
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical activation of disulfide-based multifunctional polymers for theranostic drug release†
Zhiyuan
Shi
 ab,
Qingchuan
Song
ab,
Robert
Göstl
*a and
Andreas
Herrmann
*abc
ab,
Qingchuan
Song
ab,
Robert
Göstl
*a and
Andreas
Herrmann
*abc
aDWI – Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52056 Aachen, Germany. E-mail: goestl@dwi.rwth-aachen.de; herrmann@dwi.rwth-aachen.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
cZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 15th December 2020
Abstract
Drug delivery systems responsive to physicochemical stimuli allow spatiotemporal control over drug activity to overcome limitations of systemic drug administration. Alongside, the non-invasive real-time tracking of drug release and uptake remains challenging as pharmacophore and reporter function are rarely unified within one molecule. Here, we present an ultrasound-responsive release system based on the mechanochemically induced 5-exo-trig cyclization upon scission of disulfides bearing cargo molecules attached via β-carbonate linker within the center of a water soluble polymer. In this bifunctional theranostic approach, we release one reporter molecule per drug molecule to quantitatively track drug release and distribution within the cell in real-time. We use N-butyl-4-hydroxy-1,8-naphthalimide and umbelliferone as fluorescent reporter molecules to accompany the release of camptothecin and gemcitabine as clinically employed anticancer agents. The generality of this approach paves the way for the theranostic release of a variety of probes and drugs by ultrasound.
Introduction
In conjunction with radiation therapy, surgery, or hyperthermia therapy, chemotherapeutic agents remain the most effective way to cure various forms of cancer.1,2 However, the systemic application of anti-cancer drugs leads to severe side effects due to the necessity to balance drug activity, selectivity, toxicity, and emerging resistances.3 Strategies to control where and when a drug is active thus arguably are promising pathways to improve the therapeutic efficiency of chemotherapeutics and simultaneously mitigate side effects. In this light, controlled drug release systems responsive to internal or external stimuli have received considerable attention.4 For internal stimuli, the targeted drug is generally equipped with a pH-5 or redox-responsive6,7 linker, while light8,9 and electromagnetic fields10 are prototypical external stimuli controlled by the physician. Ultrasound (US) is a clinically widespread external stimulus to release drugs from carriers, such as micelles, liposomes, or microbubbles,11,12 or to synergistically increase drug efficacy.13,14 Recently we showed that US in the context of polymer mechanochemistry15,16 can address force-responsive molecular moieties (mechanophores)17,18 embedded in macromolecular structures to activate and release drugs with reasonable selectivity.19 Thereby, we reported the polymer mechanochemical scission of disulfides20–25 that underwent an intermolecular Michael addition to Diels–Alder adduct conjugates with furan-bearing drugs spawning the subsequent release of the latter.19Here we broaden the scope of polymer mechanochemically induced drug release to a wide range of amino- and hydroxy-terminated drugs and incorporate the drug payload molecules on β-carbonate linkers adjacent to the mechanoresponsive disulfide within the same macromolecule. We use ultrasound to initiate disulfide scission spurring an intramolecular 5-exo-trig cyclization26 which subsequently releases the cargo molecules (Scheme 1).
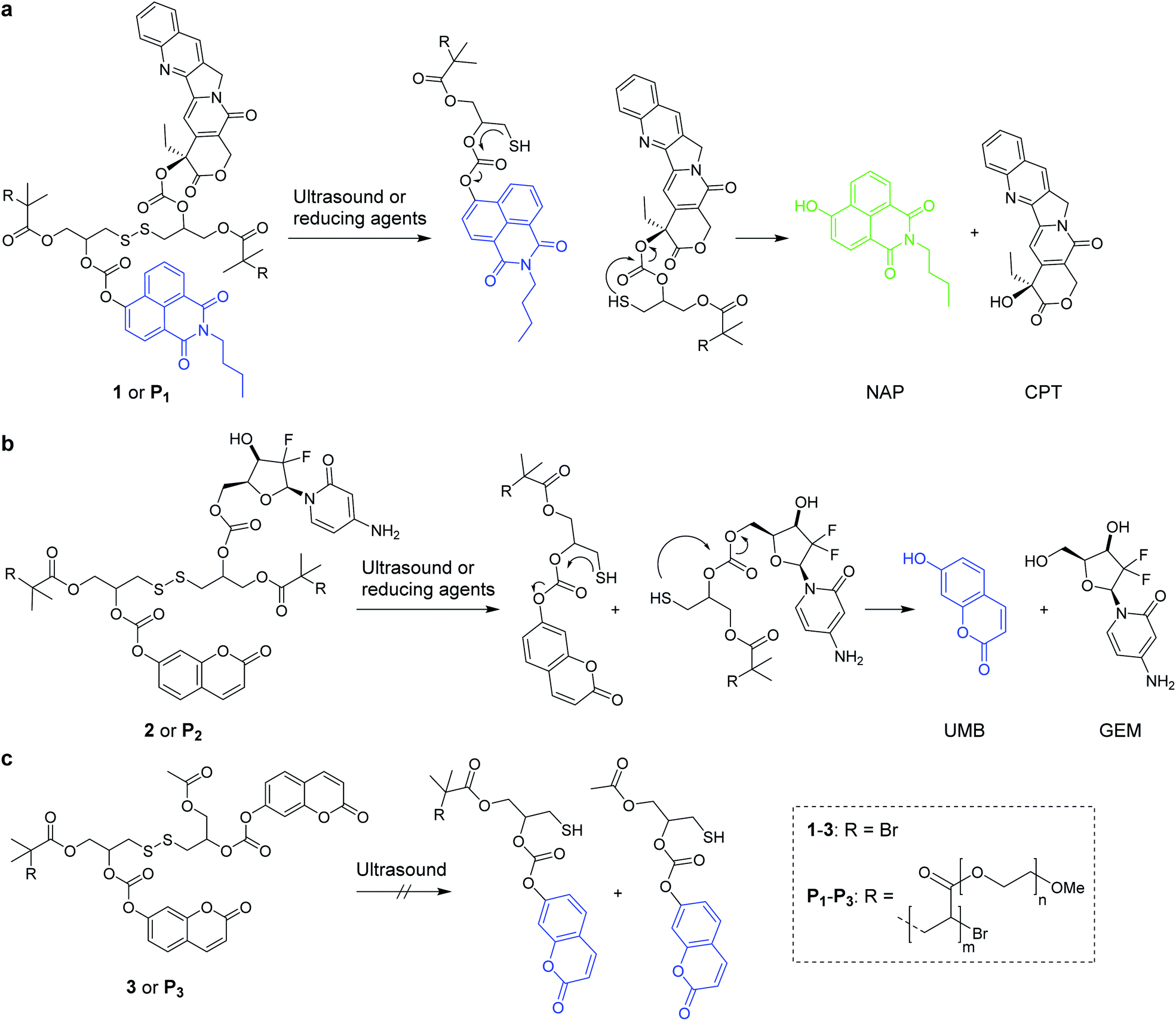 | ||
| Scheme 1 Reaction mechanisms of the mechanochemical and reductive scission of disulfide-centered polymers with adjacent β-carbonate linked drugs and fluorophores that are released by a subsequent 5-exo-trig cyclization. (a) N-Butyl-4-hydroxy-1,8-naphthalimide (NAP) was released in conjunction with camptothecin (CPT) from 1 or P1; (b) umbelliferone (UMB) together with gemcitabine (GEM) from 2 and P2; (c) small molecule 3 and terminally functionalized polymer P3 controls did not lead to UMB release by ultrasonication. | ||
As the assessment whether a drug is released and taken up is generally indirectly inferred from measuring proliferation or cell adhesion,27 it is difficult to determine precisely when, where, and to which extent the pharmacologically active agents are delivered to the target cell. To tackle this issue, we here use bifunctional polymers that release a fluorophore alongside the drug molecule in a theranostic approach. We release N-butyl-4-hydroxy-1,8-naphthalimide (NAP) and umbelliferone (UMB) as fluorescent probes simultaneously with the chemotherapeutics camptothecin (CPT) and gemcitabine (GEM) for non-invasive imaging of drug delivery and uptake (Scheme 1).
We investigated two different theranostic agents of which one bore NAP as reporter fluorophore and CPT as drug (Scheme 1a) and the other UMB as fluorescent probe and GEM as active component (Scheme 1b). The corresponding disulfide-centered derivatives 1 and 2 bearing two α-bromoisobutyryl moieties served both as bifunctional initiator for subsequent polymerization towards telechelic polymers P1 and P2 and as small molecular control substances. The synthetic pathway towards 1 and 2 is presented in Scheme S1† and the resulting compounds were verified by 1H- and 13C-nuclear magnetic resonance (NMR) spectroscopy as well as electrospray ionization mass spectrometry (ESI-MS) (Fig. S1–S23†).
From these initiators, polymers were obtained by Cu0-mediated controlled radical polymerization of oligo(ethylene glycol)methyl ether acrylate (OEGMEA) yielding disulfide-centered, water-soluble POEGMEA P1 (Mn = 140.9 kDa, ĐM = 1.65) and P2 (Mn = 146.3 kDa, ĐM = 1.69) (Fig. S29†). Moreover, the controls 3 and terminally functionalized polymer P3 (Mn = 109.7 kDa, ĐM = 1.45) were synthesized according to the synthetic pathway in Scheme S2† to confirm the mechanochemical origin of the observed phenomena (Fig. S24–S28 and S30†).
Results and discussion
Both 1 and P1 (Scheme 1a) were composed of CPT, which acts as inhibitor of topoisomerase I,28 a disulfide mechanophore,19–25 and NAP yielding a considerably bathochromic shift in absorption and emerging fluorescence at ca. 550 nm upon release from its corresponding β-carbonate (Fig. 1a and b).29,30 Note that residual NAP fluorescence, likely due to incomplete fluorophore quenching in the carbonate form, was observable but consistent with previous reports.29–31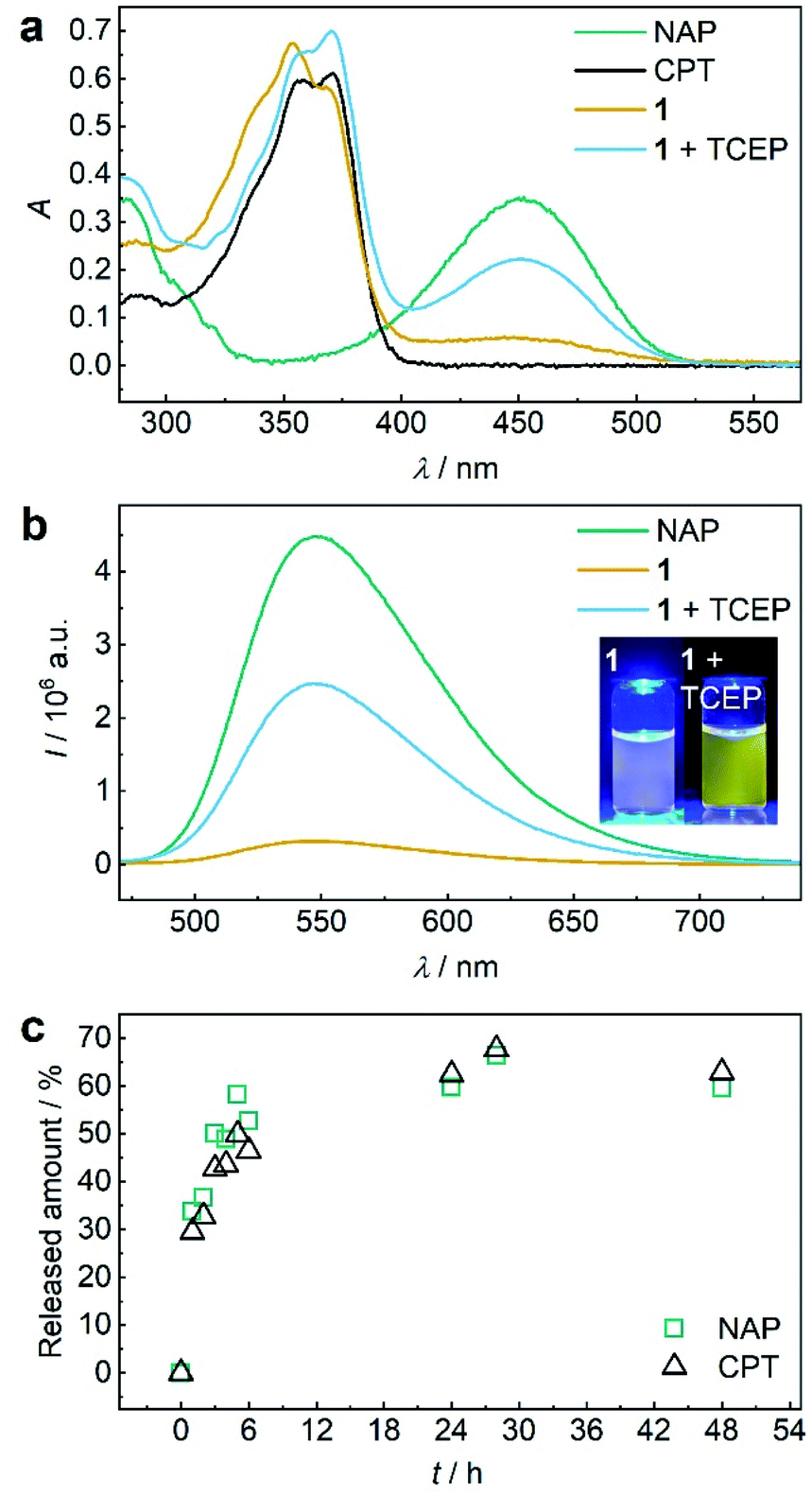 | ||
Fig. 1 TCEP-induced release of CPT and NAP from 1. (a) UV-vis spectra (c = 25 μM) of NAP, CPT, 1, and 1 treated with 20 eq. TCEP at 37 °C for 48 h. (b) Fluorescence spectra (c = 5 μM, λexc = 450 nm, spectral bandwidths 3 nm) of NAP, 1, and 1 treated with 20 eq. TCEP at 37 °C for 48 h. Inset: photograph of the solutions under UV-light (λexc = 455 nm). (c) Released amount of NAP and CPT over reaction time calculated from normalized integrated areas of the PDA detector signals of UPLC elugrams (Fig. S32a†). All spectra were recorded in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 2:3 (v/v) at room temperature. :H2O = 2:3 (v/v) at room temperature. | ||
Initially, we verified the ability to release both drug and reporter molecules from their β-carbonate linkers upon disulfide bond scission and subsequent intramolecular 5-exo-trig cyclization using small molecule control 1.
Therefore, we reduced 1 with tris(2-carboxyethyl)phosphine (TCEP) and monitored UV-vis absorption and fluorescence (Fig. 1a and b). We calculated a released fraction of 66.5% NAP via its molar absorptivity at 450 nm using UV-vis spectroscopy and 60.5% via calibration curve using fluorescence emission at 550 nm after reduction at 37 °C for 48 h (Fig. S31†).
For the theranostic approach it is paramount to correlate the release of the probe NAP to the release of the drug CPT. For this purpose, we monitored the reductive release by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS), where the integration of peak areas measured by the UV-vis diode array detector (DAD) allowed quantification (Fig. S26a†) and the MS detector allowed identification of the corresponding NAP and CPT peaks (Fig. S32b and c†). We normalized the integrated DAD signals from UPLC-MS measurements to the maximum obtainable released fraction of 66.5% as largest value (Fig. 1c). By this, we showed that NAP release proceeded at a rate comparable to CPT release thus underlining the suitability of fluorophore NAP as a diagnostic probe for the release of the drug CPT. We then confirmed that in addition to small molecule 1, polymer P1 could also release NAP and CPT by reduction with TCEP. UV-vis absorption and fluorescence spectroscopy as well as UPLC-MS conclusively proved this result (Fig. S33†).
Encouraged by these findings, we turned towards the mechanochemical activation of CPT and NAP from the disulfide-centred polymer P1. Therefore, P1 was subjected to sonication using an immersion probe sonicator (f = 20 kHz, IP = 15.84 W cm−2). Samples collected over the course of the sonication for UV-vis and fluorescence spectroscopy indicated successful disulfide scission culminating in the release of NAP and CPT (Fig. 2a). This led us to a calculated released amount of 56% NAP (and therewith CPT) from P1 after 3 h of sonication (Fig. 2b). Notably, small molecule control 1 only showed around 4% non-specific release under the same conditions verifying the mechanochemical origin of the observed effects (Fig. 2b and S34†).
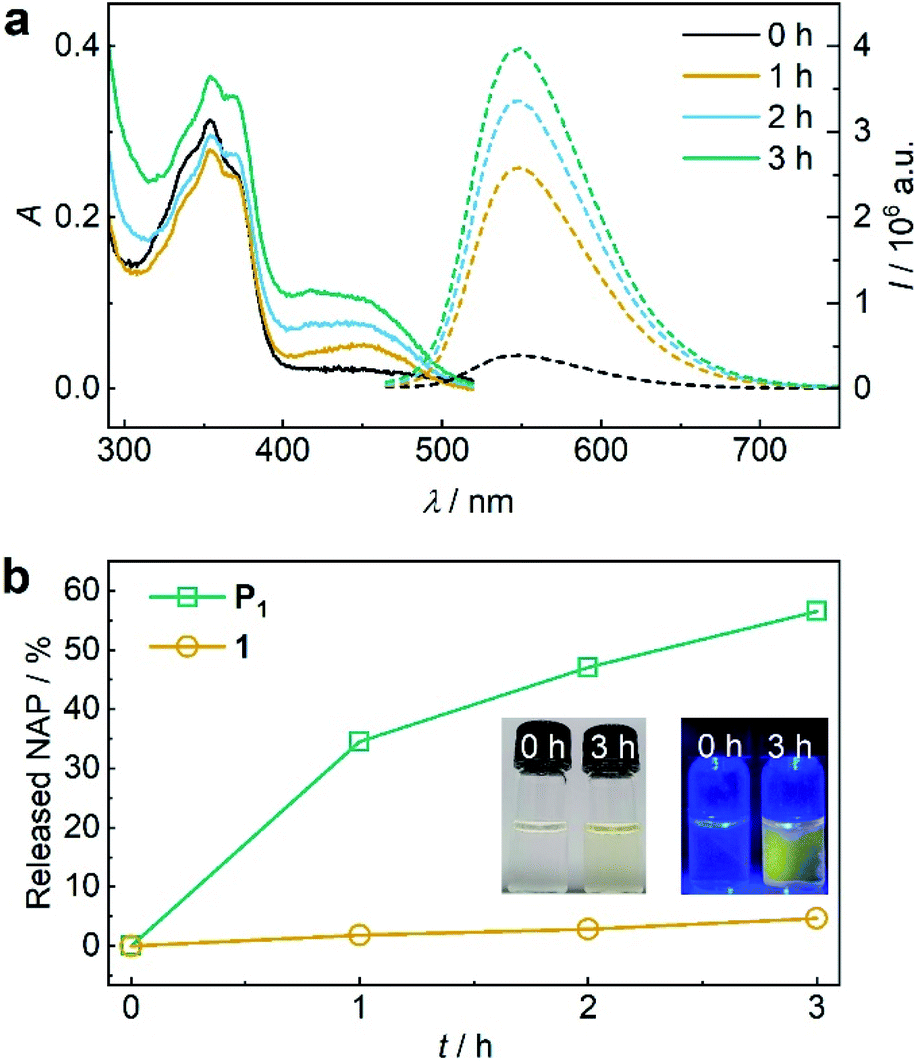 | ||
| Fig. 2 US-induced release of NAP from β-carbonate disulfide polymer P1 (11.6 μM, 3 mg mL−1) and small molecule control 1 (25 μM) in MeCN:H2O = 2:3 (v/v). (a) UV-vis and fluorescence (λexc = 450 nm) spectra of P1 during sonication. (b) Released NAP from P1 and control 1 during sonication calculated from fluorescence peak intensity of NAP at λem = 550 nm. Insets: photographs of the solutions under daylight and UV-light (λexc = 455 nm). | ||
Hereafter, we investigated the diagnostic behaviour of US-activated P1 in HeLa cells by imaging using confocal laser scanning microscopy (CLSM). For this purpose, we stained the HeLa cells with sonicated P1 after confirming, once again, successful NAP release by UPLC-MS (Fig. S35†) and assuring cell viability (Fig. S36†). NAP fluorescence was visible as opposed to non-sonicated P1 or untreated control cells (Fig. 3a). This indicated the successful mechanochemically-induced release of NAP quickly diffusing into the cell (Fig. S37†), which would not be observed for P1 alone due to the hindered cellular internalization of macromolecules.32 To quantitatively confirm CLSM measurements, fluorescence-activated cell sorting (FACS) using the NAP emission was carried out (Fig. 3b). The population of HeLa cells stained with US-activated P1 showed fluorescence ca. one order of magnitude higher than those treated with pristine P1 or the control sample. This underlined the potential use of this theranostic system for monitoring drug release.
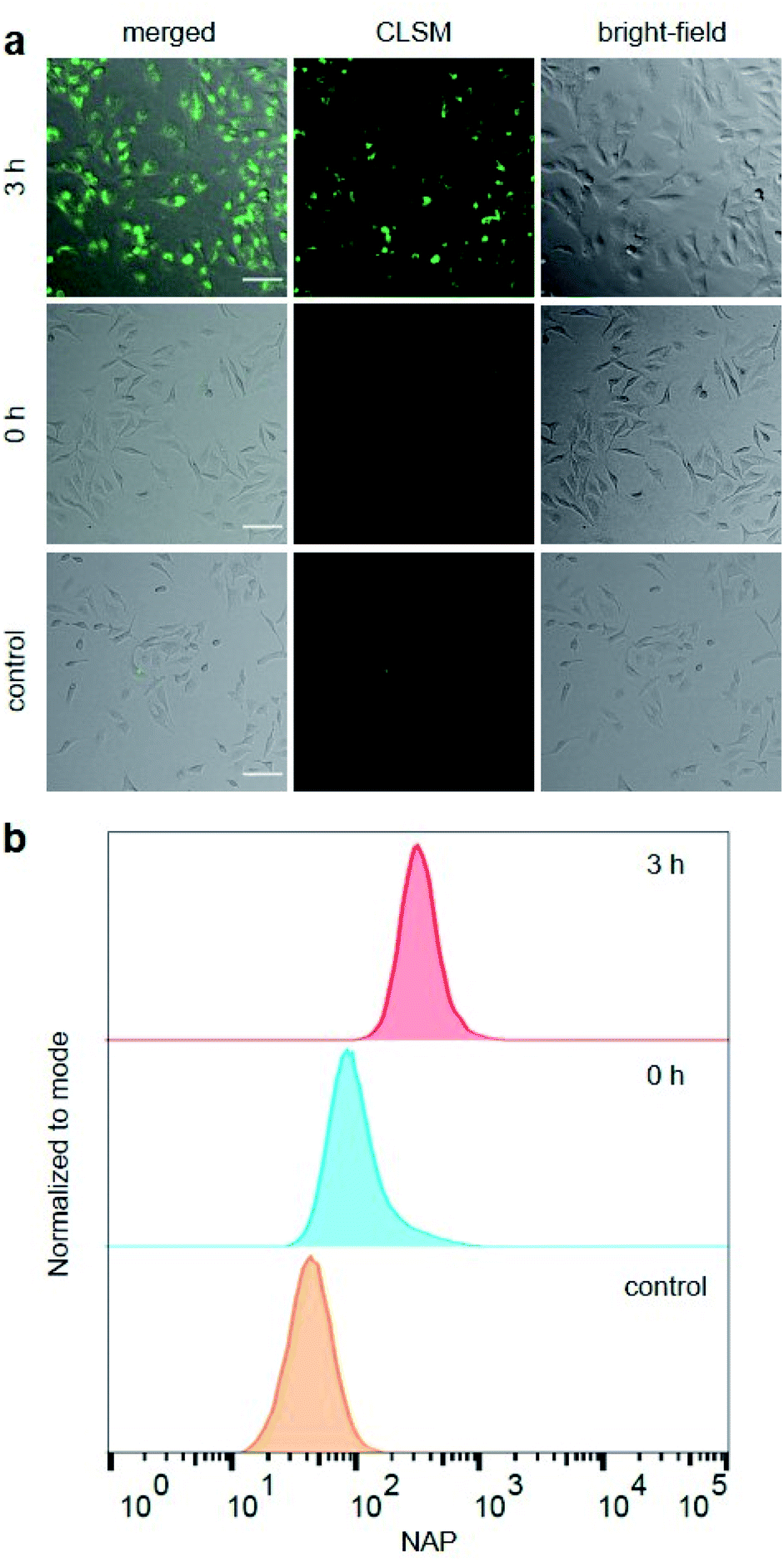 | ||
| Fig. 3 HeLa cells stained for 30 min with sonicated and non-sonicated P1 as well as untreated control. (a) Bright-field, CLSM, and merged micrographs (scale bar: 100 μm). Enlarged micrographs are shown in Fig. S37.† (b) Flow cytometry analysis using NAP fluorescence. | ||
The activity of theranostic agent P1 was then investigated with MTS proliferation assays after treatment of HeLa cells. Control experiments showed that pristine POEGMEA and NAP both had no cytotoxic effect at the applied conditions while the IC50 value of small molecule 1 resembled that of pristine CPT (Fig. 4a). Notably, CPT-bearing polymer P1 was shielded from intracellular access, thiol exchange, or disulfide reduction from macromolecular thiolated proteins, such as HSA, under physiological conditions within its hydrodynamic coil and exhibited greatly diminished cytotoxicity compared to 1 and pure CPT. While this is a routine measure exploited for designing macromolecular drug carriers,32,33 it is unprecedented for a mechanoresponsive prodrug system and a considerable advance compared to the intermolecular mechanochemical drug release system relying on drugs bound to small molecules reported by us recently19 as these were taken up by cells and proved to be prone to endogenous macromolecular thiols.
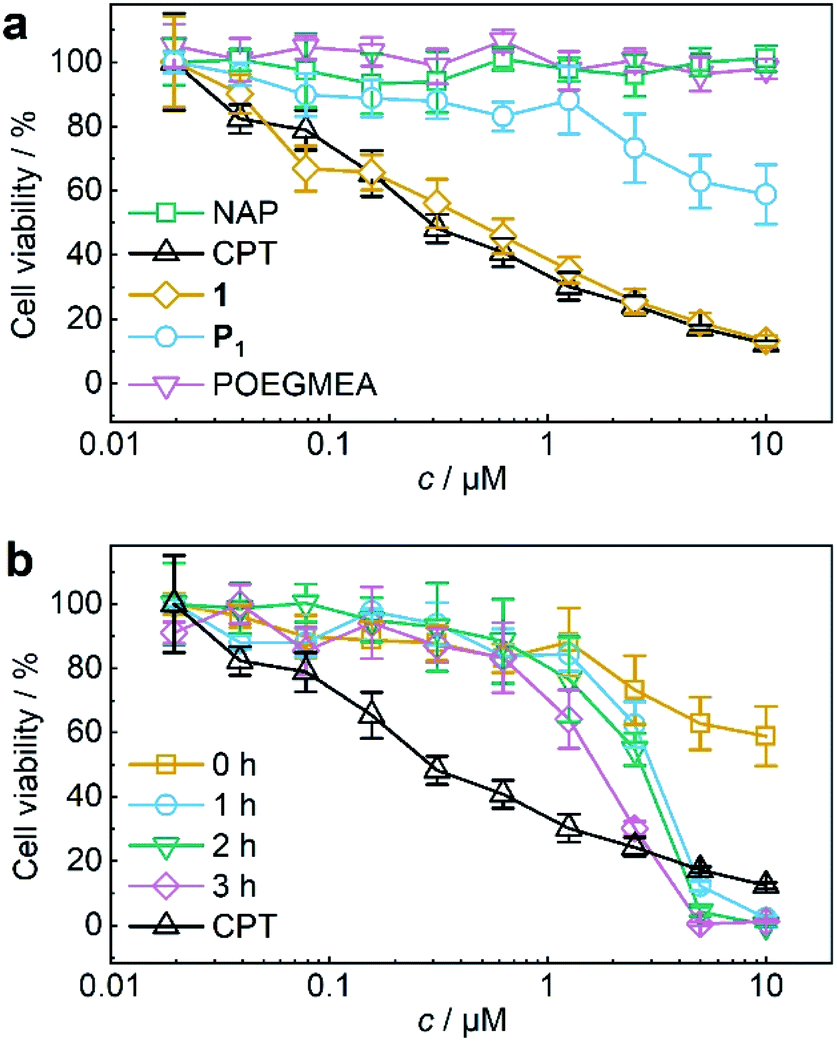 | ||
| Fig. 4 US-induced release of CPT from P1 (11.6 μM, 3 mg mL−1 in MeCN:H2O = 2:3 (v/v)) for MTS proliferation assays. (a) Control MTS proliferation assays of NAP, CPT, 1, P1, and pristine POEGMEA. (b) MTS proliferation assay of P1 with progressing ex situ sonication. Assay of pure CPT is shown again for comparison. Mean values ± SD from the mean, N = 3 independent experiments. | ||
For the mechanochemical activation of CPT release by irradiation with US, samples of P1 taken over the course of its sonication were freeze-dried, dissolved in cell culture medium, and after CPT release was confirmed once more by UPLC-MS (Fig. S38†) incubated with HeLa cells for 48 h. The decrease in cell viability with progressing ex situ sonication became visible and indicated the successful US-activation of P1 spawning the release of CPT (Fig. 4b). The diagnostic CLSM and FACS data together with the MTS proliferation assays demonstrated the potential of using the NAP-CPT couple for the mechanochemically induced activation of theranostic functionality.
We then investigated the generality of this theranostic approach by exchanging both the diagnostic fluorophore NAP and the drug CPT against UMB and GEM, respectively. GEM is a nucleoside analogue used for the treatment of pancreatic, breast, and ovarian cancers. GEM replaces cytidine during DNA replication and interacts with ribonucleotide reductase resulting in cell apoptosis.10,26,27 However, rapid enzymatic deamination to inactive 2′-deoxy-2′,2′-difluorouridine (dFdU),34 short plasma half-life, and adverse toxicity10,35 limit its therapeutic efficacy. Hence GEM would benefit from a controlled delivery approach in a theranostic combination with a reporter fluorophore. Moreover, protecting GEM within the random coil of a polymer chain is a promising strategy to decrease renal clearance and enhance its circulation half-life.36
Both 2 and P2 (Scheme 1b) contained GEM and UMB bound to the β-carbonate functionality alongside the disulfide mechanophore. We reasoned that upon activation of P2, the disulfides centred at the polymer chain were readily cleaved and liberated GEM and UMB from the carbonates analogously to P1. Note that again residual UMB fluorescence was visible, most likely stemming from the diminished fluorescence quantum yield of UMB in the carbonate form compared to the free hydroxide.37
Again, we initially verified the capability to release GEM and UMB from small molecule control 2. Therefore, we reduced 2 with glutathione (GSH) overnight at room temperature and monitored UV-vis absorption and fluorescence (Fig. S39†). The release of UMB was confirmed by its characteristically emerging absorption and fluorescence bands. UPLC-MS measurements supported this observation detecting both UMB and GEM with their corresponding retention times and mass spectra after GSH reduction (Fig. S40†). In addition to small molecule 2, polymer P2 could also release UMB and GEM by TCEP reduction, as proven by UV-vis absorption and fluorescence spectroscopy as well as UPLC-MS measurements (Fig. S41†). We then investigated the mechanochemical activation of GEM and UMB from the disulfide-centred polymer P2. Therefore, P2 was subjected to ultrasonication using the same setup described for P1. UV-vis and fluorescence spectroscopy over the course of the sonication indicated the successful release of UMB and GEM (Fig. 5a). By calibrating UMB fluorescence in dependence of its concentration by fluorescence spectroscopy (Fig. S42†), we calculated an approximate released amount of 55% UMB after 4 h of sonication (Fig. 5b). Importantly, small molecule control 2 as well as terminally substituted polymer P3 showed considerably reduced non-specific release under the same conditions verifying the mechanochemical origin of the observed effects (Fig. 5b, S43 and S44†).
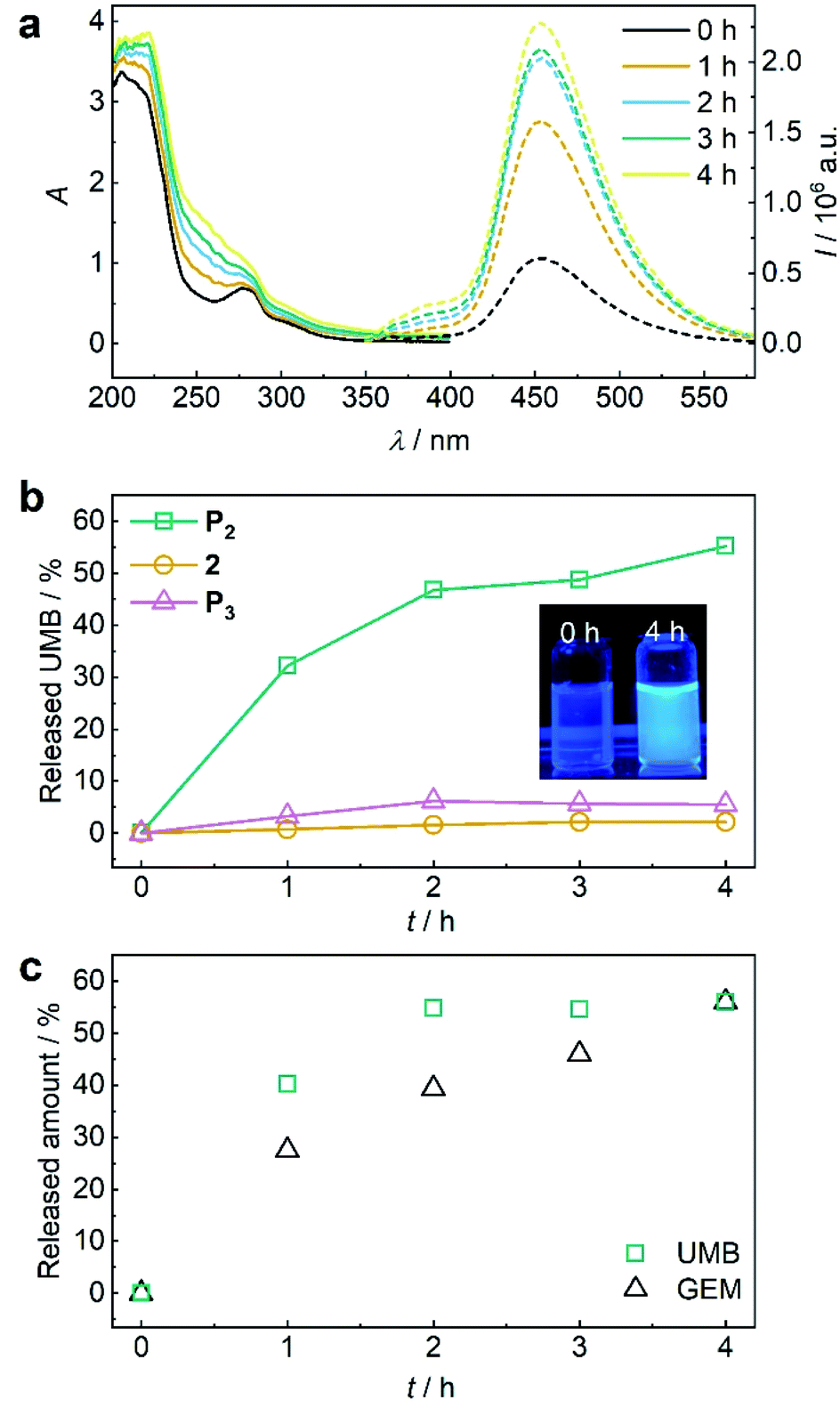 | ||
| Fig. 5 US-induced release of UMB and GEM from β-carbonate disulfide polymer P2 (3 mg mL−1) and control 2 (25 μM) in MeCN:H2O = 2:3 (v/v). (a) UV-vis and fluorescence (λexc = 325 nm) spectra of P2 during sonication. (b) Released UMB from P2 and controls 2 and P3 during sonication calculated from fluorescence peak intensity of UMB at λem = 450 nm. Inset: photograph of the solutions under UV-light (λexc = 365 nm). (c) Released amount of UMB and GEM over time visualized by PDA detector signal intensities from UPLC elugrams (Fig. S46†). | ||
While the mechanochemically induced release of UMB was readily visible, GEM release was confirmed by UPLC-MS (Fig. S45†). We again normalized the integrated DAD signals from UPLC-MS measurements during GEM and UMB release to the maximum obtainable released fraction UMB of 55% as largest value (Fig. 5c and S46†). While UMB release proceeds at an increased rate compared to GEM, the suitability of UMB as a diagnostic probe for the drug GEM was still underlined by these results.
Conclusions
In conclusion, we here presented a versatile method to mechanochemically induce theranostic drug activation where a pharmacologically active moiety alongside an indicative fluorescent reporter molecule is released. We demonstrated the versatility of this platform with the drug-probe combinations camptothecin and N-butyl-4-hydroxy-1,8-naphthalimide as well as gemcitabine and umbelliferone. We anticipate that the cargo attachment process via β-carbonate linkers accompanied by a central disulfide bond in principle is useful for many drug-probe combinations containing nucleophilic hydroxy- or amino-groups. In the future, this novel methodology will be developed towards the compatibilization with clinically established US techniques38,39 and energy doses,40 the convergence of mechanochemical scission rates and yields (i.e. released amount of drugs) with medically tolerable timeframes avoiding US-specific side effects,41 and the stability and inertness42 of the carrier systems towards small endogenous reactants, e.g. GSH.19Author contributions
Z. S., R. G., and A. H. conceived the project. Z. S., R. G., and A. H. wrote the manuscript. Z. S. and Q. S. performed synthesis and analysis experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Union (European Research Council Advanced Grant SUPRABIOTICS, No. 694610). R. G. is grateful for support by a Freigeist-Fellowship of the Volkswagen Foundation (No. 92888). Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine-Westphalia (No. 300088302). Financial support is acknowledged from the European Commission (EUSMI, No. 731019).Notes and references
- R. Jones, Medicine, 2016, 44, 25–29 CrossRef.
- C. Twelves, M. Jove, A. Gombos and A. Awada, Crit. Rev. Oncol. Hematol., 2016, 100, 74–87 CrossRef PubMed.
- S. Kunjachan, B. Rychlik, G. Storm, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2013, 65, 1852–1865 CrossRef CAS PubMed.
- C. M. Lopes, P. Barata and R. Oliveira, in Drug Targeting and Stimuli Sensitive Drug Delivery Systems, Elsevier, 2018, pp. 155–209 Search PubMed.
- E. Galbis, M.-V. De Paz, N. Iglesias, R. Lucas and J. A. Galbis, J Drug Des. Res., 2017, 4, 1047 Search PubMed.
- X. Chuan, Q. Song, J. Lin, X. Chen, H. Zhang, W. Dai, B. He, X. Wang and Q. Zhang, Mol. Pharm., 2014, 11, 3656–3670 CrossRef CAS PubMed.
- Y. C. Wang, F. Wang, T. M. Sun and J. Wang, Bioconjugate Chem., 2011, 22, 1939–1945 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567–4572 CrossRef CAS PubMed.
- S. E. Ahmed, A. M. Martins and G. A. Husseini, J. Drug Targeting, 2015, 23, 16–42 CrossRef CAS PubMed.
- F. Kiessling, S. Fokong, P. Koczera, W. Lederle and T. Lammers, J. Nucl. Med., 2012, 53, 345–348 CrossRef CAS PubMed.
- K. R. Lattwein, H. Shekhar, J. J. P. Kouijzer, W. J. B. van Wamel, C. K. Holland and K. Kooiman, Ultrasound Med. Biol., 2020, 46, 193–215 CrossRef PubMed.
- I. Rosenthal, J. Z. Sostaric and P. Riesz, Ultrason. Sonochem., 2004, 11, 349–363 CrossRef CAS PubMed.
- G. Cravotto, E. C. Gaudino and P. Cintas, Chem. Soc. Rev., 2013, 42, 7521–7534 RSC.
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS PubMed.
- C. L. Brown and S. L. Craig, Chem. Sci., 2015, 6, 2158–2165 RSC.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1418 CrossRef CAS.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS PubMed.
- W. Li and F. Gräter, J. Am. Chem. Soc., 2010, 132, 16790–16795 CrossRef CAS PubMed.
- P. Dopieralski, J. Ribas-Arino, P. Anjukandi, M. Krupicka, J. Kiss and D. Marx, Nat. Chem., 2013, 5, 685–691 CrossRef CAS PubMed.
- B. Lee, Z. Niu, J. Wang, C. Slebodnick and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 10826–10832 CrossRef CAS PubMed.
- F. Wang, M. Burck and C. E. Diesendruck, ACS Macro Lett., 2017, 6, 42–45 CrossRef CAS.
- U. F. Fritze, S. L. Craig and M. von Delius, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1404–1411 CrossRef CAS.
- U. F. Fritze and M. Von Delius, Chem. Commun., 2016, 52, 6363–6366 RSC.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed.
- R. P. Hertzberg, M. J. Caranfa and S. M. Hecht, Biochemistry, 1989, 28, 4629–4638 CrossRef CAS PubMed.
- M. H. Lee, J. H. Han, P. S. Kwon, S. Bhuniya, J. Y. Kim, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 1316–1322 CrossRef CAS PubMed.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668–12674 CrossRef CAS PubMed.
- T. D. Nalder, T. D. Ashton, F. M. Pfeffer, S. N. Marshall and C. J. Barrow, Biochimie, 2016, 128–129, 127–132 CrossRef CAS PubMed.
- R. Zhang, X. Qin, F. Kong, P. Chen and G. Pan, Drug Delivery, 2019, 26, 328–342 CrossRef CAS PubMed.
- D. Zhang, L. Li, X. Ji and Y. Gao, New J. Chem., 2019, 43, 18673–18684 RSC.
- J. H. Beumer, J. L. Eiseman, R. A. Parise, E. Joseph, J. M. Covey and M. J. Egorin, Clin. Cancer Res., 2008, 14, 3529–3535 CrossRef CAS PubMed.
- J. M. Reid, W. Qu, S. L. Safgren, M. M. Ames, M. D. Krailo, N. L. Seibel, J. Kuttesch and J. Holcenberg, J. Clin. Oncol., 2004, 22, 2445–2451 CrossRef CAS PubMed.
- R. Moog, A. Burger, M. Brandl, J. Schüler, R. Schubert, C. Unger, H. Fiebig and U. Massing, Cancer Chemother. Pharmacol., 2002, 49, 356–366 CrossRef CAS PubMed.
- J. J. Aaron, M. Buna, C. Parkanyi, M. S. Antonious, A. Tine and L. Cisse, J. Fluoresc., 1995, 5, 337–347 CrossRef CAS PubMed.
- C. P. Phenix, M. Togtema, S. Pichardo, I. Zehbe and L. Curiel, J. Pharm. Pharm. Sci., 2014, 17, 136–153 Search PubMed.
- F. Kiessling, S. Fokong, J. Bzyl, W. Lederle, M. Palmowski and T. Lammers, Adv. Drug Delivery Rev., 2014, 72, 15–27 CrossRef CAS PubMed.
- S. Mitragotri, Nat. Rev. Drug Discovery, 2005, 4, 255–260 CrossRef CAS PubMed.
- W. L. Nyborg, Ultrasound Med. Biol., 2001, 27, 301–333 CrossRef CAS PubMed.
- Y. Zhou, S. Huo, M. Loznik, R. Göstl, A. J. Boersma and A. Herrmann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010324.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic, spectroscopic, kinetic, and cell culture procedures. See DOI: 10.1039/d0sc06054b |
| This journal is © The Royal Society of Chemistry 2021 |