
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bifunctional lead–iron oxyfluoride, PbFeO2F, that functions as a visible-light-responsive photoanode and an electrocatalyst for water oxidation†
Ryusuke Mizuochi a,
Kazunari Izumib,
Yoshiyuki Inagumab and
Kazuhiko Maeda*a
a,
Kazunari Izumib,
Yoshiyuki Inagumab and
Kazuhiko Maeda*a
aDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maedak@chem.titech.ac.jp
bDepartment of Chemistry, Faculty of Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan
First published on 23rd July 2021
Abstract
The oxyfluoride PbFeO2F was investigated as a photoanode material and as an electrocatalyst for water oxidation. PbFeO2F powder, which was synthesized by a high-pressure method and had an estimated bandgap of 2.1 eV, was deposited onto a fluorine-doped tin oxide (FTO) substrate. Mott–Schottky plot measurements for the PbFeO2F/FTO electrode showed n-type semiconductivity of PbFeO2F, with a flat-band potential of +0.53 ± 0.05 V vs. reversible hydrogen electrode (RHE). The PbFeO2F/FTO electrode, which was modified with a conductive TiO2 layer and a cobalt phosphate water-oxidation cocatalyst, showed a clear anodic photocurrent in aqueous K3PO4 solution under visible-light irradiation (λ < 600 nm). The PbFeO2F/FTO electrode without any modification functioned as a stable water-oxidation electrocatalyst to form O2 with a faradaic efficiency of close to unity. This study demonstrates that PbFeO2F is a bifunctional material, serving as a water-oxidation photoanode under a wide range of visible-light wavelengths and as an electrocatalyst that operates at a relatively low overpotential for water oxidation.
Introduction
Hydrogen is expected to be used as a renewable energy carrier. Water splitting using semiconductor photoelectrodes or photocatalysts has attracted attention as a method of generating clean hydrogen using solar energy.1–6 Titanium-based metal oxides (e.g., TiO2 (ref. 7) and SrTiO3 (ref. 8)) have been developed as stable photoanode materials for solar water oxidation but are not capable of efficiently utilizing visible light, which represents the majority of solar energy, because of their wide bandgaps (>3 eV). By contrast, visible-light-responsive metal oxides (e.g., α-Fe2O3 (ref. 9–11) and BiVO4 (ref. 12–14)) unavoidably require an additional electrochemical (or external) bias for operation because their conduction-band minimum (CBM) is more positive than the H+/H2 reduction potential [0 V vs. standard hydrogen electrode (SHE) at pH 0].Compared with the aforementioned oxide materials, mixed-anion compounds such as oxynitrides and oxysulfides have relatively small bandgaps and negative CBM potentials, making them good candidate photoanode materials for water oxidation under visible light.3,15–17 Some of them (e.g., TaON18,19) theoretically enable water splitting to be driven under visible light without requiring an external bias because the CBM and the valence-band maximum (VBM) straddle the water reduction/oxidation potentials. However, the N 2p orbitals that constitute the VBM of oxynitrides are less stable than the O 2p orbitals, undergoing self-oxidation by holes generated during visible-light irradiation. This self-oxidation occurs with oxysulfides, in which the VBM is formed by S 3p orbitals. Thus, although the high-energy p-orbitals of anions are essential for providing small bandgaps, they are the main factor preventing more stable water oxidation by mixed-anion compounds.
Recently, the oxyfluoride Pb2Ti2O5.4F1.2, which is a mixed-anion compound, has been reported as a visible-light-absorbing photocatalyst with a narrow bandgap (∼2.4 eV) and n-type semiconductivity.20–22 Pb2Ti2O5.4F1.2 has a CBM at approximately −1.0 ± 0.1 V vs. SHE, which is sufficiently more negative than the water reduction potential.21 Thus, this oxyfluoride can drive standalone visible-light water splitting. Moreover, both the F 2p and O 2p orbitals are essentially stable toward self-oxidation by holes generated during visible-light irradiation of the oxyfluoride. Indeed, a Pb2Ti2O5.4F1.2 photoanode with visible-light responsivity and a relatively negative photocurrent onset potential has been reported.23 However, the literature includes only one example of a visible-light-responsive oxyfluoride photoelectrode (i.e., Pb2Ti2O5.4F1.2) that can utilize a limited portion of visible light at wavelengths as long as 500 nm. Therefore, exploration of a new oxyfluoride photoelectrode that can absorb a greater range of visible-light wavelengths is important for enabling the design of visible-light-responsive photoelectrode materials.
Fe(III)-containing materials are potentially useful as not only semiconductor photoanodes9,11 but also catalysts for water oxidation.24–27 The use of earth-abundant elements such as Fe is important for the development of water-oxidation photoanodes and/or catalysts not based on expensive metals, even if the performance of these materials initially found is moderate. Recently, oxyfluorides Co3Sb4O6F6,28 NiFe2F4.4O1.8,29 and CoFe2F6.6O0.7 (ref. 29) have been reported as electrocatalysts for water oxidation.
In the present work, the oxyfluoride PbFeO2F is examined as an electrode material for water splitting with and without irradiation. PbFeO2F, which can be synthesized by a high-pressure method,30 is an anion-disordered cubic perovskite with space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (Fig. 1, drawn by the VESTA program31). PbFeO2F has been reported to exhibit antiferromagnetic behavior.32 It has also been reported to exhibit a yellow colour and is therefore expected to function as a photoelectrode material under visible light. In addition, the fact that PbFeO2F contains iron, an element that may provide active sites for water oxidation, suggests another functionality of electrocatalyst. Herein, we report that PbFeO2F can indeed function as both a semiconductor photoanode and electrocatalyst for water oxidation.
m (Fig. 1, drawn by the VESTA program31). PbFeO2F has been reported to exhibit antiferromagnetic behavior.32 It has also been reported to exhibit a yellow colour and is therefore expected to function as a photoelectrode material under visible light. In addition, the fact that PbFeO2F contains iron, an element that may provide active sites for water oxidation, suggests another functionality of electrocatalyst. Herein, we report that PbFeO2F can indeed function as both a semiconductor photoanode and electrocatalyst for water oxidation.
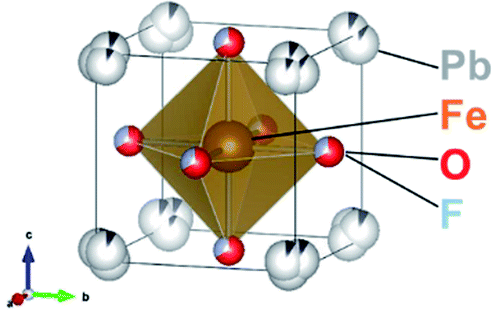 | ||
| Fig. 1 Crystal structure of PbFeO2F. | ||
Experimental
Synthesis of PbFeO2F
PbFeO2F powder was synthesized via solid-state reaction under high pressure using a mixture of PbO (99.9%, Kanto Chemical), PbF2 (99.999%, Soekawa Chemical) and Fe2O3 (99.99%, Rare Metallic).32 A stoichiometric mixture of the starting materials was dried under reduced pressure at ∼573 K overnight. The mixture was sealed in an Au capsule (0.2 mm thick, 3.1 mm inner diameter and 3.2 mm depth), and the loaded capsule was subsequently inserted into a NaCl sleeve. The sleeve and capsule were inserted into a pyrophyllite cube block (one side 13 mm) with a cylindrical graphite heater. The mixture was reacted in a TRY cubic multianvil-type high-pressure apparatus (NAMO 2001) at 6.0 GPa and 1173 K for 30 min and was subsequently quenched to room temperature.Fabrication of PbFeO2F/FTO electrodes
The PbFeO2F electrodes were fabricated using an electrophoretic deposition method.33 Electrophoretic deposition was performed in a 50 mL acetone solution (>99.5%, Kanto Chemical) containing 100 mg of PbFeO2F powder and 20 mg of I2 (>99.8%, FUJIFILM Wako Pure Chemical). Two parallel fluorine-doped tin oxide (FTO) glass substrates were immersed ∼15 mm apart in the solution, and a potential of 30 V was applied for 30 s using a stabilized DC power supply (PSW 80-13.5, GW Instek). The as-fabricated electrodes were then heated at 573 K for 1 h in air in the case of electrodes not subjected to subsequent modifications.Ti(OCH(CH3)2)4 treatment and Co–Pi electrodeposition for PbFeO2F/FTO electrodes
According to the previously reported method,34 Ti(OCH(CH3)2)4 treatment was carried out by dipping the PbFeO2F/FTO electrode in an ethanol solution of 0.1 M Ti(OCH(CH3)2)4 (>97%, Kanto Chemical), followed by drying on a hotplate at ∼423 K. The procedure was repeated five times. Finally, the electrode was heated in air at 573 K for 1 h. Cobalt phosphate (Co–Pi) cocatalyst, known as a water-oxidation promoter,35 was then electrodeposited onto the TiO2/PbFeO2F/FTO electrode.35,36 A three-electrode cell was used with the TiO2/PbFeO2F/FTO as the working electrode, a Ag/AgCl electrode as the reference electrode and Pt wire as the counter electrode. An electrochemical bias of +1.0 V vs. Ag/AgCl was applied to the working electrode in 0.1 M potassium phosphate buffered at pH 7 and containing 0.5 mM cobalt nitrate (98%, FUJIFILM Wako Pure Chemical) until the charge passing through the outer circuit reached 100 mC unless otherwise stated. The pH of the phosphate solution was controlled by mixing KH2PO4 (>98.0%, Kanto Chemical), K2HPO4 (>98.0%, Kanto Chemical) and/or K3PO4 (≥98%, Sigma-Aldrich), where the concentration was maintained at 0.1 M in total.Characterization
A crystalline phase of the PbFeO2F powder was confirmed by X-ray diffraction (XRD) measurements with a Malvern Panalytical X'Pert3 powder diffractometer (monochromated Cu Kα). The light-absorption properties of the PbFeO2F powder were characterized via UV-vis diffuse-reflectance spectroscopy (DRS) with a JASCO V-565 spectrophotometer. Scanning electron microscopy (SEM) observations combined with energy-dispersive X-ray spectroscopy (EDS) measurements were conducted on a HITACHI S4700 equipped with an EDAX Genesis apparatus at the Materials Analysis Division, Open Facility Center, Tokyo Institute of Technology. Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were conducted with a 5100 VDV apparatus (Agilent Technologies). Measurements for Mott–Schottky plots were carried out using a BAS ALS/CHI760E electrochemical analyser. Electrochemical impedance spectroscopy measurements were performed using a potentiostat (pocketSTAT, Ivium Technologies). Mott–Schottky plots were recorded at a frequency of 100 Hz with a three-electrode-type system using the PbFeO2F/FTO as the working electrode, a Ag/AgCl electrode as the reference electrode (in saturated KCl aqueous solution) and Pt wire as the counter electrode in 0.1 M aqueous potassium phosphate solutions. The solutions were stirred and purged with Ar gas for 30 min before the measurements were conducted.Photoelectrochemical measurements
The photoelectrochemical measurements were performed with a potentiostat (HSV-110, Hokuto Denko) and an electrochemical cell with a three-electrode configuration using the as-prepared PbFeO2F working electrode, an Ag/AgCl reference electrode and a Pt-coil counter electrode. The cell was made of Pyrex glass. An aqueous solution of 0.1 M K3PO4 (≥98%, Sigma-Aldrich) was used as the electrolyte, which was stirred and purged with Ar gas for 30 min before the measurements were conducted. It is known that coexistence of phosphate ions in an electrolyte solution has a positive effect on electrochemical water oxidation activity of the Co–Pi catalyst,35,37 that and basic conditions are generally preferable for water oxidation. The light source was a 300 W Xe lamp (PE300BF, Cermax) fitted with an L42, Y48, O54, O58, R62 or R70 cutoff filter (HOYA) to emit visible light of each wavelength range. The irradiation area was 3 cm2. The light intensity was approximately 0.31 W cm−2 in the wavelength range 350–700 nm unless otherwise stated. The potentials measured against the Ag/AgCl reference (saturated KCl aqueous solution) were converted to potentials vs. RHE (ERHE = EAg/AgCl + 0.059 pH + 0.197 at 298 K).Incident photon to current conversion efficiency (IPCE) was measured in a similar manner using the same 300 W xenon lamp fitted with an L38 cutoff filter and a band-pass filter centred at 420 nm (HOYA). The IPCE was calculated by the following equation:
| IPCE (%) = 1240 × i (mA cm−2)/(λ (nm) × φ (mW cm−2)) × 100 |
Quantifying electrochemical O2 evolution
To quantify the O2 evolved during controlled-potential electrolysis, electrochemical measurements were performed in a gastight H-type electrochemical cell with two chambers divided by a perfluorinated membrane (Nafion 117, Sigma-Aldrich). The PbFeO2F/FTO working electrode and an Ag/AgCl reference electrode were separated from a Pt-wire counter electrode in each chamber. The other conditions were identical to those mentioned in the description of the photoelectrochemical measurements. The evolved O2 was detected using a gas chromatograph (MGC3000A, Inficon) equipped with a thermal conductivity detector and an MS-5A column. Ar gas was used as the carrier gas.Results and discussion
Light absorption behaviour and flat-band potential of PbFeO2F
The single-phase production of the as-synthesized PbFeO2F was confirmed by XRD measurement (Fig. S1†). SEM observations show that the synthesized PbFeO2F consisted of 0.1–10 μm particles (Fig. S2†). The UV-vis DRS spectra of the PbFeO2F (Fig. 2) indicate that the material has an absorption edge at ∼600 nm and substantial absorption in the longer-wavelength region, which might be attributable to anionic defects. As reported for α-Fe2O3, the longer-wavelength absorption band is assigned to oxygen vacancies.38,39 The bandgap of the PbFeO2F was estimated to be 2.1 eV on the basis of the onset wavelength in the UV-visible DRS spectra. The previously reported PbFeO2F exhibited a yellow colour,30 whereas the as-prepared PbFeO2F in the present work was yellow-brown. This difference in colour originates from different concentrations of anionic defects (i.e., different concentrations of reduced metal ions), which commonly affect the appearance of powders.40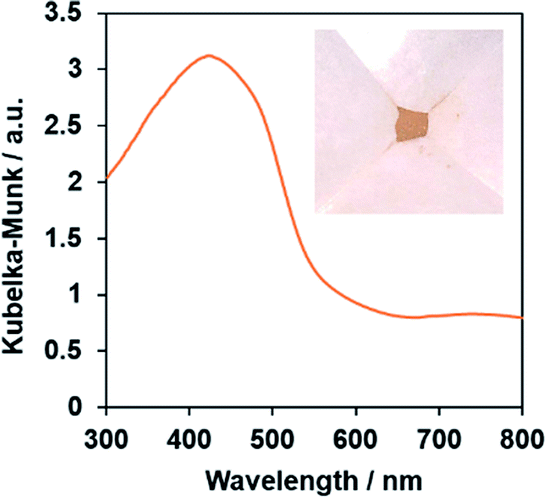 | ||
| Fig. 2 UV-vis diffuse-reflectance spectrum of the as-prepared PbFeO2F. The inset shows a photograph of the same material. | ||
The as-synthesized PbFeO2F was deposited onto a FTO substrate via electrophoretic deposition. As shown in Fig. 3, the thickness of the deposited PbFeO2F particles was 1–2 μm. In the electrophoresis method, colloidal particles suspended in liquid migrate in an electric field between two electrodes, undergoing deposition onto one side of the two electrodes.33 Therefore, light-weight, smaller particles are preferentially deposited onto the electrode. Therefore, it is considered that the size of the deposited PbFeO2F particles (0.1–2 μm) were smaller than the as-synthesized PbFeO2F ones (0.1–10 μm).
 | ||
| Fig. 3 SEM images of the PbFeO2F/FTO electrode. | ||
To determine the flat-band potential (EFB) of PbFeO2F, Mott–Schottky plots of the PbFeO2F/FTO electrode were recorded in aqueous phosphate solutions with different pH values under dark conditions. As shown in Fig. 4A, the Mott–Schottky plots show positive slopes irrespective of pH, which indicates n-type semiconducting behaviour of the PbFeO2F. The EFB values, which were obtained by extrapolation of the linear portion to the x-axis intercept, were negatively shifted with increasing electrolyte pH. The negative EFB shift corresponds to approximately −0.0591 V per pH, indicating Nernstian behaviour (Fig. 4B). Thus, the EFB of PbFeO2F was determined to be +0.53 ± 0.05 V vs. RHE. The CBM of an n-type semiconductor depends on its conductivity and lies at 0.1–0.3 V negative relative to the EFB.41 Assuming that the difference between the CBM and the EFB of PbFeO2F was 0.2 V because of the unclarified conductivity of PbFeO2F, the CBM is estimated to be +0.33 ± 0.05 V vs. RHE. This potential is more positive than the H+/H2 reduction potential (0 V vs. RHE), as displayed in Fig. 4C. On the basis of the bandgap of PbFeO2F (2.1 eV), the VBM of PbFeO2F was determined to be +2.43 ± 0.05 V, which is more positive than the water oxidation potential (+1.23 V vs. RHE).
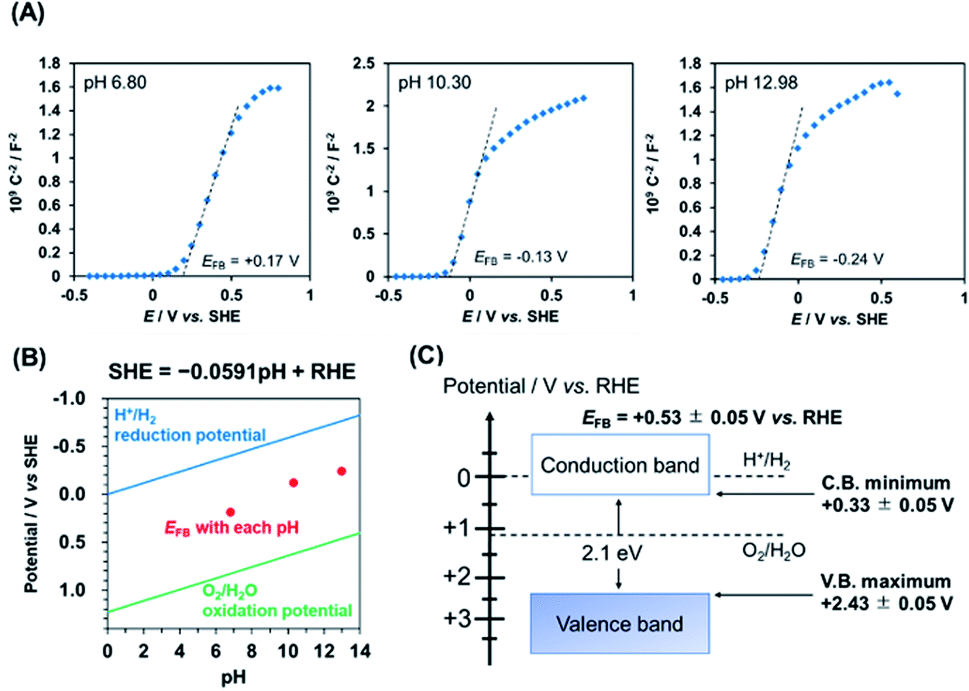 | ||
| Fig. 4 (A) Mott–Schottky plots for a PbFeO2F/FTO electrode recorded at 100 Hz in 0.1 M aqueous phosphate solutions with different pH values. These measurements were conducted in nonacidic solutions because of the potential dissolution of PbFeO2F for a strong acid. (B) pH dependence of the EFB of PbFeO2F, along with water reduction/oxidation potentials. (C) Conduction and valence band-edge potentials of PbFeO2F, as determined from the Mott–Schottky plots and the UV-vis diffuse-reflectance spectra. | ||
Photoelectrochemical response
The as-deposited PbFeO2F/FTO electrode was subjected to a post-necking treatment with an ethanol solution of titanium(IV) isopropoxide [Ti(OCH(CH3)2)4], followed by heating at 573 K for 1 h in air. This treatment resulted in the deposition of a TiO2 layer onto the PbFeO2F/FTO electrode (Fig. 5), which is expected to contribute to an enhanced photocurrent because of the reduced resistance of the electrode, as demonstrated in previous works.34,42 A Co–Pi cocatalyst, known to function as a water-oxidation promoter,35 was then electrodeposited onto the as-prepared TiO2/PbFeO2F/FTO electrode.36 The deposited Co–Pi was observed as islands on the TiO2/PbFeO2F/FTO electrode surface (Fig. 6A). EDS spectra also show the presence of P and Co species on the electrode surface (Fig. 6B). Peaks of P at 2.0 keV and Co at 6.9 keV were observed, the latter of which was overlapped with an Fe peak at 7.1 keV. In addition, EDS spot analysis demonstrated that the islands contain more P and Co species than the region surrounding the islands (Fig. S3†). | ||
| Fig. 5 (A) SEM images of the TiO2/PbFeO2F/FTO electrode. (B) EDS mapping analysis for the TiO2/PbFeO2F/FTO electrode. | ||
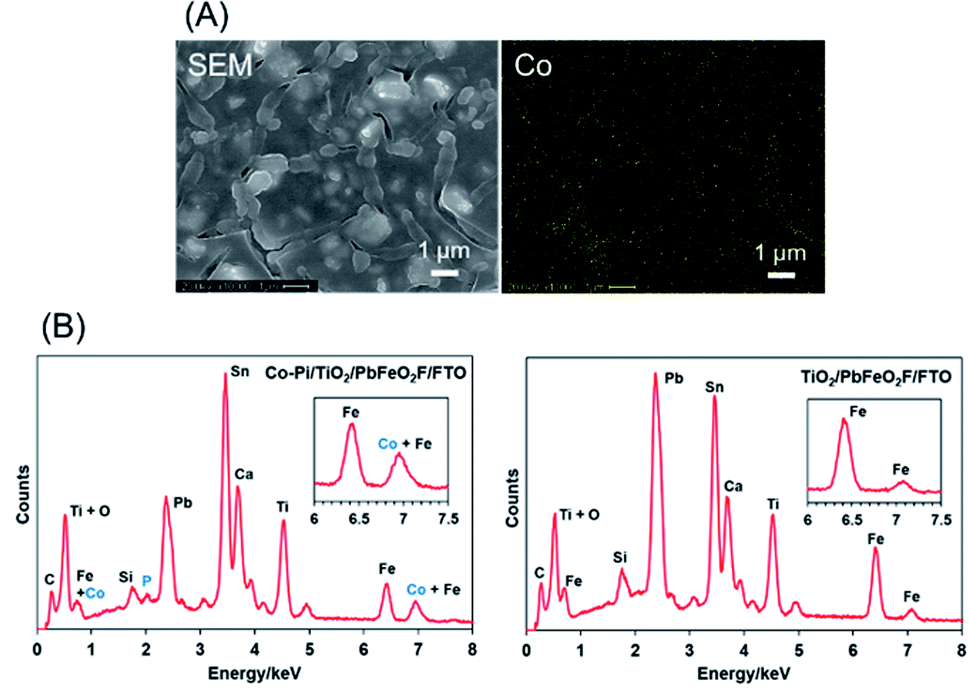 | ||
| Fig. 6 (A) EDS mapping analysis for the Co–Pi/TiO2/PbFeO2F/FTO electrode. (B) EDS spectra for (left) the Co–Pi/TiO2/PbFeO2F/FTO electrode and (right) the TiO2/PbFeO2F/FTO electrode. | ||
Cyclic voltammetry (CV) of the modified electrodes was conducted in 0.1 M K3PO4 solution under dark conditions (Fig. 7). A TiO2/FTO electrode, which was prepared by Ti(OCH(CH3)2)4 treatment of an FTO substrate, showed little dark current in the examined potential range. By contrast, the PbFeO2F/FTO and Co–Pi/TiO2/PbFeO2F/FTO electrodes exhibited dark current with a redox peak in the range from 0 to +0.6 V vs. RHE. This dark current might be attributable to a redox reaction involving Fe cations in PbFeO2F because the dark current was observed when PbFeO2F was present. A similar CV profile has been reported for an α-Fe2O3 photoanode in the same potential range investigated in the present study.43 For the PbFeO2F/FTO electrode, a dark current with an irreversible wave in the range from +1.4 V vs. RHE was observed, attributable to the oxygen evolution reaction and/or the oxidation of Fe3+ to Fe4+.43 The oxidation of Pb2+ to Pb4+ can contribute to the dark current as well.44 The Co–Pi/TiO2/PbFeO2F/FTO electrode also gave a dark current with a redox peak in the range from +0.9 to +1.8 V. The dark current originated from Co species on the Co–Pi electrodeposited electrode.35,45,46 This interpretation is supported by an increase in the dark current for electrodes that contained more Co–Pi cocatalyst (Fig. S4†). Given these results, photoelectrochemical measurements were conducted in the potential range from +0.5 to +1.3 V to avoid the dark current during the positive sweep scan. The feasibility of using PbFeO2F as a water-oxidation electrocatalyst will be discussed in a later section.
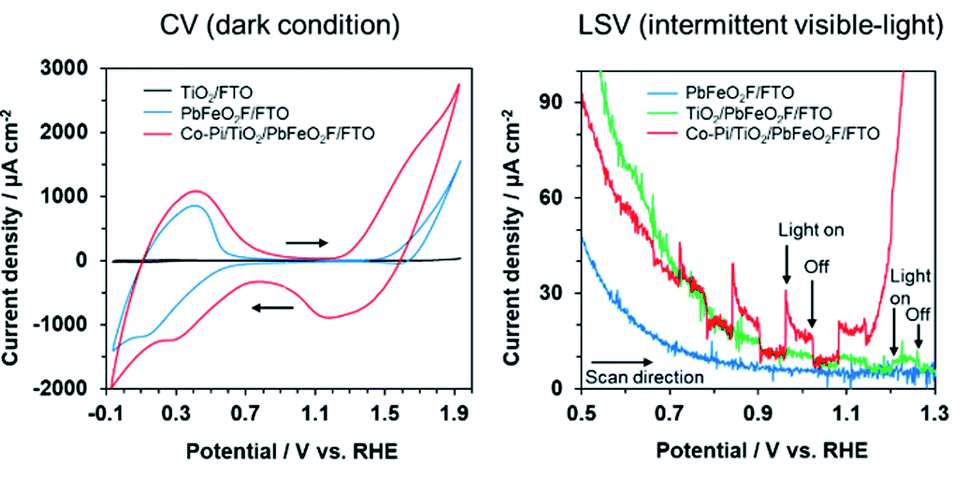 | ||
| Fig. 7 Current–potential curves for the modified PbFeO2F electrodes in aqueous 0.1 M K3PO4 solution (pH 12.4), as recorded at a sweep rate of (left) 100 mV s−1 under dark conditions and (right) 10 mV s−1 under intermittent visible-light irradiation. Light source: 300 W xenon lamp fitted with a Y48 cutoff filter (λ > 460 nm, 0.26 W cm−2). | ||
Linear-sweep voltammetry of the modified electrodes in 0.1 M K3PO4 solution was conducted under intermittent visible-light irradiation (Fig. 7). The PbFeO2F/FTO electrode exhibited no photocurrent response. By contrast, a slight photocurrent response was observed with the TiO2/PbFeO2F/FTO electrode, primarily because of reduced interparticle resistance in the electrode.34,47 As displayed in Fig. S5,† electrochemical impedance spectroscopy confirmed that the charge-transfer resistance of the TiO2-deposited electrode was smaller than that of the electrode without TiO2, as indicated by the smaller arc of the semicircle in the Nyquist plots. The reduction of the charge-transfer resistance originates from improved conductivity of the electrode, which was achieved as a result of the TiO2 treatment.
As previously mentioned, the CBM of PbFeO2F was located at +0.33 ± 0.05 V vs. RHE (Fig. 4), which is more positive than the reported CBM of TiO2 (−0.04 V vs. RHE).48 Therefore, charge transfer from the CBM of PbFeO2F to that of TiO2 is apparently not efficient. However, TiO2 has midgap states that originate from defective sites (∼0.4 V below the CBM).49 In the shallow defect states, the trapped electrons can be thermally detrapped and exhibit high mobility.50 This effect is inferred to have improved interparticle conductivity, which is known to function as an electron trapping–detrapping effect in TiO2-based dye-sensitized solar cells.51
A clear anodic photocurrent was observed for the Co–Pi/TiO2/PbFeO2F/FTO electrode (Fig. 7). Loading the Co–Pi cocatalyst resulted in improved rate of charge transfer at the electrode interface for water oxidation and in charge separation from the surface to the bulk.35,52 This effect was confirmed by electrochemical impedance spectroscopy measurements (Fig. S5†). In addition, the anodic photoresponse again showed n-type semiconducting character of PbFeO2F, with a photocurrent onset potential of +0.7 V vs. RHE, although an accurate determination was difficult because of an overlap of dark current. The photocurrent onset potential of the Co–Pi/TiO2/PbFeO2F/FTO electrode, which can be regarded as the flat-band potential of PbFeO2F, was slightly more positive than that determined from the corresponding Mott–Schottky plot (+0.53 ± 0.05 V, Fig. 4). This result implies that charge recombination in the illuminated PbFeO2F surface was substantial and concealed the real flat-band potential, similar to the case of α-Fe2O3 photoanodes.9,53
Photoelectrochemical activity under a wide range of visible light
Fig. 8 demonstrates anodic photocurrent densities of the Co–Pi/TiO2/PbFeO2F/FTO electrode at +1.0 V vs. RHE in aqueous 0.1 M K3PO4 solution (pH 13.4) under irradiation with light of different wavelengths, which was controlled using different cutoff filters. The UV-vis DRS spectrum of the PbFeO2F powder is also shown in Fig. 8. The photocurrent densities decreased with increasing cutoff wavelength and became almost zero under >600 nm irradiation. This change in the anodic photocurrent corresponded to the light-absorption properties of PbFeO2F, indicating that the PbFeO2F photoanode operated under visible-light irradiation with wavelengths as long as ∼600 nm and that the anodic photoresponse occurred by light absorption of PbFeO2F itself. It was also confirmed that no photocurrent was generated from Co–Pi/TiO2/FTO electrode (Fig. S6†).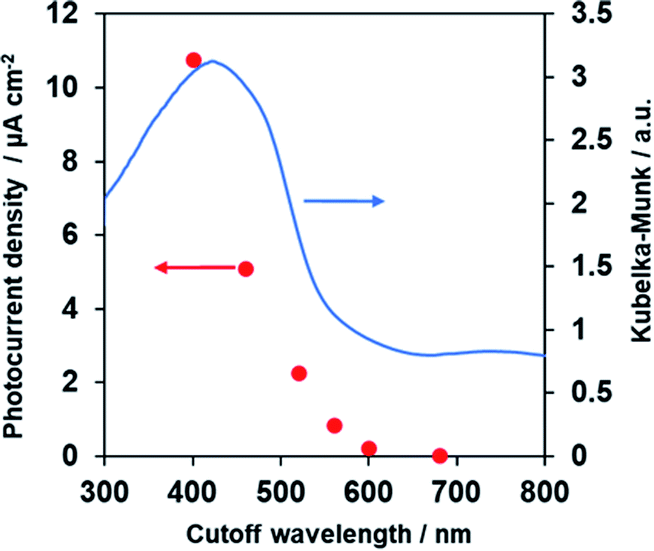 | ||
| Fig. 8 Photocurrent densities of the Co–Pi/TiO2/PbFeO2F/FTO electrode at +1.0 V vs. RHE in aqueous 0.1 M K3PO4 solution (pH 13.4) as a function of the cutoff wavelength of the incident light. Light source: 300 W xenon lamp fitted with cutoff filters (L42, Y48, O54, O58, R62 or R70). The diffuse reflectance spectrum of PbFeO2F powder is also shown. | ||
The results also indicate that an absorption band of longer wavelengths than 600 nm does not contribute to the generation of anodic photocurrent. As previously mentioned, the absorption band at longer wavelengths originates from anionic defects in PbFeO2F, as reported in α-Fe2O3.38,39 Lifetimes of charge carriers generated at defect states in a semiconductor are generally short,54,55 which could be the reason for the negligible photoresponse of PbFeO2F under >600 nm irradiation.
The stability of the anodic photocurrent was examined via controlled-potential photoelectrolysis at +1.0 V vs. RHE (Fig. S7A†). This measurement shows that the anodic photocurrent gradually decayed with increasing reaction time. The O2 evolution could not be quantified because of the small current that flowed during the photoelectrolysis. Nevertheless, the fact that Co–Pi (a well-known water oxidation promoter) improved the anodic photocurrent density of the PbFeO2F electrode (Fig. 7) strongly suggests the oxidation of water to O2. As previously mentioned, the oxidation of metal cations in the PbFeO2F/FTO electrode was suggested;43,44 however, the concentration of metal cations found in the electrolyte solution by ICP-OES was negligible (below ppm level).
It has been reported that stability of a photoanode for water oxidation is influenced by various factors (e.g., cocatalyst, electrolyte pH, operating potential and so on).18,47 It is therefore expected that optimizing these factors will improve photoelectrochemical stability of PbFeO2F and also photocurrent density, although it is beyond the scope of this work, which aimed at developing a new electrode material based on oxyfluorides. Nevertheless, more stable, larger photocurrent from the TiO2/PbFeO2F/FTO electrode was observed in the presence of I− as a reversible electron donor upon visible light than in aqueous solution without I− (Fig. S7B†). This indicates that PbFeO2F is essentially stable toward the photooxidation reaction.
Electrochemical water oxidation by PbFeO2F in the dark
The ability of PbFeO2F to function as an electrocatalyst for water oxidation was investigated by controlled-potential electrolysis using the as-prepared PbFeO2F/FTO electrode at +1.7 V vs. RHE under dark conditions (Fig. 9). Although the onset potential of water oxidation current by the PbFeO2F/FTO electrode was more negative than +1.7 V (see Fig. 7), we conducted the electrolysis experiment at +1.7 V in order to obtain more O2 gas for reliable quantification. GC analysis of the evolved O2 gas shows that the PbFeO2F functions as an electrocatalyst to stably produce O2. The amount of O2 evolved at the initial stage of the electrolysis was slightly smaller than one-fourth the amount of electrons that flowed to the outer circuit. This result is attributed primarily to a time lag of gas diffusion from the solution to the gas chromatograph. In fact, the total O2 evolved reached the value expected on the basis of the reaction stoichiometry, giving a high faradaic efficiency of 97%. This result indicates that water oxidation was the major path at the PbFeO2F/FTO electrode. Under the same condition, an α-Fe2O3/FTO electrode, prepared in a similar manner, did not produce appreciable current, indicative of its large overpotential for water oxidation.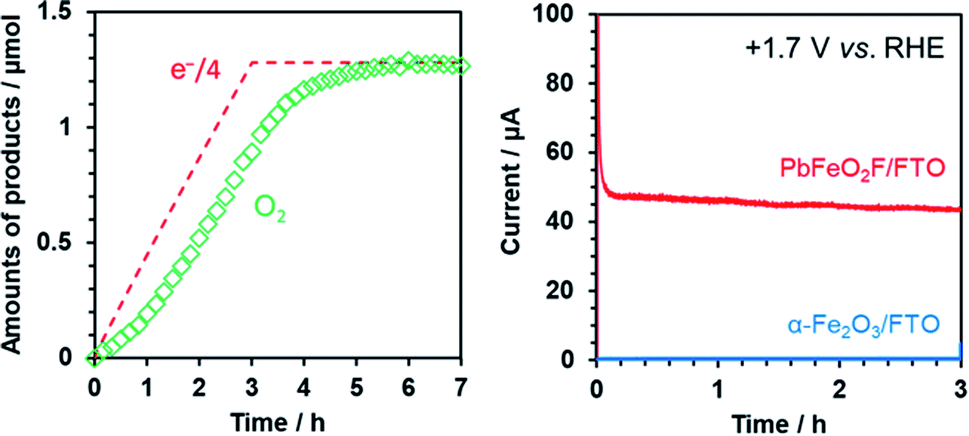 | ||
| Fig. 9 (left) Time course of O2 evolution during controlled-potential electrolysis at +1.7 V vs. RHE in aqueous 0.1 M K3PO4 solution (pH 12.9) for the PbFeO2F/FTO electrode under dark conditions. (right) The corresponding current–time curve for the electrode over a span of 3 h. Data for α-Fe2O3 is also shown for comparison. | ||
Conclusions
PbFeO2F synthesized by a high-pressure method had grain sizes ranging from 0.1 to 10 μm and an estimated bandgap of 2.1 eV. The Mott–Schottky plot measurements showed n-type semiconductivity of PbFeO2F with a flat-band potential of +0.53 ± 0.05 V vs. RHE. The PbFeO2F electrode modified with a conductive TiO2 layer and a Co–Pi water-oxidation cocatalyst exhibited a clear anodic photocurrent in aqueous K3PO4 solution under visible-light irradiation (λ < 600 nm).At present, the performance of the PbFeO2F photoanode is not satisfactory; IPCE at 420 nm was 0.14% at +1.0 V vs. RHE. Nevertheless, it is expected that photoelectrochemical performance of PbFeO2F will be improved by further development in materials synthesis and post modification technologies for PbFeO2F, as we can learn from the history of the α-Fe2O3 photoanode.9–11
Meanwhile, a PbFeO2F/FTO electrode without the modifications exhibited anodic current and O2 evolution in aqueous K3PO4 solution at +1.7 V vs. RHE, where water oxidation did not proceed for an α-Fe2O3 electrode. The present study reveals that PbFeO2F becomes a bifunctional material—that is, a photoanode material that can function under a wide range of visible-light wavelengths and as an electrocatalyst at a relatively low overpotential for water oxidation.
Author contributions
R. M. performed most of the experiments and wrote the manuscript with K. M. K. I. and Y. I. synthesized the PbFeO2F powder. K. M. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Area “Mixed Anion (Project JP16H06439 and JP16H06441)” (JSPS). K. M. wishes to acknowledge the support from a Grant-in-Aid for Scientific Research (B) (JP19H02511) (JSPS).Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Solar Water Splitting Cells, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- P. Xu, N. S. McCool and T. E. Mallouk, Water splitting dye-sensitized solar cells, Nano Today, 2017, 14, 42–58 CrossRef CAS.
- J. Seo, H. Nishiyama, T. Yamada and K. Domen, Visible-Light-Responsive Photoanodes for Highly Active, Stable Water Oxidation, Angew. Chem., Int. Ed., 2018, 57, 8396–8415 CrossRef CAS PubMed.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Semiconductor polymeric graphitic carbon nitride photocatalysts: the “holy grail” for the photocatalytic hydrogen evolution reaction under visible light, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- K. Maeda and T. E. Mallouk, Two-dimensional metal oxide nanosheets as building blocks for artificial photosynthetic assemblies, Bull. Chem. Soc. Jpn., 2019, 92, 38–54 CrossRef CAS.
- Q. Wang and K. Domen, Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. J. Ingler, Efficient photochemical water splitting by a chemically modified n-TiO2, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- Y. Ham, T. Minegishi, T. Hisatomi and K. Domen, A SrTiO3 photoanode prepared by the particle transfer method for oxygen evolution from water with high quantum efficiencies, Chem. Commun., 2016, 52, 5011–5014 RSC.
- K. Sivula, F. Le Formal and M. Gratzel, Solar water splitting: progress using hematite (α-Fe2O3) photoelectrodes, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- S. Shen, S. A. Lindley, X. Chen and J. Z. Zhang, Hematite heterostructures for photoelectrochemical water splitting: rational materials design and charge carrier dynamics, Energy Environ. Sci., 2016, 9, 2744–2775 RSC.
- P. Sharma, J. W. Jang and J. S. Lee, Key Strategies to Advance the Photoelectrochemical Water Splitting Performance of α-Fe2O3 Photoanode, ChemCatChem, 2018, 11, 157–179 CrossRef.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, Photoelectrochemical Decomposition of Water into H2 and O2 on Porous BiVO4 Thin-Film Electrodes under Visible Light and Significant Effect of Ag Ion Treatment, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- H. L. Tan, R. Amal and Y. H. Ng, Exploring the Different Roles of Particle Size in Photoelectrochemical and Photocatalytic Water Oxidation on BiVO4, ACS Appl. Mater. Interfaces, 2016, 8, 28607–28614 CrossRef CAS PubMed.
- R. Abe, Recent progress on photocatalytic and photoelectrochemical water splitting under visible light irradiation, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Expanding frontiers in materials chemistry and physics with multiple anions, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- A. Miyoshi and K. Maeda, Recent Progress in Mixed-Anion Materials for Solar Fuel Production, Sol. RRL, 2021, 5, 2000521 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, Highly Stable Water Splitting on Oxynitride TaON Photoanode System under Visible Light Irradiation, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
- S. S. Gujral, A. N. Simonov, M. Higashi, X.-Y. Fang, R. Abe and L. Spiccia, Highly Dispersed Cobalt Oxide on TaON as Efficient Photoanodes for Long-Term Solar Water Splitting, ACS Catal., 2016, 6, 3404–3417 CrossRef CAS.
- K. Oka, H. Hojo, M. Azuma and K. Oh-ishi, Temperature-Independent, Large Dielectric Constant Induced by Vacancy and Partial Anion Order in the Oxyfluoride Pyrochlore Pb2Ti2O6−δF2δ, Chem. Mater., 2016, 28, 5554–5559 CrossRef CAS.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, A Stable, Narrow-Gap Oxyfluoride Photocatalyst for Visible-Light Hydrogen Evolution and Carbon Dioxide Reduction, J. Am. Chem. Soc., 2018, 140, 6648–6655 CrossRef CAS PubMed.
- H. Wakayama, K. Utimula, T. Ichibha, R. Kuriki, K. Hongo, R. Maezono, K. Oka and K. Maeda, Light Absorption Properties and Electronic Band Structures of Lead Titanium Oxyfluoride Photocatalysts Pb2Ti4O9F2 and Pb2Ti2O5.4F1.2, J. Phys. Chem. C, 2018, 122, 26506–26511 CrossRef CAS.
- N. Hirayama, H. Nakata, H. Wakayama, S. Nishioka, T. Kanazawa, R. Kamata, Y. Ebato, K. Kato, H. Kumagai, A. Yamakata, K. Oka and K. Maeda, Solar-Driven Photoelectrochemical Water Oxidation over an n-Type Lead-Titanium Oxyfluoride Anode, J. Am. Chem. Soc., 2019, 141, 17158–17165 CrossRef CAS PubMed.
- D. Hong, Y. Yamada, T. Nagatomi, Y. Takai and S. Fukuzumi, Catalysis of Nickel Ferrite for Photocatalytic Water Oxidation Using [Ru(bpy)3]2+ and S2O82−, J. Am. Chem. Soc., 2012, 134, 19572–19575 CrossRef CAS PubMed.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Redox-inactive metal ions modulate the reactivity and oxygen release of mononuclear non-haem iron(III)–peroxo complexes, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- T. Kanazawa and K. Maeda, Chromium-substituted hematite powder as a catalytic material for photochemical and electrochemical water oxidation, Catal. Sci. Technol., 2017, 7, 2940–2946 RSC.
- W. L. Kwong, C. C. Lee, A. Shchukarev, E. Björn and J. Messinger, High-performance iron (III) oxide electrocatalyst for water oxidation in strongly acidic media, J. Catal., 2018, 365, 29–35 CrossRef CAS.
- H. Svengren, S. Hu, I. Athanassiadis, T. M. Laine and M. Johnsson, An Oxofluoride Catalyst Comprised of Transition Metals and a Metalloid for Application in Water Oxidation, Chem.–Eur. J., 2015, 21, 12991–12995 CrossRef CAS PubMed.
- K. Lemoine, J. Lhoste, A. Hémon-Ribaud, N. Heidary, V. Maisonneuve, A. Guiet and N. Kornienko, Investigation of mixed-metal (oxy)fluorides as a new class of water oxidation electrocatalysts, Chem. Sci., 2019, 10, 9209–9218 RSC.
- I. O. Troyanchuk, N. V. Kasper, O. S. Mantytskaya and E. F. Shapovalova, High-pressure synthesis of some perovskite — Like compounds with a mixed anion type, Mater. Res. Bull., 1995, 30, 421–425 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y. Inaguma, J.-M. Greneche, M.-P. Crosnier-Lopez, T. Katsumata, Y. Calage and J.-L. Fourquet, Structure and Mössbauer Studies of F–O Ordering in Antiferromagnetic Perovskite PbFeO2F, Chem. Mater., 2005, 17, 1386–1390 CrossRef CAS.
- L. Besra and M. Liu, A review on fundamentals and applications of electrophoretic deposition (EPD), Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- N. Nishimura, B. Raphael, K. Maeda, L. Le Gendre, R. Abe, J. Kubota and K. Domen, Effect of TiCl4 treatment on the photoelectrochemical properties of LaTiO2N electrodes for water splitting under visible light, Thin Solid Films, 2010, 518, 5855–5859 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. K. Zhong, M. Cornuz, K. Sivula, M. Grätzel and D. R. Gamelin, Photo-assisted electrodeposition of cobalt–phosphate (Co–Pi) catalyst on hematite photoanodes for solar water oxidation, Energy Environ. Sci., 2011, 4, 1759–1764 RSC.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Cobalt–phosphate oxygen-evolving compound, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- Z. Zhang, I. Karimata, H. Nagashima, S. Muto, K. Ohara, K. Sugimoto and T. Tachikawa, Interfacial oxygen vacancies yielding long-lived holes in hematite mesocrystal-based photoanodes, Nat. Commun., 2019, 10, 4832 CrossRef PubMed.
- L. A. Marusak, R. Messier and W. B. White, Optical absorption spectrum of hematite, α-Fe2O3 near IR to UV, J. Phys. Chem. Solids, 1980, 41, 981–984 CrossRef CAS.
- K. Maeda, H. Terashima, K. Kase, M. Higashi, M. Tabata and K. Domen, Surface Modification of TaON with Monoclinic ZrO2 to Produce a Composite Photocatalyst with Enhanced Hydrogen Evolution Activity under Visible Light, Bull. Chem. Soc. Jpn., 2008, 81, 927–937 CrossRef CAS.
- Y. Matsumoto, Energy Positions of Oxide Semiconductors and Photocatalysis with Iron Complex Oxides, J. Solid State Chem., 1996, 126, 227–234 CrossRef CAS.
- J. L. Ong, L. C. Lucas, G. N. Raikar and J. C. Gregory, Electrochemical corrosion analyses and characterization of surface-modified titanium, Appl. Surf. Sci., 1993, 72, 7–13 CrossRef CAS.
- H. Bülter, G. Denuault, S. Mátéfi-Tempfli, M. Mátéfi-Tempfli, C. Dosche and G. Wittstock, Electrochemical analysis of nanostructured iron oxides using cyclic voltammetry and scanning electrochemical microscopy, Electrochim. Acta, 2016, 222, 1326–1334 CrossRef.
- E. E. Abd El Aal, S. Abd El Wanees and A. Abd El Aal, Anodic behaviour and passivation of a lead electrode in sodium carbonate solutions, J. Mater. Sci., 1993, 28, 2607–2614 CrossRef CAS.
- E. R. Young, R. Costi, S. Paydavosi, D. G. Nocera and V. Bulović, Photo-assisted water oxidation with cobalt-based catalyst formed from thin-film cobalt metal on silicon photoanodes, Energy Environ. Sci., 2011, 4, 2058–2061 RSC.
- D. Wang, R. Li, J. Zhu, J. Shi, J. Han, X. Zong and C. Li, Photocatalytic Water Oxidation on BiVO4 with the Electrocatalyst as an Oxidation Cocatalyst: Essential Relations between Electrocatalyst and Photocatalyst, J. Phys. Chem. C, 2012, 116, 5082–5089 CrossRef CAS.
- S. S. Gujral, A. N. Simonov, M. Higashi, R. Abe and L. Spiccia, Optimization of Titania Post-Necking Treatment of TaON Photoanodes to Enhance Water-Oxidation Activity under Visible-Light Irradiation, ChemElectroChem, 2015, 2, 1270–1278 CrossRef CAS.
- J. M. Bolts and M. S. Wrighton, Correlation of Photocurrent-Voltage Curves with Flat-Band Potential for Stable Photoelectrodes for the Photoelectrolysis of Water, J. Phys. Chem., 1976, 80, 2641–2645 CrossRef CAS.
- S. Ikeda, N. Sugiyama, S.-Y. Murakami, H. Kominami, Y. Kera, H. Noguchi, K. Uosaki, T. Torimoto and B. Ohtani, Quantitative analysis of defective sites in titanium(IV) oxide photocatalyst powders, Phys. Chem. Chem. Phys., 2003, 5, 778–783 RSC.
- A. Yamakata, J. J. M. Vequizo and H. Matsunaga, Distinctive Behavior of Photogenerated Electrons and Holes in Anatase and Rutile TiO2 Powders, J. Phys. Chem. C, 2015, 119, 24538–24545 CrossRef CAS.
- A. V. Barzykin and M. Tachiya, Mechanism of Charge Recombination in Dye-Sensitized Nanocrystalline Semiconductors: Random Flight Model, J. Phys. Chem. B, 2002, 106, 4356–4363 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Photoelectrochemical and Impedance Spectroscopic Investigation of Water Oxidation with “Co–Pi”-Coated Hematite Electrodes, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS PubMed.
- D. A. Grave, N. Yatom, D. S. Ellis, M. C. Toroker and A. Rothschild, The “Rust” Challenge: On the Correlations between Electronic Structure, Excited State Dynamics, and Photoelectrochemical Performance of Hematite Photoanodes for Solar Water Splitting, Adv. Mater., 2018, 30, 1706577 CrossRef PubMed.
- D. Kan, T. Terashima, R. Kanda, A. Masuno, K. Tanaka, S. Chu, H. Kan, A. Ishizumi, Y. Kanemitsu, Y. Shimakawa and M. Takano, Blue-light emission at room temperature from Ar+-irradiated SrTiO3, Nat. Mater., 2005, 4, 816–819 CrossRef CAS.
- T. Feng, Anomalous photoelectronic processes in SrTiO3, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 25, 627–642 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data for structural characterization and electrochemical measurements. See DOI: 10.1039/d1ra04793k |
| This journal is © The Royal Society of Chemistry 2021 |