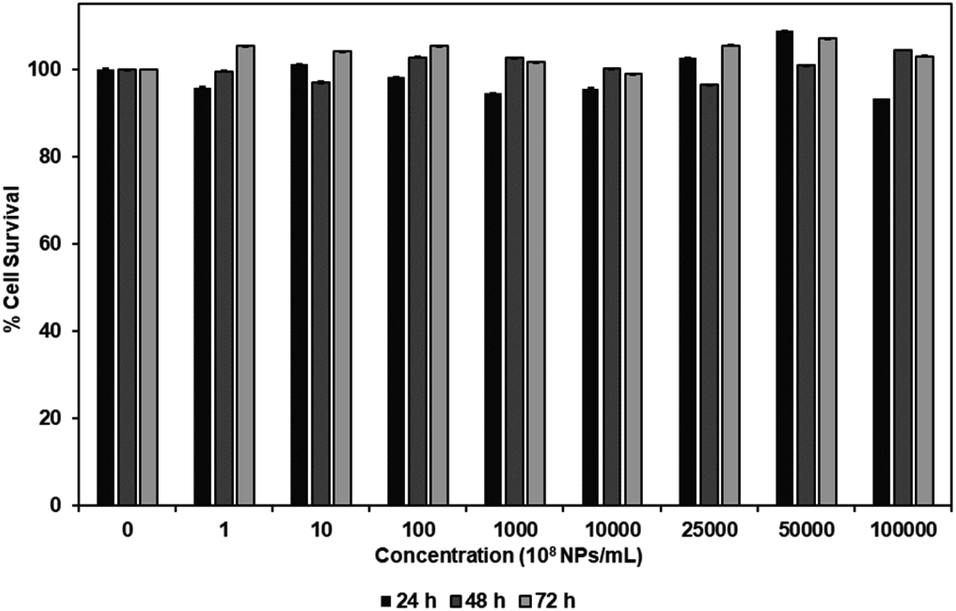 | ||
| Fig. 4 Cytotoxicity, as determined by MTS assay, of carboxylated PEG-GNPs in various concentrations (0, 108, 109, 1010, 1011, 1012, 2.5 × 1012, 5 × 1012, and 1013 NPs per mL) after incubation with L929 at 24, 48, and 72 h. | ||
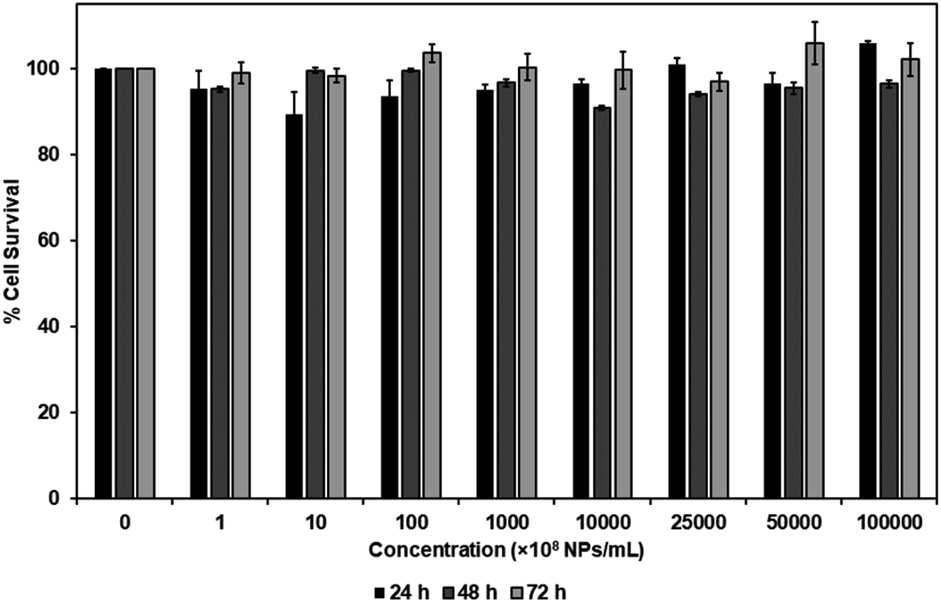 | ||
| Fig. 5 Cytotoxicity, as determined by MTS assay, of 127I-MoM-PEG-GNPs in various concentrations (0, 108, 109, 1010, 1011, 1012, 2.5 × 1012, 5 × 1012, and 1013 NPs per mL) after incubation with L929 at 24, 48, and 72 h. | ||
Inhibition assay
After performing cytotoxicity testing in normal fibroblast cell, L929, concentration of 127I-MoM-PEG-GNPs at 2 × 1011 NPs per mL was selected to observe the inhibition assay in neuroblastoma cell, SH-SY5Y. The cellular uptake of 127I-MoM-PEG-GNPs by SH-SY5Y cells in the presence of NET inhibitor (100 nM desipramine) was determined using ICP-MS. Here, as shown in Fig. 6, the cellular uptake of 127I-MoM-PEG-GNPs was 1.45 fg per cell, while 0.56 fg per cell was detected in the cells incubated with the NET inhibitor for 24 h prior (p < 0.05).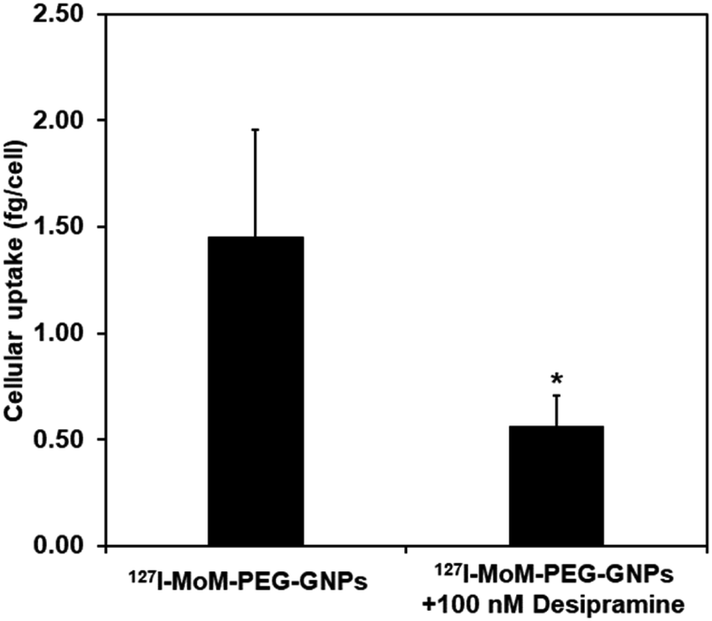 | ||
| Fig. 6 Cellular uptake of 127I-MoM-PEG-GNPs into SH-SY5Y cells in the presence of 100 nM desipramine compared to no desipramine. Error bars represent standard deviation. *p < 0.05. | ||
Basically, there are two main processes to uptake mIBG, called ‘uptake-1’ and ‘uptake-2’. Uptake-1 is an ATP-dependent and specific process to transport mIBG into the cells, while uptake-2 is an energy-independent, passive, and non-specific diffusion. Previous studies found that the majority of mIBG is internalized to the neuroblastoma cells by uptake-1, which is 50 times more efficient than uptake-2.20,67–69 The position of the guanidinomethyl group of mIBG plays an important role in terms of cellular uptake. The guanidinomethyl group at the 4-position (C-4) maintains the biological properties of mIBG. In contrast, the 5-position exhibits only nonspecific binding.42–45 Therefore, in this study, the modification was made at C-4 of mIBG to negate the affinity binding to NET. To investigate the transportation of 127I-MoM-PEG-GNPs into the neuroblastoma cells, desipramine (a tricyclic antidepressant) was utilized as a competitive inhibitor to NET.20,70 As mentioned earlier, the concentration of gold detected in the cells treated with 100 nM desipramine for 24 h prior was decreased significantly. This suggested that the majority of 127I-MoM-PEG-GNPs was internalized specifically via uptake-1, while the small amount of gold detected in desipramine-treated cells was likely transported passively into the cells via uptake-2. Therefore, 127I-MoM-PEG-GNPs could selectively target the NET of neuroblastoma cells.
Conclusions
In this study, 127I-MoM-PEG-GNPs were novelly synthesized to utilize as a model of 131I-mIBG treatment in term of radioenhancement in neuroblastoma patients. We found that 49.28% of 127I-MoM-Boc bound to carboxylated PEG-GNPs. The conjugation of 127I-MoM-Boc onto the surface of the particles caused a slight red shift of the absorption peak using UV-vis spectrophotometry. The zeta potential of the 127I-MoM-Boc PEG-GNPs was less negative due to the absence of carboxyl groups, while after deprotection the zeta potential became more positive due to the presence of free guanidine groups. EDX analysis demonstrated % mass of iodine after the conjugation which help confirm the conjugation of 127I-MoM to carboxylated PEG-GNPs. Moreover, 127I-MoM-PEG-GNPs were proved to be stable after 4 week storage due to PEG modification. Cytotoxicity testing suggested that the model did no harm to the normal fibroblast cells, even at the highest concentration tested. Cellular uptake of 127I-MoM-PEG-GNPs into the neuroblastoma cell line, SH-SY5Y, was evaluated in the presence and absence of a NET inhibitor. With NET inhibitor, the uptake of the model was decreased suggesting that the model was internalized using uptake-1, via specific binding to NET. Hence, 127I-MoM-PEG-GNPs selectively targets the NET expressed in 90% of neuroblastoma cells. According to the properties mentioned above, 127I-MoM-PEG-GNPs could be used as a model for the application of targeted nanomaterial for neuroblastoma treatment. However, the radioenhancement effect of the product after 131I substitution is required for further study. In addition, in vivo experiments are also needed to investigate the biodistribution and excretion of the product.Author contributions
Kween, Khomson, Duangporn, Abdelhamid, Suradej, Putthiporn and Kulachart contributed to the design, analyze, summary the data, and discussion. Kween, Narongpol, Eknarin, Yodsathorn, and Rujira perform the experiment. Kween, Duangporn and Kulachart drafted article and revised critically and final approval of the version to be published.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by Mahidol University and the Office of National Higher Education Science Research and Innovation Policy Council (NXPO), Thailand, through Program Management Unit for Competitiveness (PMUC) (C10F630292), and the Royal Golden Jubilee PhD Program (grant number: PHD/0007/2560), of the Thailand Research Fund.Notes and references
- N. C. Colon and D. H. Chung, Adv. Pediatr., 2011, 58, 297–311 CrossRef PubMed.
- J. R. Park, A. Eggert and H. Caron, Pediatr. Clin., 2008, 55, 97–120 Search PubMed.
- K. T. Vo, K. K. Matthay, J. Neuhaus, W. B. London, B. Hero, P. F. Ambros, A. Nakagawara, D. Miniati, K. Wheeler, A. D. Pearson, S. L. Cohn and S. G. DuBois, J. Clin. Oncol., 2014, 32, 3169–3176 CrossRef PubMed.
- T. Monclair, G. M. Brodeur, P. F. Ambros, H. J. Brisse, G. Cecchetto, K. Holmes, M. Kaneko, W. B. London, K. K. Matthay, J. G. Nuchtern, D. von Schweinitz, T. Simon, S. L. Cohn, A. D. Pearson and I. T. Force, J. Clin. Oncol., 2009, 27, 298–303 CrossRef PubMed.
- S. L. Cohn, A. D. Pearson, W. B. London, T. Monclair, P. F. Ambros, G. M. Brodeur, A. Faldum, B. Hero, T. Iehara, D. Machin, V. Mosseri, T. Simon, A. Garaventa, V. Castel, K. K. Matthay and I. T. Force, J. Clin. Oncol., 2009, 27, 289–297 CrossRef PubMed.
- K. K. Matthay, J. M. Maris, G. Schleiermacher, A. Nakagawara, C. L. Mackall, L. Diller and W. A. Weiss, Nat. Rev. Dis. Primers, 2016, 2, 16078 CrossRef PubMed.
- K. Kramer, B. Kushner, G. Heller and N. K. Cheung, Cancer, 2001, 91, 1510–1519 CrossRef CAS PubMed.
- N. R. Pinto, M. A. Applebaum, S. L. Volchenboum, K. K. Matthay, W. B. London, P. F. Ambros, A. Nakagawara, F. Berthold, G. Schleiermacher, J. R. Park, D. Valteau-Couanet, A. D. Pearson and S. L. Cohn, J. Clin. Oncol., 2015, 33, 3008–3017 CrossRef CAS PubMed.
- J. M. Maris, N. Engl. J. Med., 2010, 362, 2202–2211 CrossRef CAS PubMed.
- V. Smith and J. Foster, Children, 2018, 5, 114 CrossRef PubMed.
- P. E. Zage, Children, 2018, 5, 148 CrossRef PubMed.
- P. E. Zage, C. U. Louis and S. L. Cohn, Pediatr. Blood Canc., 2012, 58, 1099–1105 CrossRef PubMed.
- S. B. Whittle, V. Smith, E. Doherty, S. Zhao, S. McCarty and P. E. Zage, Expert Rev. Anticancer Ther., 2017, 17, 369–386 CrossRef CAS PubMed.
- T. I. Kang, P. Brophy, M. Hickeson, S. Heyman, A. E. Evans, M. Charron and J. M. Maris, J. Pediatr. Hematol. Oncol., 2003, 25, 769–773 CrossRef PubMed.
- S. G. Dubois, E. Geier, V. Batra, S. W. Yee, J. Neuhaus, M. Segal, D. Martinez, B. Pawel, G. Yanik, A. Naranjo, W. B. London, S. Kreissman, D. Baker, E. Attiyeh, M. D. Hogarty, J. M. Maris, K. Giacomini and K. K. Matthay, Int. J. Mol. Imaging, 2012, 2012, 250834 Search PubMed.
- S. G. DuBois and K. K. Matthay, Q. J. Nucl. Med. Mol. Imaging, 2013, 57, 53–65 CAS.
- S. Mastrangelo, A. Tornesello, L. Diociaiuti, A. Pession, A. Prete, V. Rufini, L. Troncone and R. Mastrangelo, Br. J. Cancer, 2001, 84, 460–464 CrossRef CAS PubMed.
- J. S. Wilson, J. E. Gains, V. Moroz, K. Wheatley and M. N. Gaze, Eur. J. Canc., 2014, 50, 801–815 CrossRef CAS PubMed.
- J. Treuner, U. Feine, D. Niethammer, W. Muller-Schaumburg, J. Meinke, E. Eibach, R. Dopfer, T. Klingebiel and S. Grumbach, Lancet, 1984, 1, 333–334 CrossRef CAS.
- K. A. Streby, N. Shah, M. A. Ranalli, A. Kunkler and T. P. Cripe, Pediatr. Blood Canc., 2015, 62, 5–11 CrossRef CAS PubMed.
- S. Bonvalot, P. L. Rutkowski, J. Thariat, S. Carrere, A. Ducassou, M. P. Sunyach, P. Agoston, A. Hong, A. Mervoyer, M. Rastrelli, V. Moreno, R. K. Li, B. Tiangco, A. C. Herraez, A. Gronchi, L. Mangel, T. Sy-Ortin, P. Hohenberger, T. de Baere, A. Le Cesne, S. Helfre, E. Saada-Bouzid, A. Borkowska, R. Anghel, A. Co, M. Gebhart, G. Kantor, A. Montero, H. H. Loong, R. Verges, L. Lapeire, S. Dema, G. Kacso, L. Austen, L. Moureau-Zabotto, V. Servois, E. Wardelmann, P. Terrier, A. J. Lazar, J. Bovee, C. Le Pechoux and Z. Papai, Lancet Oncol., 2019, 20, 1148–1159 CrossRef CAS PubMed.
- M. Fernandes, K. R. B. Singh, T. Sarkar, P. Singh and R. P. Singh, Adv. Mater. Lett., 2020, 11, 20081543 CrossRef CAS.
- V. Nayak, K. R. B. Singh, A. K. Singh and R. P. Singh, New J. Chem., 2021, 45, 2849–2878 RSC.
- Y. H. Gholami, R. Maschmeyer and Z. Kuncic, Sci. Rep., 2019, 9, 14346 CrossRef PubMed.
- J. F. Dorsey, L. Sun, D. Y. Joh, A. Witztum, G. D. Kao, M. Alonso-Basanta, S. Avery, S. M. Hahn, A. Al Zaki and A. Tsourkas, Transl. Cancer Res., 2013, 2, 280–291 CAS.
- X. Zhang, Cell Biochem. Biophys., 2015, 72, 771–775 CrossRef CAS PubMed.
- R. Cao-Milan and L. M. Liz-Marzan, Expet Opin. Drug Deliv., 2014, 11, 741–752 CrossRef CAS PubMed.
- K. Sztandera, M. Gorzkiewicz and B. Klajnert-Maculewicz, Mol. Pharm., 2019, 16, 1–23 CrossRef CAS PubMed.
- J. F. Hainfeld, F. A. Dilmanian, D. N. Slatkin and H. M. Smilowitz, J. Pharm. Pharmacol., 2008, 60, 977–985 CrossRef CAS PubMed.
- S. Jain, D. G. Hirst and J. M. O'Sullivan, Br. J. Radiol., 2012, 85, 101–113 CrossRef CAS PubMed.
- K. Haume, S. Rosa, S. Grellet, M. A. Smialek, K. T. Butterworth, A. V. Solov'yov, K. M. Prise, J. Golding and N. J. Mason, Cancer Nanotechnol., 2016, 7, 8 CrossRef PubMed.
- W. Ngwa, R. Kumar, S. Sridhar, H. Korideck, P. Zygmanski, R. A. Cormack, R. Berbeco and G. M. Makrigiorgos, Nanomedicine, 2014, 9, 1063–1082 CrossRef CAS PubMed.
- M. Le Goas, M. Paquet, A. Paquirissamy, J. Guglielmi, C. Compin, J. Thariat, G. Vassaux, V. Geertsen, O. Humbert, J. P. Renault, G. Carrot, T. Pourcher and B. Cambien, Int. J. Nanomed., 2019, 14, 7933–7946 CrossRef CAS PubMed.
- M. Le Goas, A. Paquirissamy, D. Gargouri, G. Fadda, F. Testard, C. Aymes-Chodur, E. Jubeli, T. Pourcher, B. Cambien, S. Palacin, J. P. Renault and G. Carrot, ACS Appl. Bio Mater., 2019, 2, 144–154 CrossRef CAS.
- S. Yook, Z. Cai, Y. Lu, M. A. Winnik, J. P. Pignol and R. M. Reilly, Mol. Pharm., 2015, 12, 3963–3972 CrossRef CAS PubMed.
- S. Rosa, C. Connolly, G. Schettino, K. T. Butterworth and K. M. Prise, Cancer Nanotechnol., 2017, 8, 2 CrossRef PubMed.
- C. J. Liu, C. H. Wang, C. C. Chien, T. Y. Yang, S. T. Chen, W. H. Leng, C. F. Lee, K. H. Lee, Y. Hwu, Y. C. Lee, C. L. Cheng, C. S. Yang, Y. J. Chen, J. H. Je and G. Margaritondo, Nanotechnology, 2008, 19, 295104 CrossRef PubMed.
- S. Shrestha, L. N. Cooper, O. A. Andreev, Y. K. Reshetnyak and M. P. Antosh, Jacobs J. Radiat. Oncol., 2016, 3, 026 Search PubMed.
- S. Schottler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- S. Ekelund, P. Nygren and R. Larsson, Biochem. Pharmacol., 2001, 61, 1183–1193 CrossRef CAS PubMed.
- D. Hadrich, F. Berthold, E. Steckhan and H. Bonisch, J. Med. Chem., 1999, 42, 3101–3108 CrossRef CAS PubMed.
- G. Vaidyanathan, S. Shankar, D. J. Affleck, K. Alston, J. Norman, P. Welsh, H. LeGrand and M. R. Zalutsky, Bioorg. Med. Chem., 2004, 12, 1649–1656 CrossRef CAS PubMed.
- G. Villaverde, A. Baeza, G. J. Melen, A. Alfranca, M. Ramirez and M. Vallet-Regi, J. Mater. Chem. B, 2015, 3, 4831–4842 RSC.
- M. Iarossi, C. Schiattarella, I. Rea, L. De Stefano, R. Fittipaldi, A. Vecchione, R. Velotta and B. D. Ventura, ACS Omega, 2018, 3, 3805–3812 CrossRef CAS PubMed.
- J. P. Oliveira, A. R. Prado, W. J. Keijok, P. W. P. Antunes, E. R. Yapuchura and M. C. C. Guimaraes, Sci. Rep., 2019, 9, 13859 CrossRef PubMed.
- L. Zhang, Y. Mazouzi, M. Salmain, B. Liedberg and S. Boujday, Biosens. Bioelectron., 2020, 165, 112370 CrossRef CAS PubMed.
- A. Popovtzer, A. Mizrachi, M. Motiei, D. Bragilovski, L. Lubimov, M. Levi, O. Hilly, I. Ben-Aharon and R. Popovtzer, Nanoscale, 2016, 8, 2678–2685 RSC.
- G. Tan, K. Kantner, Q. Zhang, M. G. Soliman, P. Del Pino, W. J. Parak, M. A. Onur, D. Valdeperez, J. Rejman and B. Pelaz, Nanomaterials, 2015, 5, 1297–1316 CrossRef CAS PubMed.
- Y. Wang, J. E. Q. Quinsaat, T. Ono, M. Maeki, M. Tokeshi, T. Isono, K. Tajima, T. Satoh, S. I. Sato, Y. Miura and T. Yamamoto, Nat. Commun., 2020, 11, 6089 CrossRef CAS.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- A. D'Souza and R. Shegokar, Expet Opin. Drug Deliv., 2016, 13, 1257–1275 CrossRef PubMed.
- A. L. Bailly, F. Correard, A. Popov, G. Tselikov, F. Chaspoul, R. Appay, A. Al-Kattan, A. V. Kabashin, D. Braguer and M. A. Esteve, Sci. Rep., 2019, 9, 12890 CrossRef PubMed.
- J. Gains, H. Mandeville, N. Cork, P. Brock and M. Gaze, Future Oncol., 2012, 8, 839–858 CrossRef CAS PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- S. K. Balasubramanian, J. Jittiwat, J. Manikandan, C. N. Ong, L. E. Yu and W. Y. Ong, Biomaterials, 2010, 31, 2034–2042 CrossRef CAS PubMed.
- G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274–280 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS.
- R. R. Arvizo, O. R. Miranda, M. A. Thompson, C. M. Pabelick, R. Bhattacharya, J. D. Robertson, V. M. Rotello, Y. S. Prakash and P. Mukherjee, Nano Lett., 2010, 10, 2543–2548 CrossRef CAS.
- P. R. Lockman, J. M. Koziara, R. J. Mumper and D. D. Allen, J. Drug Target., 2004, 12, 635–641 CrossRef CAS PubMed.
- S. Ding, A. I. Khan, X. Cai, Y. Song, Z. Lyu, D. Du, P. Dutta and Y. Lin, Mater. Today, 2020, 37, 112–125 CrossRef CAS PubMed.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, in Metal Nanoparticles in Pharma, ed. P. D. M. Rai and P. D. R. Shegokar, Springer International Publishing, Cham, 2017, pp. 419–436, DOI:10.1007/978-3-319-63790-7_18.
- E. Caballero-Díaz and M. Valcárcel, in Comprehensive Analytical Chemistry, ed. M. Valcárcel and Á. I. López-Lorente, Elsevier, 2014, vol. 66, pp. 207–254 Search PubMed.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, ACS Omega, 2019, 4, 242–256 CrossRef CAS.
- G. Liu, Y. Li, L. Yang, Y. Wei, X. Wang, Z. Wang and L. Tao, RSC Adv., 2017, 7, 18252–18259 RSC.
- H. Bönisch and M. Brüss, in Neurotransmitter Transporters, ed. H. H. Sitte and M. Freissmuth, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 485–524, DOI:10.1007/3-540-29784-7_20.
- L. L. Iversen, Br. J. Pharmacol. Chemother., 1963, 21, 523–537 CrossRef CAS PubMed.
- R. J. Mairs, S. H. Cunningham, J. Russell, A. Armour, J. Owens, K. McKellar and M. N. Gaze, J. Nucl. Med., 1995, 36, 1088–1095 CAS.
- L. A. Smets, C. Loesberg, M. Janssen, E. A. Metwally and R. Huiskamp, Cancer Res., 1989, 49, 2941–2944 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04054e |
| This journal is © The Royal Society of Chemistry 2021 |