
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstitution reactivity and structural variability induced by tryptamine on the biomimetic rhenium tricarbonyl complex†
Frederick J. F. Jacobs a,
Gertruida J. S. Ventera,
Eleanor Fouriea,
Robin E. Kroonb and
Alice Brink*a
a,
Gertruida J. S. Ventera,
Eleanor Fouriea,
Robin E. Kroonb and
Alice Brink*a
aDepartment of Chemistry, University of the Free State, PO Box 339, Bloemfontein 9300, South Africa. E-mail: brinka@ufs.ac.za
bDepartment of Physics, University of the Free State, Bloemfontein 9300, South Africa
First published on 13th July 2021
Abstract
A series of seven fac-[Re(CO)3(5Me-Sal-Trypt)(L)] complexes containing tryptamine on the N,O 5-methyl-salicylidene bidentate ligand backbone and where L is MeOH, Py, Imi, DMAP, PPh3 coordinated to the 6th position have been studied, including the formation of a dinuclear Re2 cluster. The crystallographic solid state structures show marked similarity in structural tendency, in particular the rigidity of the Re core and the hydrogen bond interactions similar to those found in protein structures. The rates of formation and stability of the complexes were evaluated by rapid time-resolved stopped-flow techniques and the methanol substitution reaction indicates the significant activation induced by the use of the N,O salicylidene bidentate ligand as manifested by the second-order rate constants for the entering nucleophiles. Both linear and limiting kinetics were observed and a systematic evaluation of the kinetics is reported clearly indicating an interchange type of intimate mechanism for the methanol substitution. The anticancer activity of compounds 1–7 was tested on HeLa cells and it was found that all compounds showed similar cytotoxicity where solubility allowed. IC50-values between ca. 11 and 22 μM indicate that some cytotoxicity resides most likely on the salicylidene–tryptamine ligand. The photoluminescence of the seven complexes is similar in maximum emission wavelength with little variation despite the broad range of ligands coordinated to the 6th position on the metal centre.
Introduction
Schiff bases have been of significant interest to chemists due to their ease of synthesis and the variable properties achieved in the final product.1 From a clinical perspective they boast a broad range of medicinal application such as antiviral,2 antibacterial3 and antimalarial4 characteristics. These characteristics are dependent on the substitutes bound to the imine functionality, while the final properties of the Schiff base can be tuned to fit a particular need. One example of this is the recent development of Schiff bases as antiviral agents.5–7The imine functional group is prevalent in nature because the formation thereof is a result of the transamination reaction with pyridoxal-5-phosphate present in many of the known forms of life8 and so the use of Schiff bases as ligand systems for potential model radiopharmaceutical compounds is a reasonable approach.
The metal chosen for creating the model radiopharmaceutical complexes presented in this paper is rhenium, which is ideally suited due to its similarities to technetium.9 In radiopharmacy technetium is used as a diagnostic agent utilizing the 99mTc isotope which decays to release gamma radiation. 99mTc is routinely used in thyroid,10 heart,11 kidney,12 brain13 and bone14 imaging; moreover the isotope is investigated for selective cancer imaging.15–17 The 99mTc isotope can be produced through the use of a 99Mo generator system.18 Similarly 188Re can be generated from a 188W generator19 whereas 186Re can be produced from a cyclotron and isolated in high purity.20 Both 186/188Re radionuclides serve as β emitters which are used as therapeutic agents in the radiopharmaceutical industry for example in cancer21 and rheumatoid arthritis treatment.22 Due to their chemical similarity, these group seven elements (Re and Tc) can be used interchangeably and has been demonstrated in literature for Schiff base Re/Tc multinuclear complexes as potential theranostic agents.23 Furthermore the use of ‘cold’ non-radioactive rhenium permits detailed chemical and reactivity analysis without the hazards of using the radio-active isotopes of 186/188Re, 99mTc and 99Tc.
Rhenium based complexes have been investigated for their applications as diagnostic agents through luminescence. Photoluminescence investigations for bipyridine and phenanthroline type rhenium tricarbonyl complexes have been studied extensively.24–26 Photoluminescent rhenium tricarbonyl complexes have been proposed to be photo-probes in biological imaging25 and depending on the charge, relative concentration of the metal complexes and functional groups present in the final complex, the location of uptake in cells can be altered.27 This tunability provides a viable means to theranostic pathways if, for example, the luminescent complexes are enriched with 186/188Re.28 The photoluminescence of rhenium tricarbonyl complexes has been correlated to in vivo imaging with the 99mTc isotope further supporting the theranostic claim for fac-[M(CO)3]+ (where M = Re, Tc) complexes.29,30 Most of these complexes utilize pyridine based bidentate ligand systems to date, while little information on N,O Schiff base ligands in rhenium tricarbonyl complexes are found in published reports. Schiff base ligands shows significant luminescent activity without metal centres present, however depending on the metal used the Schiff base complex could exhibit variation in luminosity.31–33 While in certain cases the luminescent properties of Schiff base ligands improve upon complexation with metals, particularly that from the lanthanide series, the cytotoxicity is not necessarily so predictable in either the ligand or metal complex of that ligand and therefore needs to be systematically evaluated. It should be noted that rhenium complexes with little to no chemotoxic activity can still be of great interest for the clinical practitioner if a radioisotope is used, for example 188Re, should the complex be intended for therapy, exploiting the high energy β particles of the decaying nuclei, i.e. for radiotoxicity.31 It is therefore crucial to evaluate both ligand and coordinated complex to determine if the possible therapeutic behaviour originates from chemo-, radio or photo-toxic activity. Furthermore in any potential radiopharmaceutical, emphasis should be placed in having some idea of how the said compound could behave in vivo. Investigating the fundamental behaviour of the compound is necessary to predict what types of reactions the potential model radiopharmaceutical would partake in and by extension knowing with which proteins and biomolecules the compound would react with in vivo. The preference of biomolecule might be very difficult to predict due to the complexity of living organisms, but the fundamental behaviour of the complex in question can be investigated through in vitro kinetic research.
Substitution kinetics in six-coordinate octahedral metal complexes can follow one of the known three distinct mechanistic pathways if a monodentate ligand is substituted. These pathways are the classic associative, interchange and dissociative mechanisms.34–36 Each of the mechanisms follows two steps, and the relative rate of each determines the type of mechanism. In an associative type mechanism, substitution occurs through the rate determining step of ligand coordination followed by the fast dissociation of the leaving group. Dissociative mechanisms have the dissociation of the bound ligand as the rate determining step and the second associative step as the fast reaction in the mechanism. Interchange mechanisms has properties of both associative and dissociative mechanisms whereby there is a subtle interchange of coordinating/leaving ligands within the outer-sphere complex to form a rapid pre-equilibrium followed by a slower rate-determining second reaction.
With radiopharmaceuticals that use the associative type of mechanism, selective protein coordination via the metal centre is in theory more likely. The dissociative mechanisms involving the protein-metal coordination is possibly non-selective as an open coordination site on the metal centre is available for a select period of time. These mechanistic studies focus on protein-metal coordination and selectivity, however it is important to note that protein–ligand (i.e. the organic ligand bound to the organometallic complex) interactions may too play a role. Each of these pathways may have a direct affect on the viability of the complex as a radiopharmaceutical and on the design of the next iteration of potential model radiopharmaceuticals. Rhenium tricarbonyl complexes tend to display interchange dissociative characteristics37–39 however mechanistic crossovers have been proposed40,41 further highlighting the importance of kinetic investigations as well as the proper design of ligand systems for these types of complexes.
This paper reports the functionalising of a rhenium tricarbonyl salicylidene complex with the biological tryptamine group. Substitution of the 6th position on the metal centre was conducted with various monodentate ligands. The solid state structure analysis was conducted and includes a dinuclear complex. The reactive mechanistic pathways which occur during substitution were evaluated. Anti-cancer evaluation of the substituted complexes was conducted as well solid state luminescence investigations which determined the effect induced by varying the substituent in the 6th position.
Experimental
Materials and methods
All reagents and solvents used during this study were of analytical grade, purchased from Sigma-Aldrich and used without further purification. Rhenium pentacarbonyl bromide was purchased from Strem Chemicals, Newburyport, Massachusetts, United States of America. fac-[NEt4]2[ReBr3(CO)3] (ReAA) was synthesised according to the procedure found in literature.42 The tetraethylammonium salt can remain as a side product from the reaction and is removed with great difficulty. Although it is stable and non-reactive with regards to further kinetic experiments, removal of the tetraethylammonium salt utilising THF or acetone/benzene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) is practiced. During the removal of the THF we are unavoidably also removing product which is partly soluble and therefore low yields are occasionally obtained. The 1H and 13C NMR spectra (150.96 and 300.13 MHz respectively) of the ligands and complexes were recorded on a Bruker AXS 600 MHz, Bruker AXS 400 MHz or on a Bruker AXS 300 MHz at 25 °C in CD3CN (1.94 ppm), (CD3)2CO (2.05 ppm), (CD3)2SO (2.50 ppm), CD3OD (3.31 ppm) and CD2Cl2 (5.32 ppm). Positive shifts are downfield. All chemical shifts are reported in ppm and the coupling constants in Hz. The tetraethylammonium salt occurs at 3.19 ppm (q, 8H, J = 7.3 Hz, CH2), 1.15 ppm (tt, 12H, J = 7.2, 3.7 Hz, CH3) for 1H NMR and at 40.1, 7.07 ppm for 13C NMR. Elemental analysis was performed in Atlantic Microlab, Inc., Norcross, GA. A Bruker Tensor 27 Standard System spectrophotometer equipped with a 4000–370 cm−1 laser range was used to record the infrared spectra. All data were collected at room temperature. A Varian Cary 50 Conc. UV-Visible Spectrophotometer was used for UV-Vis spectra data collections which were conducted in a 1.000 ± 0.001 cm quartz cuvette cell. The UV-Vis spectrophotometer was equipped with a Julabo F12-mV temperature cell regulator, which is accurate to 0.1 °C.
:9) is practiced. During the removal of the THF we are unavoidably also removing product which is partly soluble and therefore low yields are occasionally obtained. The 1H and 13C NMR spectra (150.96 and 300.13 MHz respectively) of the ligands and complexes were recorded on a Bruker AXS 600 MHz, Bruker AXS 400 MHz or on a Bruker AXS 300 MHz at 25 °C in CD3CN (1.94 ppm), (CD3)2CO (2.05 ppm), (CD3)2SO (2.50 ppm), CD3OD (3.31 ppm) and CD2Cl2 (5.32 ppm). Positive shifts are downfield. All chemical shifts are reported in ppm and the coupling constants in Hz. The tetraethylammonium salt occurs at 3.19 ppm (q, 8H, J = 7.3 Hz, CH2), 1.15 ppm (tt, 12H, J = 7.2, 3.7 Hz, CH3) for 1H NMR and at 40.1, 7.07 ppm for 13C NMR. Elemental analysis was performed in Atlantic Microlab, Inc., Norcross, GA. A Bruker Tensor 27 Standard System spectrophotometer equipped with a 4000–370 cm−1 laser range was used to record the infrared spectra. All data were collected at room temperature. A Varian Cary 50 Conc. UV-Visible Spectrophotometer was used for UV-Vis spectra data collections which were conducted in a 1.000 ± 0.001 cm quartz cuvette cell. The UV-Vis spectrophotometer was equipped with a Julabo F12-mV temperature cell regulator, which is accurate to 0.1 °C.
Substitution kinetics
The kinetic measurements were conducted on a Varian Cary 50 Conc UV-Visible Spectrophotometer fitted with a Julabo F12-mV temperature cell regulator, which is accurate to 0.1 °C, in a 1.000 ± 0.001 cm quartz cuvette cell. The Varian Cary 50 Conc UV-Visible Spectrophotometer was used for slow kinetic reactions. The Hi-Tech FS-61DX2 Stopped-Flow instrument that is equipped with a Julabo F12-mV temperature cell regulator was used for all fast kinetic reactions and can be set up to use a Diode-Array or Photo-Multiplier detector. The Diode-Array is used to determine the best wavelength of the absorbance change of a kinetic reaction. Once a suitable wavelength has been determined the Photo-Multiplier detector is used for the kinetic reactions due to its greater accuracy. The Stopped-Flow instrument is operated by Kinet Asyst Stopped-Flow Kinetic Studio software43 on a Microsoft Windows operating system. All kinetic reactions were conducted under pseudo first-order conditions wherein the substituting ligand's concentration was at least ten times greater than the total rhenium metal complex concentration. Each kinetic trace was collected in triplicate. Once the data had been collected the information was processed by using Microsoft Office Excel 200744 and Scientist 2.01 developed by MicroMath.45 Reproducibility of the kinetic results was confirmed.
X-ray structure determination
The reflection data were collected on Bruker X8 Apex II 4K CCD and Bruker D8 Quest diffractometers.46,47 The diffractometers were equipped with a graphite monochromator using a Mo-Kα X-ray generator with a wavelength of λ = 0.71073 Å. Data was collected utilizing both phi and omega scans at a temperature of 100 K. COSMO48 was utilized for multiple hemisphere data collection of the reciprocal space. Bruker SAINT-Plus49 and XPREP49 were employed for frame integration and data reduction respectively. SADABS50 was used for phase correction through the multi-scan method. SHELXS51 was applied to solve the crystal structures, ab initio methods for the ligand systems and Patterson phasing for the crystals containing heavy metal centres. WinGX52 and SHELXL-2018/351 was used for the refinement of the crystal structures. The DIAMOND 3.053 software was utilized for the generation of images. Thermal ellipsoids are drawn with 50% probability level unless stated otherwise. Non-hydrogen atoms were refined with anisotropic displacement parameters. Methyl, methylene and aromatic hydrogens were constrained by riding them on the parent atoms in geometrically idealised positions with (C–H = 0.95–0.98 Å) and (Uiso(H) = 1.5Ueq(C) and 1.2Ueq(C)).
Luminescence
Solid state photoluminescent studies were conducted via an Edinburgh Instruments FLS980 series Fluorescence Spectrometer. A 450 W continuous xenon lamp (Xe1), filtered by a double monochromator, was used to excite the samples which were packed uniformly in a 0.3 mm demountable quartz cuvette. Quantum yield experimentation was done using an integrating sphere accessory.Cancer cell testing
Synthesis
5Me-SalH-Trypt (1),31 fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2)55 and fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3)55 were synthesised as described by literature methods.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.98 (s, 1H, Ar), 7.84 (s, 1H, Ar), 7.62 (t, 2H J = 6.7, 8.0 Hz, Ar), 7.35 (d, 1H J = 8.0 Hz, Ar), 7.21 (s, 1H, Ar), 7.08 (t, 1H J = 7.1, 1.1, 0.8 Hz, Ar), 7.05 (d, 1H J = 2.2 Hz, Ar), 6.99 (t, 1H J = 7.1, 6.8, 1.1, 0.8 Hz, Ar), 6.94 (s, 1H, Ar), 6.88 (d, 1H J = 7.9 Hz, Ar), 6.56 (s, 1H, Ar), 6.30 (dd, 1H J = 6.6, 1.4 Hz, Ar), 4.23–4.19 (m, 1H, CH2), 4.08 (q, 1H J = 11.0, 9.4, 7.5 Hz, CH2), 3.18–3.13 (m, 1H, CH2), 3.02–2.96 (m, 1H, CH2), 2.16 (s, 3H, CH3). 13C NMR (600 MHz, DMSO-d6): δ 165.74 (NCH), 164.75, 144.87, 137.95, 136.20, 135.29, 135.14, 128.08, 126.93, 123.34, 121.52, 121.08, 118.41, 118.33, 117.990, 117.93, 115.85, 111.45, 110.46 (Ar), 69.24 (N–CH2), 27.46 (CH2), 21.390 (CH3). Anal. calc.: C, 42.46%; H, 6.32%; N, 14.38%; found: C, 42.46%; H, 7.24%; N, 14.21%.CH), 7.93 (d, 2H J = 7.4 Hz, Ar), 7.58 (d, 1H J = 7.8 Hz, Ar), 7.34 (d, 1H J = 8.2 Hz, Ar), 7.07 (t, 2H J = 7.0, 7.1 Hz, Ar), 6.99 (t, 1H J = 7.0 Hz, Ar), 6.91 (d, 1H J = 7.9 Hz, Ar), 6.61 (d, 2H J = 7.4 Hz, Ar), 6.58 (s, 1H, Ar), 6.32 (d, 1H J = 6.7 Hz, Ar), 4.26–4.22 (m, 1H, CH2), 4.15–4.09 (m, 1H, CH2), 3.17–3.13 (m, 1H, CH2), 2.98–2.95 (m, 1H, CH2), 2.93 (s, 6H, CH3), 2.16 (s, 3H, CH3). 13C NMR (600 MHz, DMSO-d6): δ 166.06, 164.64, 154.49, 150.11, 145.12, 136.34, 135.38, 126.95, 123.38, 121.67, 121.15, 118.46, 118.31, 118.01, 116.22, 111.50, 110.43, 107.71 (Ar), 69.19 (CH2), 51.42 (CH2), 27.52 (CH3), 21.45 (CH3). Anal. calc.: C, 45.51%; H, 6.39%; N, 9.19%; found: C, 45.28%; H, 6.47%; N, 9.41%.N), 69.43 (CH2), 65.58 (CH2), 21.41 (CH3). Anal. calc.: C, 48.36%; H, 4.78%; N, 7.83%; found: C, 48.16%; H, 4.03%; N, 7.84%.
CH), 7.98 (s, 1H, Ar), 7.84 (s, 1H, Ar), 7.62 (t, 2H J = 6.7, 8.0 Hz, Ar), 7.35 (d, 1H J = 8.0 Hz, Ar), 7.21 (s, 1H, Ar), 7.08 (t, 1H J = 7.1, 1.1, 0.8 Hz, Ar), 7.05 (d, 1H J = 2.2 Hz, Ar), 6.99 (t, 1H J = 7.1, 6.8, 1.1, 0.8 Hz, Ar), 6.94 (s, 1H, Ar), 6.88 (d, 1H J = 7.9 Hz, Ar), 6.56 (s, 1H, Ar), 6.30 (dd, 1H J = 6.6, 1.4 Hz, Ar), 4.23–4.19 (m, 1H, CH2), 4.08 (q, 1H J = 11.0, 9.4, 7.5 Hz, CH2), 3.18–3.13 (m, 1H, CH2), 3.02–2.96 (m, 1H, CH2), 2.16 (s, 3H, CH3). 13C NMR (600 MHz, DMSO-d6): δ 165.74 (NCH), 164.75, 144.87, 137.95, 136.20, 135.29, 135.14, 128.08, 126.93, 123.34, 121.52, 121.08, 118.41, 118.33, 117.990, 117.93, 115.85, 111.45, 110.46 (Ar), 69.24 (N–CH2), 27.46 (CH2), 21.390 (CH3). Anal. calc.: C, 42.46%; H, 6.32%; N, 14.38%; found: C, 42.46%; H, 7.24%; N, 14.21%.CH), 7.93 (d, 2H J = 7.4 Hz, Ar), 7.58 (d, 1H J = 7.8 Hz, Ar), 7.34 (d, 1H J = 8.2 Hz, Ar), 7.07 (t, 2H J = 7.0, 7.1 Hz, Ar), 6.99 (t, 1H J = 7.0 Hz, Ar), 6.91 (d, 1H J = 7.9 Hz, Ar), 6.61 (d, 2H J = 7.4 Hz, Ar), 6.58 (s, 1H, Ar), 6.32 (d, 1H J = 6.7 Hz, Ar), 4.26–4.22 (m, 1H, CH2), 4.15–4.09 (m, 1H, CH2), 3.17–3.13 (m, 1H, CH2), 2.98–2.95 (m, 1H, CH2), 2.93 (s, 6H, CH3), 2.16 (s, 3H, CH3). 13C NMR (600 MHz, DMSO-d6): δ 166.06, 164.64, 154.49, 150.11, 145.12, 136.34, 135.38, 126.95, 123.38, 121.67, 121.15, 118.46, 118.31, 118.01, 116.22, 111.50, 110.43, 107.71 (Ar), 69.19 (CH2), 51.42 (CH2), 27.52 (CH3), 21.45 (CH3). Anal. calc.: C, 45.51%; H, 6.39%; N, 9.19%; found: C, 45.28%; H, 6.47%; N, 9.41%.N), 69.43 (CH2), 65.58 (CH2), 21.41 (CH3). Anal. calc.: C, 48.36%; H, 4.78%; N, 7.83%; found: C, 48.16%; H, 4.03%; N, 7.84%.Results and discussion
X-ray structure determination
The crystal structures of fac-[Re(CO)3(5Me-Sal-Trypt)(PPh3)] (5) and the dinuclear cluster fac-[Re(CO)3(5Me-Sal-Trypt)]2 (7) were determined by single crystal X-ray diffraction and compared to the previous reported structure55 of fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3). The geometric parameters, basic crystallographic data and data collection parameters are summarised in Table 1. Table 2 contains selected bond angles and distances of (3), (5) and (7). Fig. 1 depicts the molecular diagrams of (3), (5) and (7). Overlays of each of the crystal structures can be seen in Fig. 2. The crystal structures of (3), (5) and (7) show typical distorted octahedral geometries for rhenium(I) tricarbonyl complexes as evident from the bite-angles (N1–Re1–O4) of 84.47(5), 84.57(8) and 80.50(7)° each. The Re1–O4 and Re1–N1 bond distances between the three crystal structures are similar and the Re1–L bond distances vary according to the atom bonded in the sixth position with (7) having the shortest distance of 2.177(2) Å, (3) with 2.227(2) Å and (5) with the longest distance of 2.509(1) Å. The consistency of the bite-angles and bond distances to the Re metal centre indicates the relative rigidity and coordination preference of the metal core. These strong interactions seem to dominate the free rotation of the tryptamine side chain in the bidentate ligand. To highlight the effect the sixth ligand has upon the freedom of rotation of the tryptamine side chain, a dihedral angle between the salicylic aromatic group and the tryptamine aromatic group can be determined and compared against the calculated cone angle of each of the monodentate ligands.| 5 | 7 | |
|---|---|---|
| Empirical formula | C39 H32 N2 O4 P Re | C48 H46 N4 O10 Re2 |
| Formula weight (g mol−1) | 809.83 | 1211.29 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 10.5060(7) Å | 7.928(1) Å |
| b (Å) | 26.6424(17) Å | 9.046(1) Å |
| c (Å) | 12.3068(8) Å | 17.152(3) Å |
| α (°) | 90° | 78.981(5)° |
| β (°) | 106.455(2)° | 89.505(5)° |
| γ (°) | 90° | 75.427(5)° |
| Volume (Å3) | 3303.6(4) | 1167.5(3) |
| Z | 4 | 1 |
| ρcalc (g cm−3) | 1.628 | 1.723 |
| Crystal colour | Yellow | Yellow |
| Crystal morphology | Plate | Plate |
| Crystal size (mm) | 0.239 × 0.137 × 0.027 | 0.529 × 0.279 × 0.101 |
| μ (mm−1) | 3.771 | 5.241 |
| F (000) | 1608 | 592 |
| θ range (°) | 1.887 to 27.999 | 4.592 to 28.000 |
| Index ranges | −13 ≤ h ≤ 13 | −10 ≤ h ≤ 10 |
| −35 ≤ k ≤ 35 | −11 ≤ k ≤ 9 | |
| −14 ≤ l ≤ 16 | −22 ≤ l ≤ 20 | |
| Reflections collected | 42725 |
23942 |
| Unique reflections | 7965 | 5614 |
| Rint | 0.0542 | 0.0358 |
| Completeness to 2 theta (°, %) | 25.24, 99.9 | 25.24, 99.2 |
| Data/restraints/parameters | 7965/0/429 | 5614/19/299 |
| GooF | 1.066 | 1.028 |
| R [I > 2σ(I)] | R1 = 0.0236 | R1 = 0.0206 |
| wR2 = 0.0540 | wR2 = 0.0388 | |
| R (all data) | R1 = 0.0290 | R1 = 0.0255 |
| wR2 = 0.0566 | wR2 = 0.0405 | |
| ρmax, ρmin (e Å−3) | 1.482 and −0.853 | 0.438 and −0.535 |
| CCDC no | 2057438 | 2057437 |
| 3 (ref. 55) | 5 | 7 | |
|---|---|---|---|
| a The planes are generated from the two aromatic groups of the 5Me-Sal-Trypt coordinated ligand in all three crystal structures the first plane is generated from C12–C13–C14–C15–C16–C17 and the second plane is generated from C21–C22–C23–C24–C25–C26–C27–C28–N2. | |||
| Re1–O4 (Å) | 2.123(1) | 2.117(2) | 2.149(2) |
| Re1–N1 (Å) | 2.197(2) | 2.183(2) | 2.185(2) |
| Re1–L (Å) | 2.227(2) | 2.509(1) | 2.177(2) |
| N1–Re1–O4 (°) | 84.47(5) | 84.57(8) | 80.50(7) |
| N1–C19–C20–C21 (°) | −178.7(2) | 179.3(3) | −173.2(2) |
| Dihedral angle (°)a | 60.89(5) | 41.17(8) | 21.59(6) |
| Effective cone angle (°) | 124.20(4) | 147.87(2) | 205.15(5) |
| Average cone angle (°) | 122.70(6) | 139.92(3) | 158.88(8) |
 | ||
| Fig. 1 Molecular diagrams of complexes (A) fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3); (B) fac-[Re(CO)3(5Me-Sal-Trypt)(PPh3)] (5); (C) of fac-[Re(CO)3(5Me-Sal-Trypt)]2 (7). All three diagrams indicate atom numbering schemes and hydrogen atoms are omitted for clarity with the thermal ellipsoids drawn at 50% probability level.55 Structure A has been relabelled to match the labelling scheme of diagram B and C for the sake of comparison. | ||
 | ||
| Fig. 2 (A) Graphical overlay of the rhenium complexes fac-[Re(CO)3(5Me-Sal-Trypt)(PPh3)] (5) in blue and fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3) in green. (B) Graphical overlay of the rhenium complexes fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3) in green and fac-[Re(CO)3(5Me-Sal-Trypt)]2 (7) in red. (C) Graphical overlay of the rhenium complexes fac-[Re(CO)3(5Me-Sal-Trypt)(PPh3)] (5) in blue and fac-[Re(CO)3(5Me-Sal-Trypt)]2 (7) in red. All overlays are drawn through the atoms of the salicylidene ligand (Re1, N1, O4, C11–C18) and the three carbonyls to allow free rotation of the tryptamine side chain and the ligand in the 6th position. RMS values of 0.683, 0.190 and 0.795 Å for (A), (B) and (C) respectively were obtained. | ||
Two distinct cone angles were calculated for each of the ligands occupying the sixth positions in the three crystal structures. The first is the effective cone angle and its calculation is derived from the Tolman cone angle using the atom that generates the largest angle between the farthest outlier from the ligand and Re–X (where X = O, N or P) bond. The second is the average cone angle where the angle is calculated as an average from two/three atoms of the ligand in a similar fashion to the effective cone angle. Both cone angles are based on the work done by Tolman56 and Bilbrey et al.57 since they incorporate similar mathematical theories. The average cone angle is used as the preference here to obtain a measurable and comparable cone angle for the dinuclear structure (7) versus the two other monomeric structures (3 and 5); both cone angles are listed Table 2. The dihedral angles decrease from 60.89(5), 41.17(8) to 21.59(6)° for (3), (5) and (7). This can directly be related to the average cone angles of the ligands coordinated to the sixth positions which are 122.70(6), 139.92(3) and 158.88(8)° for (3), (5) and (7) respectively. The tryptamine side chain, with the advantage of the ethyl link, can rotate freely to accommodate the incoming ligands of varying sterics in the sixth position. In all three crystal structures the N2 atom and its accompanying hydrogen atom takes part in H-bonding interactions. In the case of (3) and (5) the hydrogen interactions occur with the acceptor O4 atom on an adjacent complex molecule through a symmetry operator appropriate to the steric bulk of the monodentate ligand occupying the 6th position and relative position of the tryptamine side chain (symmetry operators: x, −1 + y, z for (3) and ½ + x, ½ − y, ½ + z for (5)). In the crystal structure of (7) the N2 atom participates in hydrogen interactions with an acceptor O01 atom of a solvate acetone molecule through its identity symmetry operator. The similar hydrogen interactions found in these crystals structures indicate that the nitrogen–hydrogen pairing in the indole functional group is potentially important in the development for future radiopharmaceuticals as this is a prime location for biological interactions. Hydrogen bonding in ligands is even more significant when the metal centre is not available to partake in protein reactions, such as in the case of (7), where ligand–protein interactions are all that is available. These type of interactions (N–H…acceptor) can be seen in protein crystal structures of various indole based molecules such as tryptamine (PDB code: 2PQL),58 tryptophan (PDB code: 5TIA)59 and serotonin (PDB code: 5MT3).60 Indole based pharmaceuticals has also been under development suggesting that these types of structures and soft interactions could be of further significance in the medicinal world.61
Substitution kinetics
The negative side effects experience by patients during chemo- and radiotherapy could be significantly reduced if site-specific coordination of the organometallic molecule was achieved after drug administration. Selectivity and stability in vivo however still remains the gold standard which has not yet been fully achieved in all treatments. To this end, the mechanistic and kinetic evaluation of the said complexes was investigated to achieve an improved understanding as to what may alter the mechanistic behaviour of the rhenium tricarbonyl complexes. In particular, whether the functionalised tryptamine could assist the biomimetic development of organometallic complexes. To achieve site specific selection the following arguments were taken into account in order to build a complete reaction mechanism for the substitution of the coordinated methanol, found in the 6th position. These arguments are based on preliminary measurements, characterisation and published literature:The fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) starting material has been fully characterised by 1H & 13C NMR, IR and UV-Vis. The final kinetic products fac-[Re(CO)3(5Me-Sal-Trypt)(L)] (where L = pyridine (Py), imidazole (Imi), 4-dimethylaminopyridine (DMAP) and triphenylphosphine (PPh3)) complexes were independently synthesised and characterised with IR, 1H, 13C and 31P NMR to ensure no alternative products could be formed (Fig. 4). The X-ray diffraction crystal structure data was available for the kinetic product fac-[Re(CO)3(5Me-Sal-Trypt)(Py)] (3)55 and fac-[Re(CO)3(5Me-Sal-Trypt)(PPh3)] (5) hence confirming final coordination and bonding mode. The substitution of methanol by the four monodentate entering ligands (Py, Imi, DMAP and PPh3) was followed with the use of UV-Vis, 1H and 31P NMR spectroscopy. UV-Vis was used to quantitatively determine the rate constants for its economical use of starting reagents. The fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) starting material was found to be stable for over 24 hours in methanol. The stability studies were conducted at 25 °C and observed with the use of UV-Vis spectroscopy. The hydrolysis (i.e. substitution of methanol with water) of fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) was tested at 25 °C in freshly dried methanol with 5 varying concentrations of water, [H2O] = 0.0 M to 1.3 × 10−3 M. It was found that no hydrolysis reaction occurred under these experimental conditions. The preliminary UV-Vis traces for the substitution of methanol by Py, Imi, DMAP and PPh3 indicated that the reactions occur too quickly for standard UV-Vis studies and as such the 25 °C and 15 °C reactions were performed on the Stopped-Flow instrument. The 5 °C study had reaction times that could be conducted using the normal UV-Vis instrument (i.e. Varian Cary 50 Conc UV-Visible Spectrophotometer). Reproducibility for the 5 °C reaction was conducted on both the standard UV-Vis and Stopped-Flow instruments. Identical reaction rates were obtained at the same ligand concentrations on both instruments. A single reaction was observed under experimental pseudo first-order conditions for the temperatures of 5 °C, 15 °C and 25 °C. The formation of a single product is also confirmed from the very good isosbestic points observed (Fig. 3). Additional experiments were conducted at 35 °C and 45 °C however a second reaction was observed at higher temperature, with increased metal concentration and after extended reaction times. It is proposed that the second reaction is the dinuclear formation that becomes a dominating reaction at these higher temperatures compared to the substitution of the methanol. Dinuclear formation is reported to be dependent on time and the metal concentration. The reaction is thermodynamically favoured at high temperatures.62 From the results obtained in this study and reported in literature an interchange dissociative mechanism for the substitution of methanol in fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) is proposed.37,38,63–65
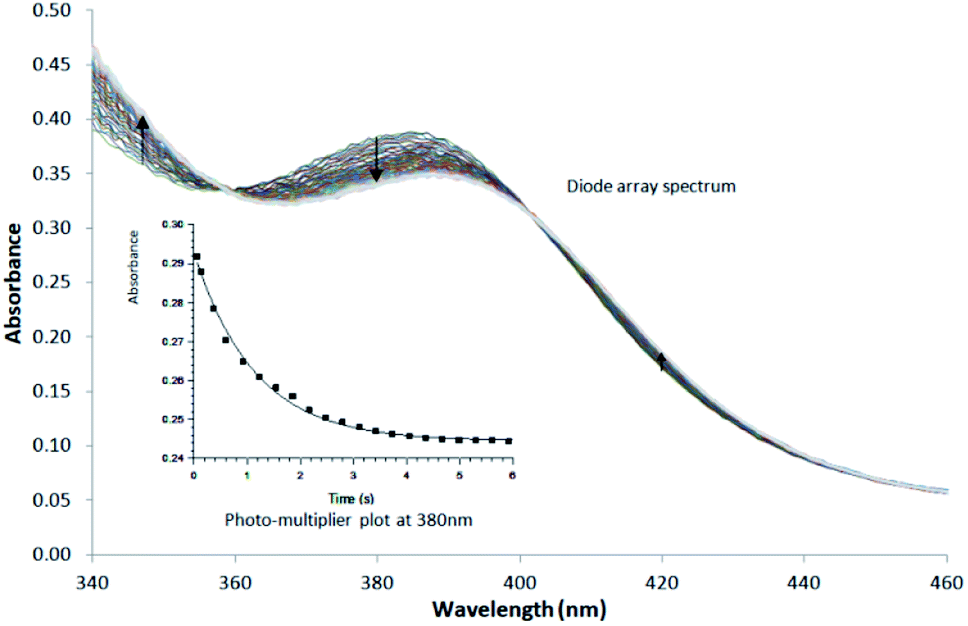 | ||
| Fig. 3 Typical Stopped-Flow UV-Vis change for the substitution of methanol in fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] by pyridine in methanol at 25 °C, [Re complex] = 7.9 × 10−4 M, [Py] = 0.40 M, Δt = 0.5 s as determined by Stopped-Flow in Diode Array mode. To accurately determine the reactant profile at 380 nm, the Photo-Multiplier setup on the Stopped-Flow was selected to produce the inserted absorbance versus time spectra. The solid line is the least-squares fit to eqn (1) obtaining kobs = 0.828 ± 0.007 s−1. Solid squares are experimentally obtained values. | ||
 | ||
| Fig. 4 Schematic diagram for the substitution of methanol by various neutral mono-dentate ligands (A) pyridine, (B) imidazole, (C) 4-dimethylaminopyridine and (D) triphenylphosphine in the fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) complex. | ||
This investigation focused on the linear relationship between concentration and absorbance for the purposes of determining the rate equation. All reactions were conducted under pseudo-first order conditions whereby [ligand] ≫ [Re complex]. After the reagents were mixed in solution the change in absorbance was recorded as a function of time. The decrease or increase in absorbance from the kinetic plots can be viewed as a representation of the change in concentration of the reagents or products of the reaction. The pseudo first-order rate constants for the reactions were determined by least-squares fitting to the equation
| Aobs = Af − (Af − Ai)e−kobst | (1) |
In eqn (1), Aobs is the observed absorbance, Af is the final absorbance, Ai the initial absorbance, kobs is the pseudo first-order rate constant and t is time. The limiting kinetic calculations i.e. the equations used and how it relates to linear kinetics (kf versus k1) can be found in a paper by Brink et al.64 as well as in the ESI† of this study.
The graphs of the pseudo first-order rate constants, kobs, versus [L] (where L = Py, Imi, DMAP and PPh3) were plotted from the UV-Vis data and yielded linear relationships with small intercepts for Py, Imi and PPh3. The rates of the reactions were collected at 5 °C, 15 °C and 25 °C and the results are represented in Fig. 5 for pyridine as a representative example. At high ligand concentrations non-linear (i.e. limiting) kinetics were obtained for DMAP (Fig. 6). The solubility of PPh3 in methanol was poor at 5 °C and 15 °C for PPh3 concentrations greater than 0.035 M. These reactions were therefore conducted at a lower concentration range of 0.002 M to 0.035 M. The solubility of Py, Imi and DMAP are high in methanol and therefore a large concentration range could be explored. The rates of the reactions generally increased by approximately a factor of two for every 10 °C increase in temperature, which is expected for most chemical reactions. The small value of k−1 indicates that there is no solvent assisted pathway present and a dominant tendency towards product formation with this specific complex in methanol under these experimental conditions further supported by the large k1 values indicating that the forward reaction of methanol substitution is the rapid and favoured reaction.
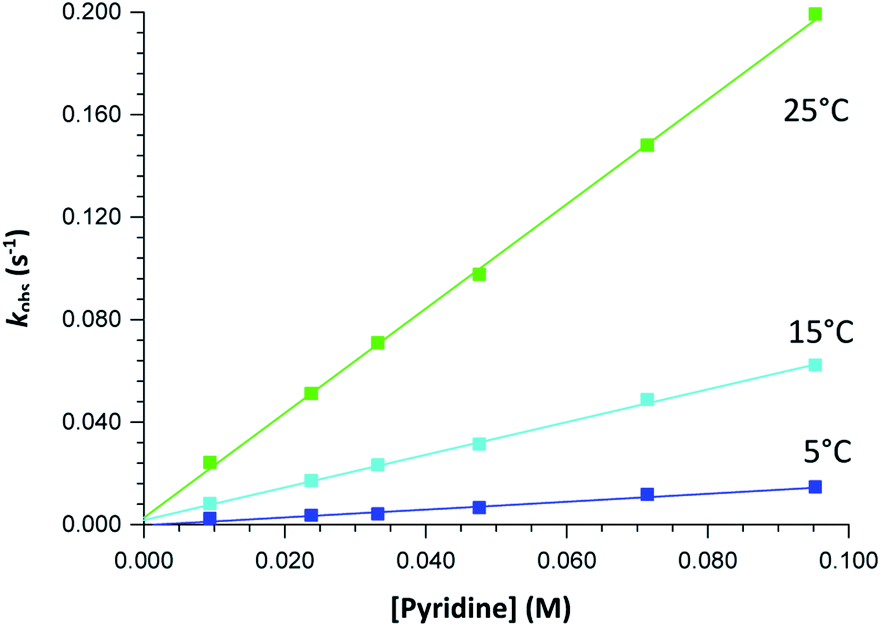 | ||
| Fig. 5 Plot of kobs versus [pyridine] for the reaction between fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) and pyridine at a range of temperatures with [Re complex] = 4 × 10−4 M and [pyridine] = 0.010 M − 0.095 M collected in methanol at 380 nm, yielding linear relationships. | ||
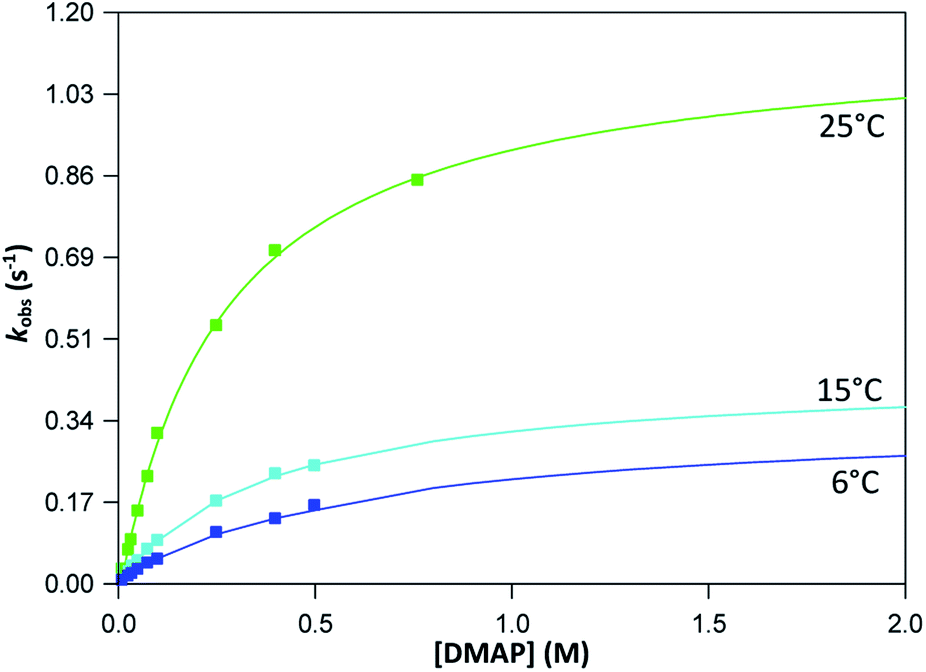 | ||
| Fig. 6 Plot of kobs versus [DMAP] for the reaction between fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) and DMAP at a range of temperatures with [Re complex] = 4 × 10−4 M and [DMAP] = 0.010 M − 0.762 M collected in methanol at 380 nm yielding non-linear, limiting kinetics. Saturation limits (i.e. the flattening of the curve to a plateau) has not yet been obtained despite a concentration range of nearly 2 orders-of-magnitude indicated by the extrapolated plot. | ||
From previous kinetic studies of rhenium salicyclidene complexes,63,64 an interchange dissociative mechanism was proposed which yields limiting (non-linear) kinetics at high ligand concentrations. At low ligand concentrations (and for ligands with low pKa values) the interchange mechanism will give linear second-order rate behaviour, i.e. kf = k1 = k3K2. Therefore the forward rate constant (kf) for an interchange mechanism is expected to be similar to the overall second-order rate constant (k1) within experimental error, especially at low ligand concentrations where saturation limits have not yet been reached (see Fig. 6 versus Fig. 7). Rate constants obtained for the reaction with DMAP illustrate the similarity in kf and k1 (within esd) (Table 3). For the sake of completion all rates were re-calculated assuming limiting kinetic traces and show a similar trend to previous reports (ESI Table 9†).
 | ||
| Fig. 7 Plot of kobs vs. ligand concentration for each ligand at 25 °C indicating linear kinetics traces for the concentration ranges of 0.01–0.1 M. | ||
| Entering ligands (L) | Temperature (°C) | k1 (M−1 s−1) | k−1 × 10−3 (s−1) | Keq × 102 (M−1) |
|---|---|---|---|---|
| a Concentration range of [Py] = 0.01–0.10 M.b [Imi] = 0.01–0.10 M.c [DMAP] = 0.01–0.10 M utilised for the calculation of linear kinetics.d [DMAP] = 0.01–0.50 M at 5.0 °C, 0.01–0.50 M at 15.1 °C and 0.01–0.76 M at 25.0 °C utilised for the calculation of limiting kinetics with line curvature.e [PPh3] = 0.0025–0.0350 M.f The forward rate constant (kf) of an interchange mechanism calculated for limiting kinetics. | ||||
| Pya | 5.0 | 0.11 ± 0.02 | 0.9 ± 0.6 | 1.1 ± 0.7 |
| 15.4 | 0.64 ± 0.01 | 1.8 ± 0.6 | 4 ± 1 | |
| 25.3 | 2.04 ± 0.03 | 3 ± 1 | 7 ± 4 | |
| Imib | 5.0 | 0.22 ± 0.01 | −0.8 ± 0.6 | −3 ± 2 |
| 15.6 | 0.62 ± 0.03 | 2 ± 1 | 3 ± 2 | |
| 25.2 | 1.66 ± 0.06 | 9 ± 3 | 1.8 ± 0.6 | |
| DMAPc,d | 5.0 | 0.56 ± 0.01 | 2 ± 0.5 | 2.6 ± 0.7 |
| 0.6 ± 0.1f | ||||
| 15.1 | 0.95 ± 0.03 | 2 ± 1 | 5 ± 4 | |
| 1.2 ± 0.1f | ||||
| 25.0 | 3.18 ± 0.08 | −8 ± 4 | −4 ± 2 | |
| 4.5 ± 0.4f | ||||
| PPh3e | 5.0 | 0.41 ± 0.03 | 0.2 ± 0.3 | 23 ± 41 |
| 15.4 | 1.87 ± 0.08 | 2 ± 1 | 8 ± 5 | |
| 25.3 | 4.1 ± 0.1 | 1 ± 1 | 34 ± 38 | |
Temperature studies to determine the activation entropy (ΔS≠) and activation enthalpy (ΔH≠) utilizing the Eyring (eqn (2)) were performed for all the reactions at temperatures ranging from ca. 5 °C to 25 °C and evaluated with the use of global fits as reported before,39,66–68
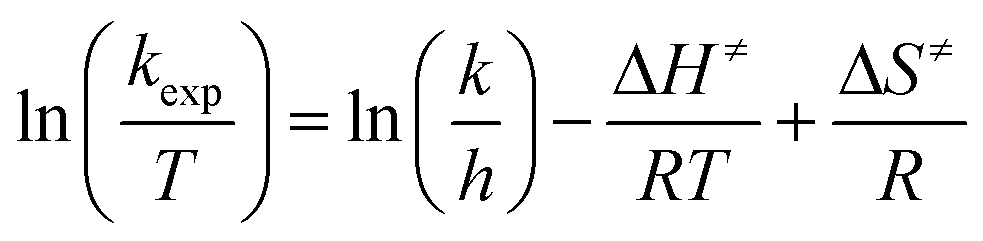 | (2) |
| ΔG≠ = ΔH≠ − TΔS≠ | (3) |
| Entering ligand (L) | k1 (M−1 s−1) | aΔG≠ (kJ mol−1) | ΔH≠ (kJ mol−1) | ΔS≠ (J K−1 mol−1) | bpKa |
|---|---|---|---|---|---|
| a Free energy of activation calculated at 25 °C.b References for pKa values.69–72c Calculation based on linear traces for [DMAP] = 0.01–0.10 M. The relative percentage contribution of TΔS≠ to ΔG≠ is 11% for Py; 1% for Imi; 6% for DMAP; 13% for PPh3. | |||||
| Py | 2.04 ± 0.03 | 71 ± 5 | 82 ± 1 | 35 ± 5 | 5.2 |
| Imi | 1.66 ± 0.06 | 72 ± 11 | 72 ± 3 | 2 ± 11 | 6.9 |
| DMAPc | 3.18 ± 0.08 | 70 ± 14 | 66 ± 4 | −13 ± 13 | 9.6 |
| PPh3 | 4.1 ± 0.1 | 70 ± 10 | 61 ± 3 | −30 ± 10 | 7.6 |
The variation in entropy (i.e. ΔS≠) are small and additional experiments such as volume of activation studies, could possibly provide ESI† for the absolute mechanism. This will be explored in the future. The relative percentage contribution of TΔS≠ to ΔG≠ is small (11% for Py; 1% for Imi; 6% for DMAP; 13% for PPh3) implying that the reactions are primarily enthalpy driven and thus more dependent on bond breaking/formation instead of ordering. The rate of methanol substitution by pyridine in various other rhenium Schiff-base complexes that have previously been conducted was compared to the results obtained in this study;63,64 fac-[Re(CO)3(N,O)(MeOH)] complexes (where N,O = 2-(m-tolyliminomethyl)phenolate, 2-(3-methylbutylimino methyl) phenolate, 2-(cyclohexyliminomethyl)phenolate, 2-(p-tolyliminomethyl)phenolate and 2-(phenyliminomethyl) phenolate). Fig. 8 graphically depicts the rates of each of the above mentioned complexes with the complex used in this study by plotting kobs versus pyridine concentration. The methyl and tryptamine functionalities in fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] (2) of this study activate the complex somewhat more than the previous complexes studied with the exception of the cyclohexylimine complex. This finding suggests that the reactivity for substitution of methanol is slightly tuneable by altering the substituents on the ligand backbone to achieve the desired reactivity for a metal complex. However no dramatic rate or mechanistic changes were observed by introducing the tryptamine functionality onto the ligand backbone. This tendency illustrates the dominant role of the N,O iminophenolate 6 membered cyclic backbone which is coordinated to the rhenium metal centre on the kinetic reactivity. The rate of methanol substitution by pyridine was conducted with N,N′, N,O and O,O′ bidentate ligand systems (1,10-phenanthroline, 2,2′-bipydine, 2-picolinate, 2-quinolinate, 2,4-dipicolinate, 2,4-diquinolinate, tribromo tropolonate and hydroxyflavonate) by Schutte-Smith et al. wherein an interchange dissociative mechanism was proposed. The reaction rates of these N,O bidentate ligand systems were found to be significantly slower than the rates of methanol substitution in fac-[Re(CO)3(5Me-Sal-Trypt)(MeOH)] with reaction rates up to three orders-of-magnitude faster which the large tryptamine side chain does not seem to hinder.37,38
 | ||
| Fig. 8 Plot of kobs versus [Pyridine] (0.01–0.40 M) for fac-[Re(CO)3(R-Sal-T)(MeOH)] complexes (where R = H, CH3 and T = m-toluidine, p-toluidine, phenylimine, 3-methylbutylimine, cyclohexylimine and tryptimine; λ = 440, 436, 441, 416, 417 and 380 nm respectively) substituted with pyridine in methanol at 25 °C. | ||
Additional investigations with the fac-[Re(CO)3(Tropolone) (MeOH)] complex substituting methanol with imidazole in high pressure kinetics studies also indicated an interchange dissociative mechanism.39 The research by Grundler et al. for water substitution in fac-[Re(CO)3(H2O)3] with monodentate ligands (acetonitrile, Br−, pyrazine, tetrahydrothiophene and dimethylsulfide) does indicate that a mechanistic changeover from an interchange dissociative to an interchange associative mechanism is possible.40,41
In conclusion an interchange dissociative mechanism is proposed based on the low, near zero values of ΔS≠; the subtle interplay of linear and limiting kinetics (particularly noticeable in the pyridine versus the DMAP); The relative consistency of k1 for all the entering nucleophiles despite the varying pKa's and steric parameters as well as the similarity of kobs behaviour (Fig. 8) to that reported in literature.63,64
Cytotoxicity studies
The bidentate ligand, 5Me-SalH-Tryptamine (1), was designed with biological activity in mind. The ligand is constructed using 4-methyl-2-hydroxybenzaldehyde and tryptamine as starting materials. The aldehyde was chosen for its similarities to aspirin which has well known bioactivity and protein interactions as seen in the protein crystallography (PDB code: 1OXR).73 Donor/acceptor interactions are observed from the protein crystal structure through the oxygen atoms and aromatic structure of the aspirin molecule. Tryptamine which is a neurotransmitter has significant biological interactions.74 From protein structure analysis (PDB code: 2PQL) donor/acceptor interactions can be observed at the amine NH2 and indole NH atoms as well as interactions with the aromatic indole functional group all of which may play a determining role in protein binding.58 Our fac-[Re(CO)3(5Me-SalH-Tryptamine)(L)] (where L = MeOH, Py, Imi, DMAP and PPh3) complexes, including the dinuclear structure (7), does lose the NH2 binding site during Schiff-base formation and ligand complexation, however the N indole, O (bidentate ligand) and carbonyl atoms are still available for donor/acceptor interactions and does appear consistently in the three small molecule crystallographic results. It is important to note that carefully thought out ligand systems and subtle variations in the final metal complex does not necessarily translate to predictably better or worse cytotoxic results and so cell line testing becomes imperative in radiopharmaceutical development.75The anticancer activity of compounds (1–7) used in this study, as well as cisplatin, was tested in vitro on HeLa cells by using the SRB assay. The half maximal inhibitory concentration (IC50) was calculated from colorimetric determinations expressed as a percentage of the control. IC50 values are summarized in Fig. 9 and all cell-growth curves are shown in the ESI† to this paper (Fig. 10).
 | ||
| Fig. 9 Graphical representation of the IC50 values (the concentration needed, in μM, to inhibit 50% of the growth of cancer cells) of each compound versus Cisplatin. Error bars and IC50 values are displayed above each bar. | ||
All of the compounds exhibited anticancer activity with an IC50 value of between 11.6 and 22 μM, except for complex (6) and (7). Compound (1) is the free ligand and complex (2), the MeOH substituted compound. Complex (3), (4) and (6) are the N-substituted complexes, (5) contains PPh3 in the 6th position and (7) is the dinuclear cluster. Comparable IC50's were obtained for (1–5), suggesting the cytotoxic capability most likely resides in the functionalized tryptamine ligand. A slightly better value is obtained for the MeOH substituted compound (2) (11.6 μM), which can be ascribed to the lability of the solvent molecule which permits for substitution and coordination once inside the cells. Compounds (6) and (7) dissolved initially but precipitated out of the aqueous solution over time, thus leading to higher IC50 values. Due to the similar anticancer activity of all compounds in this study, including the free functionalized tryptamine ligand it is postulated that the ligand possesses the dominant anticancer activity observed. This is supported by findings of Cerchiaro et al. who found anticancer activity for oxoindole type ligands.76 Results also suggest that little or no anticancer activity is based on the Re-metal. Binkley et al. indicated that [Re(TAME)(CO)3]X complexes (where TAME = trisaminomethylethane; X = Br, Cl, NO3, PF6, ClO4) showed little or no toxicity as tested against HeLa cells,77 while Marker et al. also showed very little toxicity by rhenium tricarbonyl complexes.15,78 This suggest that as used for specific labelling the rhenium tricarbonyl complexes is ideal since the chemical toxicity is nearly negligible, and only the radio-toxicity should be considered when 186/188Re is utilised.
Photoluminescence
Photoluminescence of each of the compounds (1–7) were conducted. Due to the limited sample mass and poor lambertian behaviour of some of the metal complexes quantum yields was only conducted on the ligand ((1), in the form of crystalline flakes) which was found to be 11% when excited at 350 nm, see Fig. 10. | ||
| Fig. 10 Absorption and emission spectrum of 5Me-SalH-Tryp (1) against a Spectralon reference, showing 11% quantum yield for (1). The figure also includes an insert of a depiction of the sample being illuminated at 350 nm. | ||
Previously ((1), in the form of powder) has been investigated and demonstrated to have a quantum yield of 1.3% and 0.9% at 390 nm and 280 nm.31 Reproducibility of the quantum yield of (1) in both forms (powder vs. crystalline) has been confirmed. The morphology of a luminescent material may have a strong effect on its emission intensity, e.g. samples of SrTiO3 with spherical morphology emitted much more intensely than cubic particles which were chemically identical.79 In the case of (1) the quantum yield of the crystalline flakes may be greater than the powder due to its lower surface-to-volume ratio (considering the surface as a crystalline defect which may result in non-radiative de-excitation) as well as the greater rigidity imposed on the individual molecules by the crystal structure that may inhibit quenching. The tryptophan and tryptamine has comparable excitation and emission properties to (1) indicating that the indole ring system plays a dominating role in the ligands photochemical characteristics.80 For comparative purposes the excitation wavelength for each of the compounds (1–7) was chosen at 350 nm and wavelengths at the maximum emission intensity for the complexes were found to be 570 nm, 550 nm, 570 nm, 570 nm, 550 nm and 550 nm for compounds 2–7 respectively. The excitation and emission spectra of (1–7) can be seen in Fig. 11. While no correlation could be observed between the 6th ligand coordinated to the metal centre and the peak emission wavelength the similarities between the wavelengths indicate that the bidentate Schiff base ligand dwarfs the contribution to luminescence with respect to the monodentate ligand. Very little information is available with respect to the luminescence of N,O Schiff base fac-[Re(CO)3]+ complexes however pyridine based bidentate ligand systems have received attention in this regard. Clède et al.81 reported a series of N,N′ neutral bidentate ligands coordinated to fac-[Re(CO)3]+ which had somewhat similar excitation and emission wavelengths of 335 nm and 527 nm each compared to our 350 nm excitation and 550 nm and 570 nm emission wavelengths. It is worth noting that Clède et al. conducted their photoluminescence studies in acetonitrile. Doleck et al.82 produced bipyridyl based systems on the fac-[Re(CO)3]+ core which showed large variations in luminescence wavelengths (225–402 nm excitation and 435–730 nm emission) when observed in acetonitrile solutions.
 | ||
| Fig. 11 Excitation (red) and emission (black) graphs of the compounds (1–7). | ||
Conclusions
Our interest is in the development of site-specific radiopharmaceuticals, in particularly how the fundamental mechanistic reactivity can assist the said development. We have synthesised a range of fac-[Re(CO)3(5Me-Sal-Trypt)(L)] complexes whereby L = MeOH, Py, Imi, DMAP, PPh3 including a the dinuclear cluster containing tryptamine on the ligand backbone. The solid state structures show marked similarity in structural tendency, in particular the rigidity of the Re core and the H-bond interactions of the N2 indole and O4 salicylidene backbone, interactions which may prove key for future protein–ligand coordination. The substitution kinetics confirms the stability of the complexes as well as the interchange dissociative mechanism. A mechanism which may not be ideal for selective coordination due to the 5 coordinate transition state intermediate which becomes available during substitution. However the increase reactivity (3 orders of magnitude) is ideal for radiopharmaceuticals wherein one must consider the half life of the radio-isotope. Additionally cell testing indicates that the complexes are not exceptionally cytotoxic and that radioactivity would be the driving force should any of these metal complexes be introduced in a clinical environment when using a suitable radioisotope. The photoluminescence between the complexes are similar in maximum emission wavelength with little variation despite the broad range of ligands coordinated to the 6th position on the metal centre.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial assistance from the University of the Free State and SASOL Pty Ltd is gratefully acknowledged. We also express our gratitude towards the South African National Research Foundation (SA-NRF). This work is based on the research supported in part by the National Research Foundation of South Africa: A. Brink (Grant no. UID 99139). The Grant-holder acknowledges that opinions, 'findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the authors and that the NRF accepts no liability whatsoever in this regard. We wish to thank Prof A Roodt for his guidance regarding the kinetic mechanistic studies.Notes and references
- J. Jiang and M. J. MacLachlan, Chem. Commun., 2009, 5695–5697 RSC.
- K. S. Kumar, S. Ganguly, R. Veerasamy and E. De Clercq, Eur. J. Med. Chem., 2010, 45, 5474–5479 CrossRef CAS PubMed.
- C. C. Login, I. Bâldea, B. Tiperciuc, D. Benedec, D. C. Vodnar, N. Decea and Ş. Suciu, Oxid. Med. Cell. Longevity, 2019, 2019, 1–11 CrossRef.
- C. M. Da Silva, D. L. Da Silva, L. V. Modolo, R. B. Alves, M. A. De Resende, C. V. B. Martins and Â. De Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- A. S. Sokolova, O. I. Yarovaya, A. V. Shernyukov, Y. V. Gatilov, Y. V. Razumova, V. V. Zarubaev, T. S. Tretiak, A. G. Pokrovsky, O. I. Kiselev and N. F. Salakhutdinov, Eur. J. Med. Chem., 2015, 105, 263–273 CrossRef CAS.
- J. M. Mir, S. A. Majid and A. H. Shalla, Rev. Inorg. Chem., 2021 DOI:10.1515/revic-2020-0020.
- M. N. Uddin, S. S. Ahmed and S. M. R. Alam, J. Coord. Chem., 2020, 73, 3109–3149 CrossRef CAS.
- R. Percudani and A. Peracchi, EMBO Rep., 2003, 4, 850–854 CrossRef CAS.
- D. Can, P. Schmutz, S. Sulieman, B. Spingler and R. Alberto, Chimia, 2013, 67, 267–270 CrossRef CAS.
- W. J. Dodds and M. R. Powell, Radiology, 1968, 91, 27–31 CrossRef CAS PubMed.
- D. Jain, Semin. Nucl. Med., 1999, 29, 221–236 CrossRef CAS.
- D. L. Nosco and J. A. Beaty-Nosco, Coord. Chem. Rev., 1999, 184, 91–123 CrossRef CAS.
- S. Jurisson, D. Berning, W. Jia and D. Ma, Chem. Rev., 1993, 93, 1137–1156 CrossRef CAS.
- K. Lisbon, E. Deutsch and B. L. Barnett, J. Am. Chem. Soc., 1980, 102, 2476–2478 CrossRef.
- K. E. Goffin, S. Joniau, P. Tenke, K. Slawin, E. A. Klein, N. Stambler, T. Strack, J. Babich, T. Armor and V. Wong, J. Nucl. Med., 2017, 58, 1408–1413 CrossRef CAS PubMed.
- L. O. F. Monteiro, R. S. Fernandes, L. C. Castro, V. N. Cardoso, M. C. Oliveira, D. M. Townsend, A. Ferretti, D. Rubello, E. A. Leite and A. L. B. de Barros, Biomed. Pharmacother., 2017, 89, 146–151 CrossRef CAS PubMed.
- X. Lin, Q. Ruan, L. Lin, X. Zhang, X. Duan, Y. Teng and J. Zhang, J. Radioanal. Nucl. Chem., 2018, 317, 1463–1468 CrossRef CAS.
- Z. B. Alfassi, F. Groppi, M. L. Bonardi and J. J. M. de Goeij, Appl. Radiat. Isot., 2005, 63, 37–40 CrossRef CAS.
- M. R. A. Pillai, A. Dash and F. F. Knapp, Curr. Radiopharm., 2012, 5, 228–243 CrossRef CAS PubMed.
- I. E. Alekseev and V. V. Lazarev, Radiochemistry, 2006, 48, 497–500 CrossRef CAS.
- J. M. Jeong and J. Chung, Cancer Biother.Radiopharm., 2003, 18, 707–717 CrossRef CAS.
- E. Deutsch, J. W. Brodack and K. F. Deutsch, Eur. J. Nucl. Med., 1993, 20, 1113–1127 CrossRef CAS PubMed.
- A. Frei, P. P. Mokolokolo, R. Bolliger, H. Braband, M. S. Tsosane, A. Brink, A. Roodt and R. Alberto, Chem. – Eur. J., 2018, 24, 10397–10402 CrossRef CAS PubMed.
- D. L. Ma, H. Z. He, K. H. Leung, D. S. H. Chan and C. H. Leung, Angew. Chem., 2013, 52, 7666–7682 CrossRef CAS.
- A. Carreño, K. Fernández, F. Sáez-Cortez, C. Otero, R. Arratia-Pérez, J. A. Fuentes and R. Polanco, J. Chil. Chem. Soc., 2019, 64, 4428–4431 CrossRef.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- J. Skiba, T. Bernás, D. Trzybínski, K. Wózniak, G. Ferraro, D. Marasco, A. Merlino, M. Z. Shafikov, R. Czerwieniec and K. Kowalski, Molecules, 2017, 22, 809 CrossRef PubMed.
- J. Yang, Q. Cao, H. Zhang, L. Hao, D. Zhou, Z. Gan, Z. Li, Y. X. Tong, L. N. Ji and Z. W. Mao, Biomaterials, 2018, 176, 94–105 CrossRef CAS PubMed.
- K. A. Stephenson, S. R. Banerjee, T. Besanger, O. O. Sogbein, M. K. Levadala, N. McFarlane, J. A. Lemon, D. R. Boreham, K. P. Maresca, J. D. Brennan, J. W. Babich, J. Zubieta and J. F. Valliant, J. Am. Chem. Soc., 2004, 126, 8598–8599 CrossRef CAS PubMed.
- T. Yoshimura, K. Nagata, T. Shiroyama, Y. Kino, T. Takayama, T. Sekine and A. Shinohara, Dalton Trans., 2018, 47, 16027–16030 RSC.
- A. Brink, R. E. Kroon, H. G. Visser, C. E. J. Van Rensburg and A. Roodt, New J. Chem., 2018, 42, 5193–5203 RSC.
- Y. W. Dong, R. Q. Fan, P. Wang, L. G. Wei, X. M. Wang, H. J. Zhang, S. Gao, Y. L. Yang and Y. L. Wang, Dalton Trans., 2015, 44, 5306–5322 RSC.
- D. Zhang, B. Gao and Y. Li, Luminescence, 2017, 32, 855–865 CrossRef CAS PubMed.
- C. H. Langford and H. B. Gray, Ligand substitution processes, W. A. Benjamin, Inc., New York, Amsterdam, 1966 Search PubMed.
- H. Elias, Berichte der Bunsengesellschaft für Phys. Chemie, 1992, 96, 638–639 CrossRef.
- T. W. Swaddle, Adv. Inorg. Bioinorg. Mech., 1983, 2, 95 CAS.
- M. Schutte, G. Kemp, H. G. Visser and A. Roodt, Inorg. Chem., 2011, 50, 12486–12498 CrossRef CAS PubMed.
- M. Schutte, A. Roodt and H. G. Visser, Inorg. Chem., 2012, 51, 11996–12006 CrossRef CAS PubMed.
- M. Schutte-Smith, A. Roodt and H. G. Visser, Dalton Trans., 2019, 48, 9984–9997 RSC.
- P. V. Grundler, B. Salignac, S. Cayemittes, R. Alberto and A. E. Merbach, Inorg. Chem., 2004, 43, 865–873 CrossRef CAS PubMed.
- P. V. Grundler, L. Helm, R. Alberto and A. E. Merbach, Inorg. Chem., 2006, 45, 10378–10390 CrossRef CAS PubMed.
- R. Alberto, R. Schibli and P. A. Schubiger, Polyhedron, 1996, 15, 1079–1089 CrossRef CAS.
- TgK Scientific Kinetic Studio, Version 1.0.8.32278, Copyright©TgK Scientific, 2008 Search PubMed.
- Microsoft Office Professional Edition 2003, Copyright©Microsoft Corporation, 2003 Search PubMed.
- MicoMath Scientist for Windows, Version 2.01, Copyright©MicroMath Inc, 1995 Search PubMed.
- Bruker APEX2, Version 1.0-27, Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed.
- Bruker D8 QUEST, Version 2019.11-RC6, Bruker AXS Inc., Madison, Wisconsin, USA, 2019 Search PubMed.
- COSMO, Version 1.48, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- SAINT-PLUS, Version 7.12 (including XPREP), Bruker AXS Inc., Madison, Wisconsin, USA, 2004 Search PubMed.
- SADABS, Version 2004/1, Bruker AXS Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- K. Brandenburg and H. Putz, DIAMOND, Release 3.2k. Crystal Impact, GbR, Bonn, Germany, 2005 Search PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS.
- A. Brink, H. G. Visser and A. Roodt, Polyhedron, 2013, 52, 416–423 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189–1197 CrossRef CAS.
- B. J. Mans, E. Calvo, J. M. C. Ribeiro and J. F. Andersen, J. Biol. Chem., 2007, 282, 36626–36633 CrossRef CAS.
- A. Lewis-Ballester, F. Forouhar, S. M. Kim, S. Lew, Y. Wang, S. Karkashon, J. Seetharaman, D. Batabyal, B. Y. Chiang, M. Hussain, M. A. Correia, S. R. Yeh and L. Tong, Sci. Rep., 2016, 6, 1–13 CrossRef.
- V. Palivec, C. M. Viola, M. Kozak, T. R. Ganderton, K. Křížková, J. P. Turkenburg, P. Halušková, L. Žáková, J. Jiráček, P. Jungwirth and A. M. Brzozowski, J. Biol. Chem., 2017, 292, 8342–8355 CrossRef CAS.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS.
- P. P. Mokolokolo, A. Frei, M. S. Tsosane, D. V Kama, M. Schutte-smith, A. Brink, H. G. Visser, G. Meola, R. Alberto and A. Roodt, Inorg. Chim. Acta, 2018, 471, 249–256 CrossRef CAS.
- A. Brink, H. G. Visser and A. Roodt, Inorg. Chem., 2013, 52, 8950–8961 CrossRef CAS.
- A. Brink, H. G. Visser and A. Roodt, Inorg. Chem., 2014, 53, 12480–12488 CrossRef CAS PubMed.
- B. Salignac, P. V. Grundler, S. Cayemittes, U. Frey, R. Scopelliti, A. E. Merbach, R. Hedinger, K. Hegetschweiler, R. Alberto, U. Prinz, G. Raabe, U. Kölle and S. Hall, Inorg. Chem., 2003, 42, 3516–3526 CrossRef CAS PubMed.
- T. N. Twala, M. Schutte-Smith, A. Roodt and H. G. Visser, Dalton Trans., 2015, 44, 3278–3288 RSC.
- H. J. Van Der Westhuizen, R. Meijboom, M. Schutte and A. Roodt, Inorg. Chem., 2010, 49, 9599–9608 CrossRef CAS.
- A. Frei, D. Sidler, P. Mokolokolo, H. Braband, T. Fox, B. Spingler, A. Roodt and R. Alberto, Inorg. Chem., 2016, 55, 9352–9360 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, Milton, 85th edn, 2004 Search PubMed.
- H. Walba and R. W. Isensee, J. Org. Chem., 1961, 26, 2789–2791 CrossRef CAS.
- K. Haav, J. Saame, A. Kütt and I. Leito, Eur. J. Org. Chem., 2012, 2012, 2167–2172 CrossRef CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- R. K. Singh, A. S. Ethayathulla, T. Jabeen, S. Sharma, P. Kaur and T. P. Singh, J. Drug Targeting, 2005, 13, 113–119 CrossRef CAS PubMed.
- M. Z. Khan and W. Nawaz, Biomed. Pharmacother., 2016, 83, 439–449 CrossRef CAS.
- A. K. Renfrew, Metallomics, 2014, 6, 1324–1335 CrossRef CAS PubMed.
- G. Cerchiaro and A. M. D. C. Ferreira, J. Braz. Chem. Soc., 2006, 17, 1473–1485 CrossRef CAS.
- S. L. Binkley, N. V. Barone, A. C. Underwood, A. Milsted, B. R. Franklin, R. S. Herrick and C. J. Ziegler, J. Inorg. Biochem., 2010, 104, 632–638 CrossRef CAS.
- S. C. Marker, S. N. MacMillan, W. R. Zipfel, Z. Li, P. C. Ford and J. J. Wilson, Inorg. Chem., 2018, 57, 1311–1331 CrossRef CAS PubMed.
- L. F. Da Silva, W. Avansi, M. L. Moreira, A. Mesquita, L. J. Q. Maia, J. Andrés, E. Longo and V. R. Mastelaro, J. Nanomater., 2012, 2012, 25–28 Search PubMed.
- J. W. Bridges and R. T. Williams, Biochem. J., 1968, 107, 225–237 CrossRef CAS PubMed.
- S. Clède, F. Lambert, R. Saint-Fort, M. A. Plamont, H. Bertrand, A. Vessières and C. Policar, Chem. - Eur. J., 2014, 20, 8714–8722 CrossRef.
- T. Doleck, J. Attard, F. R. Fronczek, A. Moskun and R. Isovitsch, Inorg. Chim. Acta, 2009, 362, 3872–3876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details include experimental values, spectra and supporting information regarding the reaction kinetics, cytotoxicity HeLa cell testing and photoluminescence. Crystal data has been deposited with the CSD with CCDC [2057438 and 2057437]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03750a |
| This journal is © The Royal Society of Chemistry 2021 |