
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe site pair matching of a tandem Au/CuO–CuO nanocatalyst for promoting the selective electrolysis of CO2 to C2 products†
Jun-Hao Zhou‡
,
Chen-Yue Yuan‡,
Ya-Li Zheng,
Hai-Jing Yin,
Kun Yuan,
Xiao-Chen Sun and
Ya-Wen Zhang *
*
Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, No. 5 Yiheyuan Road, Haidian District, Beijing 100871, China. E-mail: ywzhang@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 29th November 2021
Abstract
Tandem catalysis, in which a CO2-to-C2 process is divided into a CO2-to-CO/*CO step and a CO/*CO-to-C2 step, is promising for enhancing the C2 product selectivity when using Cu-based electrochemical CO2 reduction catalysts. In this work, a nanoporous hollow Au/CuO–CuO tandem catalyst was used for catalyzing the eCO2RR, which exhibited a C2 product FE of 52.8% at −1.0 V vs. RHE and a C2 product partial current density of 78.77 mA cm−2 at −1.5 V vs. RHE. In addition, the C2 product FE stably remained at over 40% over a wide potential range, from −1.0 V to −1.5 V. This superior performance was attributed to good matching in terms of the optimal working potential and charge-transfer resistance between CO/*CO-production sites (Au/CuO) and CO/*CO-reduction sites (CuO). This site pair matching effect ensured sufficient supplies of CO/*CO and electrons at CuO sites at the working potentials, thus dramatically enhancing the formation rate of C2 products.
Introduction
The electrochemical CO2 reduction reaction (eCO2RR), which can convert CO2 into valuable products, is a promising technique for lowering the concentration of atmospheric CO2, recycling and reusing CO2 to offer carbon-neutral chemical feedstocks, and storing green electricity generated using renewable energy such as from wind and solar sources.1–3 In recent years, enormous progress has been achieved in terms of catalyst design to promote the two-electron reduction of CO2 to produce 2e− products (CO or formate).4–8 However, 2e− products are much less attractive due to their limited market size and low market price.9 Multi-carbon products (C2H4, C2H5OH, CH3COOH, etc.), which are more valuable and have higher energy concentrations, have drawn widespread attention recently.10–14 Theoretical and experimental evidence has illustrated that CO2 is first adsorbed on the surface of the electrode in the form of *COOH, and it is then converted into *CO, followed by further reduction to hydrocarbons and oxygenates.15–17 C–C coupling and the formation of C2 products can only happen when the *CO intermediate is moderately adsorbed on the surface of the electrode, and Cu surfaces have the optimal binding strength for *CO.18,19 As a result, Cu-based electrocatalysts are almost the only materials capable of catalyzing the reduction of CO2 to C2 products.Therefore, various catalyst design principles have been implemented to improve the catalytic activity and selectivity for C2 products of Cu-based electrocatalysts.20 The most common approach involves alloying Cu with a heterometal to tune the binding strengths of specific intermediates, using oxidation and reduction treatments to modify Cu surfaces, utilizing morphology control to expose surfaces with abundant low-coordinated sites, and so on.21–25 However, the improvements in catalytic performance remain unsatisfactory, and this can mostly be attributed to the fact that simultaneously optimizing the binding strengths of *COOH and *CO on one type of reactive site of a Cu-based electrocatalyst is quite challenging.26 As a result, tandem catalysis, in which the CO2-to-C2 process is divided into a CO2-to-CO/*CO step and a CO/*CO-to-C2 step, and the formation of CO/*CO and the reduction of CO/*CO are catalyzed by different active sites, thus modifying the binding strengths of *COOH and *CO simultaneously, is much easier, making it a promising approach for further promoting the formation of C2 products.27 Several kinds of tandem eCO2RR catalysts have been reported very recently. Such catalysts usually consist of CO/*CO-production sites (for example, Ag or Au, single-atom catalysts, complexes) and CO/*CO-reduction sites (usually Cu).28–30 For example, Yang's group reported a Cu–Ag tandem catalyst which could achieve a 4-fold enhancement in the multi-carbon formation rate, while Jaramillo's group found that a tandem Au-on-Cu electrocatalyst was over 100 times more selective for the formation of C2+ products over methane or methanol.31,32
When it comes to Au–Cu systems, many excellent studies have obtained high C2+ product selectivity, and the reasons for these performance improvements have been explained. Zhang's group developed a general strategy for synthesizing Au–Cu Janus nanocrystals, and the obtained catalyst attained 46.4% faradaic efficiency (FE) in relation to C2 products at −0.98 V vs. reverse hydrogen electrode (RHE) (all potentials hereafter are provided with respect to RHE, unless otherwise stated).33 Grätzel's group designed CuAu bimetallic catalysts through galvanic exchange between Au3+ and Cu2O nanowires, and 70.1% FE with respect to C2+ products was obtained at −1.05 V.34 Altogether, these excellent electrocatalytic performances have been attributed to appropriate spatial distances and high local *CO coverages at interfaces.
However, in all of these studies, researchers only focused on fabricating tandem catalysts, and they did not pay attention to the conditions under which tandem catalysis could occur and proceed most effectively and efficiently. In a significant number of publications reporting Au–Cu and Ag–Cu eCO2RR catalysts, tandem catalysis did not occur at all, and 2e− products were almost exclusively produced.35–38 Therefore, merely combining components with CO/*CO-production abilities and CO/*CO-reduction abilities does not necessarily result in a tandem eCO2RR catalyst. There must be some conditions that limit the occurrence of a tandem catalysis process. If we can solve this problem, we could develop tandem catalysts with satisfactory performance more predictably.
Herein, we synthesized a nanoporous hollow Au/CuO–CuO tandem catalyst via annealing Au/CuC2O4/C in air at 400 °C. The Au/CuO–CuO tandem catalyst exhibited a C2 FE of 52.8% at −1.0 V and a C2 partial current density of 78.77 mA cm−2 at −1.5 V. Furthermore, the C2 FE was stably maintained at above 40% over a wide potential range from −1.0 V to −1.5 V. These results represent a significant improvement over a CuO catalyst synthesized through a similar procedure, and the electrochemical CO reduction reaction (eCORR) results could confirm the existence of a tandem catalysis process. In particular, we observed that a catalyst obtained via the physical mixing of Au and CuO catalysts prepared under the same procedure only produces C1 products, providing an ingenious comparison method for inferring the required conditions for the occurrence of a tandem reaction. More experiments elucidate the conditions required for tandem catalysis. The CO/*CO-production sites (Au/CuO) and CO/*CO-reduction sites (CuO) matched well in terms of the optimal working potential and charge-transfer resistance, ensuring that a sufficient amount of CO/*CO was produced and that there was a sufficient electron supply at CuO sites at potentials more negative than −1.0 V, thus further promoting the formation rate of C2 products to a satisfactory level.
Experimental section
Chemicals
HAuCl4·4H2O (A.R., Beijing Chemical Works, China), CuCl2·2H2O (≥99%, Xilong Scientific Co., Ltd, China), Na2C2O4 (A.R., Beijing Chemical Works, China), HCl solution (36%, A.R., Xilong Scientific Co., Ltd, China), KOH (≥99.999%, Aladdin), IrO2 (≥85.5%, Sun Chemical Technology (Shanghai) Co., Ltd, China), cupric oxide (99.5%, particle size: 100–200 nm, Macklin), isopropanol (A.R., Xilong Scientific Co., Ltd, China), Nafion perfluorinated resin solution (5 wt% in lower aliphatic alcohols and water, Sigma-Aldrich), D2O (A.R., Sigma-Aldrich), dimethyl sulfoxide (≥99.8%, Aladdin), and deionized water (Millipore, 18.2 MΩ cm) were obtained from the listed sources.N2 (99.99%) and compressed air (extra dry) were purchased from Beijing Haike Yuanchang Practical Gases Co., Ltd; H2 (99.999%) was purchased from Beijing AP BAIF Gases Industry Co., Ltd; CO2 (99.999%) was purchased from Beijing Beiwen Gases Co., Ltd; and CO (99.9%) was purchased from Beijing Nanfei Gases Co., Ltd.
Synthesis of Au/CuC2O4/C nanoparticles, CuC2O4/C nanoparticles, and Au/C nanoparticles
Au/CuC2O4/C nanoparticles were synthesized through a one-pot method, including a co-precipitation step and reduction step. Specifically, 240 mg of carbon black was dispersed in 20 mL of deionized water in a 100 mL flask, followed by the addition of 1 mL of HCl solution (36%). Then, 2 mmol of CuCl2·2H2O was added into the flask under vigorous stirring. After 10 min, 30 mL of 0.1 M Na2C2O4 aqueous solution was added into the flask and the mixture was kept under stirring for 60 min. Then the solution was heated to 80 °C and 5 mL of 0.02 M HAuCl4 aqueous solution was introduced into the flask. The mixture was kept at 80 °C for 4 h. The product was collected via centrifugation at 9500 rpm for 10 min and washed repeatedly with deionized water using a redispersion–centrifugation method, followed by drying at 50 °C overnight.CuC2O4/C nanoparticles were synthesized through a similar procedure, except that after the mixture was heated to 80 °C, no HAuCl4 precursor was added.
Au/C nanoparticles were synthesized through a similar procedure, except that at the beginning of synthesis, no CuCl2 precursor was added.
Annealing of Au/CuC2O4/C nanoparticles, CuC2O4/C nanoparticles, and Au/C nanoparticles
In a typical procedure, appropriate amounts of as-synthesized Au/CuC2O4/C nanoparticles, CuC2O4/C nanoparticles, and Au/C nanoparticles were annealed at 400 °C for 4 h in air at a heating rate of 10 °C min−1, individually, and then cooled down to room temperature. The obtained samples were named AuCu400, Cu400, and Au400, respectively.Synthesis of Au400 + Cu400 and Au400 + NPCuO samples
Appropriate amounts of Au400 and Cu400 were mixed together, and the obtained sample was named Au400 + Cu400. Appropriate amounts of Au400, commercial CuO NPs (NPCuO), and carbon black were mixed together, and the obtained sample was named Au400 + NPCuO. The amounts of Au400, Cu400, and NPCuO was weighed precisely based on the ratio of Cu and Au in AuCu400 obtained from ICP-OES results, so the ratios of Cu and Au in both Au400 + Cu400 and Au400 + NPCuO were nearly the same as that in AuCu400.Preparation of working electrodes
In this step, AuCu400, Cu400, Au400, AuCu (electrodes containing Au/CuC2O4/C), Au400 + Cu400, Au400 + NPCuO, and NPCuO were treated in the same way. For convenience, we take AuCu400 as an example. In a typical procedure, 5 mg of AuCu400 powder was dispersed in 1000 μL of 0.5 wt% Nafion solution via sonication for 1 h to form a homogeneous ink. The mixture was then sprayed onto carbon paper with a size of 2 cm × 0.5 cm with a gas diffusion layer and dried using a heating plate, yielding the working electrode.Preparation of counter electrodes
In a typical procedure, 2.5 mg of purchased IrO2 powder was dispersed in 1000 μL of 0.5 wt% Nafion solution via sonication for 1 h to form a homogeneous ink. The mixture was then sprayed onto carbon paper with a size of 2 cm × 0.5 cm with a gas diffusion layer and dried using a heating plate, yielding the counter electrode.Electrochemical measurements and product analysis
All electrolysis experiments involving CO2 and CO were conducted in a gas diffusion flow cell. In this work, 1 M KOH was used as the electrolyte.39–41 The gas flow rate during electrolysis was controlled using a mass flow controller at 10 sccm, while the electrolyte flow rates in the cathode and anode channels were managed using a peristaltic pump at 5 mL min−1. The out-flowing electrolyte was fed back to the cell circularly. During all electrolysis experiments, the cathode and anode were separated by a Fumasep anion exchange membrane. Ag/AgCl (saturated KCl) was used as the reference electrode. During chronoamperometry, effluent gas from the cell was introduced directly to the gas sample loop of a gas chromatograph (GC), which was equipped with two Porapak-N MS-13X and HP-AL/S columns for quantification. The gas-phase products were analyzed via GC every 13 min. CO in the gas products was analyzed using a thermal conductivity detector (TCD) with flowing H2 as a carrier gas, H2 in the gas products was analyzed using another TCD with flowing N2 as a carrier gas, and hydrocarbons in the gas products were analyzed using a flame ionization detector (FID) with flowing N2 as a carrier gas. The liquid products were characterized via 1H nuclear magnetic resonance (NMR) spectroscopy. 0.5 mL of electrolyte, after electrolysis, was mixed with 0.1 mL of D2O, and 0.05 μL of dimethyl sulfoxide (DMSO) was added as an internal standard. One-dimensional 1H spectra were measured with water suppression, implementing a solvent pre-saturation technique.Linear sweep voltammetry (LSV) was carried out at a scan rate of 5 mV s−1.
To rule out the possibility that C-based products could be produced via the decomposition of electrolyte (CO2 reacts with KOH to form K2CO3 or KHCO3), electrolysis with AuCu400 using N2 as the feed gas was performed.
The electrochemical impedance spectroscopy (EIS) analysis of samples was carried out using an AC voltage with an amplitude of 5 mV at −1.0 V vs. RHE within the frequency range from 0.01 Hz to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 Hz.
000000 Hz.
The electrochemically active surface area (ECSA) was determined via measuring the double-layer capacitance (Cdl), which was derived from cyclic voltammetry (CV) curves obtained in the narrow potential range of 0.0–0.1 V (vs. Ag/AgCl) at scan rates of 2, 4, 6, 8, 10, 20, 40, 60, 80, and 100 mV s−1. 1 M KOH was used as the electrolyte and N2 was used as the feed gas.
The adsorption of OH− as a surrogate of CO2˙− was examined based on a cathodic LSV scan between −0.4 V and 0.0 V (vs. Ag/AgCl) in N2-saturated aqueous 1 M KOH solution at a scan rate of 20 mV s−1.
Characterization of materials
Transmission electron microscopy (TEM), selected area electron diffraction (SAED) high-resolution transmission electron microscopy (HRTEM), high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), and energy dispersive X-ray spectroscopy (EDS) were performed using FEG-TEM apparatus (JEM-2100F, JEOL, Japan) operated at 200 kV. Scanning electron microscopy (SEM) analysis was performed using a field-emission scanning electron microscope (FESEM) (Hitachi S-4800, Japan). Powder X-ray diffraction (PXRD) analysis was carried out with a D/MAX-2000 diffractometer (Rigaku, Japan) using Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) analysis was conducted using an Axi Ultra imaging photoelectron spectrometer (Kratos, UK). Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed using a Profile Spec ICP-OES spectrometer (Leeman, USA). All GC spectrometry experiments were carried out and recorded using GC-2014 apparatus (Shimadzu, Japan). 1H-NMR spectroscopy analysis was conducted using a Bruker-500 MHz NMR spectrometer (Bruker, USA). Nitrogen adsorption–desorption isotherms were obtained using Micromeritics ASAP 2020 nitrogen adsorption apparatus (USA). Pore-size distributions were determined via the Barrett–Joyner–Halenda (BJH) method using the adsorption data.Results and discussion
Morphology and structural analysis of the nanoporous hollow Au/CuO–CuO catalyst
Au/CuC2O4/C was firstly synthesized using a one-pot method. A composite of carbon black and Cu(II) oxalate was first prepared via co-precipitation, and Au was then introduced onto the surface of CuC2O4 through the reduction of excess C2O42− in solution (see the Experimental section for more details). The results of transmission electron microscopy (TEM) and scanning electron microscopy (SEM) analysis (Fig. S1a–d and S2†) confirm the nanoporous hollow structure of Au/CuC2O4/C with a diameter of 200–500 nm. Energy dispersive X-ray spectroscopy (EDS) line scan measurements (Fig. S3†) further confirm this hollow structure. Selected area electron diffraction (SAED), high angle annular dark field scanning TEM (HAADF-STEM), and EDS mapping images (Fig. S1e–k†) demonstrate that most of the nanoporous hollow particles in Au/CuC2O4/C were composed of CuC2O4, while some CuC2O4 nanoparticles were covered with Au nanoparticles (Au/CuC2O4 composite). The powder X-ray diffraction (XRD) pattern (Fig. S4†) is highly consistent with that of a mixture of CuC2O4·xH2O and Au. For comparison, we also obtained CuC2O4/C and Au/C through similar methods, except that the addition of the Au precursor was omitted in the preparation process of CuC2O4/C and the addition of the Cu precursor was omitted in the preparation process of Au/C. Related characterization of CuC2O4/C and Au/C (Fig. S5 and S6†) illustrates the formation of CuC2O4 in CuC2O4/C and Au in Au/C. X-ray photoelectron spectroscopy (XPS) analysis of Au/CuC2O4/C, CuC2O4/C, and Au/C (Fig. S7 and S8†) shows the existence of Au0 and Cu2+ in Au/CuC2O4/C, Cu2+ in CuC2O4/C, and Au0 in Au/C.Thermogravimetric analysis (TGA) results (Fig. S9†) display that CuC2O4 in Au/CuC2O4/C decomposed at a temperature between 275 °C and 300 °C; thus, the pyrolysis temperature of Au/CuC2O4/C was set as 400 °C, and the obtained sample was named AuCu400. The results from TEM, SEM, SAED, HAADF-STEM, EDS mapping, EDS line scanning, and XRD analysis (Fig. 1 and S10a and b†) confirm that CuC2O4 in Au/CuC2O4/C decomposed to small CuO nanoparticles (20–50 nm in diameter), while Au/CuC2O4 decomposed to Au/CuO nanoparticles. The nanoporous hollow structure (formed via the accumulation of CuO or Au/CuO nanoparticles, 200–500 nm in diameter) was still retained after annealing in air. N2 adsorption–desorption isotherms and pore-size distributions were employed to analyze the pore structure of AuCu400. The obvious hysteresis loop between the adsorption and desorption branches and the peaks at around 3 nm, 10 nm, and 30 nm in the size distribution curve demonstrate the existence of piled mesopores, which is in good agreement with TEM and SEM imaging (Fig. S10c and d†).42 In addition, this conclusion can be verified based on the pore volume of AuCu400 being 0.263 cm3 g−1, which is about 17 times that of Au400 + NPCuO (0.015 cm3 g−1).
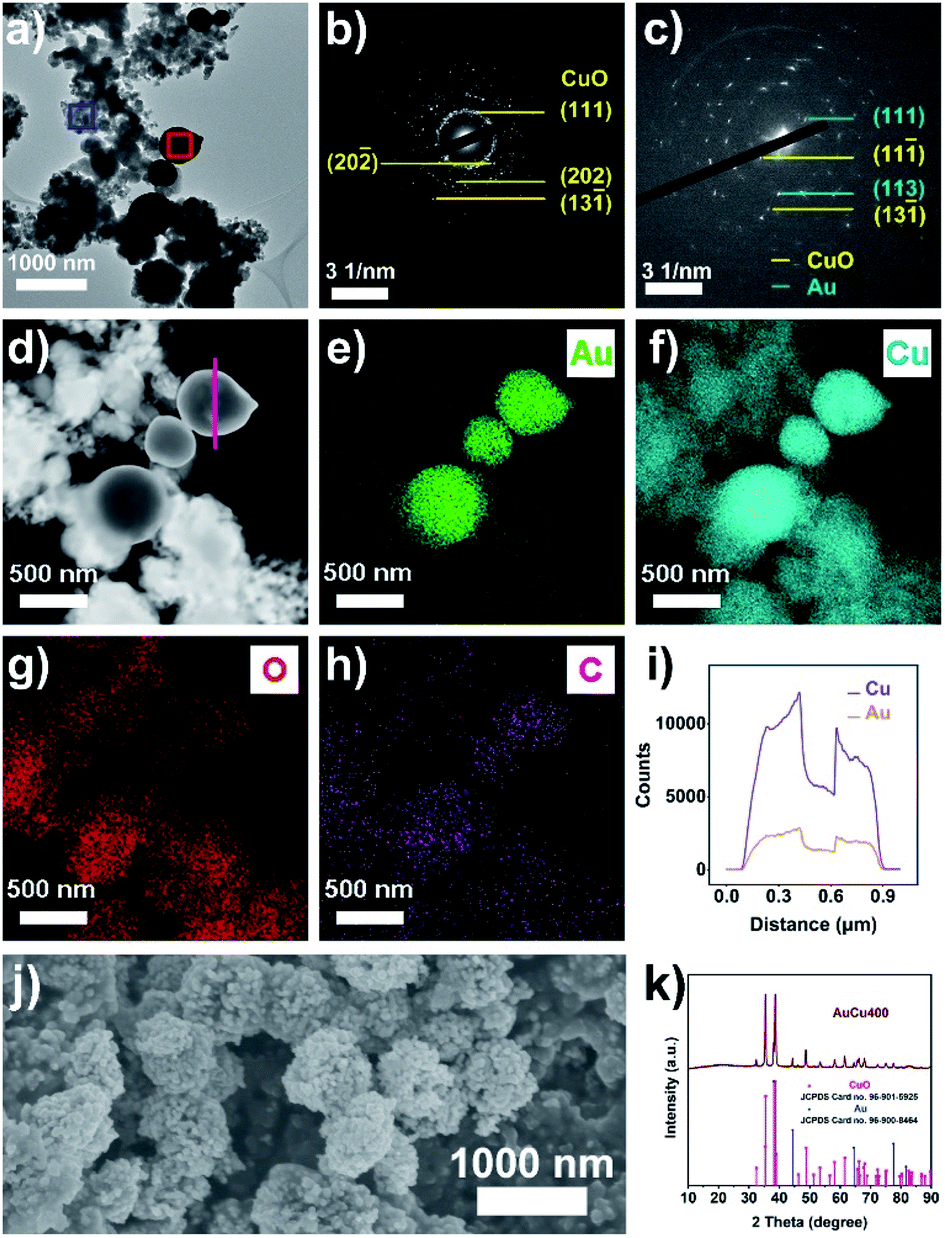 | ||
| Fig. 1 (a) A TEM image of AuCu400; SAED patterns from the (b) blue square and (c) red square from (a); (d) an HAADF-STEM image and (e–h) EDS elemental mapping images of AuCu400; (i) the EDS line scanning result for AuCu400 (the scanning area is the pink line in (d)); (j) an SEM image of AuCu400; and (k) the XRD pattern of AuCu400. | ||
In comparison, Cu400 and Au400 were also prepared through a similar annealing procedure based on CuC2O4/C and Au/C, respectively, and characterization shows that Cu400 was composed of CuO while Au400 was composed of Au (Fig. S11–S14†). XPS spectra of AuCu400, Cu400, and Au400 (Fig. S15 and S16†) also show the existence of Au0 and Cu2+ in AuCu400, Cu2+ in Cu400, and Au0 in Au400.
Evaluation of the electrocatalytic activity and selectivity during the eCO2RR
AuCu400 was ultrasonically mixed with Nafion ionomer solution in isopropanol, and the resulting suspension was sprayed onto carbon paper with a gas diffusion layer to prepare an AuCu400 electrode. In comparison, electrodes containing Cu400, Au400, and Au/CuC2O4/C were also prepared via the same method, and they are denoted as Cu400, Au400, and AuCu electrodes, respectively. eCO2RR measurements were performed in a flow cell using IrO2/C as the counter electrode and 1 M KOH aqueous solution as the electrolyte. Linear sweep voltammetry (LSV) was first conducted to investigate the electrochemical activities of the four samples. As seen in Fig. 2a, we find that at potentials more negative than −0.35 V, current densities followed the order: AuCu400 > Cu400 > Au400, meaning that a combination of Au and Cu could ensure a larger current density. The electrochemically active surface areas (ECSAs) of AuCu, Cu400, and AuCu400 were measured based on CV curves (Fig. S17a–c†), and similar slopes were obtained (Fig. S17f†), indicating that the larger current density in the case of AuCu400 is not caused by an increase in the ECSA. In addition, at potentials more positive than −0.7 V, AuCu exhibited the largest current density of the four samples, maybe resulting from the decomposition of CuC2O4.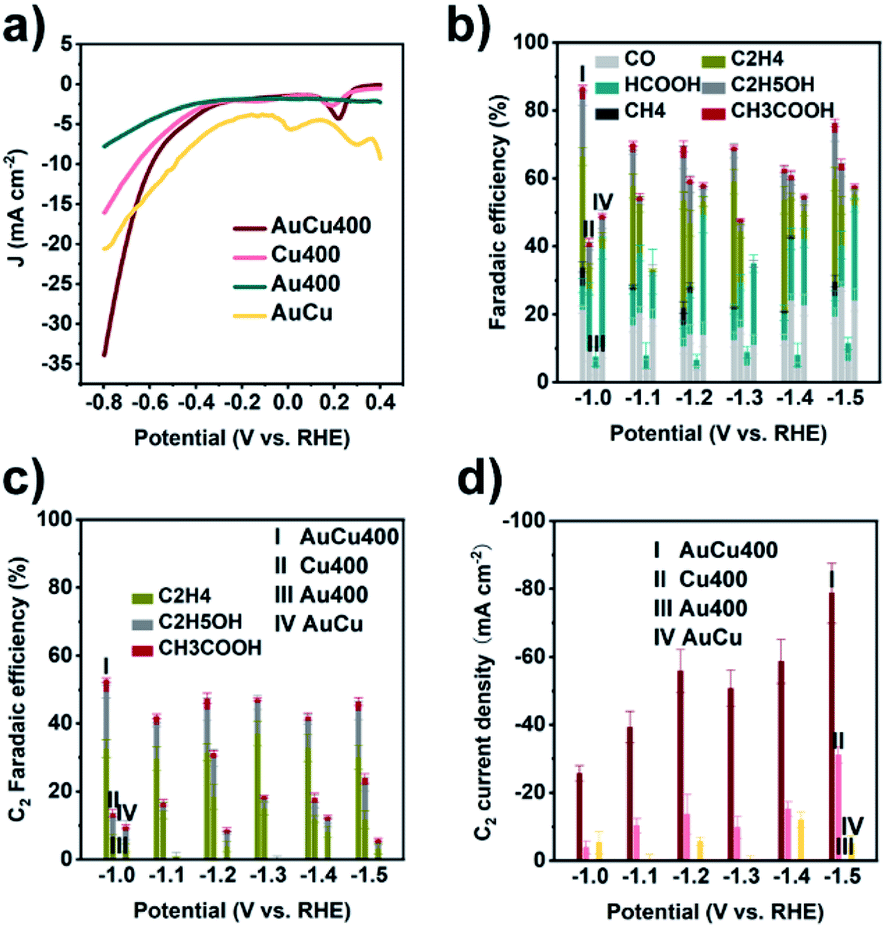 | ||
| Fig. 2 (a) LSV curves, (b) CO2 reduction product FE values, (c) C2 product FE values, and (d) C2 product partial current densities from the four samples, using CO2 as the feed gas. In (b), “I”, “II”, “III”, and “IV” refer to AuCu400, Cu400, Au400, and AuCu, respectively. | ||
The four samples were then examined as electrocatalysts for the eCO2RR based on the controlled-potential electrolysis method. Fig. 2b and Table S1† provide the faradaic efficiency (FE) values for all products using the four electrodes. At all applied potentials, AuCu400 exhibited the highest CO2 reduction product selectivity and the lowest HER activity among the four catalysts. It could reach a maximum CO2 reduction product FE of 86.7% at −1.0 V. Fig. 2c and d show the C2 product FE and C2 product partial current density values, respectively, of the four electrodes at different potentials, from which we could find that AuCu400 exhibited superior C2 product activity and selectivity. At −1.0 V, the C2 product FE of AuCu400 reaches its maximum value of 52.8%, and the C2 product FE could remain above 40% over a wide potential range, from −1.0 V to −1.5 V. At −1.5 V, AuCu400 exhibited the largest C2 product partial current density of 78.77 mA cm−2. The catalytic performance of AuCu400 towards the eCO2RR is compared with other values in the literatures, as listed in Table S2.† AuCu400 showed comparable C2 FE values to other Au–Cu-based tandem catalysts (Table S2-1†) and the widest potential window for C2-dominated selectivity. In contrast, its counterparts Cu400 and AuCu could only reduce CO2 into C2 products with poor FEs (<20%) and small partial current densities (<40 mA cm−2) at all applied potentials, while Au400 produced no C2 products at all. We note the poor stability of AuCu400 over long periods of electrolysis (Fig. S18†), which is due to commonly reported stability issues related to flooding in carbon-paper-based gas diffusion electrodes.43–46 To rule out the possibility that C-based products were produced upon the decomposition of electrolyte (CO2 reacts with KOH to form K2CO3 or KHCO3), electrolysis with AuCu400 using N2 as the feed gas was performed; the much lower current density and lack of detection of C-based products confirm that C-based products are produced from the reduction of CO2 (Fig. S19 and S20†). Significantly, although N2 LSV also showed a modest HER onset peak at the same potential, the H2 product FE of AuCu400 at −1.0 V is only 4.8% (Table S1†). Therefore, it is reasonable to assume that AuCu400 would show a more preferential catalytic effect toward the eCO2RR than the HER when using CO2 as the feed gas.
The conditions required for eCO2RR tandem catalysis
In order to investigate the formation mechanism of C2 products over AuCu400, eCORR measurements were performed over AuCu400 and Cu400. A comparison of the results (Fig. 3a) demonstrates that AuCu400 exhibited no enhancement in C2+ formation compared to Cu400 under CO-reducing conditions at three applied potentials, which illustrates that nanoporous hollow Au/CuO in AuCu400 did not influence CuO active sites and that only the CuO component was responsible for the eCORR in the cases of both AuCu400 and Cu400. To further illustrate the reaction process, we also examined the CO production rates (Fig. S21a†). As expected, AuCu400 has a greater CO reduction rate than Cu400, which means that the introduction of Au/CuO sites generates more CO. In addition, the higher ratio of C2+ products to C1 products can imply that the enhancement is due to an increase in the rate of C–C coupling (Fig. S21b†).29 Our experiments strongly support the conclusion that the nanoporous hollow Au/CuO and CuO active sites in AuCu400 operated independently. Au/CuO produced CO/*CO from CO2, and this generated CO/*CO, in addition to CO/*CO generated on CuO itself, was further utilized for multi-carbon production over the Cu surface (Fig. 4a).47 Compared to bare CuO sites, nanoporous hollow Au/CuO is much more suitable for the formation of CO/*CO due to its more optimal binding strength toward *COOH, which endows AuCu400 with higher C2 product selectivity.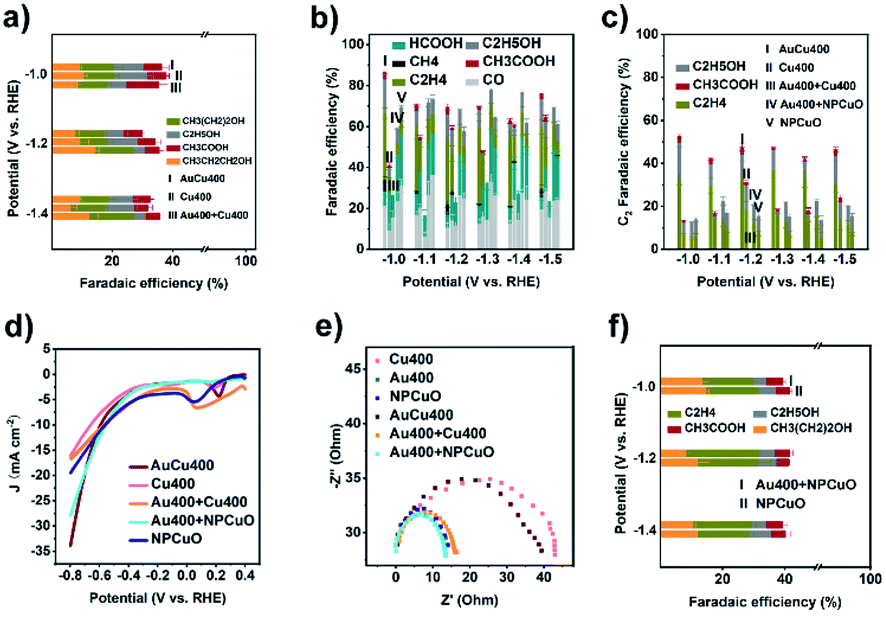 | ||
| Fig. 3 (a) C2+ product FEs of AuCu400, Cu400, and Au400 + Cu400 using CO as the feed gas; (b) CO2 reduction product FEs, (c) C2 product FEs; (d) LSV curves of AuCu400, Cu400, Au400 + Cu400, Au400 + NPCuO, and NPCuO using CO2 as the feed gas; (e) Nyquist plots for Cu400, Au400, AuCu400, Au400 + Cu400, Au400 + NPCuO, and NPCuO; and (f) C2 product FEs of Au400 + NPCuO and NPCuO using CO as the feed gas. In (b), “I”, “II”, “III”, “IV”, and “V” refer to AuCu400, Cu400, Au400 + Cu400, Au400 + NPCuO, and NPCuO, respectively. | ||
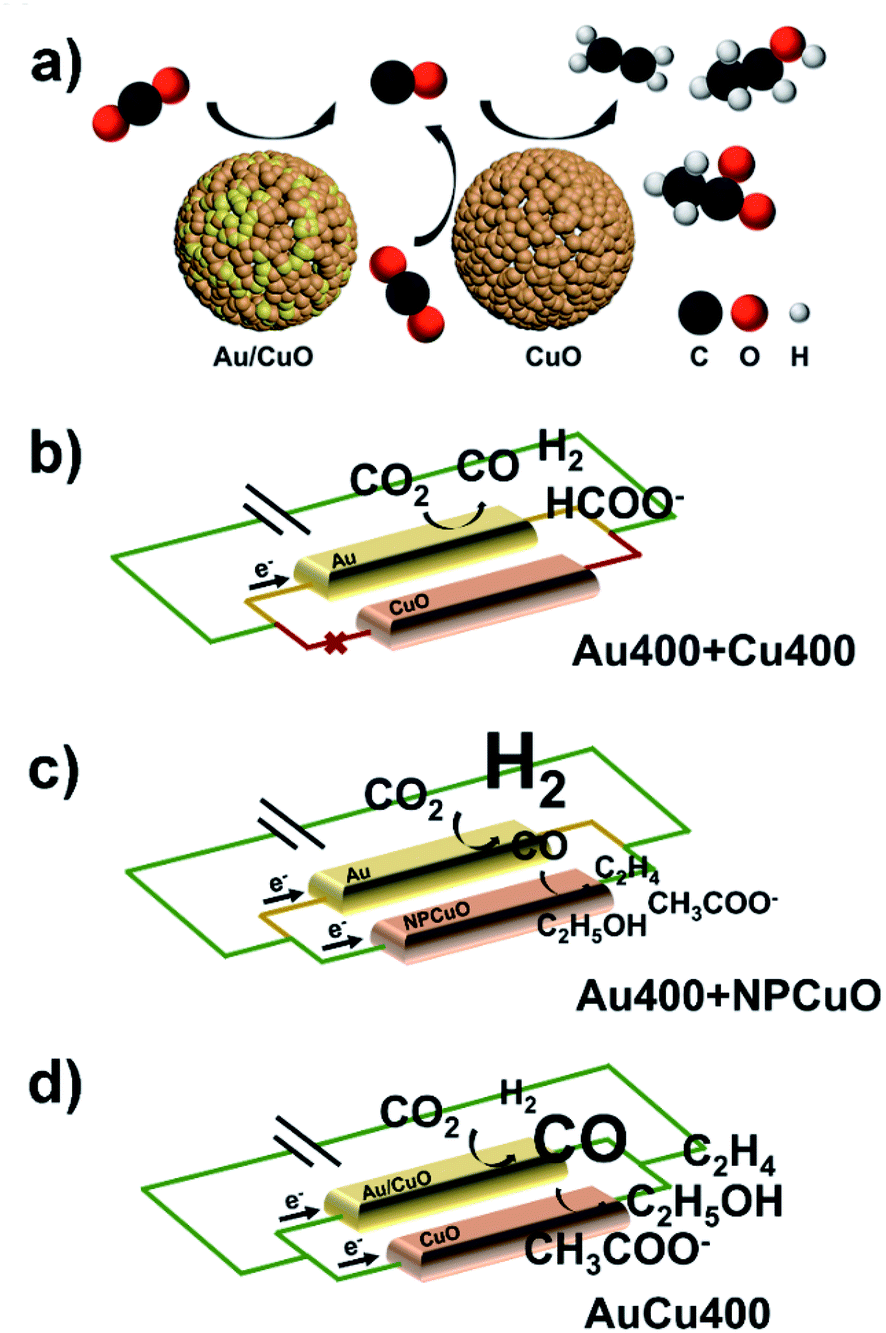 | ||
| Fig. 4 (a) The formation mechanism of C2 products on AuCu400; and the proposed origin of the different C2 FE performances of (b) Au400 + Cu400, (c) Au400 + NPCuO, and (d) AuCu400. Only tandem catalysis processes are shown in (b)–(d). | ||
In some published literature reports, this kind of catalytic mechanism, which is known as “tandem catalysis”, has been reported. However, most of these research reports focus on the fabrication of tandem catalysts via electrochemical deposition methods or physical mixing processes, and they have not explored the conditions under which a tandem catalysis process will proceed most effectively and efficiently. To solve this problem, we prepared Au400 + Cu400 (Fig. S22†) via physically mixing the above-mentioned Au400 and Cu400 materials at the same Au/Cu mass ratio found in AuCu400 (determined via ICP-OES to be 1:5.59). Unexpectedly, the catalytic performance data from the eCO2RR using AuCu400, Cu400, and Au400 + Cu400, shown in Fig. 3b–d and Table S1,† illustrates that Au400 + Cu400 only produced CO, formate, and H2 at all applied potentials, which is completely different from the other samples containing Cu, and it also exhibited a relatively low current density. However, eCORR measurements of Au400 + Cu400 showed almost the same results as for AuCu400 and Cu400 (Fig. 3a), proving that CuO, the source of CO/*CO-reduction sites in Au400 + Cu400, is still effective under CO-reducing conditions. But in the eCO2RR, Cu400, which is dominant in Au400 + Cu400, seemed to be “silenced” by the minor component Au400. Considering that the CuO sites in AuCu400 are not influenced by Au/CuO sites in the reaction process, we speculate that there are some undetected factors leading to the differences in reaction pathways, which are manifested in the failure of CuO sites in Au400 + Cu400 to be involved in the reaction where, in other words, tandem catalysis did not occur.
In order to explore these factors, electrochemical impedance spectroscopy (EIS) studies were then conducted on Au400, Cu400, AuCu400, and Au400 + Cu400 to analyze the electron transfer process. The obtained Nyquist plots (Fig. 3e) indicate that the interfacial charge-transfer resistance of Au400 was much lower than that of Cu400, and this may result from the high conductivity of Au and the low conductivity of CuO. In the catalytic process involving Au400 + Cu400, almost all of the electrons are transferred to Au sites in Au400 due to the huge difference in charge-transfer resistance between Au400 and Cu400; therefore, the CuO sites in Cu400 were “short circuited” and failed to play any role in the reduction of CO2 (Fig. 4b). The small difference in charge-transfer resistance between Au400 and Au400 + Cu400 also supports this reasoning. Although we failed to synthesize a pure Au/CuO phase (because once the proportion of Au was further increased, Au formed an independent phase in the form of larger Au nanoparticles instead of Au/CuO), we can also infer that the electrical conductivity of nanoporous hollow Au/CuO is not far apart from that of CuO because the EIS plot diameter of AuCu400 is close to that of Cu400. Therefore, as a necessary condition for the occurrence of tandem catalysis, the matching of charge-transfer resistance between CO/*CO-production sites and CO/*CO-reduction sites must be present to ensure the high C2 product selectivity of AuCu400.
To check this point, we then prepared Au400 + NPCuO via physically mixing Au400 and commercial CuO nanoparticles (NPCuO) at a Au/Cu ratio of 1:5.59. As a control sample for Cu400, NPCuO shows a similar size, XRD pattern, ECSA, and Cu 2p XPS spectrum to Cu400 (Fig. S17c and d and S23†). More importantly, as for Au400 + NPCuO, the small difference in charge-transfer resistance between Au400 and NPCuO (Fig. 3e) could ensure that the Au sites and CuO sites both gain enough electrons during the reduction process, which provides the possibility for the occurrence of tandem catalysis.
We then compared the catalytic performance towards the eCO2RR of AuCu400, Cu400, Au400 + NPCuO, and NPCuO. Au400 + NPCuO could convert CO2 into C2 products at most applied potentials. At potentials more positive than −1.0 V, the C2 product FEs of these four catalysts followed the order: AuCu400 > Cu400 > Au400 + NPCuO > NPCuO or AuCu400 > Au400 + NPCuO > Cu400 > NPCuO (Fig. 3b and c and Table S1†), which shows that the mixing of Au400 and NPCuO could enhance the selectivity for C2 products of NPCuO, while AuCu400 still exhibited the best C2 product FE. The ECSAs of Au400 + NPCuO and NPCuO were also similar (Fig. S17d–f†), indicating that the introduction of Au could also enhance the current density of NPCuO (Fig. 3d). In addition, it should be noted that the two mixed samples, Au400 + Cu400 and Au400 + NPCuO, show similar large current densities at potentials more positive than −0.7 V, which may be due to the role of Au400 component. Combining this information with the eCORR performance test results of Au400 + NPCuO and NPCuO (Fig. 3f), we could point out that a tandem catalysis mechanism also exists in the eCO2RR catalytic process when using Au400 + NPCuO as the electrode, which could account for the enhancement of the C2 product FE of Au400 + NPCuO versus NPCuO. This further proves that the matching of charge-transfer resistance is a necessary condition for tandem catalysis.
The improvement of the C2 product selectivity of AuCu400 versus Cu400 and Au400 + NPCuO versus NPCuO seems to provide evidence that the introduction of CO/*CO production sites to CuO sites would absolutely enhance the selectivity for C2 products. However, at all applied potentials, the C2 product FEs of Au400 + NPCuO were much lower than those of AuCu400, especially when focusing on the point that the extent of the C2 FE increase of Au400 + NPCuO compared with NPCuO is much lower than that of AuCu400 compared with Cu400; we attribute this to a mismatch of the optimal working potentials of Au sites and CuO sites. Most research expounds that pristine Cu or CuO NPs promote C–C coupling most effectively at potentials more negative than −1.0 V (this conclusion is also shown based on the catalytic performances of Cu400 and NPCuO towards the eCORR in this work, as shown in Fig. S24;† the C2 product FEs are significantly lower at −0.8 V), but pristine Au NPs can only convert CO2 to CO at potentials in the small range of −0.6 V to −0.8 V; at potentials more negative than −1.0 V, the formation of H2 is much more favorable on the surface of Au (this conclusion is also shown based on the catalytic performance of Au400 towards the eCO2RR in this work, as shown in Fig. S25;† the CO product FEs are significantly higher at potentials more negative than −1.0 V). As a result, at potentials more negative than −1.0 V, the amount of CO that Au could produce was relatively small, which restricted the further formation of C2 products at CuO sites (Fig. 4c). This may be the reason why the majority of tandem catalysts show high selectivity for C2 products only in a narrow potential window (about 0.2 V) near −1.0 V.
To further verify this inference, we probed OH− adsorption as a surrogate for CO2˙− adsorption through oxidative LSV scans in N2-saturated 1 M KOH solution (Fig. S26†). Since the protonation of adsorbed CO2˙− is generally considered to be the rate-determining step in CO/*CO production, the potential for surface OH− adsorption can characterize the capabilities for stabilizing CO2˙− intermediates and producing CO/*CO in the reaction path towards C2+ products.48–51 The results reveal that AuCu400 shows the lowest potential, 37 mV, 51 mV, and 67 mV lower than NPCuO, Au400, and Cu400, respectively. Based on this information, we could infer that nanoporous hollow Au/CuO in AuCu400 might be capable of converting CO2 into CO at potentials more negative than −1.0 V much more selectively and effectively than Au in Au400 (and in Au400 + NPCuO) because of changes to the intrinsic electronic properties, and the matching of the optimal working potentials of Au/CuO and CuO in AuCu400 ensured an effective and efficient tandem catalysis process, thus endowing AuCu400 with satisfactory C2 product activity and selectivity (Fig. 4d) over a wide potential range.
Conclusions
In summary, we have developed a nanoporous hollow Au/Cu–CuO tandem catalyst, which exhibited a C2 product FE of 52.8% at −1.0 V and a C2 product partial current density of 78.77 mA cm−2 at −1.5 V. In addition, the C2 product FE remained stably above 40% over a wide potential range from −1.0 V to −1.5 V. The CO/*CO production sites (Au/CuO) and CO/*CO-reduction sites (CuO) showed good matching in terms of their optimal working potentials and charge-transfer resistance values, ensuring that a sufficient amount of CO was produced and there was sufficient electron supply to CuO sites at potentials more negative than −1.0 V, thus further promoting the formation rate of C2 products to a satisfactory level. This work not only successfully fabricated a Au/CuO–CuO tandem nanocatalyst, but it also revealed the conditions under which CO/*CO-production and CO/*CO-reduction site pairing would cooperate most efficiently and under which the tandem catalysis process would proceed most effectively; this may guide us in the development of new tandem eCO2RR catalysts with satisfactory performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21832001 and 21771009) and Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202104).Notes and references
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquid, Science, 2016, 353, 467 CrossRef CAS PubMed
.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: insights into materials design, Science, 2017, 355, 4998 CrossRef PubMed
- J. W. Ager and A. A. Lapkin, Chemical storage of renewable energy, Science, 2018, 360, 707 CrossRef CAS PubMed
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Atomically dispersed Ni(I) as the active site for electrochemical CO2 reduction, Nat. Energy, 2018, 3, 140–147 CrossRef CAS
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, Design of single-atom Co-N5 catalytic site: A robust electrocatalyst for CO2 reduction with nearly 100% CO selectivity and remarkable stability, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed
- J. H. Zhou, K. Yuan, L. Zhou, Y. Guo, M. Y. Luo, X. Y. Guo, Q. Y. Meng and Y. W. Zhang, Boosting electrochemical reduction of CO2 at a low overpotential by amorphous Ag-Bi-S-O decorated Bi0 nanocrystals, Angew. Chem., Int. Ed., 2019, 58, 14197–14201 CrossRef CAS PubMed
- J. Wang, S. Kattel, C. J. Hawxhurst, J. H. Lee, B. M. Tackett, K. Chang, N. Rui, C. J. Liu and J. G. Chen, Enhancing activity and reducing cost for electrochemical reduction of CO2 by supporting palladium on metal carbides, Angew. Chem., Int. Ed., 2019, 58, 6271–6275 CrossRef CAS PubMed
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Exclusive formation of formic acid from CO2 electroreduction by a tunable Pd-Sn alloy, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS PubMed
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. Garcia de Arquer, P. Yang, D. Sinton and E. H. Sargent, A surface reconstruction route to high productivity and selectivity in CO2 electroreduction toward C2+ hydrocarbons, Adv. Mater., 2018, 30, e1804867 CrossRef PubMed
- J. M. Spurgeon and B. Kumar, A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products, Energy Environ. Sci., 2018, 11, 1536–1551 RSC
- M. Liu, M. Liu, X. Wang, S. M. Kozlov, Z. Cao, P. De Luna, H. Li, X. Qiu, K. Liu, J. Hu, C. Jia, P. Wang, H. Zhou, J. He, M. Zhong, X. Lan, Y. Zhou, Z. Wang, J. Li, A. Seifitokaldani, C. T. Dinh, H. Liang, C. Zou, D. Zhang, Y. Yang, T.-S. Chan, Y. Han, L. Cavallo, T.-K. Sham, B.-J. Hwang and E. H. Sargent, Quantum-dot-derived catalysts for CO2 reduction reaction, Joule, 2019, 3, 1703–1718 CrossRef CAS
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, Enhancing CO2 reduction by suppressing hydrogen evolution with polytetrafluoroethylene protected copper nanoneedles, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- R. P. S. Chaplin and A. A. Wragg, Effects of process conditions and electrode material on reaction pathways for carbon dioxide electroreduction with particular reference to formate formation, J. Appl. Electrochem., 2003, 33, 1107–1123 CrossRef CAS
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050 RSC
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in electrochemical reduction of CO2 at metal-electrodes in aqueous hydrogencarbonate solution, Chem. Lett., 1985, 1695–1698 CrossRef CAS
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction, Chem, 2018, 4, 1809–1831 CAS
- C. W. Li and M. W. Kanan, CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, Prism-shaped Cu nanocatalysts for electrochemical CO2 reduction to ethylene, ACS Catal., 2018, 8, 531–535 CrossRef CAS
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, Plasma-activated copper
nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed
- D. Gao, F. Scholten and B. Roldan Cuenya, Improved CO2 electroreduction performance on plasma-activated Cu catalysts via electrolyte design: Halide effect, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS
- C. W. Lee, K. D. Yang, D. H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Defining a materials database for the design of copper binary alloy catalysts for electrochemical CO2 conversion, Adv. Mater., 2018, 30, e1704717 CrossRef PubMed
- A. A. Peterson and J. K. Nørskov, Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS
- Y. Lum and J. W. Ager, Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu, Energy Environ. Sci., 2018, 11, 2935–2944 RSC
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Enhancing CO2 electroreduction to methane with a cobalt phthalocyanine and zinc-nitrogen-carbon tandem catalyst, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed
- H. Zhang, X. Chang, J. G. Chen, W. A. Goddard III, B. Xu, M. J. Cheng and Q. Lu, Computational and experimental demonstrations of one-pot tandem catalysis for electrochemical carbon dioxide reduction to methane, Nat. Commun., 2019, 10, 3340 CrossRef PubMed
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, Elucidating the facet-dependent selectivity for CO2 electroreduction to ethanol of Cu-Ag tandem catalysts, ACS Catal., 2021, 11, 4456–4463 CrossRef CAS
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Cu-Ag tandem catalysts for high-rate CO2 electrolysis toward multicarbons, Joule, 2020, 4, 1688–1699 CrossRef CAS
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst, Nat. Catal., 2018, 1, 764–771 CrossRef CAS
- H. Jia, Y. Yang, T. H. Chow, H. Zhang, X. Liu, J. Wang and C. Y. Zhang, Symmetry-broken Au-Cu heterostructures and their tandem catalysis process in electrochemical CO2 reduction, Adv. Funct. Mater., 2021, 31, 2101255 CrossRef CAS
- J. Gao, D. Ren, X. Guo, S. M. Zakeeruddin and M. Grätzel, Sequential catalysis enables enhanced C-C coupling towards multi-carbon alkenes and alcohols in carbon dioxide reduction: a study on bifunctional Cu/Au electrocatalysts, Faraday Discuss., 2019, 215, 282–296 RSC
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Norskov and P. Yang, Electrochemical activation of CO2 through atomic ordering transformations of AuCu nanoparticles, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed
- M. B. Ross, C. T. Dinh, Y. Li, D. Kim, P. De Luna, E. H. Sargent and P. Yang, Tunable Cu enrichment enables designer syngas electrosynthesis from CO2, J. Am. Chem. Soc., 2017, 139, 9359–9363 CrossRef CAS PubMed
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold-copper bimetallic nanoparticles, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed
- J. H. Zhou, D. W. Lan, S. S. Yang, Y. Guo, K. Yuan, L. X. Dai and Y. W. Zhang, Thin-walled hollow Au-Cu nanostructures with high efficiency in electrochemical reduction of CO2 to CO, Inorg. Chem. Front., 2018, 5, 1524–1532 RSC
- W. Luo, J. Zhang, M. Li and A. Züttel, Boosting CO production in electrocatalytic CO2 reduction on highly porous Zn catalysts, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and P. Berlinguette Curtis, Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- B. Liu, C. Cai, B. Yang, K. Chen, Y. Long, Q. Wang, S. Wang, G. Chen, H. Li, J. Hu, J. Fu and M. Liu, Intermediate enrichment effect of porous Cu catalyst for CO2 electroreduction to C2 fuels, Electrochim. Acta, 2021, 388, 138552 CrossRef CAS
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783 CrossRef CAS PubMed
- Y. Wang, H. Shen, K. J. T. Livi, D. Raciti, H. Zong, J. Gregg, M. Onadeko, Y. Wan, A. Watson and C. Wang, Copper nanocubes for CO2 reduction in gas diffusion electrodes, Nano Lett., 2019, 19, 8461–8468 CrossRef CAS PubMed
- W. Luc, X. Fu, J. Shi, J. J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Two-dimensional copper nanosheets for electrochemical reduction of carbon monoxide to acetate, Nat. Catal., 2019, 2, 423–430 CrossRef CAS
- M. Jouny, W. Luc and F. Jiao, High-rate electroreduction of carbon monoxide to multi-carbon products, Nat. Catal., 2018, 1, 748–755 CrossRef CAS
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2-CO co-feeds on Cu and Cu-tandem electrocatalysts, Nat. Nanotechnol., 2019, 14, 1063 CrossRef CAS PubMed
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Metallic tin quantum sheets confined in graphene toward high-efficiency carbon dioxide electroreduction, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed
- H. Won da, C. H. Choi, J. Chung, M. W. Chung, E. H. Kim and S. I. Woo, Rational design of a hierarchical tin dendrite electrode for efficient electrochemical reduction of CO2, ChemSusChem, 2015, 8, 3092–3098 CrossRef PubMed
- L.-P. Yuan, W.-J. Jiang, X.-L. Liu, Y.-H. He, C. He, T. Tang, J. Zhang and J.-S. Hu, Molecularly engineered strong metal oxide-support interaction enables highly efficient and stable CO2 electroreduction, ACS Catal., 2020, 10, 13227–13235 CrossRef CAS
- S. Zhang, P. Kang and T. J. Meyer, Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: More TEM and SEM images, PXRD and XPS analyses, and electrocatalytic measurements of the Au/CuO–CuO nanocatalyst. See DOI: E-mail: 10.1039/d1ra07507a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |