
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient Ru(II)–alkylidene based Hoveyda–Grubbs catalysts for ring-closing metathesis reactions†
Mariam Y. Al-Enezi,
Elizabeth John,
Yehia A. Ibrahim and
Nouria A. Al-Awadi *
*
Department of Chemistry, Kuwait University, P. O. Box 5969, Safat, 13060, Kuwait. E-mail: n.alawadi@ku.edu.kw
First published on 24th November 2021
Abstract
Three novel phosphine-free Ru-alkylidenes (7a–7c) have been synthesized and utilized as efficient catalysts for ring closing metathesis (RCM) reaction. Spectroscopic data, i.e. NMR and HRMS, along with single crystal X-ray diffraction analysis, were used to confirm their chemical structures. The tosylated carbenoid 7b showed the highest efficiency in cyclizing different acyclic diene substrates. RCM of various (un)substituted N,N-diallylaniline derivatives and stereoselective RCM of different macromolecular dienes were well tolerated using only a catalytic amount (0.5–2.0 mol%) of the additive catalyst (7b) as compared to the well-known Grubbs (II) and Hoveyda–Grubbs (II) catalysts.
Introduction
Grubbs (1a & 1b) and Hoveyda–Grubbs (2a & 2b) ruthenium-based carbenoids (Fig. 1) have been established as efficient catalysts for carbon–carbon double bond formation with unparallel openings for synthetic chemists to explore their utility in the synthesis of medium to large ring systems via the remarkable olefin reaction, ring-closing metathesis (RCM).1,2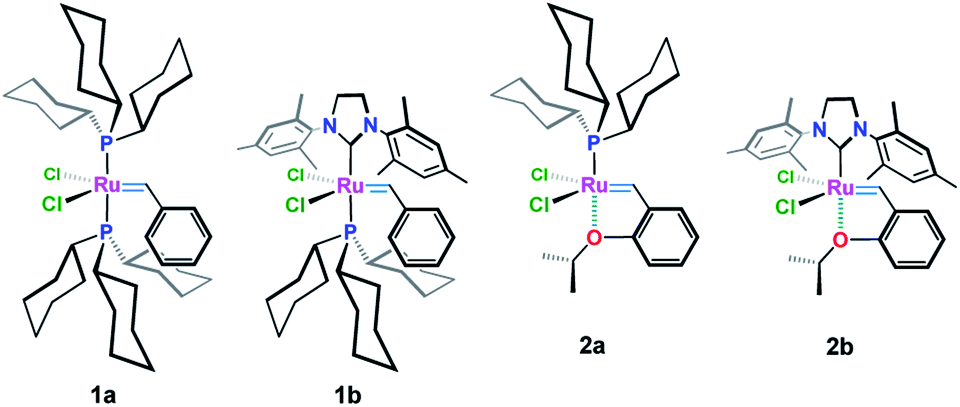 | ||
| Fig. 1 Grubbs' (1a & 2a) and Hoveyda–Grubbs’ (1b & 2b) catalysts. | ||
Chemical modifications of these organometallics upon varying the type and number of the legating sites around the Ru(II) center have many beneficial aspects that are crucial for developing more efficient catalysts with high activity and stability along with excellent tolerance behavior toward other functionalities.3–10 For example, Ru-complex 3a (ref. 3) (Table 1, entry 1), with a bulky phenyl substituent on the ortho-position, showed an increase initiation rate for different olefin reactions.4 Also an enhanced reactivity has been attained in case of electron withdrawing groups containing catalysts, i.e. 3b (ref. 5–7) and 3c (ref. 8) (Table 1, entries 2 & 3), were subjected. The presence of a bulky o-phenyl or electron withdrawing groups, e.g. –NO2 and –SO2NMe2, about the Ru(II) ion, has considerable effect in destabilizing the so-called oxygen–ruthenium interaction, leading to an increase in the catalytic activity toward C–C π-bonds, that enhanced formation of 14-electron Ru-carbene species.9,10
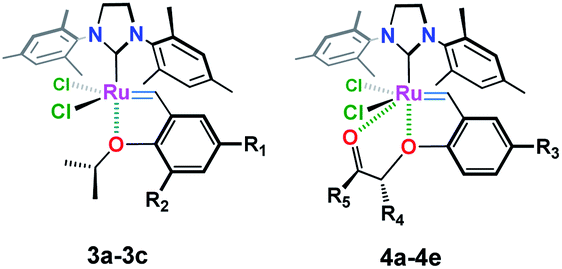
In an attempt to improve the initiation rate, as well as the stability of Hoveyda–Grubbs Ru(II)-catalysts, a number of oxygen chelated Ru(II)-alkylidenes bearing extra carbonyl groups such as ketonic 4a (ref. 3), ester 4b–4d (ref. 3, 6, 11 and 12) and amide 4e (ref. 6) functionalities, have been made (Table 1, entries 4–8). Most of these complexes, which have been verified by X-ray single crystal, shows an extra coordination with the carbonyl oxygen which result in an additional protection of the metal site, and hence, exhibiting a higher activity and stability than those achieved earlier.11–13
In this work, we describe the synthesis with full structural characterization by NMR and HRMS spectral data along with single X-ray diffraction analysis of three novel phosphine-free Ru-alkylidene complexes (7a–7c) (Fig. 2). Complete data are included in the ESI section.† Also, the catalytic activity of the new catalysts toward ring-closing metathesis (RCM) reaction has been examined in which various (un)substituted N,N-diallylanilines and stereoselective RCM of different macromolecular dienes were well tolerated using only 0.5–2.0 mol% of the additive catalyst, i.e. 7b, as compared to the well-known Ru-carbenoids, Grubbs (II) and Hoveyda–Grubbs (II) catalysts.
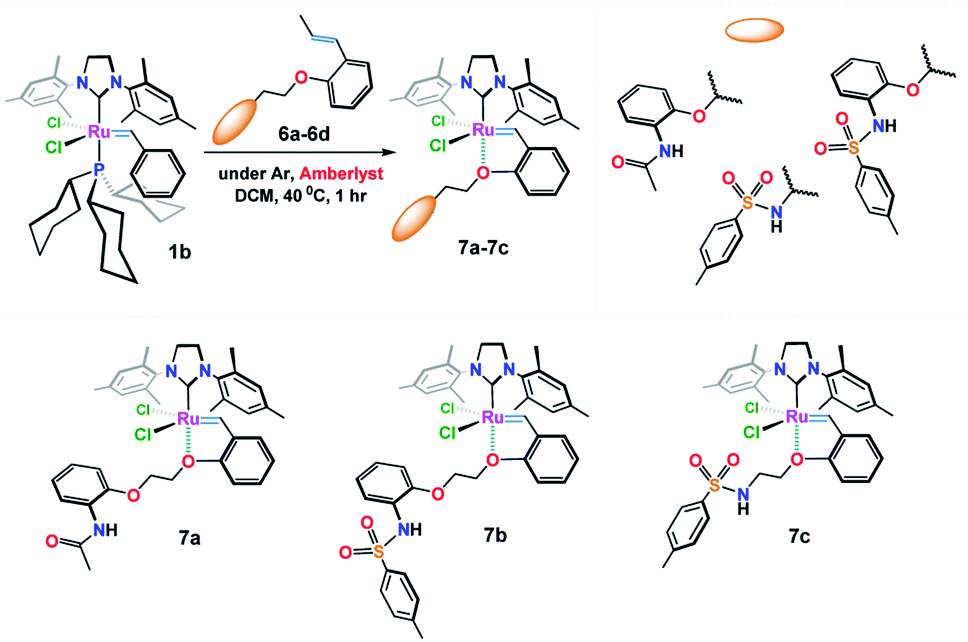 | ||
| Fig. 2 The structure of the novel Ru-alkylidene catalysts 7a–7c. | ||
The target structures (7a–7c) were obtained from the reaction of freshly prepared vinylbenzene substrates (6a–6c) with Grubbs (II) catalyst 1b as shown in Fig. 2. While Ru(II) ions in 7a and 7b complexes are surrounded by 2-(N-acetamide) and 2-(N-4-methylbenzenesulfonamide) based phenoxy substituents, catalyst 7c composes of 4-methylbenzenesulfonamide moiety around the Ru(II) central core.
Result and discussion
Synthesis of Ru-based catalysts 7a–7c
Scheme 1 illustrates the synthetic procedure for the synthesis of the Ru-alkylidenes 7a–7c starting from the appropriate vinylbenzene derivatives 6a–c. Compound 5 was prepared following the synthetic procedure described in literature.14 Substitution reaction of compound 5 with 2-acetamidophenol and p-toluenesulfonamide in the presence of potassium carbonate in refluxing acetonitrile, resulted in the formation of vinylbenzenes 6a, and 6c, respectively, whereas vinyl derivative 6b was prepared upon deprotecting the amino acetyl group of 6a in acidic media, followed by treating the resulted compound with tosyl chloride in presence of pyridine. The complementary 1-propenyls 6a–6c were then reacted with Grubbs (II) catalyst 1b in the presence of dry Amberlyst-15 hydrogen form in refluxing dichloromethane to afford Ru(II)-complexes 7a–7c in good yields (ESI†).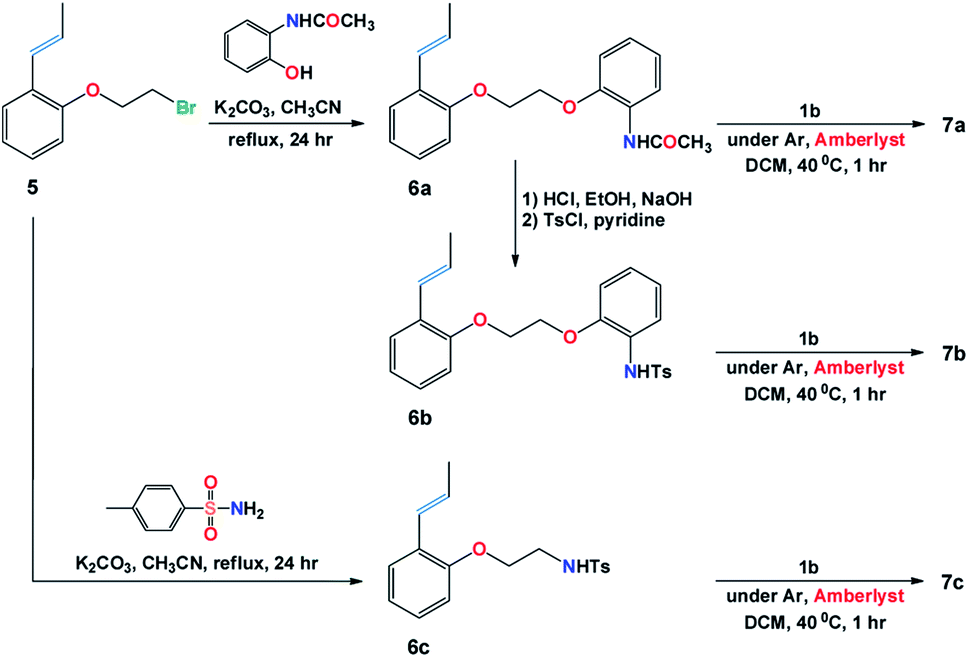 | ||
| Scheme 1 Synthesis of Ru(II)-alkylidene complexes 7a–7c. | ||
Single crystal X-ray diffraction study of the complexes 7a–7c
Single crystals of 7a–7c structures were successfully grown by solvent diffusion method and their corresponding structural details were investigated by X-ray diffraction analysis (Fig. 3).15 Strong non-bonding interactions among the adjacent atoms in these complexes making them pack efficiently to form stable crystal networks (Fig. 4). These novel structures consistent of a coordination number 5 around the Ru(II) center. The 5-coordination geometry is providing a distorted trigonal bipyramid shape to all complexes at the Ru(II) site. However, for both 7a and 7b, there is an oxygen atom available in the flexible spacer fragment of the structure which are positioned at a suitable distance (about 3.5 A) to engage a coordination bond with the Ru(II) metal ion through its lone pair of electrons. As a result, a distorted octahedral geometry could also be possible with hexa-coordinated Ru(II) ion for 7a and 7b (ESI†). Similarly, an additional coordination is possible through the nitrogen atom in complex 7c, though, the orientation of its N–H bond might inhibit the possibility of such a N–Ru coordination.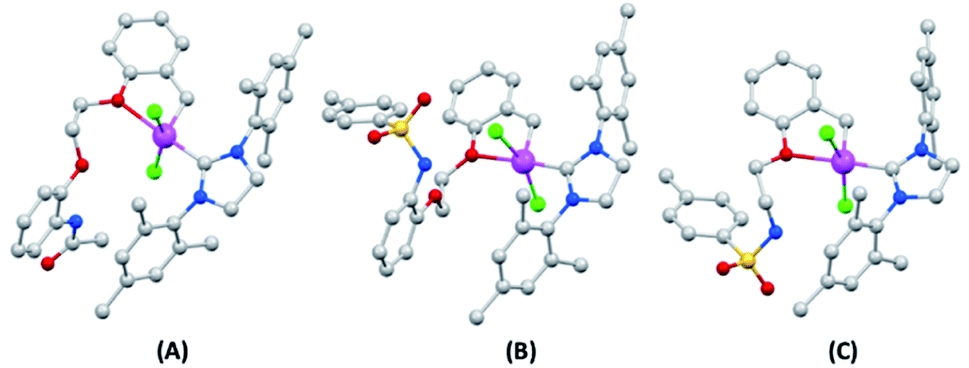 | ||
| Fig. 3 Crystal structure of (A) 7a; (B) 7b and (C) 7c; color code gray-carbon; red-oxygen; blue-nitrogen; green-chlorine; yellow-sulfur; pink-ruthenium. Hydrogen atoms are hided for clarity. | ||
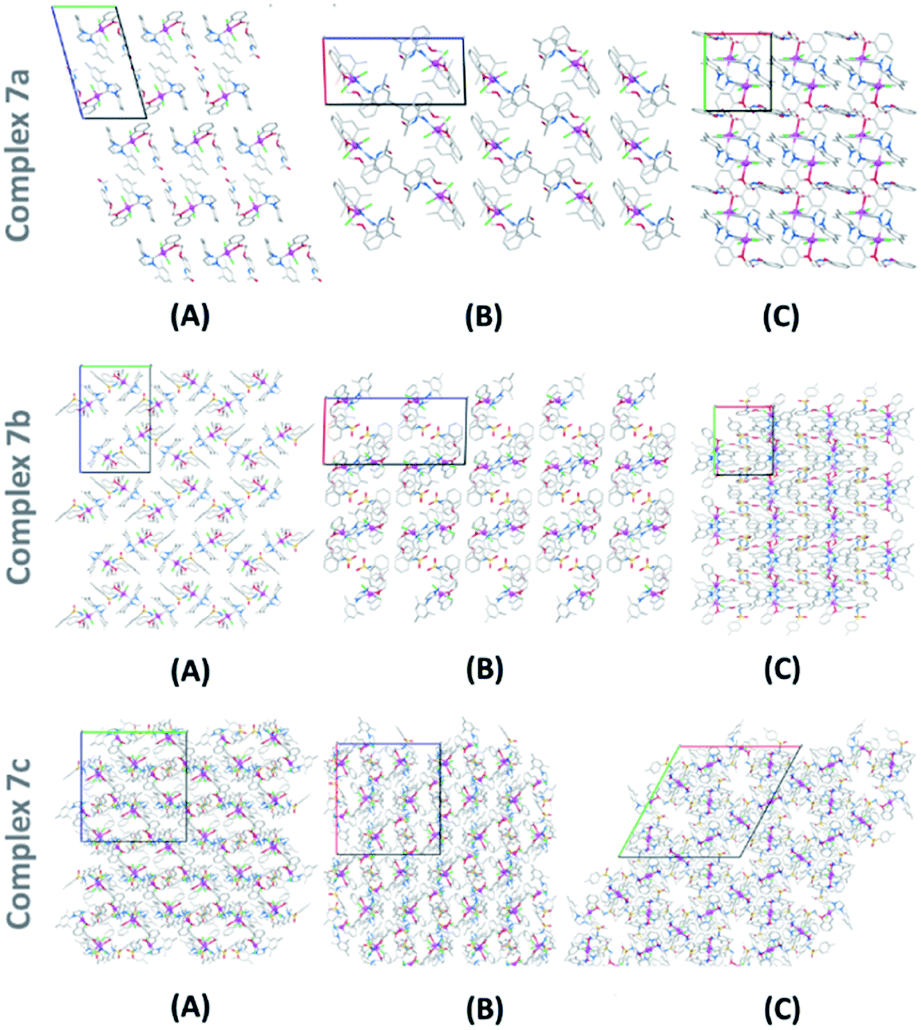 | ||
| Fig. 4 Packing pattern of complexes 7a–7c in their crystal network; view along the (A) a-direction; (B) b-direction and (C) c-direction; colour code: red-oxygen; blue-nitrogen; grey-carbon; and pink-zinc (hydrogens has been hided for clarity). | ||
Optimizing ring-closing metathesis catalysed by Ru(II)-complexes (7a–7c), Grubbs (1b) and Hoveyda–Grubbs (2b) catalysts
The ring-closing metathesis (RCM) was investigated to evaluate the catalytic activity of the novel complexes (7a–7c) as compared to both Grubbs 1b and Hoveyda–Grubbs 2b catalysts. For optimization, RCM reactions of N,N-diallyl-4-methylbenzenesulfonamide 8a (ref. 9) (0.1 mmol) with the additive catalysts (0.5 mol%) were carried out only in 10 min in 1 mL dichloromethane at 25 °C (Fig. 5).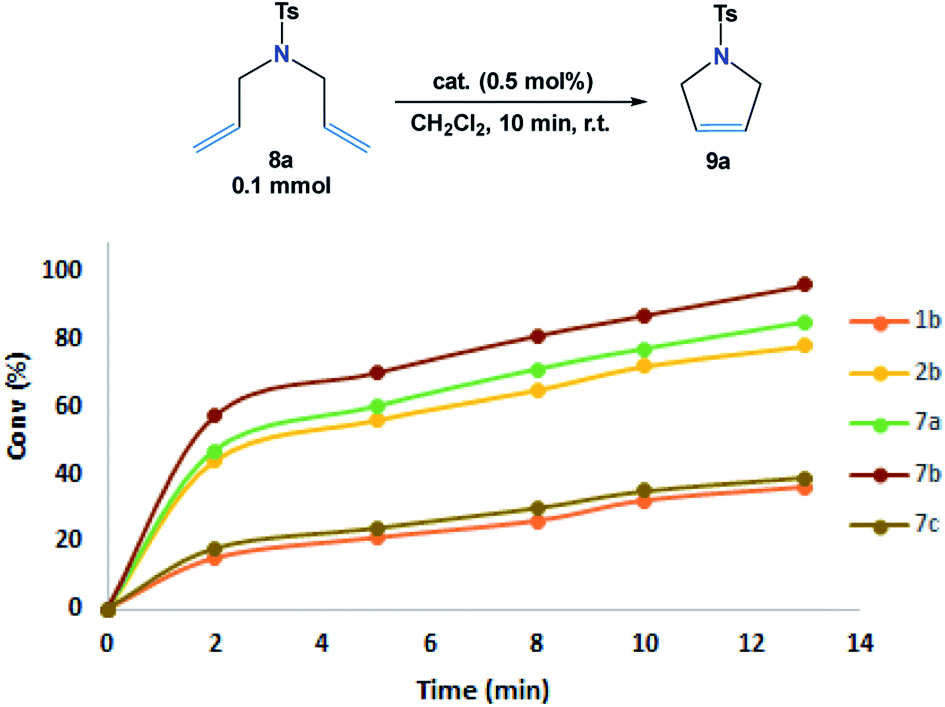 | ||
| Fig. 5 Conversion (%) of cyclic alkene 8a in 10 min at 25 °C with the additive catalysts (0.5 mol%). Determined by 1H NMR. | ||
From the results shown in Fig. 5, it was observed that the RCM reactions of the studied diene 8a were poorly progressed in the presence of 7c and Grubbs 1b catalysts (<40% conv.), whereas, the cyclization of 8a was enhanced to 78% conv. when Hoveyda–Grubbs 2b was subjected. Remarkably, in the reactions catalyzed by 7a and 7b, conversion yields of 84% and 96% were obtained. Such results can be possibly explained from the crystal structure demonstrated in Fig. 3. The crystal structures of 7a and 7b shows an open area in between the dichloro ruthenium fraction and two parallelly oriented flexible aromatic substituents. These aromatic substituents could provide a delocalized electronic flux in this open area to accommodate the approaching diene guest during the catalytic reaction (Fig. 6).
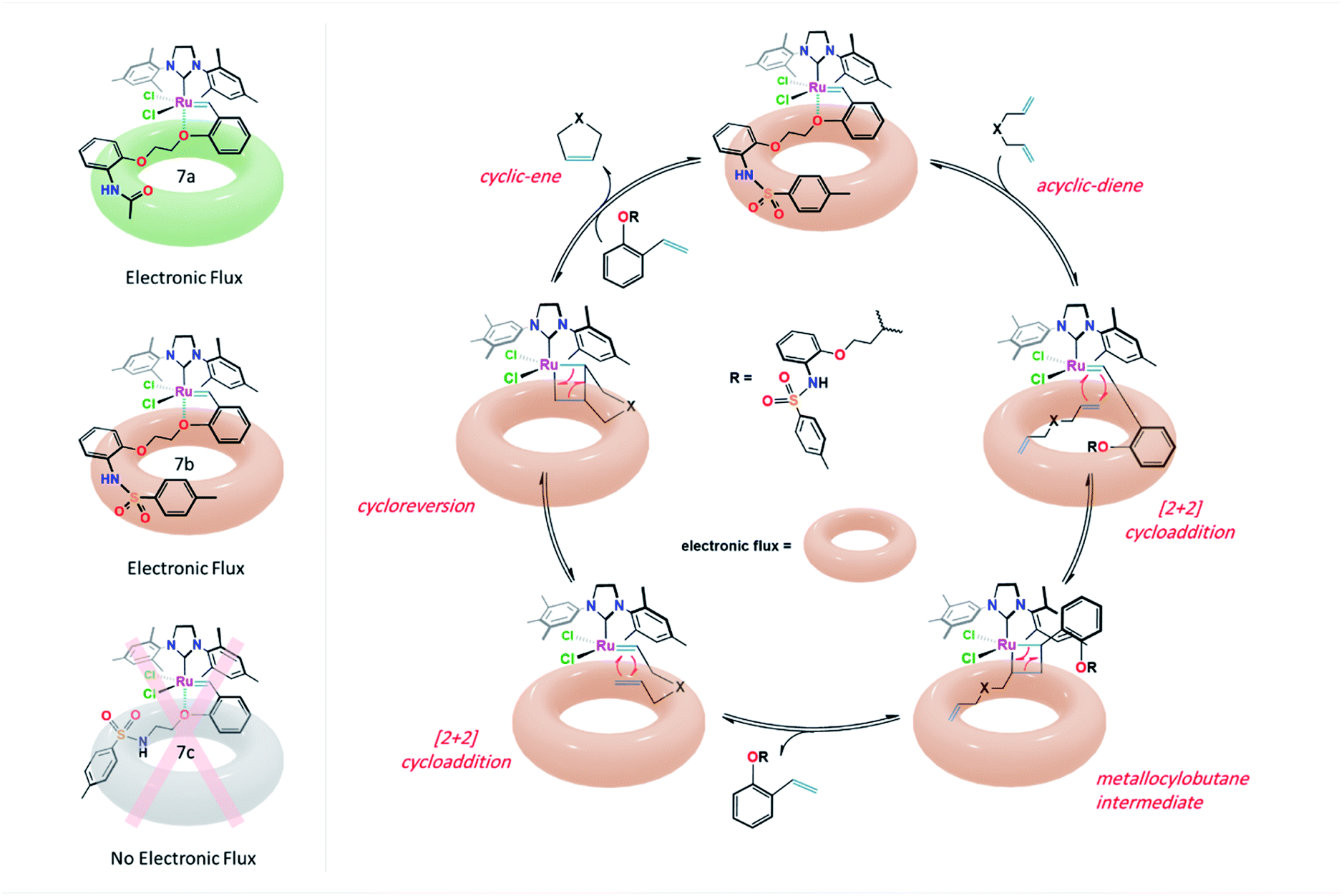 | ||
| Fig. 6 Electronic flux in complexes 7a & 7b and proposed ring-closing metathesis (RCM) reaction mechanism catalyzed by catalyst 7b. | ||
Moreover, 7b complex, amongst the catalysts, was used for further investigations. The catalytic efficiency along with the initiation rate of 7b as compared to Hoveyda–Grubbs 2b toward the olefination reaction of diene 8b was determined by 1H NMR, by monitoring the allyloxy –OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2– protons (H3) before and after the pyrrole 9a formation, at different reaction time, of 2, 4, 6, 8 and 10 min (Fig. 7). Both reactions were performed in a 600 MHz NMR instrument under the following reaction condition; diene 8a (0.1 mmol) with the additive catalysts (0.5 mol%) in 1 mL CD2Cl2 at 25 °C. Notably, it was determined from the time profile introduced in Fig. 7A that the cyclization process of 8a was effectively proceeded by 7b catalyst in which ∼80% conversion was obtained only in 2 min as compared to <80% conversion achieved in 10 min when Hoveyda–Grubbs 1b was subjected (Fig. 7B).
CH2– protons (H3) before and after the pyrrole 9a formation, at different reaction time, of 2, 4, 6, 8 and 10 min (Fig. 7). Both reactions were performed in a 600 MHz NMR instrument under the following reaction condition; diene 8a (0.1 mmol) with the additive catalysts (0.5 mol%) in 1 mL CD2Cl2 at 25 °C. Notably, it was determined from the time profile introduced in Fig. 7A that the cyclization process of 8a was effectively proceeded by 7b catalyst in which ∼80% conversion was obtained only in 2 min as compared to <80% conversion achieved in 10 min when Hoveyda–Grubbs 1b was subjected (Fig. 7B).
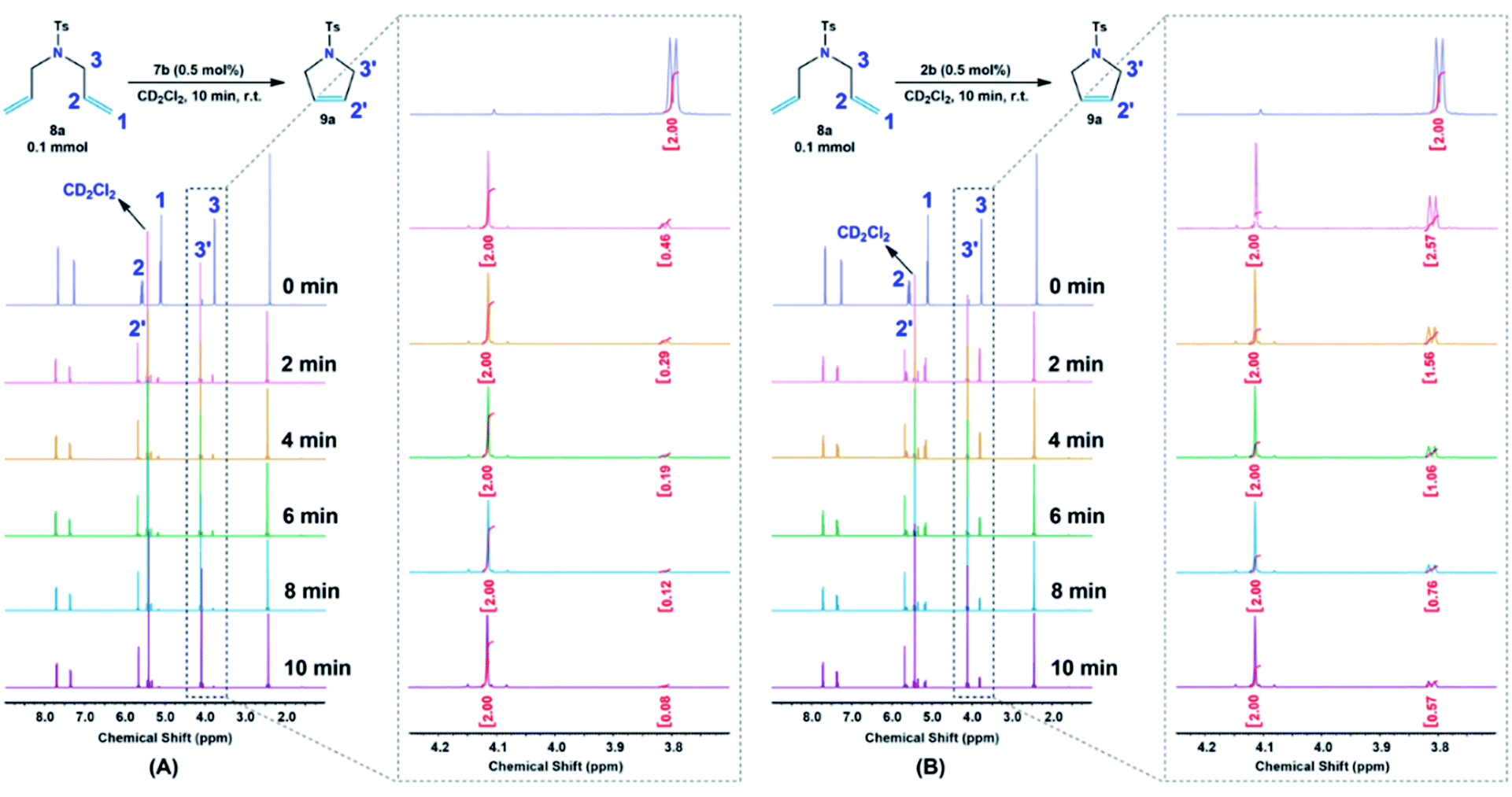 | ||
| Fig. 7 Time profile of the RCM reaction of diene 8a catalyzed by (A) 7b and (B) 2b as monitored by 1H NMR at different reaction time. | ||
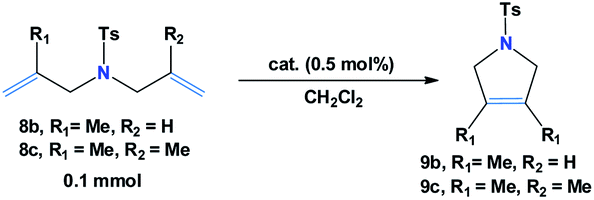
Next, the capability of catalyst 7b in catalyzing acyclic diene moieties was examined toward N-allyl-4-methyl-N-(2-methylallyl) 8b and 4-methyl-N,N-bis(2-methylallyl) 8c benzenesulfonamides and results are tubulated in Table 2. These results indicate that the reaction of diene 8b in dichloromethane was well tolerated in the presence of catalyst 7b (0.5 mol%) in which a stoichiometric quantity of 9b was obtained in 30 min at 25 °C (Table 2, entry 1). However, no reaction took place in case of diene 8c under similar reaction condition (Table 2, entry 2). Likewise, an insignificant improvement in pyrrole 9c formation was observed when the RCM reaction was performed at a higher reaction temperature (40 °C, 14% yield), though, elevating the temperature to 80 °C by replacing dichloromethane with toluene as a solvent media, resulted in the desired pyrrole product 9c in good yield (70%) within 24 h.
Synthesis of (un)substituted N,N-diallyl aniline derivatives (8d–8s)
To further scope the ability of 7b in catalyzing RCM reactions, a series of (un)substituted N,N-diallylaniline derivatives (8d–8s) have been prepared. All allyl containing anilines were attained in good to high yields from the substitution reaction of (un)substituted aniline substrates (1.0 mmol) with allyl bromide (3.0 mmol and additional 1.0 mmol was added every 12 h) in the presence of potassium carbonate. All reactions were carried out in refluxing acetonitrile for 2 days and results are presented in Table 3. The structure of all novel dienes (8f, 8i–8m, 8p–8r) were confirmed from their respective NMR and HRMS spectral data (ESI†).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | R3 | R4 | Yieldb (%) |
| a Reaction condition: aniline substrates (1.0 mmol), allyl bromide (3.0 mmol), potassium carbonate (4.0 mmol), acetonitrile (20 mL), reflux, 2 days, an additional allyl bromide (1.0 mmol) was added to the reaction mixture every 12 h.b Isolated yield by chromatography. | ||||||
| 1 | 8d | H | H | H | H | 70 |
| 2 | 8e | OMe | H | H | H | 50 |
| 3 | 8f | H | OMe | H | H | 64 |
| 4 | 8g | H | H | OMe | H | 44 |
| 5 | 8h | H | H | Me | H | 86 |
| 6 | 8i | Me | Me | H | H | 50 |
| 7 | 8j | H | Me | H | Me | 65 |
| 8 | 8k | F | H | H | H | 45 |
| 9 | 8l | H | Br | H | H | 55 |
| 10 | 8m | Cl | H | H | H | 63 |
| 11 | 8n | H | H | Cl | H | 40 |
| 12 | 8o | F | H | F | H | 63 |
| 13 | 8p | H | F | F | H | 40 |
| 14 | 8q | F | H | Cl | H | 46 |
| 15 | 8r | Me | Cl | H | H | 54 |
| 16 | 8s | H | H | NO2 | H | 40 |
Scoping the RCM reaction of (un)substituted N,N-diallyl-anilines (8d–8s) using catalyst 7b
Next, we explored the RCM reaction of the obtained dienes (8d–8s) catalyzed by complex 7b. It was found that the reactions were not competent using 0.5 mol% of additive catalyst in dichloromethane at 25 °C. However, better results were efficiently obtained by increasing the catalytic amount of 7b to 1.0 mol% in refluxing dichloromethane and results are presented in Table 4. The structures of all new dienes (8i, 8k, 8o & 8q) were confirmed using NMR and HRMS spectroscopic techniques (ESI†).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
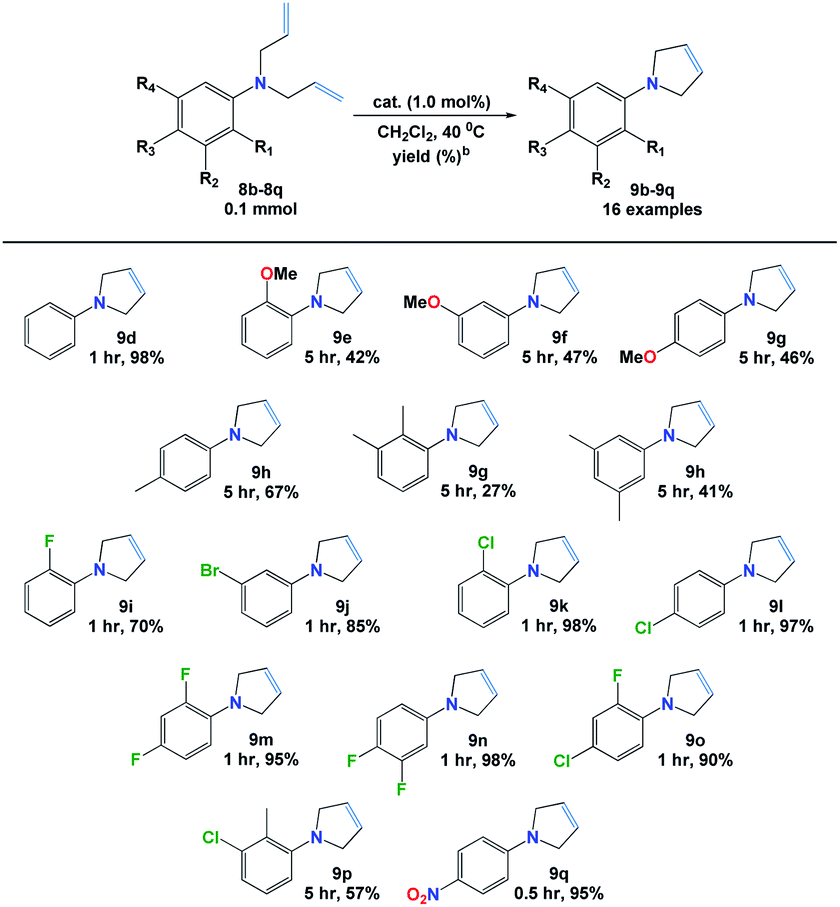
From the results, it was observed that the reaction of diene 8d was efficiently proceeded in only 1 h to give pyrrole 9d in excellent yield (98%). However, lower yields (27–67%) along with longer reactions time, i.e. 5 h, were attained when electron-donor groups containing dienes (8e–8h) were used. Further, the cyclization process occurred with ease (>90% yields) in 1 h when halogenated dienes (8j–8o) were applied with an exception of 2-fluoro N,N-diallylaniline (8i) which was gained in 70% yield. Similarly, the strongly electron withdrawing group (–NO2) presented in 8q was found to be the most reactive diene toward RCM in which 95% of pyrrole 9q was achieved only in 30 min. Still, the presence of both electron donor (–Me) and electron with drawing (–Cl) groups, on 8p core, resulted in 9p in 57% in 5 h. All novel pyrroles (9f, 9i, 9j, 9l, 9m, 9o, 9q & 9r) were fully utilized from their respective NMR and HRMS spectral data (ESI†).
Synthesis of unsaturated macrocyclic structures via RCM reaction using catalyst 7b
RCM reaction has been applied as a key step synthetic process for macrocyclization reactions.16 Hence, from our published data based on the construction of macrocycles 11a–11c catalyzed by Grubbs (I) 1a, complex 7b has been used for comparison (Scheme 2).17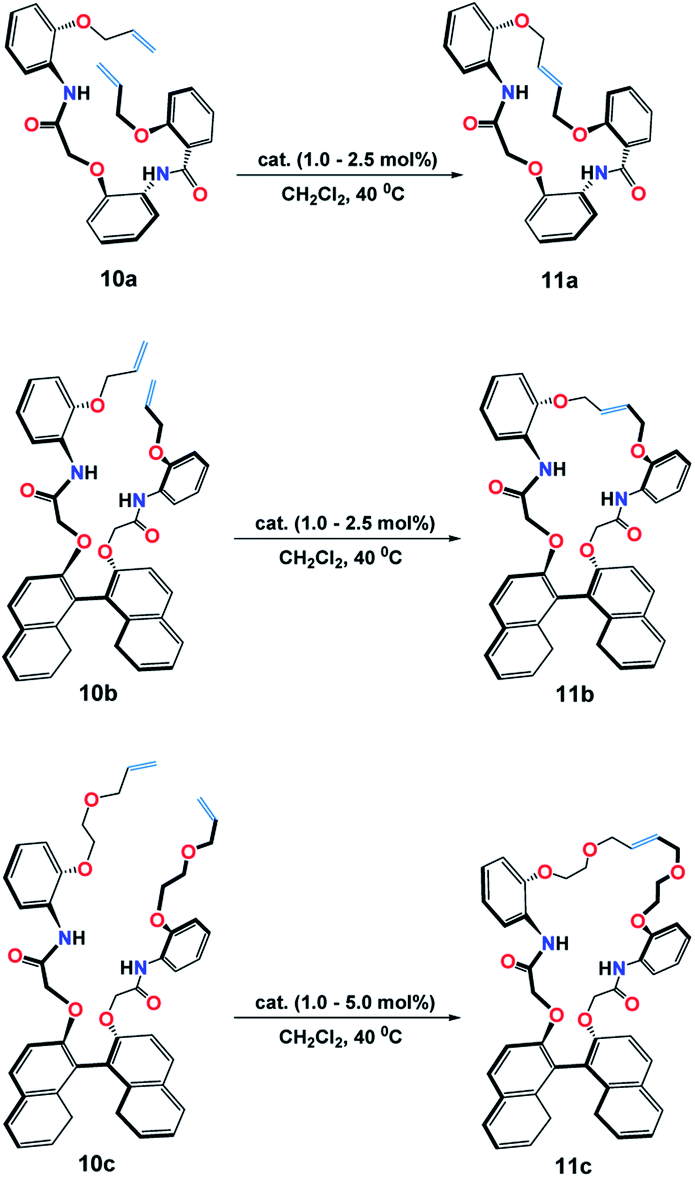 | ||
| Scheme 2 RCM of dienes 10a–10c catalysed by complex 7b and Grubbs (I) 1a in the synthesis of macrocycles 11a–11c. | ||
Following the typical procedure reported: macrocyclic dienes (0.2 mmol), DCM (25 mL), 40 °C in 24 h (Table 5), it was found that the macrocyclization of diene 10a catalyzed by complex 7b (1.0 mol%) in the formation of crown 11a was achieved in 98% yield as compared to 92% yield when Grubbs 1a (2.5 mol%) was used (Table 5, entries 1 & 2), whereas, 1.0 mol% of catalyst 7b was capable to catalyze the RCM reaction of dienes 10b and 10c to yield macrocycles 11b and 11c in 98%.and 70%, respectively (Table 5, entries 3–6).
a
| Entry | Product | Cat./(mol%) | Yieldb (%) | Product E:Z ratiob |
|---|---|---|---|---|
| a Reaction condition: acyclic macrodienes (0.2 mmol), DCM (25 mL), 40 °C.b Determined by 1H NMR. | ||||
| 1 | 11a | 1a, 2.5 mol% | 92 | 78:22 |
| 2 | 11a | 7b, 1.0 mol% | 98 | 100:0 |
| 3 | 11b | 1a, 2.5 mol% | 91 | 75:25 |
| 4 | 11b | 7b, 1.0 mol% | 98 | 100:0 |
| 5 | 11c | 1a, 5.0 mol% | 97 | 75:25 |
| 6 | 11c | 7b, 1.0 mol% | 70 | 92:8 |
Interestingly, a control of the stereoselectivity, that Grubbs (I) lacks, was feasibly obtained when catalyst 7b was applied in which both 10a and 10b products were attained in their E-configurations, while macrocyclic crown 10c was afforded, mainly, in its E-configuration, i.e. E (92%):Z (8%). These results were determined upon comparing the 1H NMR spectra (400 MHz, CDCl3, at 25 °C) of the macrocyclic crowns (11a–11c) and their corresponding macrodienes (10a–10c) before and after the RCM reactions (Fig. 8).
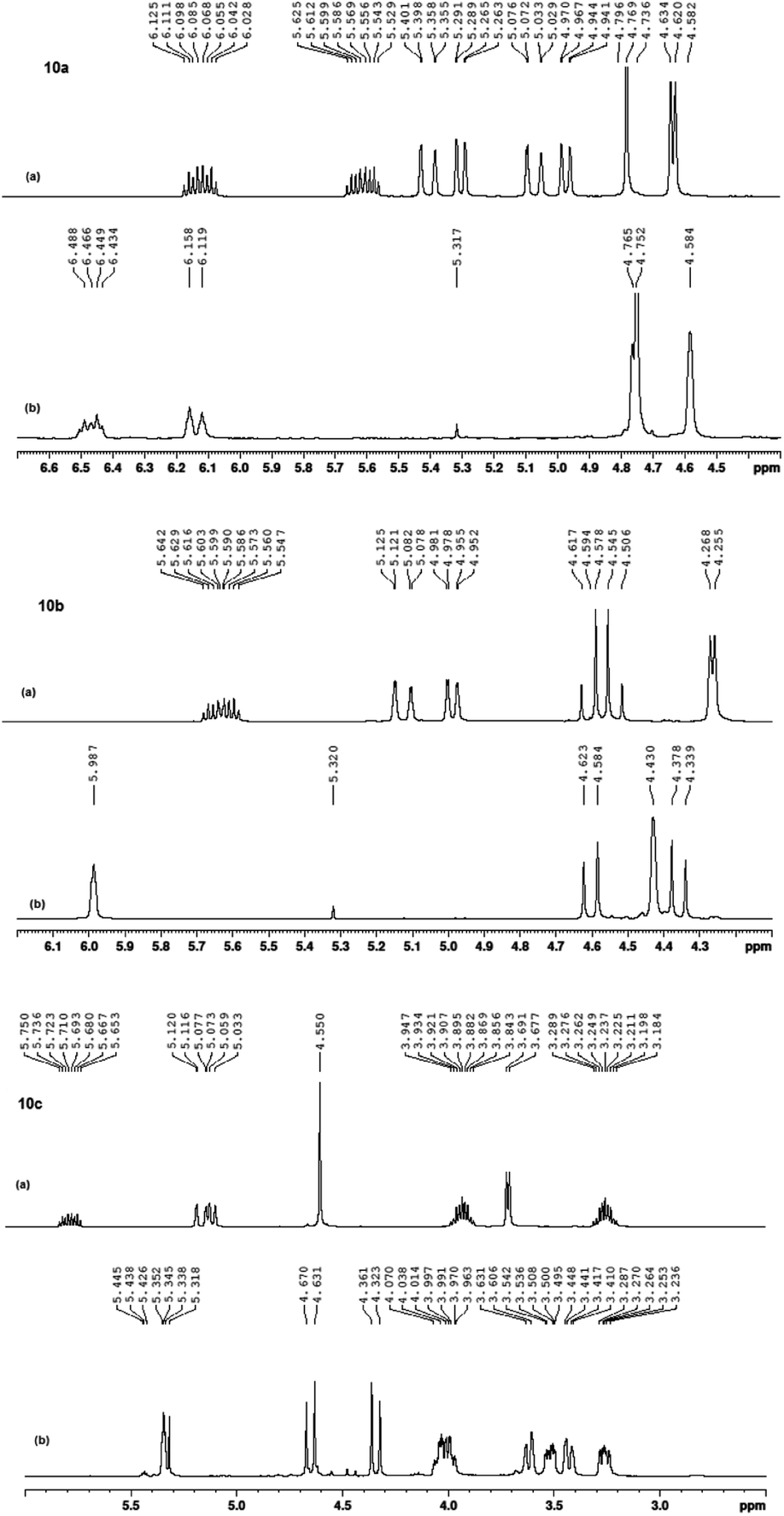 | ||
| Fig. 8 1H NMR (400 MHz, 600 MHz, CDCl3) spectra of 10a, 10b and 10c (a) before RCM and (b) after RCM. | ||
The disappearance of the terminal allyloxy protons (–OCH2CHCH2, H1 in 10a–10c & H1/H7 in 10a), i.e. the disappearance of: [H1 in 10a, Fig. 8A; two sets of doublet of a doublet at 4.94–4.97 ppm (dd, J = 1.3, 10.5 Hz, 1H) and at 5.27–529 ppm (dd, J = 1.3, 10.5 Hz, 1H), H7 in 10a, Fig. 8A; two sets of doublet of a doublet at 5.03–5.07 ppm (dd, J = 1.5, 17.3 Hz, 1H) and at 5.36–5.40 ppm (dd, J = 1.3, 17.3 Hz, 1H); H1 in 10b, Fig. 8C; two sets of doublet of a doublet at 4.96–4.98 ppm (dd, J = 1.3, 10.5 Hz, 2H) and at 5.08–5.13 ppm (dd, J = 1.3, 17.3 Hz, 2H); H1 in 10c, Fig. 8E; two sets of doublet of a doublet at 5.03–5.06 ppm (dd, J = 0.8, 10.5 Hz, 2H) and at 5.08–5.12 ppm (dd, J = 1.5, 17.3 Hz, 2H)] and the shift of the internal allyl protons (–OCH2CHCHCH2O–, H2 in 11a–11c & H6 in 11c), i.e. [H2 in 11a, Fig. 8B; two sets of triplets at 6.12 ppm (t, J = 4.5 Hz, 1H) and at 6.16 ppm (t, J = 4.5 Hz, 1H), H6 in 11a, Fig. 8B; two sets of triplets at 6.45 ppm (t, J = 6.5 Hz, 1H) and at 6.49 ppm (t, J = 4.5 Hz, 1H), H2 in 11b, Fig. 8D; a triplet at 5.99 ppm (t, J = 3.0 Hz, 2H), H2 in 11c, Fig. 8F; a triplet at 5.35 ppm for the E-configuration (t, J = 3.0 Hz, 2H, 92%) and a triplet at 5.44 ppm for the Z-configuration (t, J = 3.0 Hz, 2H, 8%)] were used to confirm the formation of the products (Fig. 9).
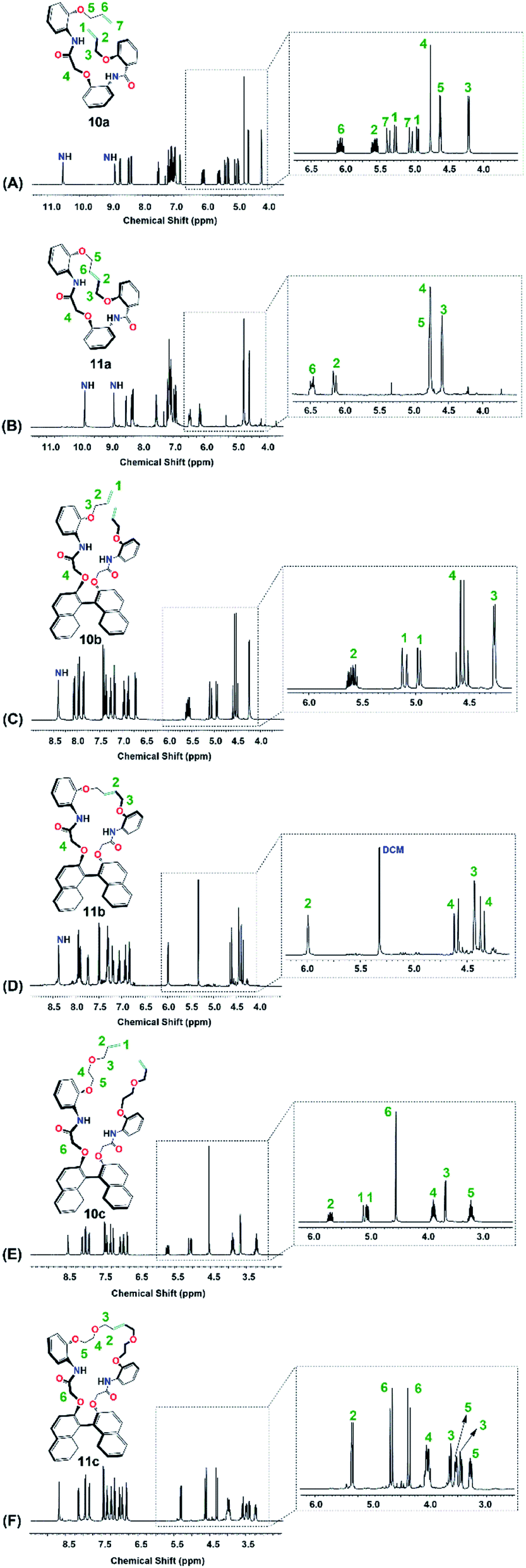 | ||
| Fig. 9 Full 1H NMR of (A) 10a; (B) 11a; (C) 10b; (D) 11b; (E) 10c and (F) 11c. | ||
Conclusion
In conclusion, three catalytic systems (7a–7c) based on Hoveyda–Grubbs complex have been successfully prepared and characterized from their respective NMR and HRMS spectral data and X-ray single structure. The synthesized Ru(II) complexes have been established as efficient catalysts for ring-closing metathesis (RCM). Complex 7b, among the other catalytic structures prepared, was found to have the highest efficiency in catalyzing different (un)substituted N,N-diallylanilines for 1H-pyrroles formation. Also, a control of the stereoselectivity, i.e. E-configuration, of macrocyclic crowns have been obtained using complex 7b.Experimental
General
All reactions were carried out under nitrogen atmosphere unless otherwise noted, all analyses determined in Research Sector Projects Unit (RSPU) at Kuwait, TLC was performed using Polygram sil G/UV 254 TLC plates and visualization was carried out by ultraviolet lights at 254 nm and 350 nm. Column chromatography was performed using Merck silica gel 60 of mesh size 0.040–0.063 mm. 1H and 13C NMR spectra were recorded using Bruker NEO 400 MHz, Bruker Avance II 600 MHz superconducting NMR spectrometers. Mass spectra were recorded with a GCMS-DFS-Thermo, High Resolution spectrometer and Bruker, ultraflextreme mass spectrometer MALDI-ToF MS with Smartbeam-II laser. The single crystal X-ray analysis was carried out on Rigaku Rapid II and Bruker X8 prospector diffractometers respectively. Melting points were determined via differential scanning calorimetry (DSC) analyses on Shimadzu DSC-50.1-(2-Bromoethoxy)-2-propenylbenzene (5)
A catalytic amount of palladium chloride (0.02 g, 0.2 mol%) was added to 50 mL DCM and 30 drops of acetonitrile. The mixture was stirred for 5 min at room temperature followed by the addition of 1-allyl-2-(2-bromoethoxy)-benzene (14 g, 58 mmol) allowing the reaction mixture to stir for 40 h at room temperature. The mixture was washed with water, extracted with DCM and passed through a short column using hexane as an eluent to give compound 5 as colorless oil in 12.6 g (90% yield); 1H NMR (400 MHz, CDCl3) δ = 1.90 (d, J = 6.5 Hz, 3H), 3.68 (t, J = 6.3 Hz, 2H), 4.30 (t, J = 6.3 Hz, 2H), 6.21–6.30 (m, 1H), 6.74 (d, J = 16.0 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.94 (t, J = 7.4 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 19.2, 29.5, 68.6, 112.8, 121.7, 125.6, 126.8, 127.1, 127.9, 127.91, 154.9; m/z (EI) 240 (M+, 75%), 242 (M+2, 74%); m/z (EI) 240.0145 (M+, C11H1379BrO requires 240.0144).14N-{2-[2-(2-Propenyl-phenoxy)-ethoxy]-phenyl}-acetamide (6a)
A mixture of compound 5 (6.0 g, 2.5 mmol), o-acetamidophenol (4.9 g, 3.2 mmol) and anhydrous potassium carbonate (4.3 g, 3.1 mmol) in dry acetonitrile (50 mL) was heated under reflux overnight. The residue was filtered off and the solvent was evaporated. The remaining crude product was recrystallized from ethanol to give compound 6a as colorless crystals in 6.6 g (85% yield); mp 143–144 °C; 1H NMR (400 MHz, DMSO-d6) δ = 1.79 (dd, J = 6.6 Hz, 3H), 2.03 (s, 3H), 4.38–4.43 (m, 4H), 6.21–6.31 (m, 1H), 6.62 (dd, J = 1.2, 14.5 Hz, 1H), 6.90–6.95 (m, 2H), 7.04–7.07 (m, 2H), 7.15–7.22 (m, 2H), 7.43 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 7.6 Hz, 1H), 8.90 (bs, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 18.7, 23.9, 66.9, 67.7, 112.7, 113.2, 120.8, 120.9, 121.9, 124.1, 125.3, 125.9, 126.1, 126.3, 127.95, 128.0, 148.6, 155.0, 168.3; m/z (EI) 311 (M+); m/z (EI) 311.1515 (M+, C19H21NO3 requires 311.1516).4-Methyl-N-{2-[2-(2-propenyl-phenoxy)-ethoxy]-phenyl}-benzenesulfonamide (6b)
A mixture of compound 6a (4 g, 1.3 mmol), and conc. HCl (4 mL) in ethanol (40 mL) was heated under reflux for 2 h. Ethanol was evaporated, ether was added to this mixture and the precipitated solid material was collected and crystallized from ethanol. The obtained phenylamine salt was stirred in NaOH solution for 10 min which was then extracted with DCM and dried over anhydrous sodium sulfate and evaporated. The obtained colorless solid, i.e. 2-[2-(2-propenylphenoxy)-ethoxy]-phenylamine (0.28 g, 1 mmol), was dissolved in dry pyridine (1.8 mL) followed by the addition of p-toluenesulfonyl chloride (0.25 g, 1.2 mmol). The mixture was stirred at 0 °C for 1 h and then kept overnight in the fridge. The precipitate obtained after addition of cold water was collected and recrystallized from ethanol to give compound 6b as colorless crystals in 0.31 g (70% yield); mp 136–137 °C; 1H NMR (400 MHz, CDCl3) δ = 1.89 (dd, J = 6.8, 1.6 Hz, 3H), 2.29 (s, 3H), 4.20 (d, J = 3.4 Hz, 4H), 6.21–6.30 (m, 1H), 6.67 (d, J = 16.0 Hz, 1H), 6.86 (dd, J = 8.0, 2.4 Hz, 2H), 6.94 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 7.05–7.11 (m, 4H), 7.23 (t, J = 7.2 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ = 19.1, 21.6, 66.9, 67.5, 112.1, 112.6, 121.8, 121.9, 125.49, 125.50, 126.6, 126.9, 127.1, 127.4, 127.8, 128.0, 129.5, 136.5, 143.7, 148.8, 155.2; m/z (EI) 423 (M+); m/z (EI) 423.1499 (M+, C24H25NO4S requires 423.1499).p-Methyl-N-[2-(2-propenyl-phenoxy)-ethyl]-benzenesulphonamide (6c)
A mixture of compound 5 (2.4 g, 1.0 mmol), p-toluenesulphonamide (5.1 g, 3.0 mmol), and anhydrous potassium carbonate (2.0 g, 1.5 mmol) in dry acetonitrile (75 mL) was heated under reflux overnight. The residue was filtered off and the solvent was evaporated in vacuo. The remaining product was purified by column chromatography using eluent pet. ether/EtOAc in the ratio of 2:1 to give compound 6c as colorless crystals in 0.83 g (25% yield); mp 102–103 °C; 1H NMR (400 MHz, CDCl3) δ = 1.92 (dd, J = 5.4, 1.2 Hz, 3H), 2.43 (s, 3H), 3.40 (m, 2H), 4.00 (t, J = 5.1 Hz, 2H), 4.94 (bs, 1H), 6.19–6.22 (m, 1H), 6.59 (dd, J = 15.6, 1.8 Hz, 1H), 6.70 (d, J = 8.4 Hz, 1H), 6.94 (t, J = 7.8 Hz, 1H), 7.13 (dt, J = 1.2, 7.2 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.39 (dd, J = 7.8, 1.2 Hz, 1H), 7.77 (d, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ = 19.2, 21.7, 42.9, 67.0, 112.5, 121.8, 125.4, 126.8, 127.18, 127.24, 127.5, 128.0, 130.0, 137.0, 143.8, 154.8; m/z (EI) 331 (M+); m/z (EI) 331.1238 (M+, C18H21NO3S requires 331.1237).
General procedure for the synthesis of the novel Ru(II)-catalysts (7a–c)
To a flame-dried Schlenk flask equipped with a Teflon-coated magnetic stir bar under argon, was charged with each of compounds 6a–c (0.03 mmol), dry Amberlyst-15 hydrogen form (0.025 g), 1b (0.035 mmol) in 2 mL DCM. The resulting mixture was stirred at 40 °C for 3–4 h till the reaction color changes from maroon to brown or green. The reaction vessel was cooled to room temperature, and the reaction mixture was filtered through a pad of cotton in a glass pipette to remove the Amberlyst-15 resin. The filtrate was then concentrated in vacuo and the crude product was precipitated by dilution with cold petroleum ether (40–60 mL) which then dried under high vacuum to obtain the desired product.Ru(II) complex 7a
Green solid in 0.021 g, (90% yield); mp 105–106 °C; 1H NMR (600 MHz, CD2Cl2) δ = 1.90 (s, 3H), 2.22 (s, 6H), 2.31 (s, 12H), 4.04 (s, 4H), 4.13 (t, J = 4.2 Hz, 2H), 4.35 (t, J = 4.2 Hz, 2H), 6.73 (dd, J = 8.4, 1.2 Hz, 1H), 6.83–6.85 (m, 5H), 6.91–6.96 (m, 3H), 7.00 (dt, J = 7.8, 1.8 Hz, 1H), 7.51 (dt, J = 8.4, 2.4 Hz, 1H), 8.23 (dd, J = 8.4, 1.8 Hz, 1H), 8.31 (s, 1H), 16.60 (s, 1H); 13C NMR (150 MHz, CD2Cl2) δ = 19.0, 20.8, 24.4, 51.8, 65.7, 68.5, 112.3, 112.8, 120.6, 121.4, 122.1, 123.3, 123.8, 129.2, 129.4, 129.8, 138.6, 138.9, 144.6, 147.0, 153.1, 168.8, 209.2, 294.2; m/z (EI) 761 (M+, 0.2%), 763 (M+2, 0.17%); m/z (EI) 761.1719 (M+, C38H4335Cl2N3O3102Ru requires 761.1719).Ru(II) complex 7b
Green crystals in 0.019 g (96% yield); mp 206–207 °C; 1H NMR (400 MHz, CD2Cl2) δ = 2.30 (s, 6H), 2.39 (s, 3H), 2.42 (s, 12H), 3.97 (t, J = 4.4 Hz, 2H), 4.05 (t, J = 4.4 Hz, 2H), 4.16 (s, 4H), 6.67 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 8.4 Hz, 2H), 6.92 (s, 2H), 7.05–7.10 (m, 6H), 7.19 (t, J = 6.8 Hz, 1H), 7.51 (d, J = 7.6 Hz, 2H), 7.59 (d, J = 8.0 Hz, 1H), 7.70 (t, J = 8.4 Hz, 1H), 7.90 (s, 1H), 16.69 (s, 1H); 13C NMR (150 MHz, CD2Cl2) δ = 19.6, 21.4, 21.8, 52.3, 65.0, 68.5, 112.2, 112.9, 121.8, 122.9, 124.5, 124.7, 126.4, 126.9, 127.8, 129.6, 130.0, 130.3, 137.9, 139.2, 139.4, 143.7, 145.0, 150.0, 153.5, 209.5, 294.9; m/z (MALDI-MS) 874.958 (M + 1, C43H4735Cl2N3O4102RuS requires 873.1702).Ru(II) complex 7c
Green crystals in 0.022 g (95% yield); mp 214–215 °C; 1H NMR (600 MHz, CD2Cl2) δ = 2.40 (s, 6H), 2.43 (s, 15H), 3.09 (m, 2H), 3.95 (t, J = 4.5 Hz, 2H), 4.21 (s, 4H), 6.42 (d, J = 8.4 Hz, 1H), 6.44 (t, J = 6.0 Hz, 1H), 6.99 (m, 2H), 7.05 (s, 4H), 7.25 (d, J = 8.4 Hz, 2H), 7.84 (m, 1H), 7.69 (d, J = 8.4 Hz, 2H), 16.55 (s, 1H); 13C NMR (150 MHz, CD2Cl2) δ = 19.6, 21.4, 21.8, 41.2, 52.2, 69.1, 113.1, 112.6, 124.3, 127.1, 130.0, 130.26, 130.3, 139.4, 139.8, 140.1, 143.6, 145.2, 153.4, 209.6, 296.1; m/z (FAB-MS) 781 (M+, C37H4335Cl2N3O3102RuS requires 781.1440).General procedure for the synthesis of N,N-diallylanilines (8d–8s)
A mixture of (un)substituted anilines (10.0 mmol), allyl bromide (30.0 mmol) and anhydrous K2CO3 (40.0 mmol) in acetonitrile (20 mL) was heated under reflux for 48 h. An additional allyl bromide (10.0 mmol) was added to the reaction mixture every 12 h. The solvent was removed in vacuo and the remaining material was purified by chromatography on silica gel using pet. ether/EtOAc (4:1) as eluent to give the desired products as colorless oil. The spectroscopic data for compounds 8d–h, 8j, 8l–n, 8p, 8r and 8s are coherent with those reported in literature.18–24
N,N-Diallyl-2,3-dimethylaniline (8i)
Colorless oil in 0.9 g (44% yield); 1H NMR (400 MHz, CDCl3) δ = 2.30 (s, 3H), 2.31 (s, 3H), 3.59 (d, J = 5.6 Hz, 4H), 5.13 (d, J = 10.0 Hz, 2H), 5.20 (d, J = 17.2 Hz, 2H), 5.79–5.89 (m, 2H), 6.92–6.95 (m, 2H), 7.07 (t, J = 7.6 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ = 14.3, 20.9, 56.3, 117.1, 120.0, 125.2, 132.9, 135.7, 138.0, 150.1; m/z (EI) 201 (M+); m/z (EI) 201.1515 (M+, C14H19N requires 201.1512).N,N-Diallyl-2-fluoroaniline (8k)
Colorless oil in 0.95 g (50% yield); 1H NMR (600 MHz, CDCl3) δ = 3.84 (d, J = 3.8 Hz, 4H), 5.19 (d, J = 6.8 Hz, 2H), 5.23 (d, J = 11.4 Hz, 2H), 5.86–5.93 (m, 2H), 6.88 (s, 1H), 6.97 (s, 1H), 6.97–7.05 (m, 2H); 13C NMR (150 MHz, CDCl3) δ = 54.3, 116.2–116.3, 117.1, 120.6, 121.1–121.0, 124.0, 134.7, 138.4, 154.5–156.1; m/z (EI) 191 (M+); m/z (EI) 191.1102 (M+, C12H14NF requires 191.1105).N,N-Diallyl-2,4-difluoroaniline (8o)
Colorless oil in 0.84 g (40% yield) 1H NMR (400 MHz, CDCl3) δ = 3.76 (d, J = 5.6 Hz, 4H), 5.17–5.24 (m, 4H), 5.81–5.91 (m, 2H), 6.75–6.85 (m, 2H), 6.90–6.96 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 55.0, 104.4–104.9, 110.3–110.6, 117.6, 122.2–122.3, 134.7, 134.9–135.0, 154.6–156.4, 157.1–158.8; m/z (EI) 209 (M+); m/z (EI) 209.1011 (M+, C12H13F2N requires 209.1011).N,N-Diallyl-4-chloro-2-fluoroaniline (8q)
Colorless oil in 1.41 g (63% yield); 1H NMR (400 MHz, CDCl3) δ = 3.79 (d, J = 5.9 Hz, 4H), 5.17–5.23 (m, 4H), 5.80–5.89 (m, 2H), 6.89 (s, 1H), 6.99–7.06 (m, 2H); 13C NMR (150 MHz, CDCl3) δ = 54.6, 117.0, 117.2, 117.7, 121.4, 124.3, 134.5, 154.2, 155.8; m/z (EI) 225 (M+); m/z (EI) 225.0718 (M+, C12H13FClN requires 225.0715).General procedure for the ring-closing metathesis in the synthesis of 1-phenyl-2,5-dihydro-1H-pyrrole derivatives (9d–9s)
Into a solution of each of the dienes 8d–s (0.1 mmol) in CH2Cl2 (1 mL), catalyst 7b (1 mg, 1.0 mol%) was added and the reaction mixture heated at 40 °C and completion of reaction was monitored by TLC. The solvent was removed in vacuo and the remaining material was purified by chromatography on silica gel using pet. ether/EtOAc (4:1) as eluent to give the desired products. The spectroscopic data for compounds 9d, 9e, 9g, 9h, 9k, 9n, 9p and 9s are coherent with those reported in literature.25,26
1-(3-Methoxyphenyl)-2,5-dihydro-1H-pyrrole (9f)
Colorless oil in 0.008 g (47% yield); 1H NMR (400 MHz, CDCl3) δ = 3.84 (s, 3H), 4.17 (s, 4H), 5.96 (s, 2H), 6.24 (s, 1H), 6.30 (d, J = 8.4 Hz, 1H), 6.37 (dd, J = 8.4, 2.0 Hz, 1H), 7.21 (t, J = 8.0H, 1H); 13C NMR (150 MHz, CDCl3) δ = 55.7, 63.2, 107.0, 113.1, 119.6, 130.5, 142.2, 160.7; m/z (EI) 175 (M+); m/z (EI) 175.0993 (M+, C11H13NO requires 175.0992).1-(2,3-Dimethylphenyl)-2,5-dihydro-1H-pyrrole (9i)
Colorless oil in 0.0046 g (27% yield); 1H NMR (400 MHz, CDCl3) δ = 2.31 (s, 3H), 2.35 (s, 3H), 4.13 (s, 4H), 5.96 (s, 2H), 6.89 (d, J = 7.2 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 15.9, 21.0, 58.3, 116.2, 123.7, 125.9, 127.2, 129.9, 138.3, 149.5; m/z (EI) 173 (M+); m/z (EI) 173.1198 (M+, C12H15N requires 173.1199).1-(3,5-Dimethylphenyl)-2,5-dihydro-1H-pyrrole (9j)
Colorless solid in 0.070 g (41% yield); mp 48–49 °C; 1H NMR (400 MHz, CDCl3) δ = 2.42 (s, 6H), 4.20 (s, 4H), 6.04 (s, 2H), 6.30 (s, 2H), 6.49 (s, 1H). 13C NMR (150 MHz, CDCl3) δ = 21.9, 54.6, 109.4, 118.0, 126.5, 139.0, 147.5. m/z (EI) 173 (M+); m/z (EI) 173.1198 (M+, C12H15N requires 173.1199).1-(3-Bromophenyl)-2,5-dihydro-1H-pyrrole (9l)
Colorless oil in 0.015 g (85% yield); 1H NMR (400 MHz, CDCl3) δ = 4.11 (s, 4H), 5.97 (s, 2H), 6.47 (dd, J = 2.0,8.0 Hz, 1H), 6.69 (s, 1H), 6.82 (d, J = 7.6 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ = 54.8, 110.2, 114.3, 118.8, 123.7, 126.4, 130.7, 148.3; m/z (EI) 223 (M+); m/z (EI) 222.9991 (M+, C10H10NBr requires 222.9991).2-Chlorophenyl-2,5-dihydro-1H-pyrrole (9m)
Colorless oil in 0.017 g (98% yield); 1H NMR (400 MHz, CDCl3) δ = 4.41 (s, 4H), 5.93 (s, 2H), 6.77 (t, J = 7.4 Hz, 1H), 6.90 (s, 1H), 7.18 (dt, J = 1.2, 7.2 Hz, 1H), 7.32 (dd, J = 1.2, 8.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ = 57.4, 117.1, 119.5, 122.3, 126.3, 127.6, 132.1, 145.5; m/z (EI) 179 (M+); m/z (EI) 179.0497 (M+, C10H10NCl requires 179.0496).2,4-Difluorophenyl-2,5-dihydro-1H-pyrrole (9o)
Colorless oil in 0.016 g (90% yield); 1H NMR (400 MHz, CDCl3) δ = 4.32 (d, J = 2.7 Hz, 4H), 5.94 (s, 2H), 6.57 (s, 1H), 6.76–6.86 (m, 1H), 6.97–7.01 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 56.7, 104.6–105.1, 110.8–111.1, 114.7–114.8, 126.3, 133.0–133.1, 149.7–152.3, 153.2–155.7; m/z (EI) 181 (M+); m/z (EI) 181.0699 (M+, C10H9NF2 requires 181.0698).1-(4-Chloro-2-fluorophenyl)-2,5-dihydro-1H-pyrrole (9q)
Colorless oil in 0.018 g (90% yield); 1H NMR (400 MHz, CDCl3) δ = 4.30 (d, J = 2.9 Hz, 4H), 5.93 (s, 2H), 6.78 (t, J = 8.8 Hz, 1H), 7.01–7.08 (m, 2H); 13C NMR (100 MHz, CDCl3) δ = 56.7, 115.4, 116.8–117.0, 120.6–120.7, 124.0, 126.1, 134.9–135.0, 149.8–152.2; m/z (EI) 197 (M+); m/z (EI) 197.0400 (M+, C10H9NFCl requires 197.0402).3-Chloro-2-methylphenyl-2,5-dihydro-1H-pyrrole (9r)
Colorless oil in 0.01 g (57% yield); 1H NMR (400 MHz, CDCl3) δ = 2.46 (s, 3H), 4.16 (s, 4H), 5.96 (s, 2H), 6.98 (d, 1H, J = 7.8), 7.05–7.12 (m, 2H); 13C NMR (150 MHz, CDCl3) δ = 17.3, 58.0, 109.3, 116.1, 122.2, 126.8, 128.1, 136.3, 150.9. m/z (EI) 193 (M+); m/z (EI) 193.0654 (M+, C11H12NCl requires 193.0653).General procedure for the ring-closing metathesis in the synthesis of macrocycles (9d–9s)
Into a solution of each of the dienes 10a–c (0.2 mmol) in CH2Cl2 (25 mL), catalyst 7b (1.0 mol%) was added and the reaction mixture heated at 40 °C and completion of reaction was monitored by TLC. The reaction mixture was mixed with silica gel (100–200 mm, 0.1 g), stirred for 30 min, filtered and the silica was washed with DCM (15 mL) to obtain the desired products as colorless solids. The spectroscopic data for compounds 11a–c are coherent with those reported in literature.17Conflicts of interest
There are no conflicts to declare.Acknowledgements
The investigators gratefully acknowledge the financial support of this project by the Research Administration of Kuwait University through research grant (SC 11/16). The analytical services provided by RSPU unit general facilities of the Faculty of Science through research grants GFS (GS 01/01, GS 01/03, GS 01/05, and GS 03/08) is highly appreciated. We are grateful to Dr Ali Husain for his valuable input in drawing the chemical structures, schemes and artwork.Notes and references
- T. M. Trnka and R. H. Grubbs, The development of L2X2Ru=CHR olefin metathesis catalysts: an organometallic success story, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed.
- A. H. Hoveyda and A. R. Zhugralin, The remarkable metal-catalysed olefin metathesis reaction, Nature, 2007, 450, 243–251 CrossRef CAS PubMed.
- A. M. Dunne, S. Mix and S. Blechert, A highly efficient olefin metathesis initiator: improved synthesis and reactivity studies, Tetrahedron Lett., 2003, 44(13), 2733–2736 CrossRef CAS.
- H. Wakamatsu and S. Blechert, A Highly Active and Air-Stable Ruthenium Complex for Olefin Metathesis, Angew. Chem., Int. Ed., 2002a, 41(5), 794–796 Search PubMed; H. Wakamatsu and S. Blechert, A New Highly Efficient Ruthenium Metathesis Catalyst, Angew. Chem., Int. Ed., 2002b, 41(13), 2403–2405 Search PubMed; K. P. Wang, S. Y. Yun, D. Lee and D. J. Wink, Structure and Reactivity of Alkyne-Chelated Ruthenium Alkylidene Complexes, J. Am. Chem. Soc., 2009, 131(42), 15114–15115 CrossRef CAS PubMed.
- K. Grela, S. Harutyunyan and A. Michrowska, A Highly Efficient Ruthenium Catalyst for Metathesis Reactions, Angew. Chem., Int. Ed., 2002, 41(21), 4038–4040 CrossRef CAS PubMed.
- S. Harutyunyan, A. Michrowska and K. Grela, Catalysts for Fine Chemical Synthesis, Metal Catalysed Carbon-Carbon Bond-Forming Reactions, ed. S. J. Connon, A. M. Dunne, S. Blechert, G. Bhaskar and B. Venkateswara Rao, John Wiley & Sons, Ltd, 2004, vol. 3, pp. 169–173 Search PubMed.
- A. Michrowska, R. Bujok, S. Harutyunyan, V. Sashuk, G. Dolgonos and K. Grela, Nitro-Substituted Hoveyda-Grubbs Ruthenium Carbenes: Enhancement of Catalyst Activity through Electronic Activation, J. Am. Chem. Soc., 2004, 126(30), 9318–9325 CrossRef CAS PubMed.
- Z.-Y. Zhan, Preparation of ruthenium complex ligand, ruthenium complexes, supported ruthenium complex catalysts for olefin metathesis, WO Patent, 2007003135, 2007 Search PubMed.
- Y. Zhang, M. Shao, H. Zhang, Y. Li, D. Liu, y. Cheng, G. Liu and J. Wang, Synthesis and reactivity of oxygen chelated ruthenium carbene metathesis catalysts, J. Organomet. Chem., 2014, 756, 1–9 CrossRef CAS.
- L. Cavallo, Mechanism of Ruthenium-Catalyzed Olefin Metathesis Reactions from a Theoretical Perspective, J. Am. Chem. Soc., 2002, 124(30), 8965–8973 CrossRef CAS PubMed.
- M. Bieniek, R. Bujok, M. Cabaj, N. Lugan, G. Lavigne, D. Arlt and K. Grela, Advanced Fine-Tuning of Grubbs/Hoveyda Olefin Metathesis Catalysts:A Further Step toward an Optimum Balance between Antinomic Properties, J. Am. Chem. Soc., 2006, 128(42), 13652–13653 CrossRef CAS PubMed.
- S.-X. Luo, K. M. Engle, X. Dong, A. Hejl, M. K. Takase, L. M. Henling, P. Liu, K. N. Houk and R. H. Grubbs, An Initiation Kinetics Prediction Model Enables Rational Design of Ruthenium Olefin Metathesis Catalysts Bearing Modified Chelating Benzylidenes, ACS Catal., 2018, 8(5), 4600–4611 CrossRef CAS PubMed.
- E. Merino, E. Poli, U. Díaz and D. Brunel, Synthesis and characterization of new ruthenium N-heterocyclic carbene Hoveyda II-type complexes. Study of reactivity in ring closing metathesis reactions, Dalton Trans., 2012, 41, 10913–10918 RSC.
- N. L. Bauld, D. Gao and J. T. Alpin, Cation radical chain cycloaddition polymerization: a fundamentally new polymerization mechanism, J. Phys. Org. Chem., 1999, 12(11), 808–818 CrossRef CAS.
- Crystallographic data of compounds 7a, 7b and 7c have been deposited with the Cambridge Data Center as supplementary publication # CCDC 2105379, 1481883, 2105381 respectively, Copies of the data can be obtained free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail E-mail: deposit@ccdc.cam.uk.
- (a) Y. A. Ibrahim, H. Behbehani and M. R. Ibrahim, Efficient atom economic approaches towards macrocyclic crownamides via ring closure metathesis, Tetrahedron Lett., 2002, 43, 4207–4210 CrossRef CAS; (b) H. Behbehani, M. R. Ibrahim and Y. A. Ibrahim, Efficient atom economic approaches towards macrocyclic crown diamides via ring-closing metathesis, Tetrahedron Lett., 2002, 43, 6421–6426 CrossRef CAS; (c) Y. A. Ibrahim, H. Behbehani and N. S. Khalil, Efficient route to 20–32 membered macrocyclic crown diamides via ring closing metathesis, Tetrahedron, 2004, 60, 8429–8436 CrossRef CAS.
- Y. A. Ibrahim and E. John, Efficient synthesis of 18–40 membered macrocyclic polyoxadiamides and polyoxatetraamides via ring closing metathesis, Tetrahedron, 2006, 62, 1001–1014 CrossRef CAS.
- W. Chen and J. Wang, Synthesis of Pyrrole Derivatives from Diallylamines by One-Pot Tandem Ring-Closing Metathesis and Metal-Catalyzed Oxidative Dehydrogenation, Organometallics, 2013, 32(6), 1958–1963 CrossRef CAS.
- N. Scalacci, G. W. Black, G. Mattedi, N. L. Brown, N. J. Turner and D. Castagnolo, Unveiling the Biocatalytic Aromatizing Activity of Monoamine Oxidases MAO-N and 6-HDNO: Development of Chemoenzymatic Cascades for the Synthesis of Pyrroles, ACS Catal., 2017, 7(2), 1295–1300 CrossRef CAS.
- B. Schmidt, S. Krehl and E. Jablowski, Assisted tandem catalytic RCM-aromatization in the synthesis of pyrroles and furans, Org. Biomol. Chem., 2012, 10, 5119–5130 RSC.
- S. J. Lia, L. Hana and S. K. Tian, 1,2-Aminohalogenation of arynes with amines and organohalides, Chem. Commun., 2019, 55, 11255–11258 RSC.
- K. H. Kang, K. Il Choi, H. Y. Koh, Y. Kim, B. Y. Chung and Y. S. Cho, Synth. Commun., 2006, 31(15), 2277–2286 CrossRef.
- T. Saitoh, S. Yoshida and J. Ichikawa, Naphthalene-1,8-diylbis(diphenylmethylium) as an Organic Two-Electron Oxidant:Benzidine Synthesis via Oxidative Self -Couplingof N,N-Dialkylanilines, J. Org. Chem., 2006, 71(17), 6414–6419 CrossRef CAS PubMed.
- C. Chiappe, P. Piccioli and D. Pieraccini, Selective N-alkylation of anilines in ionic liquids, Green Chem., 2006, 8(3), 277–281 RSC.
- L. A. Gharat, 3-Azabicyclo[3.1.0]hexane derivatives as vanilloid receptor ligands, pharmaceutical compositions containing them and process for their preparation, PCT Int. Appl., WO2008/010061A2, 2008.
- M. Fuji, J. Chiwata, M. Ozaki, S. Aratani and Y. Obora, In Situ-Generated Niobium-Catalyzed Synthesis of 3-Pyrroline Derivatives via Ring-Closing Metathesis Reactions, ACS Omega, 2018, 3(8), 8865–8873 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2105379, 1481883 and 2105381. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07428h |
| This journal is © The Royal Society of Chemistry 2021 |