
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh pressure as a novel tool for the cationic ROP of γ-butyrolactone
Roksana Bernat ab,
Paulina Maksym*bc,
Magdalena Tarnackabd,
Katarzyna Malarzbd,
Anna Mrozek-Wilczkiewiczbd,
Tadeusz Bielae,
Sylwia Golbabc,
Ewa Kamińskaf,
Marian Paluchbd and
Kamil Kamińskibd
ab,
Paulina Maksym*bc,
Magdalena Tarnackabd,
Katarzyna Malarzbd,
Anna Mrozek-Wilczkiewiczbd,
Tadeusz Bielae,
Sylwia Golbabc,
Ewa Kamińskaf,
Marian Paluchbd and
Kamil Kamińskibd
aInstitute of Chemistry, University of Silesia in Katowice, Szkolna 9, 40-007 Katowice, Poland
bSilesian Centre for Education and Interdisciplinary Research, University of Silesia in Katowice, 75 Pułku Piechoty 1A, 41-500 Chorzów, Poland. E-mail: paulina.maksym@us.edu.pl
cInstitute of Materials Engineering, University of Silesia in Katowice, 75 Pułku Piechoty 1, 41-500 Chorzów, Poland
dChelkowski Institute of Physics, University of Silesia in Katowice, 75 Pułku Piechoty 1, 41-500 Chorzów, Poland
eDepartment of Polymer Chemistry, Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363, Lódź, Poland
fDepartment of Pharmacognosy and Phytochemistry, Faculty of Pharmaceutical Sciences in Sosnowiec, Medical University of Silesia in Katowice, Jagiellońska 4, 41-200 Sosnowiec, Poland
First published on 28th October 2021
Abstract
In this study, we report the acid-catalyzed and high pressure assisted ring-opening polymerization (ROP) of γ-butyrolactone (GBL). The use of a dually-catalyzed approach combining an external physical factor and internal catalyst (trifluoromethanesulfonic acid (TfOH) or p-toluenesulfonic acid (PTSA)) enforced ROP of GBL, which is considered as hardly polymerizable monomer still remaining a challenge for the modern polymer chemistry. The experiments performed at various thermodynamic conditions (T = 278–323 K and p = 700–1500 MPa) clearly showed that the high pressure supported polymerization process led to obtaining well-defined macromolecules of better parameters (Mn = 2200–9700 g mol−1; Đ = 1.05–1.46) than those previously reported. Furthermore, the parabolic-like dependence of both the molecular weight (MW) and the yield of obtained polymers on variation in temperature and pressure at either isobaric or isothermal conditions was also noticed, allowing the determination of optimal conditions for the polymerization process. However, most importantly, this strategy allowed to significantly reduce the reaction time (just 3 h at room temperature) and increase the yield of obtained polymers (up to 0.62 gPGBL/gGBL). Moreover, despite using a strongly acidic catalyst, synthesized polymers remained non-toxic and biocompatible, as proven by the cytotoxicity test we performed in further analysis. Additional investigation (including MALDI-TOF measurements) showed that the catalyst selection affected not only MW and yield but also the linear/cyclic form content in obtained macromolecules. These findings show the way to tune the properties of PGBL and obtain polymer suitable for application in the biomedical industry.
1. Introduction
Aliphatic polyesters are one of the most important groups of polymers that find many applications in various kinds of industries.1 Importantly, due to their enhanced biocompatibility and biodegradability with respect to the other macromolecules, they caught the attention of researchers dealing with the development of new materials for biomedical applications such as scaffolds, implants, tissue engineering, etc. Having this fact in mind, a large emphasis is put to work out an effective and non-toxic polymerization strategy to obtain polymers of well-defined parameters, including molecular weight (Mn), dispersity (Đ), topology, or chain-end fidelity. Briefly, there are two main approaches (i) polycondensation and (ii) ring-opening polymerization (ROP), characterized by a completely different reaction mechanism allowing the production of polyesters. Unfortunately, the first method suffers from several limitations, such as difficulty in water removal and the occurrence of side reactions, i.e., cyclization. As a consequence, it does not warrant good control over the process, yielding macromolecules of low/moderate molecular weight and relatively high Đ. On the other hand, the ROP of cyclic esters allows the production of high Mn polymers (>105 g mol−1)2 with better precision than polycondensation, and more importantly, with much higher control over polymer architecture, topology, morphology, dispersity, and chain-end fidelity. However, it should be reminded that achieving better control over ROP, especially through the suppression of back-biting reactions that form undesirable cyclic products, strictly depends on the applied reaction conditions and initiating/catalytic species. What is more, the high effectiveness of ROP is very often gained at the expense of the purity, the biocompatibility of the final product.Herein, it is worth emphasizing that depending on the monomer, initiating/catalytic system and the nature of the formed active species, ring-opening may proceed via covalent, coordination, cationic or anionic, metathetic, enzymatic and radical mechanism of the reaction. Among them, only anionic polymerization can fulfill the criteria of the living process involving the synthesis of polyesters of precisely controlled Mn, Đ and the structure of end groups. Unfortunately, living anionic and ‘pseudo-living’ coordination ROP processes require use of electron transfer agents/organometallic compounds as initiating/catalytic systems simultaneously keeping the harsh conditions of the reaction, where even traces of oxygen, water and protic impurities (i.e., rigorous reagents purification, handling of reagents in vacuum or inert gas) may completely deactivate the polymerizing system. Interestingly, over the years, organometallic-based catalysts have been replaced with organocatalysts and enzymes, providing several advantages, including the most obvious synthesis under mild conditions and the absence of the costly removal of metal impurities from the final products. Unfortunately, these strategies turned out to be not suitable enough to limit the participation of many side reactions, e.g., inter- and intramolecular transesterification. This problem is particularly important in the case of biocompatible and biodegradable polyesters3 intended for biomedical applications (e.g., polylactide (PLA), poly(ε-caprolactone) (PCL), poly(glycolic acid) (PGA), poly(hydroxybutyrate) (PHB) and related copolymers), where ultra-high product purity and precisely defined macromolecular parameters are highly required. Moreover, the biodegradability of these materials is strictly connected to their Mn, Đ, chain-end fidelity, composition and topology (the content of linear and cyclic products).4 Finally, it is worth mentioning that the most convenient and simpler way to synthesize polyesters involves organic and inorganic acids. However, due to poor control over the polymerization process, low Mn and relatively high Đ of produced polymers, cationic ROP is of marginal importance. It is also considered unsuitable for the synthesis of special materials characterized by specific properties and functionalities.
Although all the above-mentioned approaches can be more or less successfully applied to polymerize most cyclic esters, they often fail in the polymerization of γ-butyrolactone. It turned out that due to the unfavorable thermodynamics of polymerization (positive enthalpy and negative entropy of the process),5 resulting from the stabilization provided by the five-membered ring, this monomer was regarded as hardly polymerizable.6,7 On the other hand, poly(γ-butyrolactone) (PGBL), which is structurally identical to poly(4-hydroxybutyrate) (P4HB), is a material characterized by one of the fastest biodegradation times among polyesters (complete biodegradability 8–52 weeks).5,8,9 Moreover, it shows full thermal recyclability, and more interestingly, its mechanical properties can be easily tuned by the content of (oligo) cyclic products. In this context, PGBL can be considered a promising replacement for common polymers characterized by a long recycling time.
Nevertheless, although such a great interest in the synthesis of PGBL, so far, only a few successful attempts to perform the ROP of GBL have been reported in the literature.10,11 However, it should be mentioned that these approaches relied on the application of heavy conditions, i.e., ultra-high pressure,12 extremely low reaction temperature,13 or a synthetic path supported by catalytic/initiating systems based on organometallic compounds, which are not only expensive but also exhibit some level of toxicity.13,14 Some organocatalytic compounds (including phosphazenes,15,16 the bicyclic guanidine derivative 1,5,7-triazabicyclo [4.4.0] dec-5-ene (TBD),13,17 N-heterocyclic carbenes,18 N-heterocyclic olefins19 and systems of ion pairs based on (thio) ureas and phosphazenes20,21/tetra-n-butyl ammonium hydroxide (TBAOH)22 or ureas and alkoxide23) turned out to be a less toxic alternative to organometallic compounds, also capable of mediating GBL polymerization. Moreover, it should be emphasized that the above examples of GBL ROP and difficulties/limitations are related to classical polymerizing systems (batch). In this context, it is worth mentioning that the polymerization process can proceed more controllably under appropriate thermodynamic conditions, avoiding undesirable side reactions.24 One should remember that macromolecules of well-defined structural properties can also be obtained when we expose the polymerizing system to the ‘external’ physical factors, such as laser, light irradiation, ultrasounds, friction, confinement and compression. This strategy often leads to strong enhancement in control over the reaction and macromolecular properties of the produced polymer with respect to the ‘internal’-catalyzed reactions, as demonstrated in our previous studies.25–27 Among these ‘external’ factors, the utilization of high pressure seems to be particularly promising since upon compression we observe the enhancement of both chain propagation and chain transfer rates and the suppression of bimolecular chain termination reactions.28,29 Importantly, many competitive reactions occurring during the polymerization process (e.g., chain transfer or transesterification) have different activation volumes at elevated pressure, what significantly affects the properties of resultant polymers (e.g., degrees of polymerization, chain branching). The application of compression also influences several other factors, such as density, viscosity, intermolecular interactions, diffusivity, thermodynamics (lowers the ceiling temperature), the rate and the equilibrium position of the polymerization process.29 Thus, it also enhances the polymerizability of monomers considered as hardly-polymerizable/unreactive at ambient pressure, including the less-activated (LAMs) and sterically hindered ones. Finally, the other crucial benefit of this approach is the possibility to reduce the content of solvent and catalyst, allowing us to obtain the material of ultra-high purity. It is also worth mentioning that this strategy is no longer considered a serious application obstacle since high-pressure conditions are successfully achievable on an industrial scale (e.g., high-pressure processing technique for food preservation that is carried out using pressure in the range of 100–1000 MPa30).
However, although such interesting observations, insufficient attention has been paid to the effect of high pressure application in the ROP of lactones and lactides. Only one paper by Yamashita et al. focused on the acid-catalyzed ROP of GBL performed at elevated pressure,12 but not detailed enough, as only two successful attempts were presented, showing poor control and yielding polymers of moderate dispersity (Đ = 1.49–1.57). This research indicates that the acid-catalyzed ROP is difficult to perform in a controlled manner, and the obtained polymers are subjected to unwanted partial degradation/uncontrolled hydrolysis. Beside this example, there are also two other studies reported by our group concerning the highly efficient water-initiated ROP of lactones (ε-caprolactone) under compression, yielding polyesters of moderate Mn values up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1 and low to moderate dispersity (Đ = 1.03–1.56) and confirming very clearly the unique properties of a high-pressure approach.31,32
700 g mol−1 and low to moderate dispersity (Đ = 1.03–1.56) and confirming very clearly the unique properties of a high-pressure approach.31,32
Our study presents how the application of high pressure allowed us to conduct the acid-catalyzed ROP of GBL under mild temperature conditions and obtain well-defined products with better parameters and efficiency than previously reported in the literature. The strategy proposed in this paper combines a dual-catalyzed process forced by high pressure and an acidic catalyst – trifluoromethanesulfonic acid (TfOH) or p-toluenesulfonic acid (PTSA). The series of reactions, varying in the composition of the reaction mixture (type of catalyst) and the reaction time were conducted under different thermodynamic conditions (pressure, temperature) to optimize the polymerization process and properties of final products. We demonstrated that compression (p = 700–1500 MPa) could force fast (3 hours!) ROP of GBL at relatively mild temperature conditions (T = 278–323 K) yielding well-defined PGBLs of absolute molecular weight in range Mn = 2200–9700 g mol−1, narrow to moderate dispersity (Đ = 1.05–1.46) and higher yields (0.09–0.62 gPGBL/gGBL) in comparison to previous scientific reports. The work was also enriched with other analyses, e.g., the characterization of basic thermal properties, toxicity test and the film-forming ability.
2. Materials and methods
2.1. Materials
All chemicals used were purchased from Sigma-Aldrich. γ-Butyrolactone (GBL) (≥99%) and trifluoromethanesulfonic acid (TfOH) (≥99%) were used as received. p-Toluenesulfonic acid monohydrate (PTSA) (≥98.5%) was dried using a Schlenk line before use. Benzyl alcohol (anhydrous, 99.8%) and toluene (anhydrous, 99.8%) were purged by passing an inert gas through them for 15 minutes before use.2.2. Procedures
After the desired amount of time, the pressure was released, and the post-reaction mixture was added to the vial filled with cold methanol. Next, the precipitate of PGBL was filtered, washed with methanol 3-times and dissolved in a small portion of chloroform. Then, an alumina column was employed to purify the polymer and remove the remaining monomer and catalyst. The polymer was isolated by precipitation into cold methanol, filtered and dried under vacuum to a constant mass.
The conversion of monomer could not be determined by 1H NMR because of the risk of depolymerization of PGBL. The reaction yield was assessed by gravimetric analysis. Purified PGBL was dissolved in DMF to perform SEC-LALLS measurements.
2.3. Instruments
:10:1 v/v. Data were acquired in continuum mode until acceptable averaged data were obtained and were analyzed using a Shimadzu Biotech Launchpad program.Tested polymers were poured (in sterile conditions) on a 12-well plate directly before the main viability assay. Then tested normal cell lines were seeded at a density of 50000 cells per well and incubated at standard growing conditions for 48 h. After this time, 200 μL CellTiter 96® AQueous One Solution – MTS (Promega) solution in 1 mL DMEM medium (without phenol red and FBS) was added to each well. After 1 h, the absorbance of the synthesized formazan (mitochondrial activity indicator) was measured at 490 nm using a multi-plate reader Synergy 4 (BioTek). The obtained results are expressed as a percentage of the reference material – uncoated well. Each polymer was tested in three independent experiments.
000 cells per cm2 directly on studied polymers and incubated at 37 °C for 48 h. After this time, the culture medium was replaced by a 5 μM dye solution – CellTracker Green CMFDA (Invitrogen) and incubated at 37 °C for 1 h. Next, polymer samples with attached cells were washed three times with PBS and then the materials were transferred to a 35 mm imaging dish with a polymer coverslip (ibidi). The visualization of cells using a 485 nm excitation laser and a 520 nm emission filter was performed using Zeiss AxioObserver Z1 inverted fluorescence microscope equipped with an AxioCam RMn camera and the corresponding software. The total cell fluorescence intensity was calculated from seven images for each material in the ImageJ software.3. Results and discussion
In the case of pressure-assisted polymerization processes, it is essential to select properly all the reactants (type of monomer, initiating/catalytic system, as well as the amount and type of solvent) and set the appropriate thermodynamic conditions (p, T). It is a direct consequence of pressure-induced phase transition (e.g., crystallization), viscosity and solubility changes that can dramatically affect the polymerization pathway and, hence the properties of the produced polymers.17 Our recent studies concerning the high-pressure ROP of CL and GBL have clearly shown that at elevated pressure, the Ist order transition (crystallization) had a significant impact on the progress of the reaction since it is considered as a competitive process to polymerization.31 Briefly, since temperatures of phase transitions (including crystallization) generally increases with compression, one has to be careful in selecting thermodynamic conditions of polymerization to avoid undesired crystallization of solvent/monomer that suppresses the progress of reactions. Strong densification also affects the viscosity of the polymerizing system leading to the retardation of the termination rate. However, both these affect the crystallization and viscosity increased, which may be minimized/eliminated in diffusion-controlled processes by using solvents of low transfer constant or conducting the reaction at higher temperature and lower pressure regimes, respectively. Notably, in a previous paper that discussed the TBD-catalyzed ROP of GBL,17 we were able to find optimal thermodynamic conditions and avoid the GBL crystallization by real-time monitoring of the polymerization progress using Broadband Dielectric Spectroscopy (BDS). We observed that for TBD-catalyzed GBL ROP performed in toluene, GBL had revealed a tendency of crystallization at low temperatures (below 248 K) and high pressure values (above 1300 MPa).As we intended to establish the most optimal conditions for the acid-catalyzed GBL ROP, we referred to the previous paper of Yamashita et al.12 In the mentioned study, it was found that acid-catalyzed polymerization occurred in the temperature range, T = 313–353 K and elevated pressure (p = 350–1000 MPa), yielding polymers of Mn = 6450–7950 g mol−1 (relative to PS standards) and moderate dispersity Đ = 1.49–1.57 after 24 h.12 However, it should be emphasized that higher yields were noted only when high-pressure conditions (p = 800–1000 MPa) were employed. Having in mind these results, we performed series of reactions at constant p = 1000 MPa and low-temperature regime T = 278–323 K (mild conditions) using monomer:initiator:catalyst ratio ([GBL]0/[I]0/[C]0 = 100/1/1), benzyl alcohol (BnOH) as the initiator, two different catalysts (TfOH or PTSA), and toluene as a solvent (50% in respect to GBL volume, please see Table 1). Reactions were conducted for 24 h. Our main goal was to check the influence of the selected conditions on polymerization yield and parameters of the obtained polyesters (Mn, Đ) (Fig. 3). The structure of the produced PGBL with the successful incorporation of initiator moiety into polymer backbone was confirmed using 1H NMR, 13C NMR and MALDI-TOF analysis. The representative 1H and 13C NMR spectra of produced PGBLs using TfOH and PTSA as a catalyst are presented in Fig. 1 (note the peaks at δ = 3.36; 3.42; 5.06; 7.29 (a) and δ = 128.32 (b) originated from the end group of benzyl alcohol; and peaks at δ = 1.90; 2.30; 4.05 (a) and δ = 23.90; 30.65; 63.53 (b) being a confirmation of the presence of –CH2 groups of PGBL units) and Fig. 2 (note the peaks at δ = 3.42; 3.62; 5.06; 7.29 (a) and δ = 61.95; 128.25 (b) from the end groups of benzyl alcohol; and peaks at δ = 1.90; 2.33; 4.06 (a) and δ = 23.99; 30.74; 63.53 (b) from –CH2 groups of PGBL units).
| Sample no. | Time [h] | Pressure [MPa] | Temperature [K] | Catalyst | Mna [g mol−1] | Đa | Yield [gPGBL/gGBL] |
|---|---|---|---|---|---|---|---|
| a Determined by SEC-LALLS (DMF as an eluent). Estimated dn/dc = 0.047. Benzyl alcohol (BnOH) as an initiator. Toluene 50%v/v as a solvent. [GBL]0/[BnOH]0/[catalyst]0 = 100/1/1.b [GBL]0/[BnOH]0/[catalyst]0 = 500/1/1. | |||||||
| 1 | 3 | 1000 | 303 | TfOH | 4800 | 1.09 | 0.21694 |
| 2 | 6 | 1000 | 303 | 6500 | 1.07 | 0.27244 | |
| 3 | 24 | 1000 | 303 | 9700 | 1.17 | 0.48280 | |
| 4 | 48 | 1000 | 303 | 8900 | 1.14 | 0.62443 | |
| 5 | 24 | 500 | 303 | — | — | — | |
| 6 | 24 | 700 | 303 | 5200 | 1.14 | 0.17515 | |
| 7 | 24 | 1500 | 303 | 3500 | 1.21 | 0.16935 | |
| 8 | 24 | 1000 | 278 | 3100 | 1.05 | 0.26701 | |
| 9 | 24 | 1000 | 288 | 3800 | 1.40 | 0.30988 | |
| 10 | 24 | 1000 | 323 | 2300 | 1.14 | 0.08674 | |
| 11b | 72 | 1000 | 303 | 7000 | 1.31 | 0.45905 | |
| 12 | 3 | 1000 | 303 | PTSA | 2900 | 1.09 | 0.21809 |
| 13 | 6 | 1000 | 303 | 3100 | 1.12 | 0.33756 | |
| 14 | 24 | 1000 | 303 | 5600 | 1.16 | 0.54026 | |
| 15 | 48 | 1000 | 303 | 5700 | 1.19 | 0.55100 | |
| 16 | 24 | 500 | 303 | — | — | — | |
| 17 | 24 | 700 | 303 | 4100 | 1.18 | 0.12750 | |
| 18 | 24 | 1500 | 303 | 2500 | 1.46 | 0.17851 | |
| 19 | 24 | 1000 | 278 | 2400 | 1.24 | 0.10087 | |
| 20 | 24 | 1000 | 288 | 4500 | 1.11 | 0.37165 | |
| 21 | 24 | 1000 | 323 | 2200 | 1.06 | 0.11449 | |
| 22b | 72 | 1000 | 303 | 4400 | 1.14 | 0.27163 | |
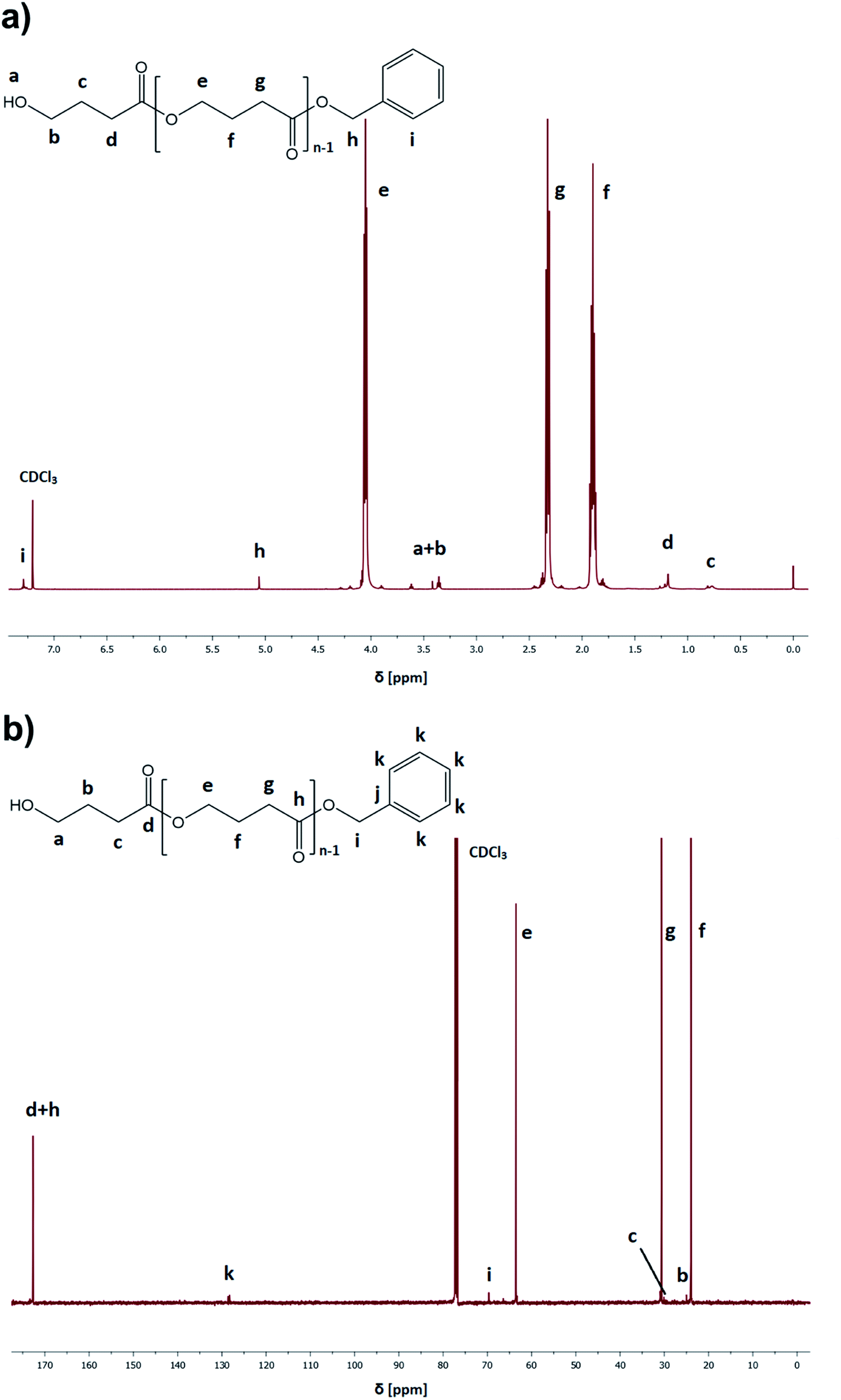 | ||
| Fig. 1 Representative 1H NMR (a) and 13C NMR (b) spectra of PGBL samples produced within TfOH-catalyzed (sample 3) high pressure assisted ROP. | ||
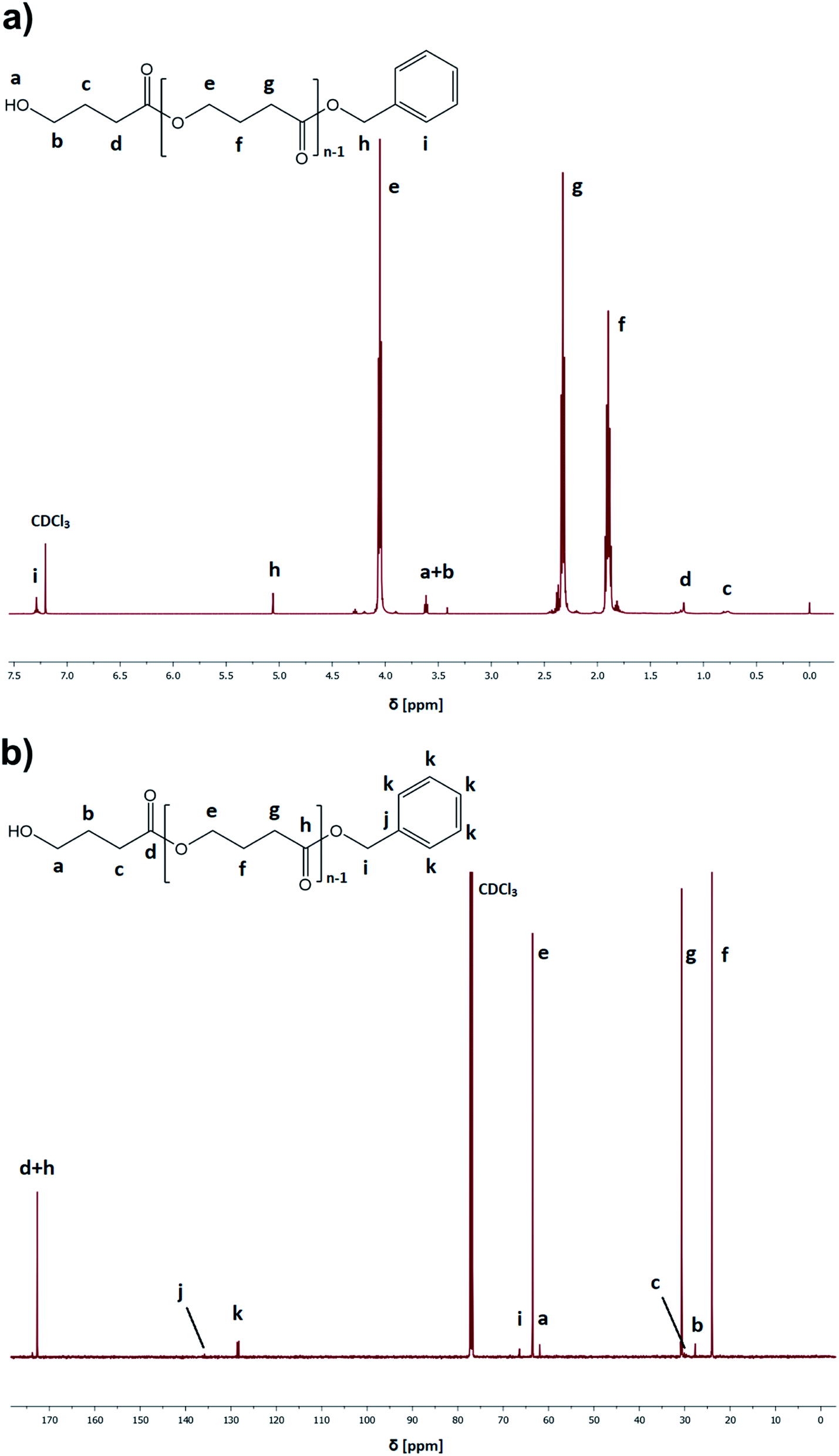 | ||
| Fig. 2 Representative 1H NMR (a) and 13C NMR (b) spectra of PGBL samples produced within PTSA-catalyzed (sample 14) high pressure assisted ROP. | ||
We noticed that processes performed at lower temperatures (T = 278–288 K) at p = 1000 MPa proceed with lower yield giving polymers of relatively low Mn and low/moderate Đ independently of the applied catalysts (please see Fig. 3, TfOH: samples 8 and 9 (a) PTSA: samples 19 and 20 (b)). A similar scenario was observed for reactions conducted at higher studied temperature T = 323 K. Interestingly, p = 1000 MPa, T = 303 K turned out to be the most optimal for GBL ROP yielding PGBL of the highest Mn (Mn = 9700; Mn = 5600 g mol−1), lowest Đ (Đ = 1.17; Đ = 1.16) and simultaneously the highest yields (0.48250; 0.54026 gPGBL/gGBL) in the case of both TfOH (sample 3) and PTSA (sample 14) catalysts, respectively. It is worth emphasizing that we were able to significantly mitigate the conditions of the cationic ring-opening polymerization of GBL to the range of room temperatures (or even lower), maintaining well-defined parameters of produced PGBL and high yield.
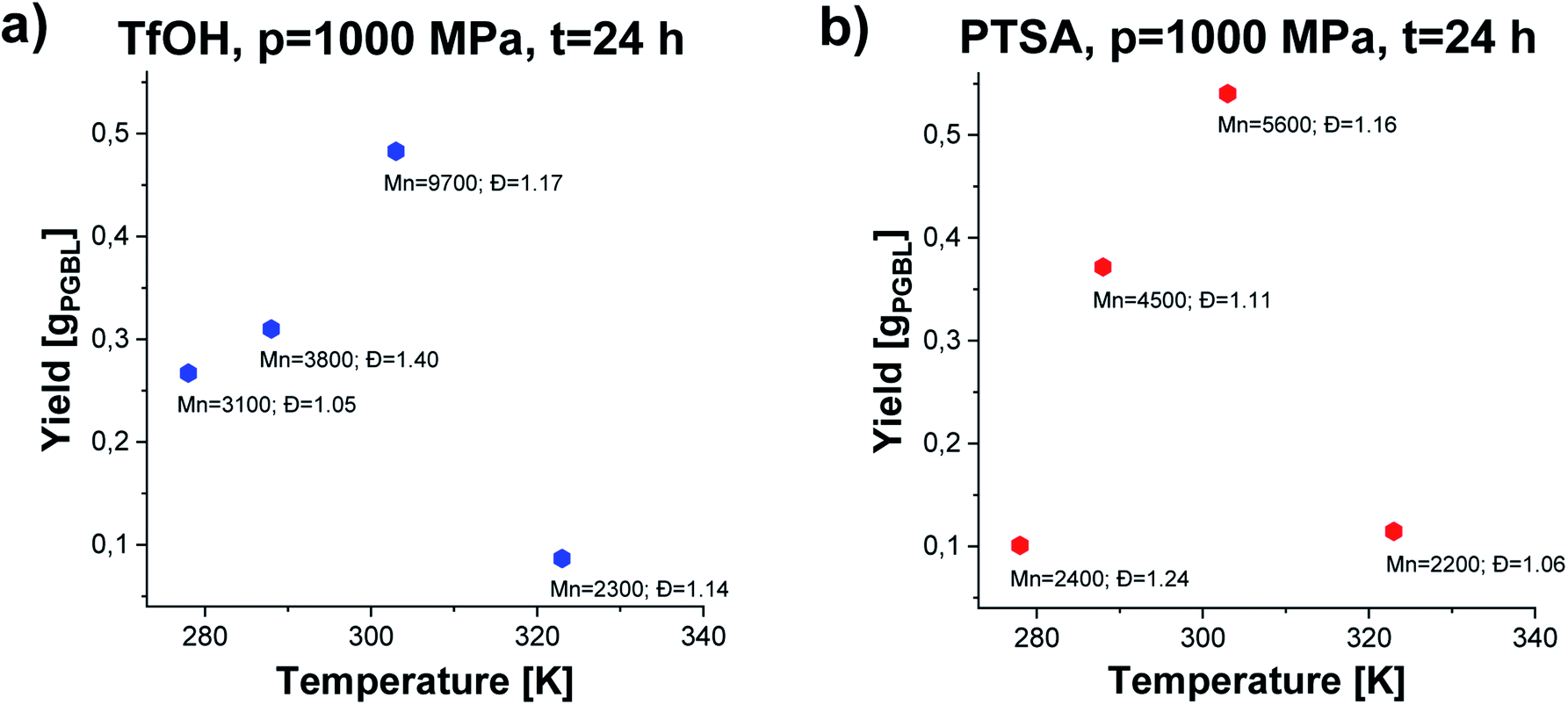 | ||
| Fig. 3 The dependence of yield vs. temperature of PGBL samples produced within TfOH-catalyzed (a) and PTSA-catalyzed (b) high pressure assisted ROP at p = 1000 MPa. | ||
In the next step, we conducted several further syntheses, varying the pressure (in the range of 500–1500 MPa) while maintaining the previously set optimal temperature (T = 303 K). Interestingly, we also recorded a clearly parabolic-like dependence of yield vs. pressure, (Fig. 4), with the maximum efficiency for the reaction conducted at p = 1000 MPa (TfOH: sample 3; PTSA: sample 14). Importantly, these isobaric investigations revealed that not only the yield but also PGBL macromolecular parameters are the most favorable (highest Mn, lowest Đ). As the pressure decreased, a drop in both the yield and Mn and no significant change in Đ values (TfOH: sample 6; PTSA: sample 17) were observed. A similar decrease in Mn and yield was noted when the synthesis was performed at p = 1500 MPa (TfOH: sample 7; PTSA: sample 18). It is worth mentioning that at p = 500 MPa (TfOH: sample 5; PTSA: sample 16) no reaction occurred in the case of both catalysts. Thus, we can assume that p = 700 MPa is a minimal pressure value required to trigger GBL ROP at room temperature. Herein, it is worth referring to the data reported by Yamashita et al.12 who also investigated the acid-catalyzed ROP of GBL (using TfOH) at various temperatures (313, 333 and 353 K) and high pressures (350, 800 and 1000 MPa). Interestingly, in this paper, Authors have demonstrated that only two syntheses performed at T = 313 K and relatively high pressures, i.e. p = 800 and p = 1000 MPa, turned out to be successful yielding PGBL with moderate molecular weights and dispersity (Mn = 6450 and Mn = 7950 g mol−1; Đ = 1.49 and Đ = 1.57, respectively) determined by the use of PS standards. It needs to be emphasized that these reactions lasted 24 hours. What is more, at lower pressures (p = 350 MPa and T = 313 K) or higher temperatures (T = 333–353 K and p = 800 MPa) only oligomeric products (theoretical Mn = 200–1400 g mol−1) not analyzed by the authors using SEC or MALDI were obtained. They also determined the efficiency of the process at 4–45% based on the amount of PGBL produced from the ether-insoluble polymer fraction (starting with γ-BL, 23.6 mmol). In our extensive research, we performed the synthesis in a much wider range of temperatures and pressures than Yamashita et al.12 did. Therefore, we were able to find the optimal conditions for the acid-catalyzed GBL ROP process and perform it even at room temperature. Note that we also extended our investigation and examined two acidic catalysts of different power/acidities: TfOH and PTSA (pKa = −14.72833 and pKa = −2.829,34 respectively). Moreover, the obtained macromolecules were thoroughly characterized by determining the absolute molecular weights as well as the structure and chain-end groups using NMR and MALDI measurements.
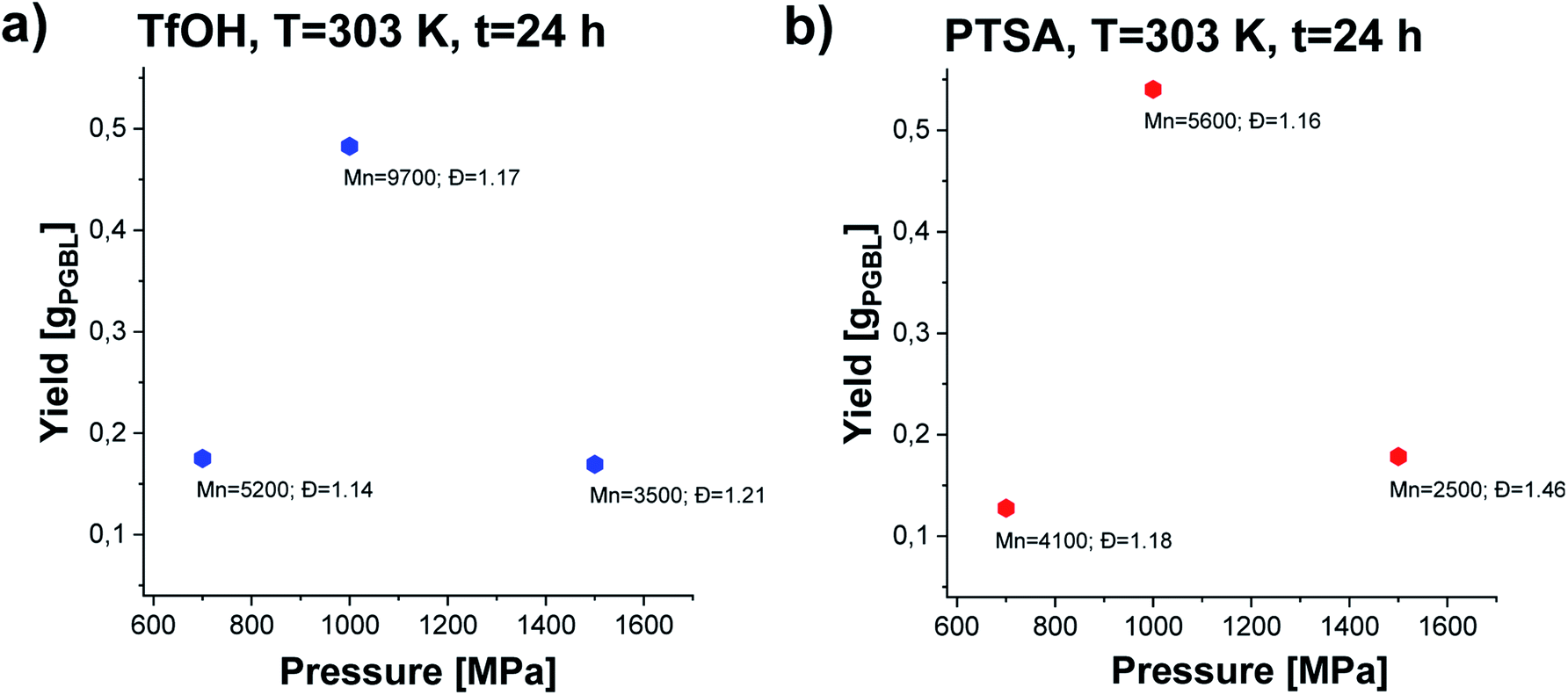 | ||
| Fig. 4 The dependence of yield vs. pressure of PGBL samples produced within TfOH-catalyzed (a) and PTSA-catalyzed (b) high pressure assisted ROP at T = 303 K. | ||
The established optimal thermodynamic conditions as T = 303 K and p = 1000 MPa were successfully adopted to further experiments to verify how extending the reaction time influences the parameters of the obtained macromolecules (Mn, Đ) (Fig. 5). As presented in Fig. 5, the molecular weight of the obtained macromolecules highly depends on the time in case of the reaction catalyzed by both acids. Firstly, it needs to be stressed that by applying high pressure we were able to obtain PGBL of moderate Mn and low Đ values after just 3 hours, while, after 24 hours, we get high-weight products but not at the expense of increasing dispersity. It is also worth emphasizing that it was possible to conduct highly efficient acid-catalyzed ROP of GBL yielding polymers of better parameters (higher absolute Mn together with lower dispersity) than previously reported in literature.12 Within this work, we also tried to carry out the experiments using 10-times less catalyst content (p = 1000 MPa, T = 303 K, 24 h) that yielded PGBL of not satisfying macromolecular properties reflected in low both Mn and yield (Mn = 2300 g mol−1, Đ = 1.86, yield = 0.2760 gPGBL/gGBL; Mn = 2600 g mol−1, Đ = 1.09; yield = 0.0299 gPGBL/gGBL; for TfOH and PTSA, respectively).
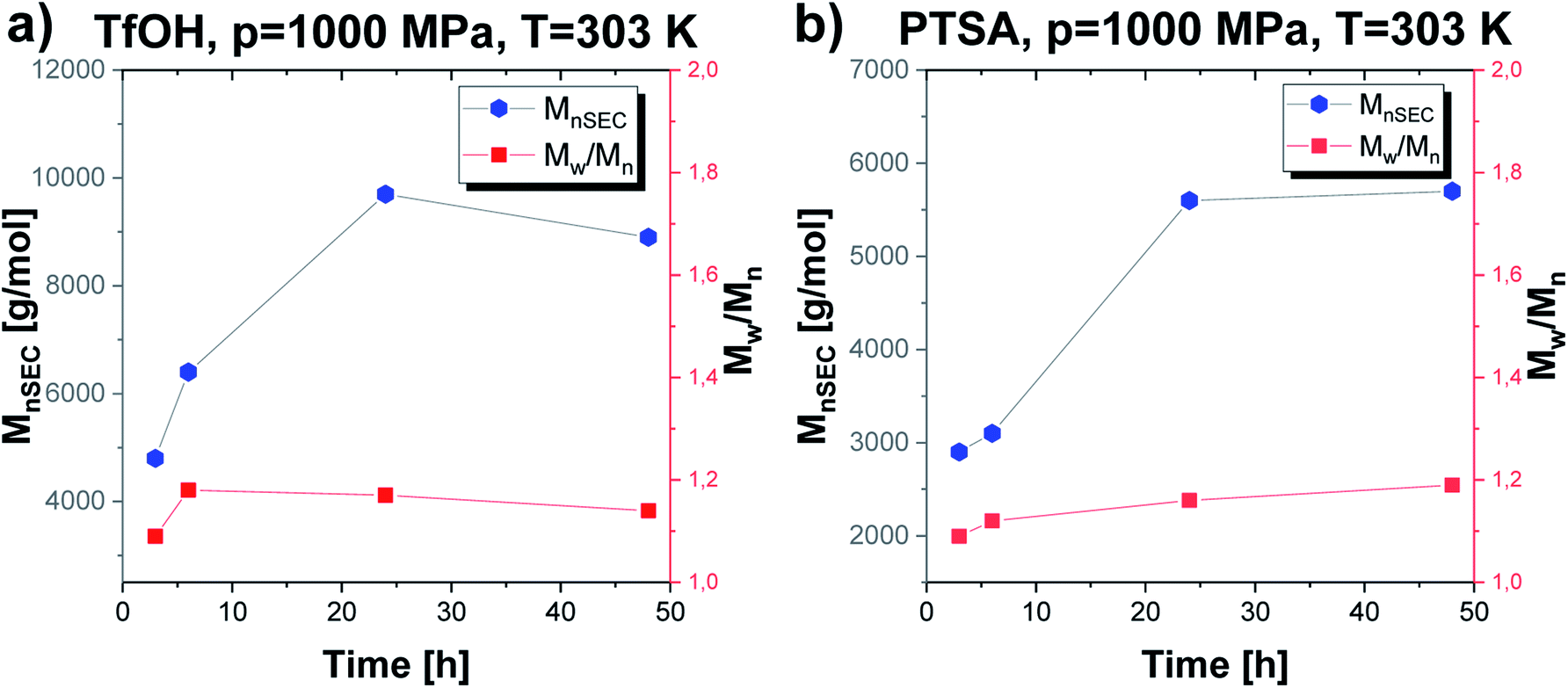 | ||
| Fig. 5 The dependence of Mn and Đ vs. time of PGBL samples produced within TfOH-catalyzed (a) and PTSA-catalyzed (b) high pressure assisted ROP. | ||
In the case of TfOH-catalyzed ROP, we synthesized polymers of higher Mn with respect to the analogues ROP of PGBL supported by PTSA (Mn max = 9700 g mol−1 vs. Mn max = 5700 g mol−1, respectively). Moreover, for PTSA-catalyzed reactions, there is a clear increase in dispersity as the polymerization time gets longer, which is consistent with the results of our group's previous study concerning TBD-mediated ROP of GBL.17 Mentioned results clearly indicated that by simple extension of the reaction time, we were able to modify the properties of the obtained polymers (e.g., increase the dispersity and number of the macrocyclic form). Interestingly, a further increase in the polymerization time up to 48 h (TfOH: sample 4; PTSA: sample 15) did not significantly affect the Mn and Đ of the resulting polymer but led to a slightly higher yield in comparison to samples obtained after 24 h. The presented data suggest that 24 h is the optimal reaction time to set the balance between Mn and Đ values, together with yield (Table 1).
Kinetic analysis was followed by carrying out SEC investigations of the obtained polymers. The measurements clearly confirmed well-defined properties of PGBL produced via high-pressure cationic-ROP reflected by the symmetric and monomodal shape of the SEC-LALLS traces (Fig. 6 and 7). Moreover, the kinetic dependence (increase in molecular weight together with increasing reaction time) (a) as well as the influence of pressure (b) and temperature (c) on the parameters of the macromolecules obtained within TfOH- and PTSA-mediated ROP of GBL are presented in Fig. 7.
 | ||
| Fig. 6 SEC-LALLS traces of PGBL samples produced within TfOH-catalyzed and PTSA-catalyzed high pressure assisted ROP (in monomer:initiator:catalyst ratio [GBL]0/[I]0/[C]0 = 500/1/1). | ||
 | ||
| Fig. 7 SEC-LALLS traces of PGBL samples produced within TfOH-catalyzed and PTSA-catalyzed high pressure assisted ROP (in monomer:initiator:catalyst ratio [GBL]0/[I]0/[C]0 = 100/1/1) in a function of the reaction time (a), pressure (b) and temperature (c). | ||
After routine SEC analysis, we conducted further, more detailed MALDI-TOF studies on the PGBL produced via TfOH-mediated and PTSA-mediated reactions to check dependency of the linear/cyclic form content on the type of applied catalyst. Fig. 8 shows mass spectra of representative polymers produced within PTSA-catalyzed (a) and TfOH-catalyzed (b) ROP of GBL. The experimental results revealed that in the investigated samples of PGBL, two main series of signals could be found and both of them confirm the presence of poly(γ-butyrolactones) chains. Based on the calculation and mechanism of the polymerization process, the structure of the macromolecules can be proposed as BnO–(γBL)n–H for linear chains (Fig. 9, noted with a triangle) or (γ-BL)n for cyclic ones (Fig. 9, noted with circle). For macromolecules to be MALDI active, ionization must occur, which is realized by potassium adduct formation in both cases of the proposed structures.
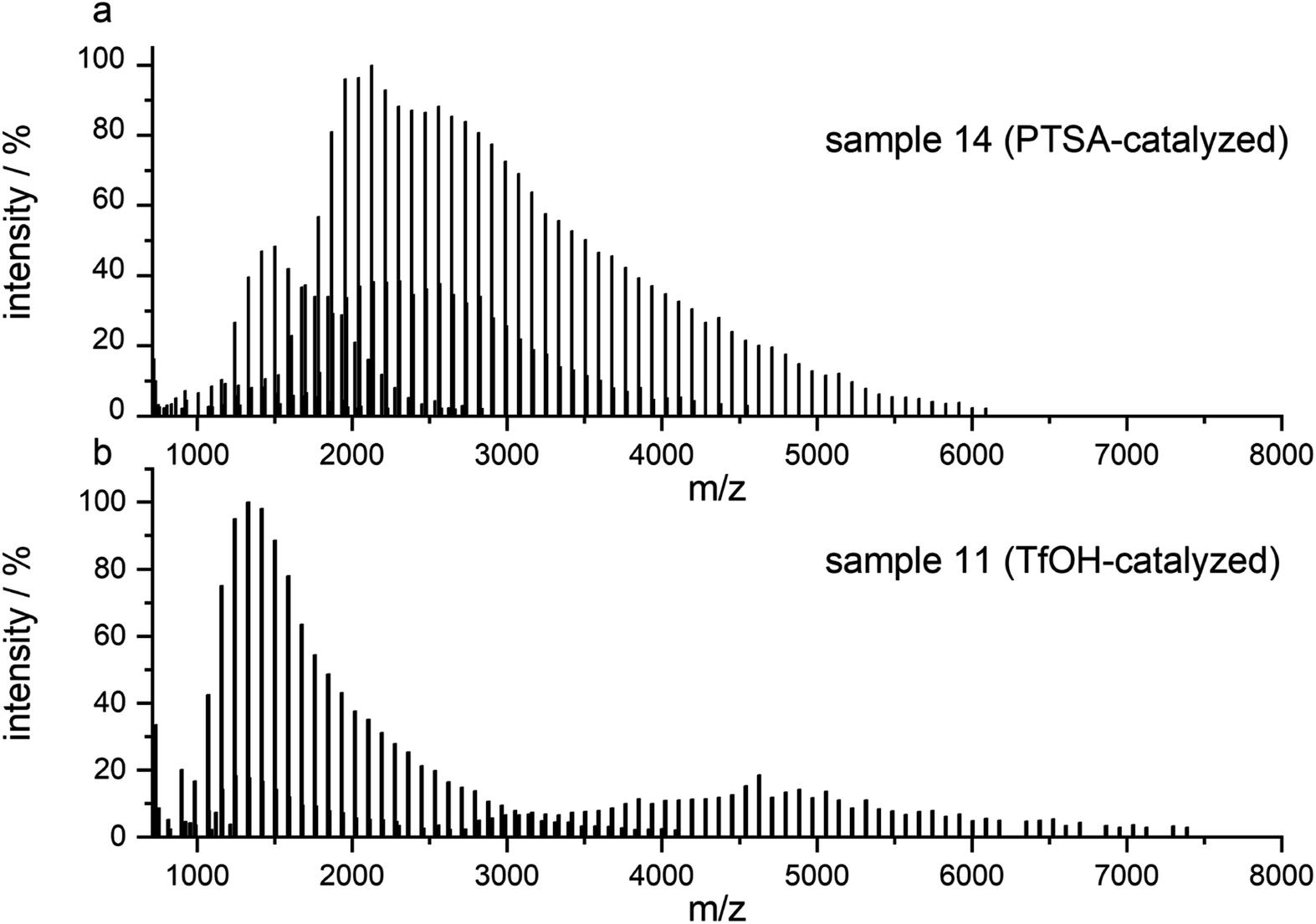 | ||
| Fig. 8 MALDI-TOF MS spectra of PGBL samples: sample 14 (a) and sample 11 (b) with the full spectra range. | ||
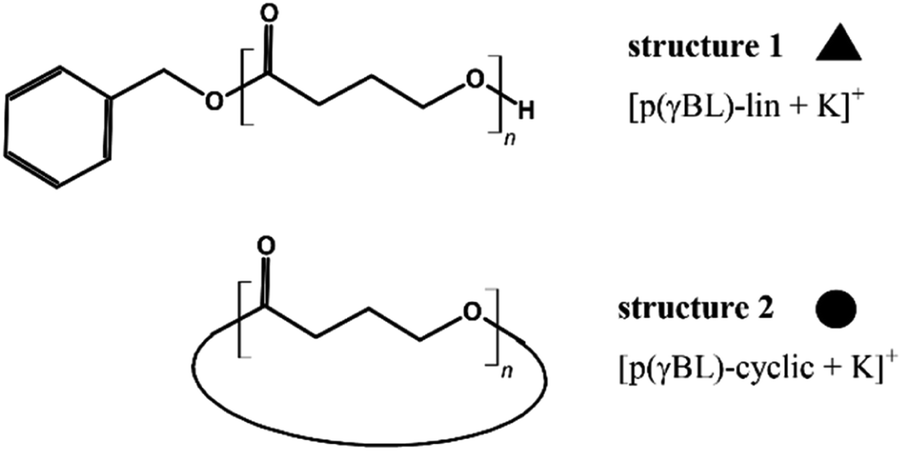 | ||
| Fig. 9 The proposed structures of the produced polymers. | ||
As presented in Fig. 8a, the dominant groups of signals for sample 14 are located at a higher mass range and represent the linear components of the samples with a peak to peak distance equal to 86.10 u reflecting the incorporation of γ-butyrolactone as mer unit. They also contain signals of cyclic components; however, their intensity is much lower in comparison to the linear ones.
On the contrary, the spectrum of sample 11 (Fig. 8b) prepared via TfOH-mediated GBL ROP reveals the presence of both linear and cyclic structures with more intense signals of the latter one.
The series of signals at the higher mass region is prescribed mainly to the linear type of the structure capable of potassium adduct formation. For example, for sample 14, peaks at 4021.46 (Mcalc: 4021.27), 4107.67 (Mcalc: 4107.36), 4193.55 (Mcalc: 4193.45), 4279.93 (Mcalc: 4279.54), 4365.92 (Mcalc: 4365.63), 4452.16 (Mcalc: 4451.72) and 4538.35 (Mcalc: 4537.81) reflect the potassium adduct of linear macromolecules with a degree of polymerization in a range from 45 to 51. They are accompanied by lower intensity series of signals corresponding to potassium adducts of cyclic macromolecules, for example, peaks at 1846.29 (Mcalc: 1846.99), 1932.48 (Mcalc: 1933.08), 2018.65 (Mcalc: 2019.17), 2104.82 (Mcalc: 2105.26). 2190.98 (Mcalc: 2191.35), 2277.03 (Mcalc: 2277.44), 2363.83 (Mcalc: 2363.53), 2449.23 (Mcalc: 2449.62), 2536.04 (Mcalc: 2535.71) reflect the macromolecules with n equal in a range from 21 to 29 (Fig. 10).
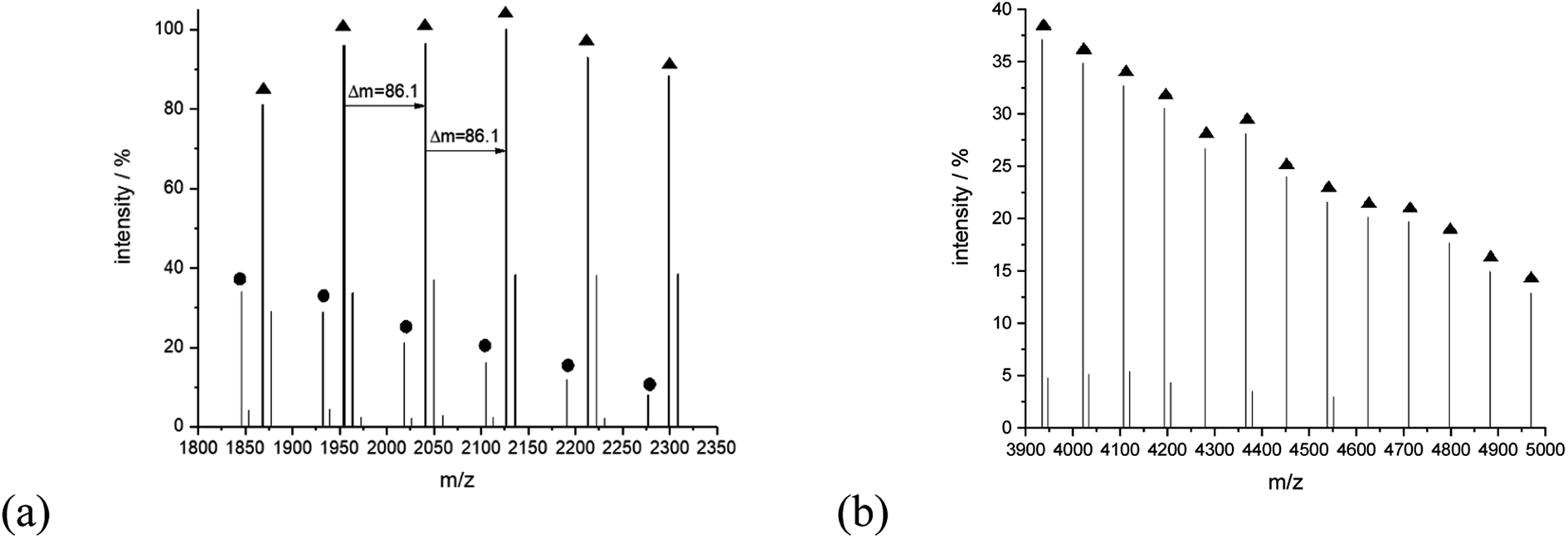 | ||
| Fig. 10 MALDI-TOF MS spectra of sample 14 with the expanded spectral region at (a) 1800–2350, (b) 3900–5000. | ||
A closer analysis of the nature of the spectrum for sample 11 allowed to assign signals to linear and cyclic macromolecules. The series of signals at the lower mass region revealed the presence of mainly potassium adducts of cyclic macromolecules, for example, peaks at 1243.75 (Mcalc: 1244.36), 1329.80 (Mcalc: 1330.45), 1415.95 (Mcalc: 1416.54). 1502.09 (Mcalc: 1502.63), 1588.20 (Mcalc: 1588.72), 1674.37 (Mcalc: 1674.81), 1760.39 (Mcalc: 1760.90) reflect the macromolecules with n in a range from 14 to 20. The range of this series extended even to 4000 u. The series corresponding to the potassium adducts of linear type macromolecules spans further with peaks at 4108.13 (Mcalc: 4107.36), 4194.29 (Mcalc: 4193.45), 4280.39 (Mcalc: 4279.54), 4365.86 (Mcalc: 4365.63), 4452.69 (Mcalc: 4451.72), 4540.60 (Mcalc: 4537.81), 4627.59 (Mcalc: 4623.90) and 4710.89 (Mcalc: 4709.99). They reflect the potassium adduct of linear macromolecules with a degree of polymerization ranging from 46 to 53 (Fig. 11).
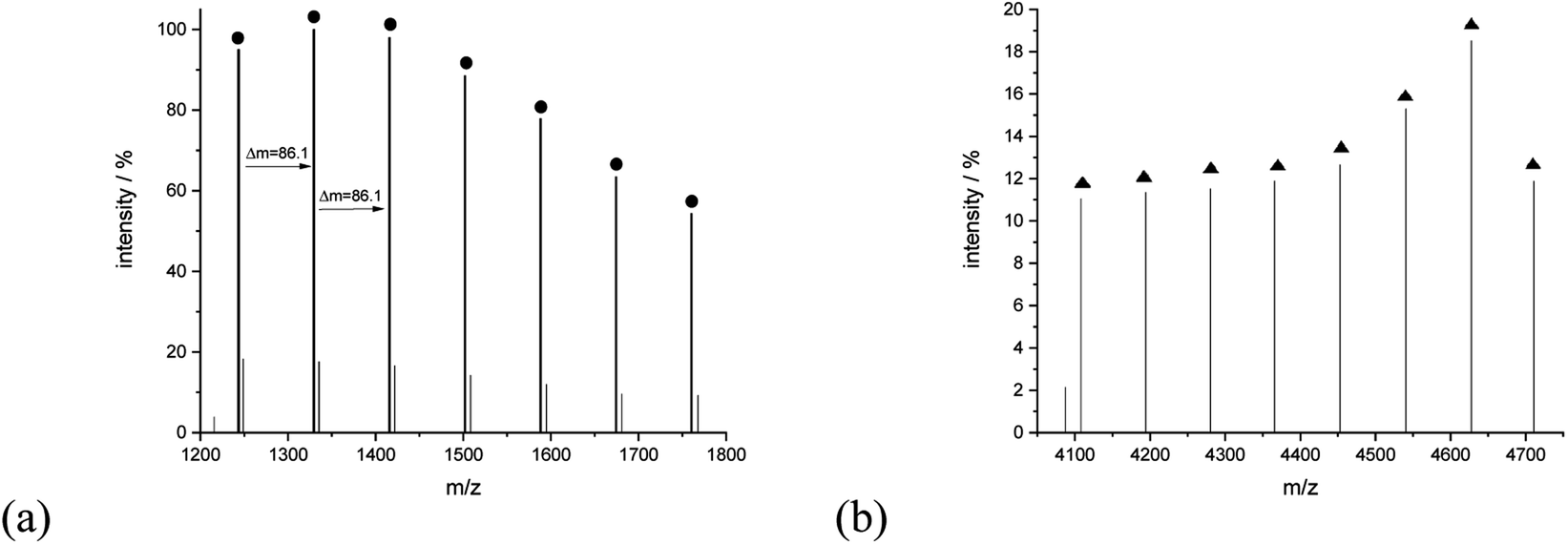 | ||
| Fig. 11 MALDI-TOF MS spectra of sample 11 with the expanded spectral region at (a) 1200–1800, (b) 4050–4750. | ||
Based on MALDI-TOF spectra analysis, one can conclude that the content of linear/cyclic forms depends on the power/acidity of the used acidic catalyst. The use of PTSA catalyst allowed obtaining samples with a predominant content of linear fraction, while the use of TfOH as a catalyst led to an increased tendency to create cyclic structures.
As a final step of our investigation, we measured the basic thermal properties of the produced PGBL. Representative DSC thermograms obtained for selected polymers are shown in Fig. 12. Interestingly, the DSC curves obtained for samples characterized by a similar Mn and Đ but containing a different number of linear and cyclic forms are clearly different. In the case of PGBL with a dominant number of cyclic macromolecules, one can observe a prominent endothermic peak related to the melting of the examined sample, that maximum is located at Tm ∼330 K. In contrast, for the sample with the predominance of linear chains, the detected melting temperature is shifted to higher temperatures (where Tm ∼340 K), see Fig. 12. Note that as cyclic chains' content increases in the examined PGBL, the observed endothermic peak becomes bimodal. On the other hand, samples characterized by a negligible number of cyclic polymers displayed only one monomodal endothermic transition, see Fig. 12. This finding seems to be in agreement with the literature data reported for PGBL homopolymers,13,35 including our own previous studies,17 which clearly show that the temperature range of observed Tm depends on the polymer structures. Note that melting temperatures of cyclic and linear polymers were observed to be located at lower (Tm ∼323 K) and higher temperature range, Tm ∼333 K, respectively.
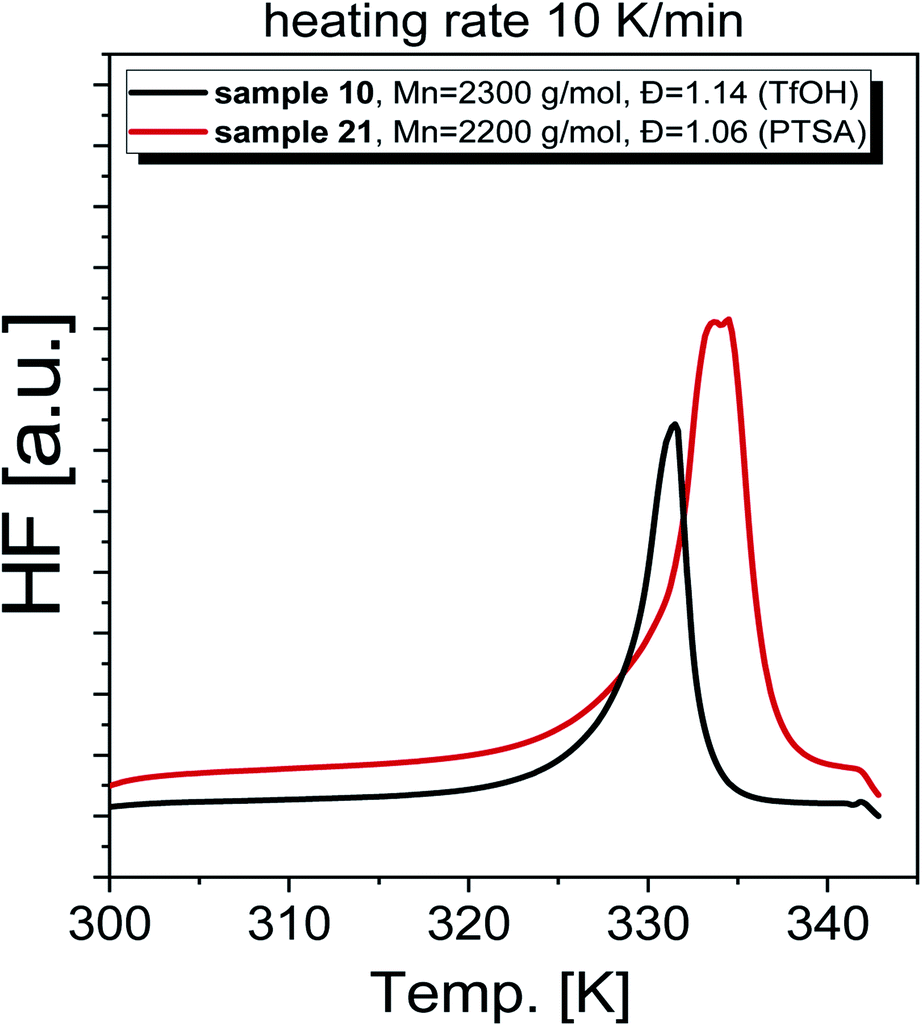 | ||
| Fig. 12 DSC thermograms recorded on chosen PGBL synthesized at high-pressure conditions. | ||
Keeping in mind that PGBL is considered a highly desirable biomaterial and that we used catalysts based on strong organic acids in current experiments, we decided to extend our research to cytotoxicity studies to check whether obtained polymer samples are suitable for potential biomedical applications. The cytotoxicity studies were conducted on fibroblast and osteoblast normal cell lines. In these experiments, we seeded the tested cell lines on the sample surfaces and monitored the proliferation process. After 48 h of incubation, the viability of cells was measured with an MTS assay. As presented in Fig. 13, in all of the tested cases, we did not note the decrease of the proliferating fraction of cells below 50%. It means that all of the tested samples did not exhibit significant toxicity toward tested cell lines. Nevertheless, some differences between the samples were registered. Namely, the best conditions for the proliferation were created on the surface of the sample 3 (produced within TfOH-catalyzed ROP of GBL) in both fibroblasts and osteoblasts. Cell viability exceed 80% (for NHDF) and 100% (for HOB). On the other hand, in sample 14 (produced within PTSA-catalyzed ROP of GBL), good cell culture properties were observed for the HOB cell line only (close to 95%). The NHDF cell line was less susceptible to growing on that surface (reaching 55%). However, we have to remember that the obtained results were compared to the cell culture Petri dish dedicated to cell culture and coated with ingredients helping cells to attach. Therefore, lower proliferating fractions of cells do not mean that the tested samples expressed toxicity. Certainly, in the case of toxicity, the surviving fraction would be much lower.
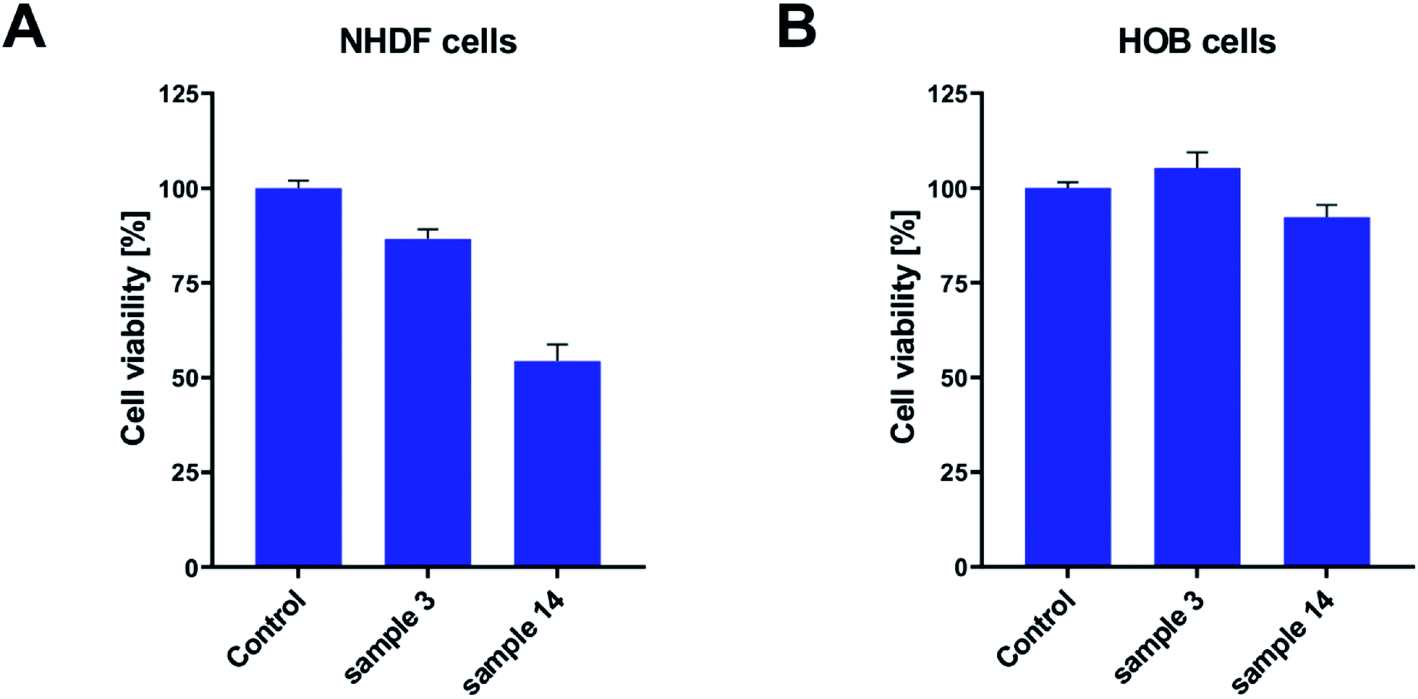 | ||
| Fig. 13 The bioactivity of PGBL samples against normal human cell lines: fibroblast – NHDF (A) and osteoblast – HOB (B). | ||
Additionally, we performed cell morphology imaging with fluorescence microscopy technique. In order to picture the shape and condition of cells, we used fluorescent dye because of the non-transparency of samples. As shown in Fig. 14, we observed proliferating osteoblasts in numbers overlapping with toxicity results. Even that the surface of the tested PGBL materials was not coated with any adhesive promoter, the HOB cell line proliferated. The cell morphology was not disturbed, and we register normal cell stretching.
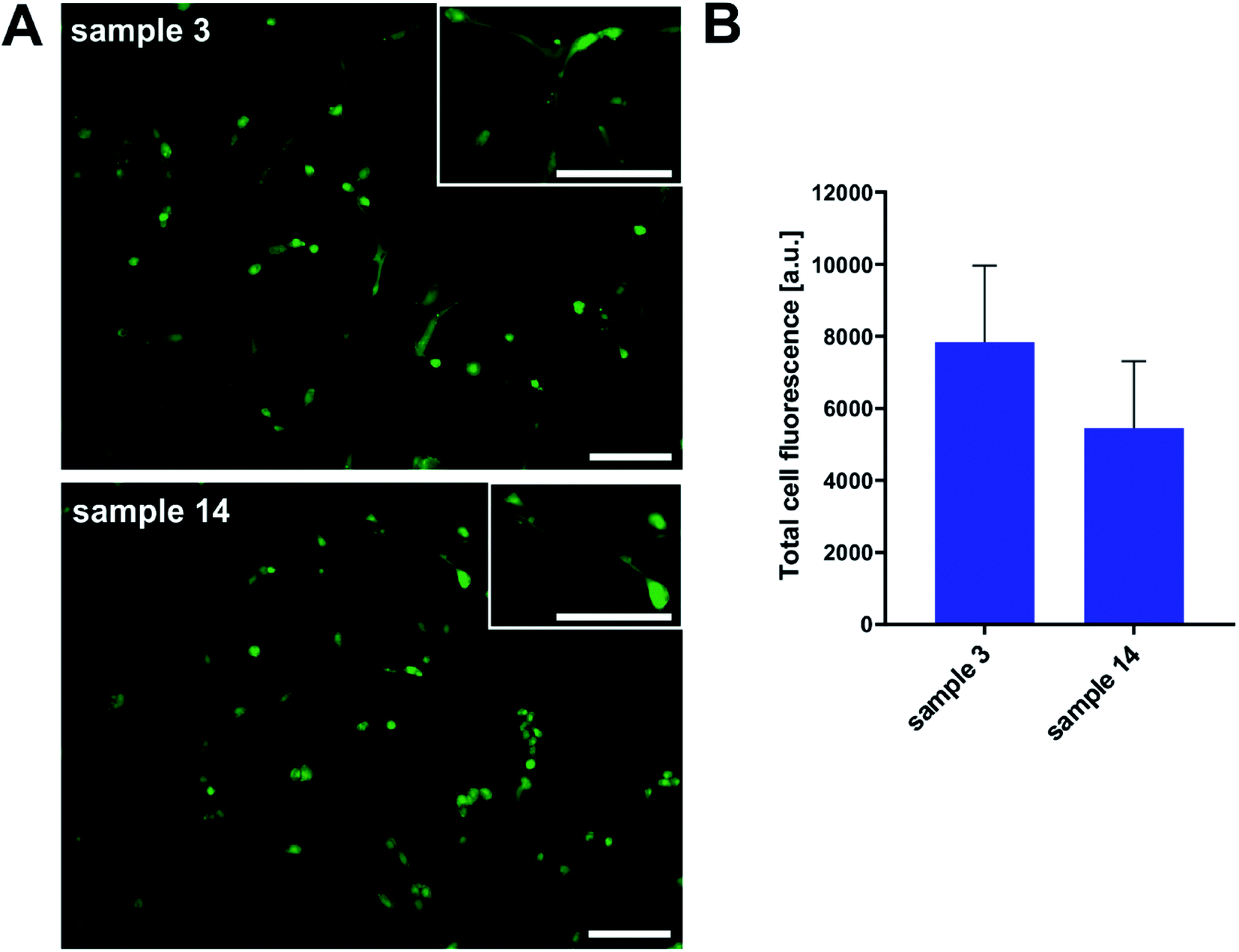 | ||
| Fig. 14 Cell adhesion of osteoblasts onto PGBL materials (A). Scale bars = 50 μm. Total cell fluorescence was obtained by measuring objects' fluorescence intensity from seven images for each sample in ImageJ 1.41 software (B). | ||
To sum up this part, we can conclude that the surface of samples produced within TfOH-catalyzed ROP of GBL is more suitable for both cell cultures' growth than films of PGBL produced within the PTSA-mediated process. Importantly, both PGBL samples did not exhibit toxicity towards tested normal cell lines. Thus, we can assume that despite using a strong acidic catalyst, the produced PGBL samples remained non-toxic materials that may have optimal biocompatibility, making them suitable for biomedical applications.
4. Conclusions
We report an acid-catalyzed ROP of GBL performed at various thermodynamic conditions (T = 278–323 K and at p = 700–1500 MPa). Our strategy was based on a dually-catalyzed process combining an external physical factor (high pressure) and an internal acidic catalyst (TfOH or PTSA). Using this approach, we have managed to obtain well-defined macromolecules of better parameters (an absolute molecular weight in range Mn = 2200–9700 g mol−1 and narrow to moderate dispersity Đ = 1.05–1.46) in comparison to previous reports concerning the high pressure assisted GBL polymerization. Moreover, we synthesized PGBL with significantly higher yields (0.09–0.62 gPGBL/gGBL) and reduced the time of reaction to just 3 hours at room temperature. Further analysis of the obtained macromolecules, including toxicity and proliferation tests, has proven that the synthesized PGBL samples are non-toxic and biocompatible materials despite using acidic catalysts. Whereas, more detailed MALDI-TOF studies indicated that the use of PTSA as catalyst led to obtaining macromolecules with a predominant content of linear fraction, while the use of the TfOH showed the tendency to create cyclic structures. These findings may be important in terms of potential application since the film-forming ability is strictly connected to the linear/cyclic form content. Concluding our research showed that the acid-catalyzed ROP of GBL enabled obtaining non-toxic and biocompatible PGBLs that can be successfully applied in many biomedical applications.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
R. B. and P. M. are thankful for financial support from the Polish National Science Centre within the SONATA project (DEC-2018/31/D/ST5/03464). K. K. acknowledges the financial assistance from the Polish National Science Centre within the SONATA BIS 5 project (DEC-2015/18/E/ST4/00320). E. K. is grateful for the financial support from the National Science Centre within the framework of the Sonata BIS project (DEC-2016/22/E/NZ7/00266).References
- D. Mondal, M. Griffith and S. S. Venkatraman, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 255–265 CrossRef CAS.
- J. Scheirs and T. E. Long, Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters – Google Books, 2003 Search PubMed.
- H. Tsuji, M. Nishikawa, Y. Osanai and S. Matsumura, Macromol. Rapid Commun., 2007, 28, 1651–1656 CrossRef CAS.
- W. George, in Handbook of Material Weathering (Sixth Edition), ed. G. Wypych, ChemTec Publishing, 2018, pp. 829–849 Search PubMed.
- D. P. Martin and S. F. Williams, Biochem. Eng. J., 2003, 16, 97–105 CrossRef CAS.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- A. Duda and S. Penczek, Biopolymers, Wiley-VCH, Weinheim, 2002 Search PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- M. Pluta, Polymer, 2004, 45, 8239–8251 CrossRef CAS.
- Q. Song, J. Zhao, G. Zhang, F. Peruch and S. Carlotti, Polym. J., 2020, 52, 3–11 CrossRef CAS.
- Y. Liu, J. Wu, X. Hu, N. Zhu and K. Guo, ACS Macro Lett., 2021, 10, 284–296 CrossRef.
- K. Yamashita, K. Yamamoto and J. I. Kadokawa, Chem. Lett., 2014, 43, 213–215 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- A. Duda, S. Penczek, P. Dubois, D. Mecerreyes and R. Jérôme, Macromol. Chem. Phys., 1996, 197, 1273–1283 CrossRef CAS.
- N. Zhao, C. Ren, H. Li, Y. Li, S. Liu and Z. Li, Angew. Chem. Int. Ed., 2017, 56, 12987–12990 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Angew. Chem. Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed.
- R. Bernat, P. Maksym, M. Tarnacka, K. Koperwas, J. Knapik-Kowalczuk, K. Malarz, A. Mrozek-Wilczkiewicz, A. Dzienia, T. Biela, R. Turczyn, L. Orszulak, B. Hachuła, M. Paluch and K. Kamiński, Polymer, 2021, 124166 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- P. Walther, W. Frey and S. Naumann, Polym. Chem., 2018, 9, 3674–3683 RSC.
- C. J. Zhang, L. F. Hu, H. L. Wu, X. H. Cao and X. H. Zhang, Macromolecules, 2018, 51, 8705–8711 CrossRef CAS.
- Y. Shen, Z. Zhao, Y. Li, S. Liu, F. Liu and Z. Li, Polym. Chem., 2019, 10, 1231–1237 RSC.
- G. Fang, W. Zhe, Z. Yanyun and W. Xiaowu, Macromol. Chem. Phys., 2020, 221, 1–11 CrossRef.
- L. Lin, D. Han, J. Qin, S. Wang, M. Xiao, L. Sun and Y. Meng, Macromolecules, 2018, 51, 9317–9322 CrossRef CAS.
- P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409–1437 CrossRef CAS.
- M. Tarnacka, A. Dzienia, P. Maksym, A. Talik, A. Ziba, R. Bielas, K. Kaminski and M. Paluch, Macromolecules, 2018, 51, 4588–4597 CrossRef CAS.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Wolnica, M. Dulski, K. Erfurt, A. Chrobok, A. Ziba, A. Brzózka, G. Sulka, R. Bielas, K. Kaminski and M. Paluch, RSC Adv., 2019, 9, 6396–6408 RSC.
- M. Tarnacka, P. Maksym, A. Ziba, A. Mielańczyk, M. Geppert-Rybczyńska, L. Leon-Boigues, C. Mijangos, K. Kamiński and M. Paluch, Chem. Commun., 2019, 55, 6441–6444 RSC.
- M. Buback, A. Kuelpmann and C. Kurz, Macromol. Chem. Phys., 2002, 203, 1065–1070 CrossRef CAS.
- M. Buback and F.-D. Kuchta, Macromol. Chem. Phys., 1995, 196, 1887–1898 CrossRef CAS.
- D. G. Yordanov and G. V. Angelova, Biotechnol. Biotechnol. Equip., 2010, 24, 1940–1945 CrossRef.
- A. Dzienia, P. Maksym, M. Tarnacka, I. Grudzka-Flak, S. Golba, A. Ziba, K. Kaminski and M. Paluch, Green Chem., 2017, 19, 3618–3627 RSC.
- A. Dzienia, P. Maksym, B. Hachuła, M. Tarnacka, T. Biela, S. Golba, A. Ziba, M. Chorażewski, K. Kaminski and M. Paluch, Polym. Chem., 2019, 10, 6047–6061 RSC.
- A. Trummal, L. Lipping, I. Kaljurand, I. A. Koppel and I. Leito, J. Phys. Chem. A, 2016, 120, 3663–3669 CrossRef CAS PubMed.
- J. P. Guthrie, Can. J. Chem., 1978, 56, 2342–2354 CrossRef CAS.
- J. Tang and E. Y. X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2271–2279 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |