
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ortho-aryl substituted DPEphos ligands: rhodium complexes featuring C–H anagostic interactions and B–H agostic bonds†
James J.
Race
 ab,
Arron L.
Burnage
c,
Timothy M.
Boyd
ab,
Alex
Heyam
b,
Antonio J.
Martínez-Martínez
b,
Stuart A.
Macgregor
*c and
Andrew S.
Weller
*a
ab,
Arron L.
Burnage
c,
Timothy M.
Boyd
ab,
Alex
Heyam
b,
Antonio J.
Martínez-Martínez
b,
Stuart A.
Macgregor
*c and
Andrew S.
Weller
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: andrew.weller@york.ac.uk
bChemistry Research Laboratories, University of Oxford, Oxford OX1 3TA, UK
cInstitute of Chemical Sciences, Heriot Watt University, Edinburgh EH14 4AS, UK. E-mail: S.A.Macgregor@hw.ac.uk
First published on 25th May 2021
Abstract
The synthesis of new Schrock–Osborn Rh(I) pre-catalysts with ortho-substituted DPEphos ligands, [Rh(DPEphos-R)(NBD)][BArF4] [R = Me, OMe, iPr; ArF = 3,5-(CF3)2C6H3], is described. Along with the previously reported R = H variant, variable temperature 1H NMR spectroscopic and single-crystal X-ray diffraction studies show that these all have axial (C–H)⋯Rh anagostic interactions relative to the d8 pseudo square planar metal centres, that also result in corresponding downfield chemical shifts. Analysis by NBO, QTAIM and NCI methods shows these to be only very weak C–H⋯Rh bonding interactions, the magnitudes of which do not correlate with the observed chemical shifts. Instead, as informed by Scherer's approach, it is the topological positioning of the C–H bond with regard to the metal centre that is important. For [Rh(DPEphos–iPr)(NBD)][BArF4] addition of H2 results in a Rh(III) iPr–C–H activated product, [Rh(κ3,σ-P,O,P-DPEphos-iPr′)(H)][BArF4]. This undergoes H/D exchange with D2 at the iPr groups, reacts with CO or NBD to return Rh(I) products, and reaction with H3B·NMe3/tert-butylethene results in a dehydrogenative borylation to form a complex that shows both a non-classical B–H⋯Rh 3c-2e agostic bond and a C–H⋯Rh anagostic interaction at the same metal centre.
Introduction
Diphosphine chelates that contain an ether linkage in their backbone, such as DPEphos and xantphos, are an important and popular class of ligand that are used in synthesis and catalysis (Fig. 1A). Initially developed as wide bite-angle, κ2-P,P-cis-coordinating, ligands for Rh-based hydroformylation catalysis,1,2 such ligands also have the ability to act in κ3-P,O,P binding modes often leading to hemilabile3 behaviour through reversible coordination of the ether linkage in response to changes in the metal coordination sphere or oxidation state. DPEphos is now widely used in a variety of catalytic settings,4–7 and the vast majority of applications make use of the commercially available phenyl phosphine derivative. Modification of aryl phosphine ligands, more generally, by introducing steric bulk using ortho-substitution has been shown to promote enantioselectivity;8 regioselectivity;9 overall efficiency and catalyst stability;10–13 as well as aryl-group restricted rotation.14 Despite these potential advantages, ortho-substituted variants of DPEphos (or xantphos) are rare, Fig. 1B, and their use limited to a handful of examples.11,15–19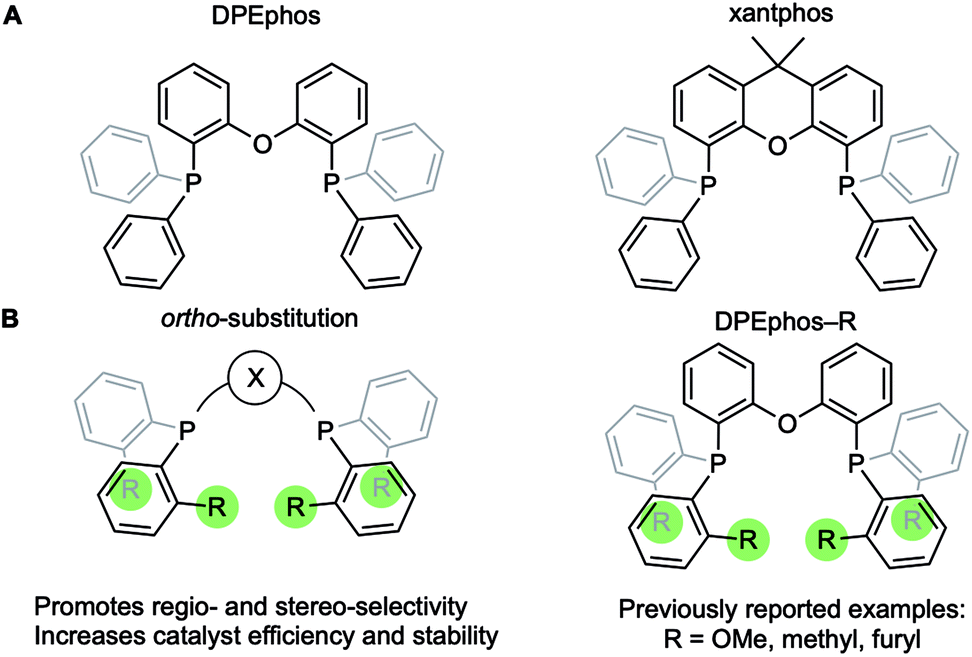 | ||
| Fig. 1 (A) Xantphos and DPEphos ligands. (B) Ortho-aryl substitution. | ||
The cationic Schrock–Osborn [Rh(chelating-phosphine)]+ system is widely used in catalysis and synthesis,20,21 and the active species are often accessed via hydrogenation of a suitable diene precursor, such as [Rh(chelating-phosphine)(NBD)][anion] (NBD = norbornadiene), in a coordinating solvent such as acetone. We have particular interest in such systems with the DPEphos ligand, with regard to their use as pre-catalysts for amine-borane dehydropolymerisation,22,23 alkene and alkyne hydroacylation,24–26 and alkyne carbothiolation,27 amongst other applications. We now report the synthesis of new Schrock–Osborn systems with ortho-substituted DPEphos ligands, including a new iPr-substituted ligand (Fig. 2). A detailed structural, variable-temperature spectroscopic, and computational study reveals these to show well-defined examples of anagostic C–H⋯Rh interactions,28,29 even for the previously-reported24 parent DPEphos complex; while a reactivity study demonstrates intramolecular C–H activation can occur after hydrogenation of the NBD ligand, that is dependent on the identity of the R-group. Reaction of such a cyclometallated complex with H3B·NMe3 leads to a dehydrogenative borylation and a complex that features both non-classical B–H 3c-2e agostic28 and anagostic C–H structural and spectroscopic features, Fig. 2B. This serves to highlight the key differences between anagostic and agostic motifs of E–H bonds with d8-metal centres in a single complex.
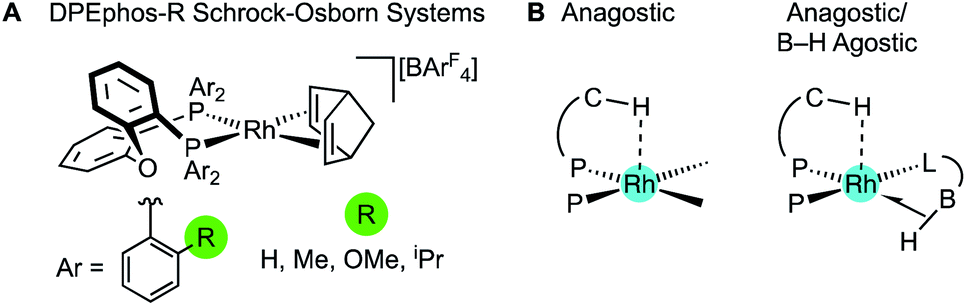 | ||
| Fig. 2 (A) [Rh(DPEphos-R)(NBD)][BArF4] systems, and (B) schematic examples of the C–H anagostic interactions and 3c-2e B–H agostic bonds, reported in this contribution. | ||
In describing the anagostic interactions in these systems we borrow from the analysis of Scherer30 who showed that axial positioning of a C–H bond at a square-planar d8 metal centre orientates it over a region of charge concentration. When the complex is then placed in a magnetic field (i.e., the NMR experiment) induced current density at the metal results in magnetic field effects that cause the signature downfield chemical shift of the anagostic proton. In our analysis we find that descriptors that define the bonding between the Rh centres and C–H bonds show no correlation with either the observed or computed chemical shifts, supporting Scherer's topological, induced current, description for anagostic interactions.
Results and discussion
Synthesis and solid-state structures of the NBD complexes
The ortho-substituted DPEphos-R ligands used in this study are shown in Fig. 3: R = H, 1-H; Me, 1-Me; OMe, 1-OMe; and iPr, 1-iPr. Ligands 1-H and 1-Me are commercially available, 1-OMe was prepared using the reported procedure.17 DPEphos-iPr, 1-iPr, is a new ligand and was prepared as an analytically pure white solid from reaction of the corresponding dichlorophosphine with ortho-isopropyl phenyl lithium (ESI†). The solid-state structure is shown in Fig. 3. In the room temperature 31P{1H} NMR spectrum a single 31P environment is observed at δ −37.6.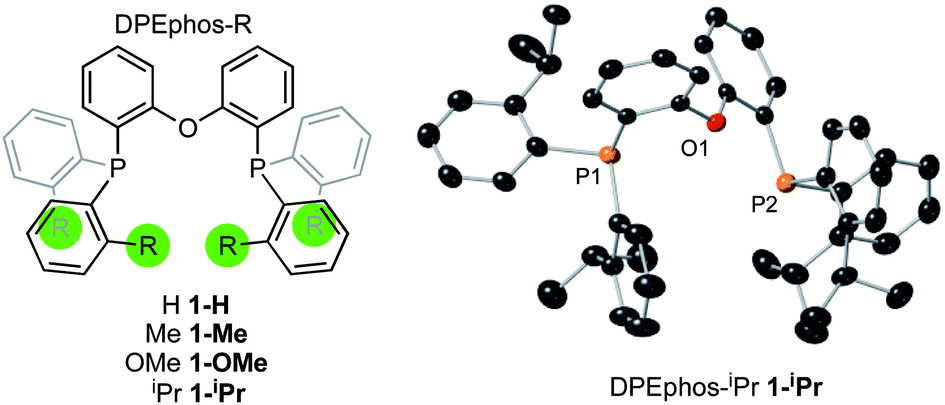 | ||
| Fig. 3 DPEphos-R ligands used in this study. Crystallographically determined structure of 1-iPr. Ellipsoids shown at the 50% probability level. Hydrogen atoms omitted for clarity. See ESI† for full details. | ||
Interestingly, the room temperature 1H NMR spectrum is rather simple with only a single (integral 24 H) environment observed for the iPr-methyl groups – despite their diastereotopic nature in the solid-state structure. This suggests inversion at P is a low energy process for free 1-iPr,31 which has been shown to be the case for other bulky iPr-substituted tris-aryl phosphines.32
The target, Schrock–Osborn, [Rh(DPEphos-R)(NBD)][BArF4] complexes [ArF = 3,5-(CF3)2C6H3] were prepared by addition of the DPEphos-R ligands to the appropriate Rh-precursor. [Rh(DPEphos-H)(NBD)][BArF4], 2-H, has already been reported to be formed from addition of 1-H to [Rh(NBD)Cl]2, using Na[BArF4] to extract the halide (Scheme 1).24 A slightly refined method, using 1,2-F2C6H4 as a solvent, was used to make [Rh(DPEphos-R)(NBD)][BArF4], R = H, 2-H; Me, 2-Me; and OMe, 2-OMe. For the bulkier ligand, 1-iPr, [Rh(NBD)2][BArF4] was used to make 2-iPr. The new complexes were isolated in moderate to good yield (65 to 85%), as crystalline solids. Fig. 4 shows the solid-state structures of the cations in these new complexes as determined by single-crystal X-ray diffraction. While 2-H is known,24 the solid-state structure had not been reported, and so is included here. All the cations have pseudo square planar Rh(I) centres, with the NBD ligands binding η2η2, and cis-κ2-P,P DPEphos-R ligands. Bond lengths and angles are generally unremarkable (ESI†). The closest Rh⋯O distance in 2-OMe is 3.081(3) Å from an axially-orientated methoxyl group – which is clearly non-bonding.
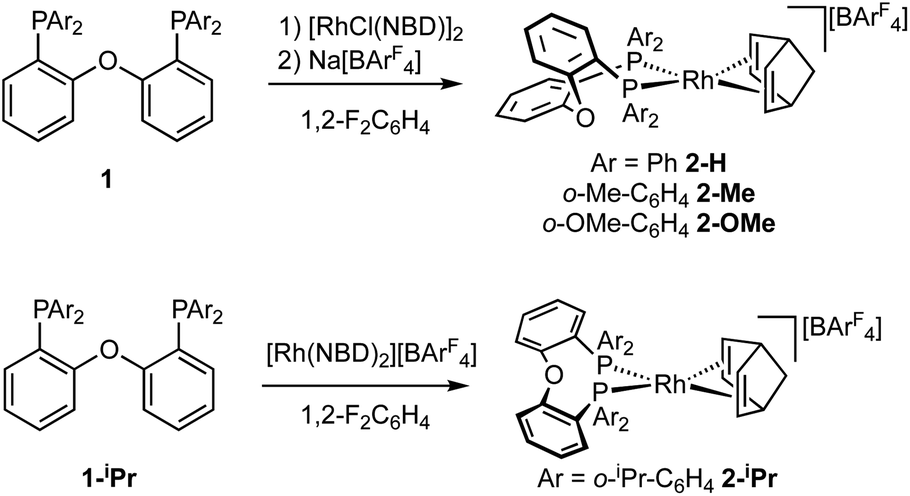 | ||
| Scheme 1 Synthesis of the new Rh-complexes. | ||
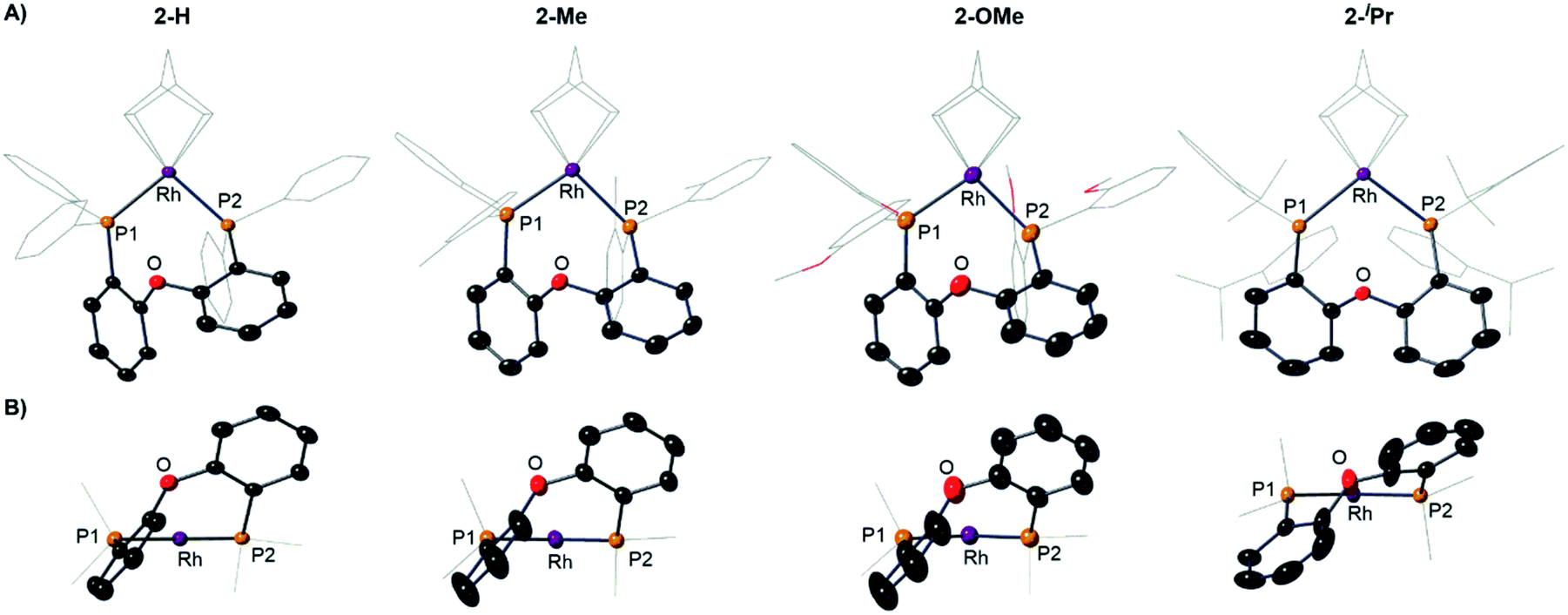 | ||
| Fig. 4 (A) Solid-state structures of the cations in 2-H, 2-Me, 2-OMe and 2-iPr as determined by single-crystal X-ray diffraction. Displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and [BArF4]− anions not shown. Selected DPEphos-R and NBD groups shown as wireframe. (B) End-on view highlighting the relative orientation of the DPEphos backbones. Bond lengths and angles are given in the ESI (Table S2).† | ||
Notable differences, however, come from the relative orientation of the DPEphos-R diphenylether backbone, Fig. 4B. For 2-H, 2-Me and 2-OMe this lies above the P–Rh–P plane sitting in an asymmetric envelope-like conformation.33 If retained in solution this would give the cation C1 symmetry (i.e. none). The iPr groups in 2-iPr force a, non-crystallographic, C2-axis. Reflecting the increase in steric bulk, the Rh–P distances are ∼0.1 Å longer and the P–Rh–P bite angle ∼ 3° wider in 2-iPr compared with the other complexes (Table S2†). In all cases the DPEphos ether oxygen atom sits distant from the Rh-centre [3.498(8)–3.5545(18) Å]. For all, there are aryl or methyl C–H bonds in the ortho-aryl groups that are axially positioned above the Rh-square plane, i.e. potential anagostic interactions. These are discussed in detail after the solution NMR spectroscopic data have been presented that signal this orientation.
Variable temperature solution NMR spectroscopy and the identification of anagostic interactions in solution and solid-state
Room temperature NMR spectra of the Rh–NBD complexes indicate fluxional behaviour in solution that is dependent on the identity of the phosphine ancillary group. For 2-H24 a very simple, sharp, set of signals is observed for the room temperature 1H NMR spectrum (i.e., a single NBD alkene environment), along with a single environment in the 31P{1H} NMR spectrum. Together these indicate time averaged C2v symmetry in solution. For 2-Me broad signals are observed in both the 1H NMR and 31P{1H} NMR spectra, with the latter showing two species: one with a single 31P environment and one with inequivalent environments. For 2-OMe the situation is similar, except that only one – very broad – environment is observed in the 31P{1H} NMR spectrum. These data, in comparison with the solid-state structures, suggest fluxional processes are operative in solution that are fast for 2-H, but slower for 2-Me and 2-OMe and also involve observable equilibrium populations of different conformers. For bulky 2-iPr the NMR spectra are again sharp, but now indicate C2, rather than C2v, symmetry for the NBD (four signals) and DPEphos-iPr (two methine, four CH3 and one 31P environment) ligands via1H and 31P{1H} NMR spectroscopy. In the low-field region of the 1H NMR spectrum of 2-iPr a distinct, relative integral 2H, signal is observed at δ 9.34 that shows coupling to P and H [J(PH) = 17, J(HH) = 7 Hz], Fig. 5A. There is no evidence for Rh–H coupling.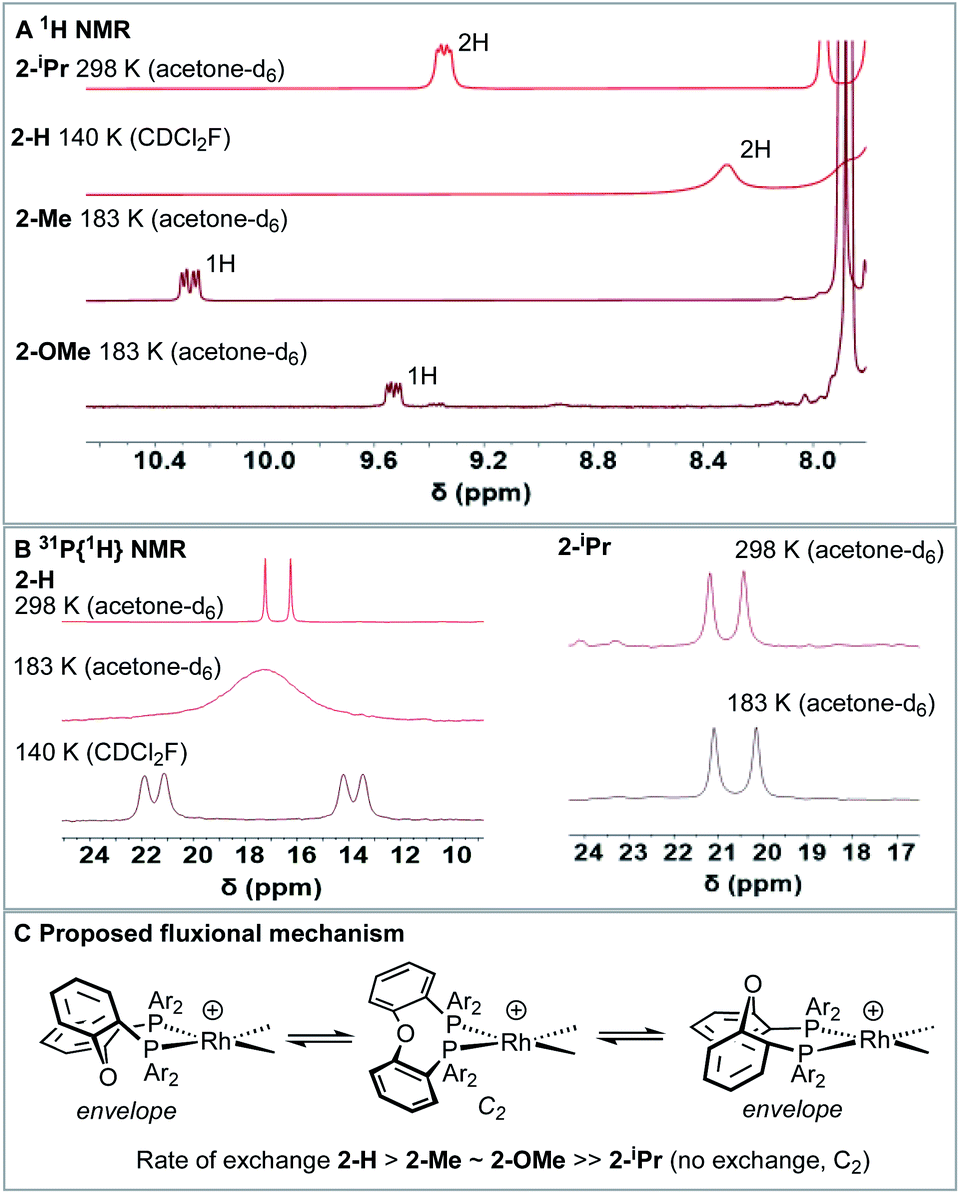 | ||
| Fig. 5 (A) Low-field (δ 7.8–10.5) region of the 1H NMR spectra for the [Rh(DPEphos-R)(NBD)][BArF4] complexes showing the shifted signals (temperature and solvent as noted) (B) 31P{1H}NMR spectra for 2-H and 2-iPr at various temperatures. (C) Proposed fluxional process. | ||
While such downfield shifted signals are not observed in the room temperature 1H NMR spectra of the other complexes, progressive cooling to much lower temperatures reveals similarly shifted peaks and corresponding changes in the 31P NMR spectra. For 2–H cooling to 183 K (acetone-d6) results in very broad signals in the 1H NMR spectrum, suggesting the low temperature limit had not been reached. By using CDCl2F34 as a solvent a 1H NMR spectrum could be obtained at 140 K in which a low-field shifted, albeit broad, signal (2H) is observed at δ 8.32. For 2-Me and 2-OMe similar behaviour is observed on cooling but now 243 K and 203 K, respectively, are sufficient to reveal downfield-shifted aromatic resonances.35 However, these integrate to only 1H each, at δ 10.27 and δ 9.53 respectively (in acetone-d6, 183 K). 2-Me also shows a downfield shifted methyl resonance at δ 3.68 (3H, 183 K). For 2-Me and 2-OMe four different NBD alkene environments are observed in the low temperature 1H NMR spectra, along with two mutually coupled signals in the corresponding 31P{1H} NMR spectra [e.g. J(PP) = 28 Hz 2-Me] that also couple to 103Rh. For 2-H these signals are broad even at 140 K (fhwm = 80 Hz) and the 31P–31P coupling is not resolved, Fig. 5B. These data point to fluxional processes that are arrested, or considerably slowed, at low temperature to give structures that are similar to those determined in the solid-state, i.e. an envelope-like conformation of the DPEphos-R ligand. On increasing the temperature, conversion between enantiomeric C1 forms via a C2 intermediate is proposed, Fig. 5C. This has been modelled for 2-Me using line-shape analysis (see ESI†). Related ring-flipping processes in POP-type ligands have been reported previously.36,37 For 2-iPr there is no change on cooling (Fig. 5B), the ∼C2-symmetric solid-state structure is retained in solution at room temperature. It is thus not fluxional. These observations are consistent with relative steric bulk of the o-substituents: 1-H < 1-Me ∼ 1-OMe ≪ 1-iPr. Downfield chemical shifts in the 1H NMR spectrum can be diagnostic of anagostic C–H interactions, which are located above a region of charge concentration at a d8 metal centre, i.e. an occupied dz2 orbital.30,38–40 These are distinct from agostic,28 3c-2e, bonds that are characterised by donation from a C–H bond into an unoccupied metal orbital and upfield chemical shifts in the 1H NMR spectrum. The fluxional processes operating at room temperature mean these characteristic signals are only resolved on cooling, apart from for 2-iPr in which the static structure makes them persistent. We next turn to inspecting the solid-state structures of the NBD adducts more closely to identify such anagostic interactions: Fig. 6 and Table 1.
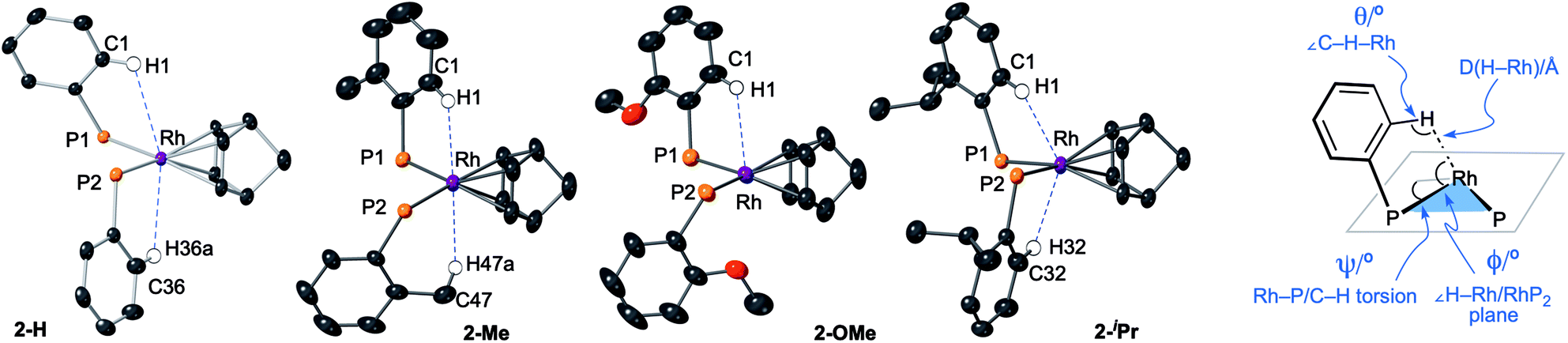 | ||
| Fig. 6 Views of the NBD complexes highlighting the close C–H⋯Rh, anagostic, interactions with selected structural markers. Diphenyl ether linkages on the DPEphos-R ligands are not shown. Hydrogen atoms are placed in calculated positions. | ||
| 2-H | 2-Me | 2-OMe | 2-iPr | |
|---|---|---|---|---|
| a See Fig. 6 for definitions. b Difference in chemical shift of H1 (500 MHz, CD2Cl2, 203 K) compared with free ligand (CD2Cl2, 295 K). c Numbers in parenthesis associated with methyl groups. d The ortho phenyl protons in DPEphos-H could not be unambiguously identified. | ||||
| Θ (°)a | 114.7, 122.6 | 129.8, (144.2)c | 121.6 | 132.7, 135.6 |
| Ψ (°)a | 42.0, 1.7 | 1.3c | −6.2 | −1.4, −8.2 |
| Φ (°)a | 63.1, 58.0 | 64.3, (69.3)c | 63.9 | 64.3, 64.3 |
| Rh⋯H1 (Å) | 2.92, 2.97 | 2.57, (2.63)c | 2.88 | 2.58, 2.47 |
| δ(H) (ppm) | 8.32 | 9.97, (3.56)c | 9.19 | 9.14 |
| Δδ(H) (ppm)b | +0.99 to 1.11d | +2.82, (+1.3)c | +2.34 | +1.85 |
| J(PH) (Hz) | Broad | 17 | 17 | 18 |
| J(HH) (Hz) | Broad | 8 | 7 | 8 |
All four complexes show relatively close C–H⋯Rh approaches from an ortho C(aryl)–H group in the phenyl phosphine (H atoms in calculated positions, see Table 2 for computational analysis). For 2-H there are two, albeit long (∼2.9 Å); for 2-iPr there are also two, but these are considerably shorter (∼2.5 Å); while 2-OMe has a single close C(aryl)–H⋯Rh distance (∼2.9 Å). 2-Me shows two different types: C(aryl)–H⋯Rh (2.57 Å), and C(Me)–H⋯Rh (2.63 Å). The phenyl rings associated with these C(aryl)–H⋯Rh contacts generally align with the associated Rh–P vector (C–H/Rh–P torsion angles, Ψ, 8.2 to 1.3°) and the C–H⋯Rh angle (θ) is rather open (121.6–144.2°). Although 2-H has one phenyl ring twisted away from this (Ψ = 42.0, θ = 114.7°), the Rh⋯H distance is similar. The number of these close C–H⋯Rh distances correlates well with relative integrals of the downfield shifted signals observed in the 1H NMR spectra: 2-H, 2H; 2-Me, 1H (aryl), 3H (methyl); 2-OMe, 1H; and 2-iPr, 2H. As there is no crystallographically imposed symmetry in the solid-state we assume any equivalent environments observed in solution arise from very low energy fluxional processes. The changes in chemical shifts of these C–H protons due to the presence of the Rh(I) centre have been experimentally determined by comparison with the free ligands, as aided by 1H/1H COSY, HMBC and HSQC experiments. While all shift downfield, the variation observed shows no strong correlation with any of the structural descriptors discussed above, as detailed in Table 1. However, in a more general sense, for all the complexes the angle formed between the RhH vector and the RhP2 plane (ø) shows the C–H proton is orientated towards the apical position (which at the limit ø = 90°). Thus, following Scherer's analysis,30 the positioning of the C–H bond over a region of charge concentration (occupied d orbitals, ø approaching 90°) induces the downfield chemical shift in the NMR spectrum that is diagnostic of an anagostic interaction. In contrast, orientation of a C–H bond toward a charge depleted region (a vacant orbital in the metal coordination plane, ø approaching 0°) results in upfield-shifted signals that are characteristic of agostic, 3c-2e, bonding. Such demarcations are not always clear-cut, however, as axial sites can also display Lewis-acidic character.29,41
| Cation | Bond path | Distance/Å | QTAIM (au) | NBO donor–acceptor interactions (kcal mol−1) | NMR/ppm | ||||
|---|---|---|---|---|---|---|---|---|---|
| ρ(r) | ∇2ρ(r) | q H | σC–H → Rhb |
|
δ(H)calcd | δ(H)exp | |||
a QTAIM and NBO data are based on the experimental crystal structures; computed chemical shifts are based on the lowest energy conformations.45
b Sum of donation into the two 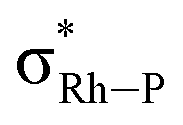 NBOs.
c Sum of donation from the Rh lone pairs and σRh–P bonding NBOs.
d Data are weighted averages taking into account all low energy conformations.
e Average of all three Me hydrogens. See ESI for full details. NBOs.
c Sum of donation from the Rh lone pairs and σRh–P bonding NBOs.
d Data are weighted averages taking into account all low energy conformations.
e Average of all three Me hydrogens. See ESI for full details.
|
|||||||||
| [2-H]+ | Rh⋯H1 | 2.83 | 0.012 | +0.036 | 0.031 | 0.57 | 1.33 | +9.5 | +8.32 |
| Rh⋯H36a | 2.87 | 0.011 | +0.030 | 0.028 | 0.52 | 1.22 | +9.1 | ||
| [2-Me]+ | Rh⋯H1 | 2.45 | 0.022 | +0.053 | 0.026 | 0.69 | 4.38 | +10.6 | +9.97 |
| Rh⋯H47a | 2.51 | 0.020 | +0.045 | 0.027 | 0.49 | 4.29 | +6.0 (+3.9e) | +3.56 | |
| [2-OMe]+ | Rh⋯H1 | 2.79 | 0.013 | +0.035 | 0.047 | 0.33 | 1.91 | +9.6 | +9.19 |
| [2-iPr]+ | Rh⋯H1 | 2.33 | 0.026 | +0.059 | 0.024 | 2.08 | 8.98 | +9.9 | +9.14 |
| Rh⋯H32 | 2.45 | 0.021 | +0.050 | 0.027 | 1.71 | 6.70 | +9.8 | ||
While with hindsight it is not surprising that the most sterically bulky ligand, DPEphos–iPr, enforces an anagostic interaction at room temperature, the presence of both aryl and, rarer,42,43 alkyl anagostic interactions in 2-Me is perhaps more notable. What was unanticipated is that in the parent DPEphos–H complex such interactions are also present – albeit only observed at very low temperature in solution. Similar properties (C–H⋯M, 2.23–3.01 Å, low-field chemical shifts and apical approaches of C–H groups to d8 metal centres), have been discussed by others, including: Bergman,44 Dyker,42 Fairlamb,45 and Sabo-Etienne,38Fig. 7.
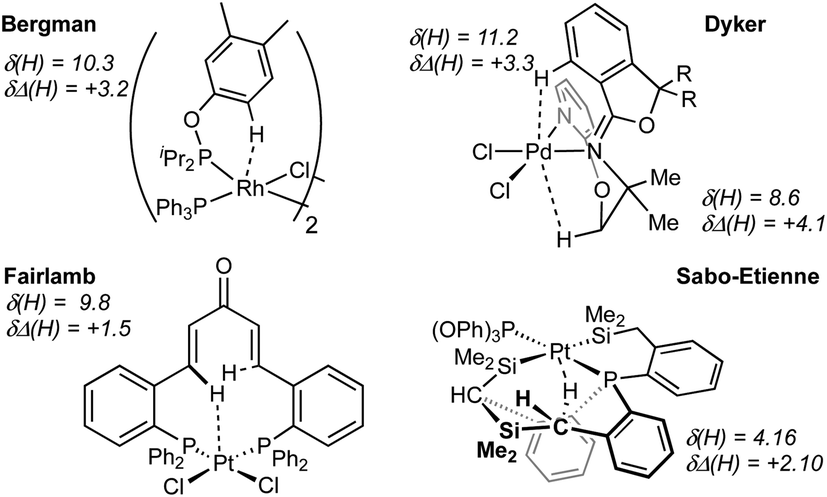 | ||
| Fig. 7 Examples of previously reported anagostic C–H⋯M interactions. | ||
So, while the presence of anagostic C–H⋯Rh(I) interactions has been demonstrated here experimentally by both structural and spectroscopic studies, the correlation between the observed chemical shifts and measured structural descriptors is less obvious. We thus turned to a computational analysis to examine the nature of these anagostic C–H⋯Rh(I) interactions more closely.
Computational studies: structures, bonding and chemical shifts
Computed metrics for the Rh⋯H–C moieties in the isolated cations of all four DPEphos-R complexes are provided in Table 2. Geometries for these analyses are based on the experimental structures with the heavy atoms fixed at their observed positions and the H atoms optimised. The calculated Rh⋯H distances are therefore ca. 0.1 Å shorter (and the C–H bonds ca. 0.15 Å longer) than those determined experimentally. Fig. 8 displays the molecular graph, the topology of the Laplacian and a non-covalent interaction (NCI) plot for the cationic portion of 2-Me, [2-Me]+, where we have chosen to showcase the system featuring both aryl- and alkyl-C–H⋯Rh anagostic interactions. The presented data are representative of all four cations and equivalent figures for the remaining systems are provided in the ESI.† The bond critical point (BCP) metrics indicate the presence of weak Rh⋯H–C interactions with low BCP electron densities, ρ(r), small positive values for the Laplacian and small, positive charges on the anagostic H atoms. In [2-Me]+ the Rh⋯H47a (alkyl) interaction is slightly weaker than the Rh⋯H1 (aryl) interaction, although this likely reflects the longer Rh⋯H47a distance rather than any intrinsic difference. Plots of ρ(r) and ∇2ρ(r) against the computed RhH distances provide excellent correlations (Fig. S42 and S43†) and the strongest Rh⋯H–C interactions are seen in [2-iPr]+. This is mirrored in the NBO 2nd order perturbation analyses that show the major component, donation, to increase upon shortening the Rh⋯H distance. σC–H → Rh donation shows a similar trend but this is minimal, even in [2-iPr]+. This weak Rh⋯H–C interaction therefore shares some characteristics of a H-bond46,47 and this is also evident in the NCI plot of [2-Me]+ where light turquoise (i.e. weakly stabilising) regions are seen along the Rh⋯H1 and Rh⋯H47a vectors.
donation, to increase upon shortening the Rh⋯H distance. σC–H → Rh donation shows a similar trend but this is minimal, even in [2-iPr]+. This weak Rh⋯H–C interaction therefore shares some characteristics of a H-bond46,47 and this is also evident in the NCI plot of [2-Me]+ where light turquoise (i.e. weakly stabilising) regions are seen along the Rh⋯H1 and Rh⋯H47a vectors.
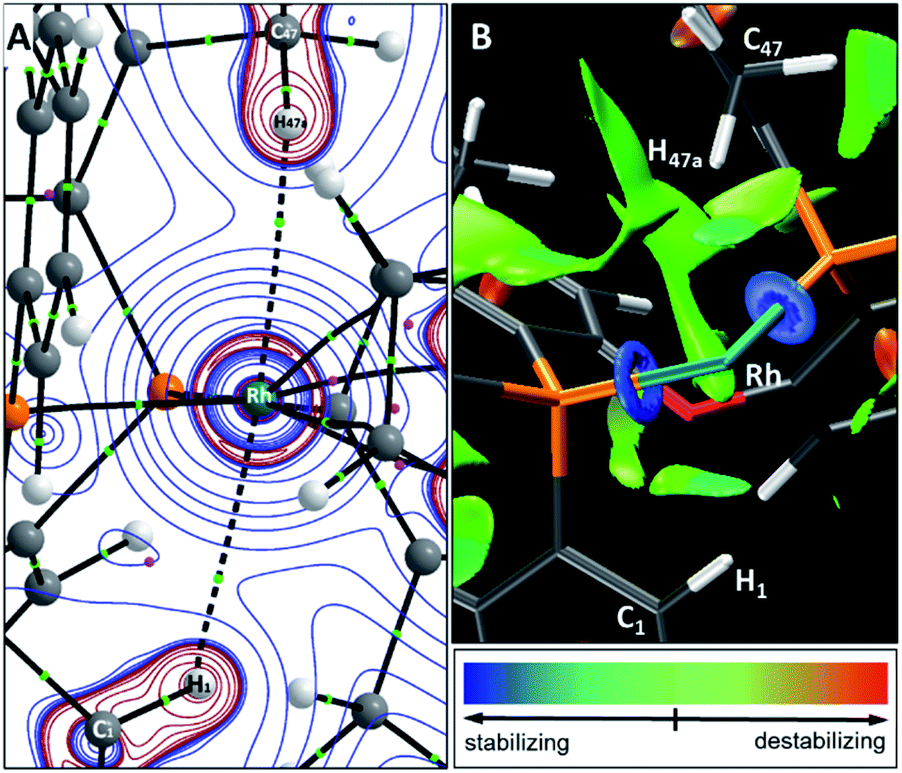 | ||
| Fig. 8 (A) Molecular graph of the [2-Me]+ cation showing the contour plot of the Laplacian in the H47aRhH1 plane. Bond critical points and ring critical points are shown as green and pink spheres respectively; blue contours show areas of charge depletion, red contours charge accumulation; (B) non-covalent interaction plot highlighting weak stabilising Rh⋯H1 and Rh⋯H47a interactions; the NBD ligand is removed for clarity and the isosurface is generated for σ = 0.3 au and −0.07 < ρ < 0.07 au. Key shows isosurface colouring. | ||
The Laplacian plot around the Rh atom in [2-Me]+ indicates that both the Rh⋯H1 and Rh⋯H47a bond paths pass through regions of axial charge concentration. Thus both C–H bonds are oriented towards areas of charge accumulation at Rh, consistent with the downfield 1H anagostic chemical shift.30 Computed 1H NMR chemical shifts reproduce these downfield shifts for all four cations. In this case the calculations were performed on the fully optimised structures to model behaviour in solution. In general, the computed chemical shifts lie further downfield than the experimental values. The largest discrepancy is for [2-H]+ and this may reflect that the low temperature limit had not been achieved experimentally. In addition, conformational searching revealed additional low energy structures that also contribute to the final observed chemical shift.48 For [2-Me]+ the static structure in the calculations reveals the large downfield shift associated with the Me proton H47a (δcalc = +6.0 ppm) while the average of all three Me protons is 3.9 ppm, in good agreement with experiment (δ 3.56) where the methyl group will be freely rotating leading to a weighted-average chemical shift.
Interestingly, although there is a clear relationship between the Rh⋯H–C distance and the computed bonding metrics, no such correlation is seen with the computed chemical shifts of the anagostic hydrogens (Table 2). Thus, the nature of the Rh⋯H–C interaction does not relate to the extent of the downfield chemical shift, suggesting the orbitals involved are not responsible for the chemical shift. Instead the situation is more consistent with Scherer's observations30 that it is the spatial positioning of the anagostic H above the d8 square-planar metal coordination plane (i.e. ø) together with the complex interplay of induced current densities that are responsible for the precise chemical shift observed. Thus while the computation of a weak M⋯H bond path and weak 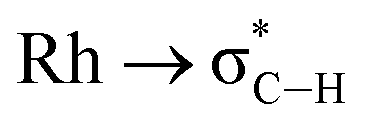 donation are usually features that are associated with an anagostic interaction,40 they are not in themselves responsible for the signature downfield chemical shifts observed in NMR spectra that signal the positioning of the C–H bond relative to the metal centre.
donation are usually features that are associated with an anagostic interaction,40 they are not in themselves responsible for the signature downfield chemical shifts observed in NMR spectra that signal the positioning of the C–H bond relative to the metal centre.
Reactivity: hydrogenation of NBD, reversible C–H activation, and a complex with both anagostic and B–H agostic motifs
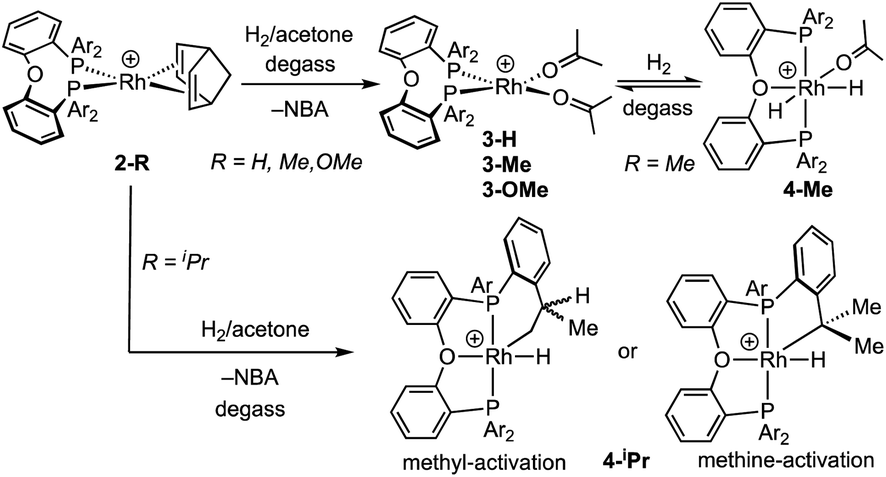
The Schrock–Osborn [Rh(DPEphos-R)(NBD)]+ complexes are precatalysts for a variety of important transformations.20 Activation is often by hydrogenation in situ in a coordinating solvent, for example acetone to form [Rh(DPEphos-R)(acetone)2]+ (3-R) and free norbornane (NBA).9,24,49 [Rh(DPEphos-H)(acetone)2][BArF4]25 has been reported using this method, and we now extend this methodology to the complexes 2-Me, 2-OMe and 2-iPr. The product of these reactions is dependent on the R-substituent, with more electron donating/bulkier substituents resulting in Rh(III) hydride products.50Addition of H2 to yellow acetone-d6 solutions of 2-Me or 2-OMe, followed by degassing, results in the hydrogenation of bound NBD and the in situ formation of the red acetone adducts493-Me and 3-OMe (Scheme 2). These adducts could not be isolated and presented broad signals at room temperature in their 1H and 31P NMR spectra. Free NBA was observed to be formed by 1H NMR spectroscopy. For R = Me, if the solution is not degassed post H2 addition, the yellow Rh(III) dihydride complex, [Rh(DPEphos-Me)(H)2(acetone)][BArF4], 4-Me, is formed quantitatively. Degassing results in loss of H2 and the formation of red 3-Me. Complex 4-Me is characterised at 298 K by the observation in the 1H NMR spectrum of a broad, relative integral 2H, hydride resonance at δ −19.5 in the region characteristic of hydride ligands, and a broad signal in the 31P{1H} NMR spectrum at δ 26. Cooling to 183 K reveals sharper signals, and thus that a fluxional process is occurring, likely reversible dissociation of acetone.51,52 A major and a minor species are observed (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) at low temperature, both with inequivalent hydrides [ca. δ −18 and −20] that integrate in total to 2H and show coupling to Rh, P and the other hydride [dddd]. In the 31P{1H} NMR spectrum signals are observed that show large J(PP) coupling [ca. 340 Hz] and small J(RhP) [ca. 117 Hz] – identifying them as being in a trans arrangement on a Rh(III) centre.53 These data, alongside selective decoupling experiments (ESI†), allow a structure to be assigned for 4-Me as shown in Scheme 2, that is similar to [Rh(κ3-P,O,P-xantphos)(H)2(acetone)][BArF4].51 The two different species observed at low temperature are assigned to conformers arising from different orientations of the ortho-Me substituted phenyl groups that undergo restricted P–C rotation.10,14
:1 ratio) at low temperature, both with inequivalent hydrides [ca. δ −18 and −20] that integrate in total to 2H and show coupling to Rh, P and the other hydride [dddd]. In the 31P{1H} NMR spectrum signals are observed that show large J(PP) coupling [ca. 340 Hz] and small J(RhP) [ca. 117 Hz] – identifying them as being in a trans arrangement on a Rh(III) centre.53 These data, alongside selective decoupling experiments (ESI†), allow a structure to be assigned for 4-Me as shown in Scheme 2, that is similar to [Rh(κ3-P,O,P-xantphos)(H)2(acetone)][BArF4].51 The two different species observed at low temperature are assigned to conformers arising from different orientations of the ortho-Me substituted phenyl groups that undergo restricted P–C rotation.10,14
 | ||
| Scheme 2 Hydrogenation of NBD adducts 2-R. [BArF4]− anions not shown. | ||
For the DPEphos-iPr ligand the product of hydrogenation in acetone is different, and a Rh(III)-hydride iPr-cyclometallated product is formed, [Rh(κ3-P,O,P-DPEphos-iPr′)(H)][BArF4] 4-iPr [DPEphos-iPr′ = (o-iPr-C6H4)2P(C6H4)O(C6H4)P(o-iPr-C6H4)(o-(CH2CH3CH)C6H4)]. 4-iPr is fluxional in solution, and is unchanged when free H2 is removed under vacuum. At room temperature in acetone-d6 solution the formation of 4-iPr is signalled by a, integral 1H, environment observed at δ −19.81 [dt, J(RhH) = 29, J(PH) 15 Hz], an alkyl region that shows a complex set of overlapping resonances (further complicated by the presence of free NBA), and a very broad 31P{1H} NMR spectrum [δ 20.8]. Warming to 338 K sharpens the 31P NMR spectrum, so a broad apparent doublet is observed at δ 21.7;54 while the 1H NMR spectrum at this temperature retains a sharp multiplet hydride signal. There is some decomposition on warming. Progressive cooling moves though a coalescence regime, ∼243 K, so that at 183 K a sharp 31P{1H} NMR spectrum is observed that shows three major sets of inequivalent phosphine environments, between δ 4 and δ 41, all with trans P–P coupling [J(PP) ∼360 Hz] and J(RhP) coupling indicative of a Rh(III) centre [J(RhP) = 112–121 Hz].53 In the 1H NMR spectrum (183 K) at least three different hydride multiplet environments are observed between δ −19.40 and −19.95 [J(RhH) = 29–31 Hz from selective decoupling], that combined integrate to a single proton.55 No H–H coupling is observed, which is different from dihydride 4-Me.
Collectively these NMR data suggest complex 4-iPr is formed as a mixture of at least three iPr-cyclometallated species, that interconvert on the NMR timescale at room temperature by a process that does not break and exchange the Rh–H bond. Reversible reductive elimination and exchange with other C–H groups in the ligand would be expected to result in loss of the hydride signal and associated coupling if it occurred on the NMR timescale.56–60 We thus propose that this fluxional process is associated with a restricted P–C rotation10,14 of the bulky iPr-aryl groups that leads to different, but exchanging, rotamers61 of the same ortho-metalled isomer. In the absence of a single-crystal X-ray structure we cannot definitively assign a structure to 4-iPr as one where the iPr methine or methyl group has undergone C–H activation, and both motifs are known.62 While we cannot unequivocally rule out a ground-state structure arising from methine-iPr activation, we favour methyl activation as the hydride peaks correlate to methyl, aromatic and methine signals in the low temperature NOESY spectrum (ESI†). Very similar spectra are obtained on hydrogenation in 1,2-F2C6H4 or o-xylene solvent (ESI†), meaning there is no evidence for significant solvent coordination at the Rh(III) centre, or agostic interactions, the latter albeit expected to be weak.63,64 The hydride is located trans to the coordinated oxygen on the basis of the observed chemical shift.65
While reversible cyclometallation of 4-iPr is not observed on the NMR timescale, it does occur on the laboratory timescale as probed by a variety of experiments, Schemes 3 and 4:
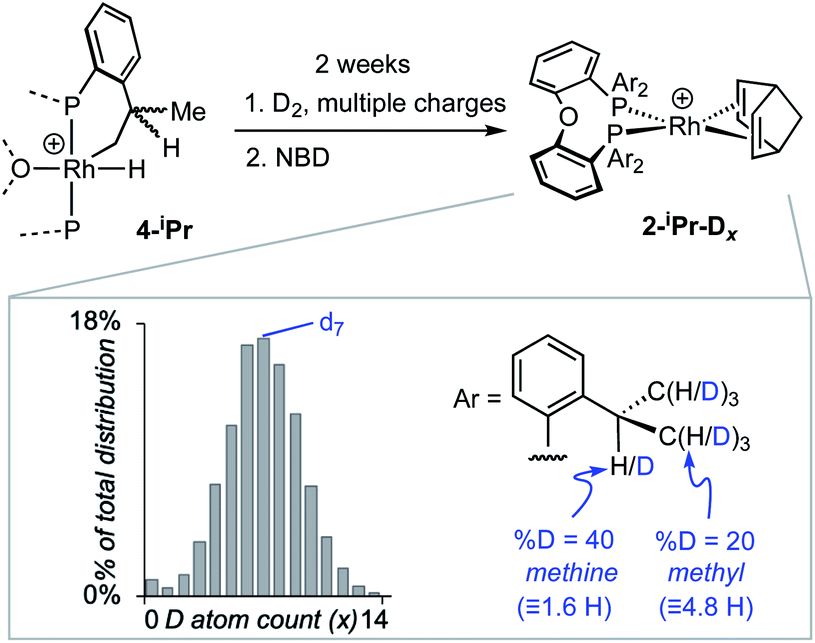 | ||
| Scheme 3 H/D exchange in 4-iPr and trapping with NBD to form 2-iPr-Dx. Inset shows the distribution of isotopologues of 2-iPr-Dx as measured by ESI-MS and analysed using an in-house Python script. [BArF4]− anions not shown. | ||
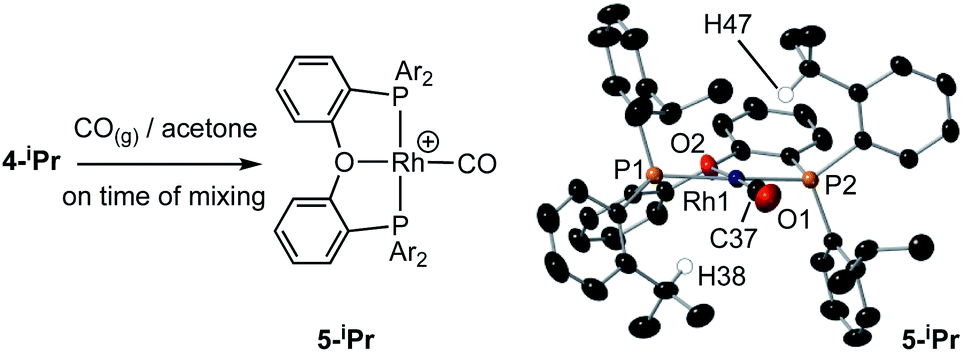 | ||
| Scheme 4 Reaction of 4-iPr with CO, and solid-state structure of 5-iPr highlighting the position of anagostic contacts. [BArF4]− anions not shown. Displacement ellipsoids are shown at the 50% probability level. Rh1–P1, 2.3145(7); Rh1–P2, 2.3027(8); Rh1–C37, 1.819(4); Rh1–O2, 2.128(3); Rh–H38, 2.821; Rh–H47, 2.627; P1–Rh1–P2, 162.41(4); O2–Rh1–C1, 177.0(1). | ||
(i) Addition of NBD quantitatively reforms 2-iPr on time of mixing.
(ii) Repeated charging of an o-xylene solution of 4-iPr over two weeks with D2 results in a significant, but slow, reduction in intensity of the hydride signal and the concomitant appearance of signals in the hydride and alkyl regions of the 2H NMR spectrum. Subsequent addition of NBD results in the formation of 2-iPr-Dx, that could be reliably analysed using electrospray ionisation mass spectrometry (ESI-MS) and NMR spectroscopy. Processing of the resulting isotope pattern for the cation in 2-iPr-Dx (ESI†) reveals a distribution of isotopologues, x = 0 to 14, centred around x = 6 to 8 (Scheme 3). That both methyl and methine C–H activation occurs is demonstrated in the 1H NMR spectrum of 2-iPr-Dx that shows a reduction in intensity for both these environments, corresponding to 20% D and 40% D incorporation respectively (4.8 D and 1.6 D respectively). No H/D exchange is observed in the C–H bonds of the aryl groups.664-Me undergoes no exchange under the same conditions.
(iii) Addition of CO to 4-iPr results in the quantitative formation of the Rh(I) complex [Rh(κ3-P,O,P-DPEphos-iPr)(CO)][BArF4], 5-iPr, the structure of which has been determined by single-crystal X-ray diffraction (Scheme 4). Complex 5-iPr has two anagostic C–H⋯Rh interactions, similar to 2-iPr, but now from two methine C–H groups (H38, 2.821 Å, ø = 59.0°; H47, 2.671 Å, ø = 64.0°). In solution at 298 K the cation displays time averaged C2v symmetry by NMR spectroscopy. Two methine environments are observed in the 1H NMR spectrum, one shifted significantly downfield from the other: δ 4.74 and 3.10 (2 H integral each), and the former signal is assigned to the anagostic pair H38/H47 (δcalc = 4.6).
The reaction of 4-iPr with CO and NBD on time of mixing indicates that this Rh(III) complex acts as a “masked”57 source of Rh(I). While this suggests a kinetically accessible Rh(I) intermediate could be in equilibrium with 4-iPr (I, Scheme 5), invoking this as the only accessible intermediate would not account for H/D exchange observed on addition of D2 nor the fluxional process observed on the NMR timescale that retains the Rh–H bond. Alternatively, ligand-assisted reductive elimination67–69 from a Rh(III) intermediate (II) could result in the direct formation of a Rh(I) product without involving I, to give 2-iPr (NBD) or 5-iPr (CO, shown).
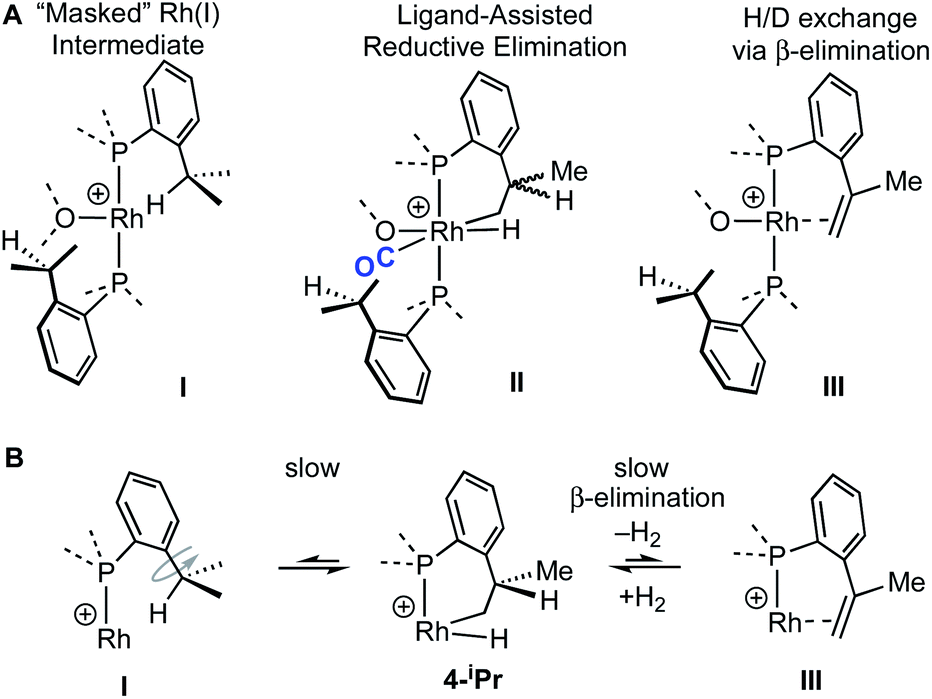 | ||
| Scheme 5 (A) Possible intermediates for the formation of Rh(I) complexes, and H/D exchange starting from 4-iPr. (B) Proposed mechanism for H/D exchange. | ||
The lack of H/D exchange for 4-Me suggests that if C–H oxidative addition does operate for this complex, subsequent exchange with D2 at the Rh(III) centre70 (e.g. via a σ-CAM process71) must be a high energy, inaccessible process. By extension, the H/D exchange observed in 4-iPr likely also proceeds by an alternative mechanism and we propose a β-elimination/dehydrogenative process via an intermediate such as III, as previously used to explain, albeit faster, well-defined reversible C–H activation processes.72,73 Subsequent addition of D2 would then provide pathways for methine and hydride D-incorporation. An additional slower, reversible, reductive elimination to form I would account for both multiple methyl H/D exchanges within one iPr group and for more than one iPr group undergoing H/D exchange (i.e., dx > 7 Scheme 3).66 Consistent with this, HD(dissolved) is also observed [δ 4.39, J(HD) = 43 Hz]. The overall very slow H/D exchange indicates relatively significant barriers operate for the formation of I, consistent with the observation of an intact Rh–H group on the NMR timescale.
While the intermediate III has not been observed, indirect evidence that it is kinetically accessible comes from the reaction of 4-iPr with H3B·NMe3 and the hydrogen acceptor tert-butyl ethene (tbe). This, slowly (7 days), but cleanly, forms a new product, in which a cyclometallated iPr-group has formally undergone a double-dehydrogenative borylation74–76 with H3B·NMe3 to form a Rh(I) vinylborane complex [Rh(κ2-P,P-(DPEphos-iPr″)-η2-BH2NMe3)][BArF4], 6-iPr, which is isolated in good yield (88%) as a green analytically pure solid. The solid-state structure of 6-iPr is shown in Scheme 6. This reveals a Rh(I) centre complexed with a chelating vinyl amine-borane [C39–C37, 1.392(6) Å] that coordinates to the Rh(I) centre through the alkene and a non-classical B–H 3c-2e agostic28 bond[Rh⋯H1B, 1.99(5); Rh⋯B, 2.391(6) Å]. This last distance is suggestive of an η2-interaction of the B–H bond with the Rh(I) centre, underscored by the rather closed Rh–H1B–B angle, 87.8(18) Å.77,78 The Rh–P bond opposite the weaker trans-influence B–H agostic bond is correspondingly shorter than that opposite the alkene. Room temperature NMR data are fully consistent with the crystallographically determined structure, showing two inequivalent, mutually coupled, environments in the 31P{1H} NMR spectrum. In the 1H{11B} NMR spectrum a relative integral 1H vinyl [δ 3.86], B–H(terminal) [δ 1.89, d, J(HH) 14 Jz; δcalc = +2.2] and agostic B–H⋯Rh [δ −7.54, J(RhH) 14, J(PH) 52, J(HH) 14 Hz; δcalc = −6.4] are observed. The agostic B–H signal is significantly upfield shifted compared to both the terminal B–H and the free vinyl borane PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh(BH2·NMe3), δ 2.40.79 The 11B{1H} NMR spectrum shows a very broad signal centred at δ −10.2 assigned to the borane. The chelating motif of the amine-borane in 6-iPr is similar to that reported for RuH2{η2,η2-HCHB(NiPr2)CH2C6H4PPh2}(PCy3), A, which also shows a similar chemical shift for the η2-M⋯H–B interaction in the 1H spectrum.80 The M⋯B distance in 6-iPr is longer however, [2.391(6) versus 2.173(3) Å] reflecting that there is no vacant p-orbital on boron available for back donation from the metal, unlike for A.
CPh(BH2·NMe3), δ 2.40.79 The 11B{1H} NMR spectrum shows a very broad signal centred at δ −10.2 assigned to the borane. The chelating motif of the amine-borane in 6-iPr is similar to that reported for RuH2{η2,η2-HCHB(NiPr2)CH2C6H4PPh2}(PCy3), A, which also shows a similar chemical shift for the η2-M⋯H–B interaction in the 1H spectrum.80 The M⋯B distance in 6-iPr is longer however, [2.391(6) versus 2.173(3) Å] reflecting that there is no vacant p-orbital on boron available for back donation from the metal, unlike for A.
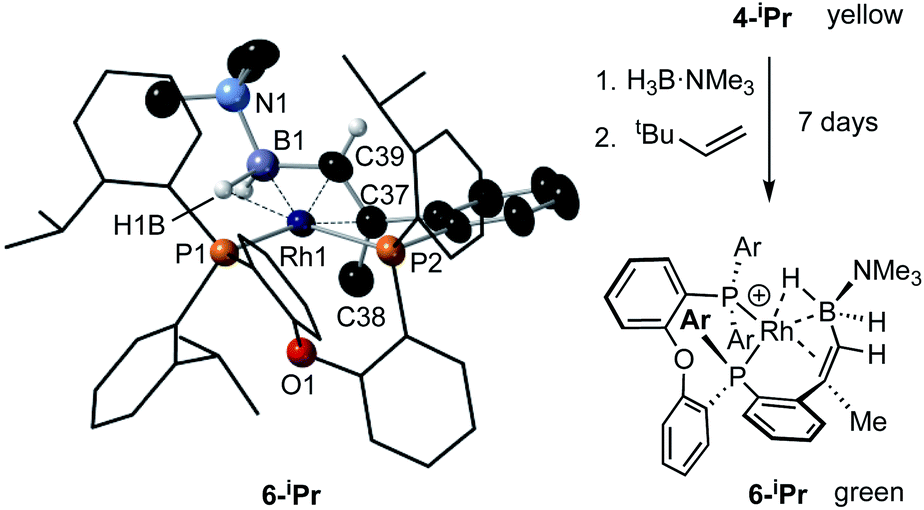 | ||
| Scheme 6 Synthesis and solid-state structure of 6-iPr. Displacement ellipsoids are shown at the 30% probability level. Selected bond distances and angles: Rh1–P1, 2.3361(9); Rh1–P2, 2.2696(11); Rh1–B1, 2.391(6); Rh1–C37, 2.266(3); Rh1–C39, 2.152(3); Rh1–H1B, 1.99(5); B1–C39, 1.557(6); C37–C38, 1.511(6); C37–C39, 1.392(6); P1–Rh1–P2, 100.59(4); B1–C39–C37, 123.2(3); Rh1–H1B–B1, 87.8(18). | ||
Particularly noteworthy in the 1H NMR spectrum of 6-iPr are two downfield shifted signals (1H relative integral each) at δ 4.92 and 4.75 (δcalc = 5.2 and 4.7), which are comparable to the signals assigned to anagostic C–H hydrogens in 5-iPr. Closer inspection of the solid-state structure shows that the methine C–H protons H49 and H46 are in close approach to the Rh(I) centre and orientated above and below the RhP2B1 plane, Fig. 9, (ø = 66.2° and 64.8°). In comparison, the upfield shifted signal, at δ −7.54, is due to the agostic 3c-2e Rh⋯H–B motif that sits squarely in the RhP2 plane (ø = 6.6°). 6-iPr thus highlights, in a single complex, the relationship between the orientation of the approaching E–H bond to the metal centre: the C–H anagostic interaction lying above the d8 metal coordination plane and the 3c-2e B–H→Rh agostic bond sitting within the coordination plane.
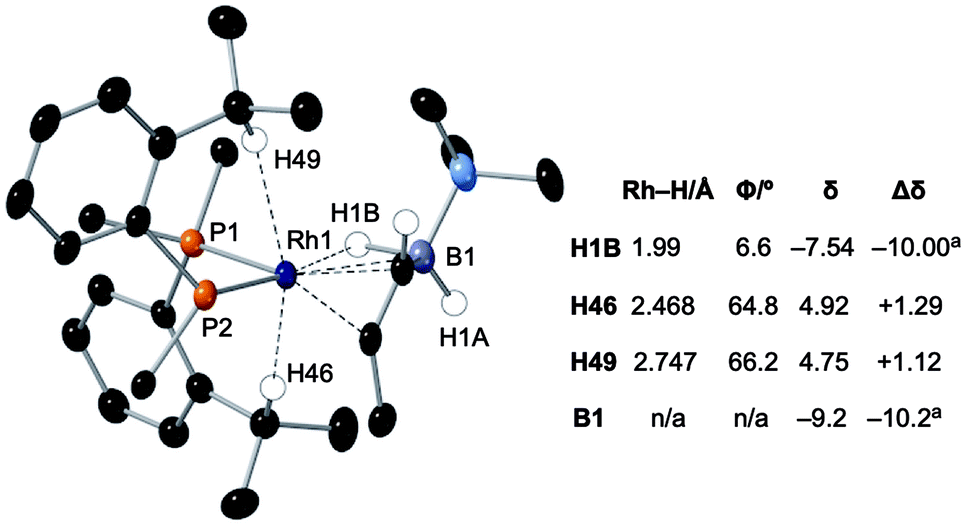 | ||
| Fig. 9 Comparison of selected structural and spectroscopic data for the anagostic/B–H agostic interactions in 6-iPr. Selected aryl groups are removed for clarity. aChemical shifts compared with the vinyl borane PhCHCPh(BH2·NMe3).79 | ||
Selected data from the computational analysis of [6-iPr]+ are shown in Table 3 and suggest the Rh⋯H46 interaction is similar in strength to the Rh⋯H1 interaction in [2-iPr]+. Both these C–H⋯Rh anagostic interactions exhibit relatively weak 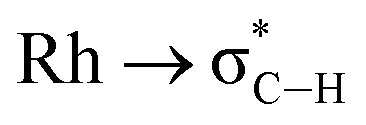 donation. In contrast the 3c-2e B–H⋯Rh agostic motif is markedly stronger and is now dominated by very strong donation from an occupied B–H orbital into an unoccupied Rh-orbital that NBO analysis quantifies at 52.4 kcal mol−1, i.e. a 3c-2e bond. This is significantly stronger than in the related [(NNN)Rh(H3BNMe3)]+ adduct (NNN = 2,6-bis-[1-(2,6-diisopropyl-phenylimino)ethyl]pyridine),77 consistent with a much shorter computed Rh⋯H distance (1.78 A cf. 1.91 A) and longer B–H distance (1.35 A cf. 1.28 A) in 6-iPr.
donation. In contrast the 3c-2e B–H⋯Rh agostic motif is markedly stronger and is now dominated by very strong donation from an occupied B–H orbital into an unoccupied Rh-orbital that NBO analysis quantifies at 52.4 kcal mol−1, i.e. a 3c-2e bond. This is significantly stronger than in the related [(NNN)Rh(H3BNMe3)]+ adduct (NNN = 2,6-bis-[1-(2,6-diisopropyl-phenylimino)ethyl]pyridine),77 consistent with a much shorter computed Rh⋯H distance (1.78 A cf. 1.91 A) and longer B–H distance (1.35 A cf. 1.28 A) in 6-iPr.
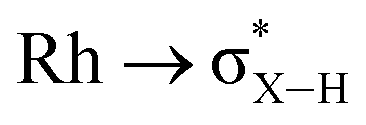
A suggested, abbreviated, mechanism for the formation of 6-iPr is shown in Scheme 7: (i) dehydrogenation of an iPr group gives intermediate III (Scheme 5),72,81 (ii) hydroboration of the alkene using H3B·NMe3;82 (iii) followed by dehydrogenation, via C–H activation/β-elimination.75,76 Throughout tbe acts as a sacrificial hydrogen acceptor. While this scheme captures the gross transformations, the precise order of events currently remains unresolved.
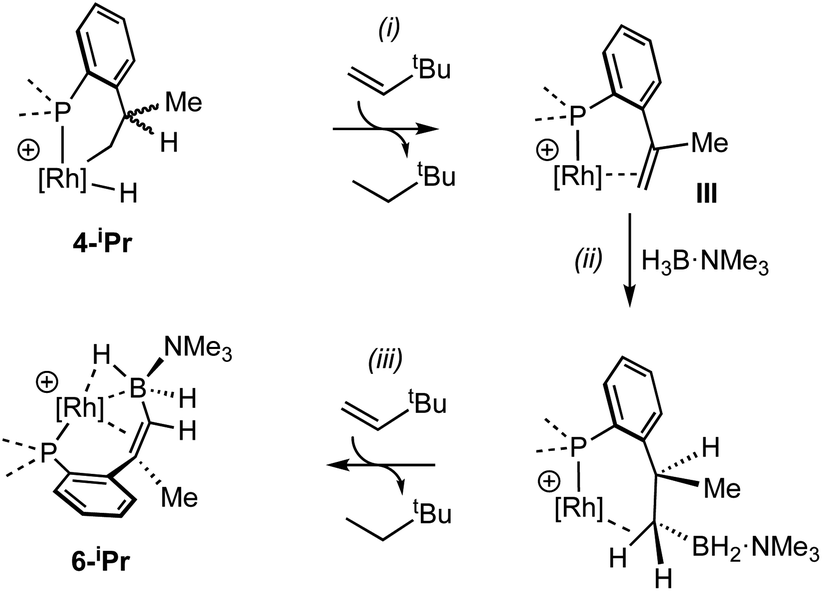 | ||
| Scheme 7 Suggested outline mechanism for the dehydrogenative borylation 4-iPr. Only key ligands shown. [BArF4]− anion omitted for clarity. | ||
Conclusions
We have shown that aryl-group ortho-substitution in [Rh(NBD)(DPEphos-R)]+ leads to differences in structures, fluxional processes and reactivities – which can be related to the steric bulk of the ortho-group. Broadly speaking, OMe and Me substituents lead to solid-state and solution structures that are not too dissimilar to parent DPEphos. With the iPr group fluxional processes in solution are retarded, and C–H activation processes occur. DPEphos-iPr thus cannot be considered an innocent ligand, this being related – more broadly – to the decomposition pathways of parent DPEphos that occur via C–O bond cleavage.27,83Common to all the Rh(I) DPEPhos-R complexes structurally described herein (with their associated NBD, CO or vinylborane co-ligands) is the observation of downfield-shifted signals in their 1H NMR spectra that signal an anagostic M⋯H–C interaction,28 for which the steric bulk of the ligand determines the temperature at which they are observed. As discussed previously,30,38,40,45 while such anagostic interactions are associated with weak 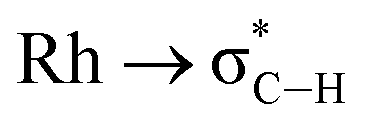 and minimal σC–H → Rh orbital donations, the driver for the downfield chemical shift of the C–H protons observed in the 1H NMR spectrum does not come from these. Instead, the positioning of the anagostic hydrogen with reference to different regions of valence shell for the d8 metal centre is important, as Scherer30 has previously elegantly described for Rh(CAAC)(CO)Cl systems (CAAC = cyclic alkyl-aminocarbene). Our observations here, on a consistent set of complexes, reinforce this analysis. Thus, when the hydrogen atoms are forced, through steric constraints, to sit in an axial position (ø approaching 90°) that places them above a region of charge concentration, the associated magnetically-induced current density results in a downfield shift in the NMR spectrum, Fig. 10A. This analysis differentiates anagostic interactions from 3c-2e agostic bonds, the latter being characterised by upfield shifts in their 1H NMR spectra due to the associated hydrogen atoms being located in a region of charge depletion in the ligand plane of a d8 ML3 type fragment (Fig. 10B, ø approaching 0°). Complex 6-iPr offers E–H bonds (E = C, B) in both these topologies, and thus shows both upfield and downfield chemical shifts in the 1H NMR spectrum. While, as for 6-iPr, any agostic bond will likely show a significantly stronger 3c-2e σX–H → Rh interaction compared to the weak
and minimal σC–H → Rh orbital donations, the driver for the downfield chemical shift of the C–H protons observed in the 1H NMR spectrum does not come from these. Instead, the positioning of the anagostic hydrogen with reference to different regions of valence shell for the d8 metal centre is important, as Scherer30 has previously elegantly described for Rh(CAAC)(CO)Cl systems (CAAC = cyclic alkyl-aminocarbene). Our observations here, on a consistent set of complexes, reinforce this analysis. Thus, when the hydrogen atoms are forced, through steric constraints, to sit in an axial position (ø approaching 90°) that places them above a region of charge concentration, the associated magnetically-induced current density results in a downfield shift in the NMR spectrum, Fig. 10A. This analysis differentiates anagostic interactions from 3c-2e agostic bonds, the latter being characterised by upfield shifts in their 1H NMR spectra due to the associated hydrogen atoms being located in a region of charge depletion in the ligand plane of a d8 ML3 type fragment (Fig. 10B, ø approaching 0°). Complex 6-iPr offers E–H bonds (E = C, B) in both these topologies, and thus shows both upfield and downfield chemical shifts in the 1H NMR spectrum. While, as for 6-iPr, any agostic bond will likely show a significantly stronger 3c-2e σX–H → Rh interaction compared to the weak 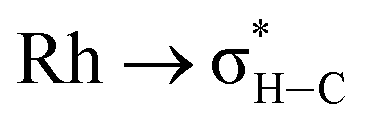 donation associated with the anagostic interactions, the relationship, if any, between these bonding descriptors and the observed chemical shift has yet to be demonstrated.
donation associated with the anagostic interactions, the relationship, if any, between these bonding descriptors and the observed chemical shift has yet to be demonstrated.
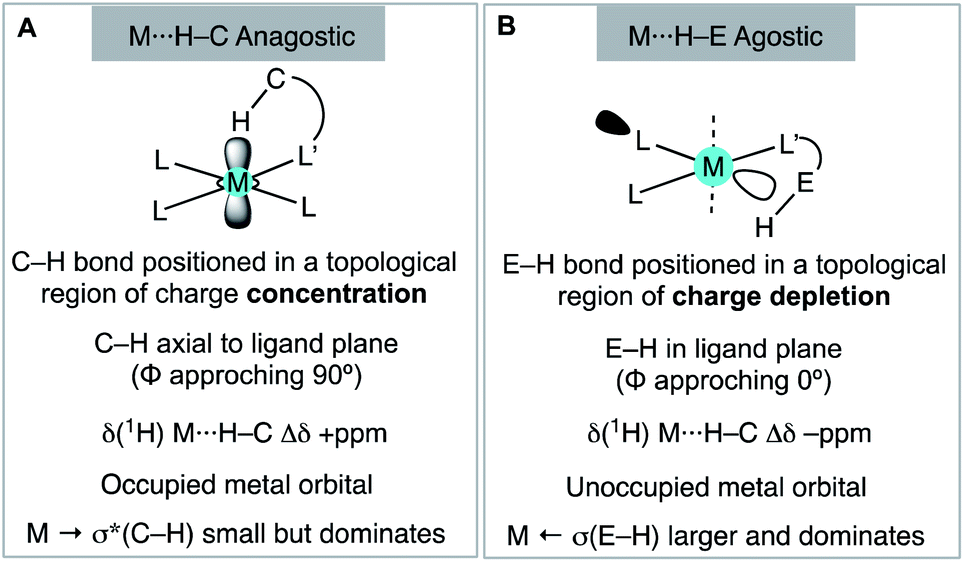 | ||
| Fig. 10 Structural, electronic and NMR properties of anagostic interactions (A) and E–H agostic bonds (B), as based upon Scherer's analysis. | ||
These observations reinforce the analysis that the chemical shift changes observed by 1H NMR spectroscopy in d8 square planar complexes with anagostic C–H bonds located above the ligand plane result from topologically enforced ring current effects, rather than signalling an interaction that has a considerable orbital contribution. In this regard they are perhaps more related to the chemical shift changes that are well-established for protons that are forced to sit in topologically distinct regions close to arenes.30,84,85 We thus suggest there is a clear demarcation between anagostic interactions, and agostic, 3c-2e, bonds; differences that arise from both the topological orientation and the nature of the orbital interactions that prevail for each.
Data availability
All data is in the ESI. There is no more to deposit.Author contributions
J. J. R.: conceptualisation, experimentation, data analysis, drafting the manuscript; A. L. B.: conceptualisation and computational analysis; T. M. B., A. H. and A. M. M.: NMR data fitting and single crystal X-ray analysis; S. A. M., A. S. W.: conceptualisation, project coordination, writing the final manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The EPSRC (M024210) and SCG Chemicals Ltd. Oxford and Heriot-Watt Universities and the EPSRC for studentship support to J. J. R., T. M. B. and A. L. B., and Professors Simon Duckett (York), Michael Willis (Oxford) and Odile Eisenstein (Montpellier) for useful discussions.Notes and references
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC.
- G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317–14328 CrossRef CAS PubMed.
- V. Khakyzadeh, Y.-H. Wang and B. Breit, Chem. Commun., 2017, 53, 4966–4968 RSC.
- K. Hori, H. Motohashi, D. Saito and K. Mikami, ACS Catal., 2019, 9, 417–421 CrossRef CAS.
- P. A. Spreider, A. M. Haydl, M. Heinrich and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 15569–15573 CrossRef CAS PubMed.
- B. D. Vineyard, W. S. Knowles, M. J. Sabacky, G. L. Bachman and D. J. Weinkauff, J. Am. Chem. Soc., 1977, 99, 5946–5952 CrossRef CAS.
- C. Gonzalez-Rodriguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 5134–5138 CrossRef CAS.
- S. J. Dossett, A. Gillon, A. G. Orpen, J. S. Fleming, P. G. Pringle, D. F. Wass and M. D. Jones, Chem. Commun., 2001, 699–700 RSC.
- J. N. L. Dennett, A. L. Gillon, K. Heslop, D. J. Hyett, J. S. Fleming, C. E. Lloyd-Jones, A. G. Orpen, P. G. Pringle, D. F. Wass, J. N. Scutt and R. H. Weatherhead, Organometallics, 2004, 23, 6077–6079 CrossRef CAS.
- N. A. Cooley, S. M. Green, D. F. Wass, K. Heslop, A. G. Orpen and P. G. Pringle, Organometallics, 2001, 20, 4769–4771 CrossRef CAS.
- L. Lavanant, A.-S. Rodrigues, E. Kirillov, J.-F. Carpentier and R. F. Jordan, Organometallics, 2008, 27, 2107–2117 CrossRef CAS.
- R. A. Baber, A. G. Orpen, P. G. Pringle, M. J. Wilkinson and R. L. Wingad, Dalton Trans., 2005, 659–667 RSC.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 3694–3703 CrossRef CAS.
- Y. Zhu and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 111–114 CrossRef CAS PubMed.
- J. Wang, G. Meng, K. Xie, L. Li, H. Sun and Z. Huang, ACS Catal., 2017, 7, 7421–7430 CrossRef CAS.
- S. Raoufmoghaddam, E. Drent and E. Bouwman, ChemSusChem, 2013, 6, 1759–1773 CrossRef CAS PubMed.
- Y. Jiao, M. S. Torne, J. Gracia, J. W. Niemantsverdriet and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2017, 7, 1404–1414 RSC.
- R. R. Schrock and J. A. Osborn, J. Am. Chem. Soc., 1976, 98, 4450–4455 CrossRef CAS.
- A. Meißner, E. Alberico, H.-J. Drexler, W. Baumann and D. Heller, Catal. Sci. Technol., 2014, 4, 3409–3425 RSC.
- G. M. Adams, D. E. Ryan, N. A. Beattie, A. I. McKay, G. C. Lloyd-Jones and A. S. Weller, ACS Catal., 2019, 9, 3657–3666 CrossRef CAS PubMed.
- D. E. Ryan, K. A. Andrea, J. J. Race, T. M. Boyd, G. C. Lloyd-Jones and A. S. Weller, ACS Catal., 2020, 10, 7443–7448 CrossRef CAS.
- G. L. Moxham, H. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem.–Eur. J., 2008, 14, 8383–8397 CrossRef CAS PubMed.
- J. Barwick-Silk, S. Hardy, M. C. Willis and A. S. Weller, J. Am. Chem. Soc., 2018, 140, 7347–7357 CrossRef CAS PubMed.
- R. J. Pawley, M. A. Huertos, G. C. Lloyd-Jones, A. S. Weller and M. C. Willis, Organometallics, 2012, 31, 5650–5659 CrossRef CAS.
- J. F. Hooper, A. B. Chaplin, C. Gonzalez-Rodríguez, A. L. Thompson, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 2906–2909 CrossRef CAS PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- W. Scherer, A. C. Dunbar, J. E. Barquera-Lozada, D. Schmitz, G. Eickerling, D. Kratzert, D. Stalke, A. Lanza, P. Macchi, N. P. M. Casati, J. Ebad-Allah and C. Kuntscher, Angew. Chem., Int. Ed., 2015, 54, 2505–2509 CrossRef CAS PubMed.
- J. E. Barquera-Lozada, A. Obenhuber, C. Hauf and W. Scherer, J. Phys. Chem., 2013, 117, 4304–4315 CrossRef CAS PubMed.
- At low temperature (203 K) the 1H and 31P{1H} NMR spectra are significantly more complex, consistent with the freezing out of a fluxional process. See ESI.†.
- R. T. Boeré, A. M. Bond, S. Cronin, N. W. Duffy, P. Hazendonk, J. D. Masuda, K. Pollard, T. L. Roemmele, P. Tran and Y. Zhang, New J. Chem., 2008, 32, 214–231 RSC.
- A. G. Orpen, Chem. Soc. Rev., 1993, 22, 191–197 RSC.
- J. S. Siegel and F. A. L. Anet, J. Org. Chem., 1988, 53, 2629–2630 CrossRef CAS.
- The low-field region of 2-OMe at 183 K shows minor signals that are assigned to an alternative isomer, the presence of which is signalled in the 31P{1H} NMR spectrum. See ESI.†.
- D. Pingen, T. Lebl, M. Lutz, G. S. Nichol, P. C. J. Kamer and D. Vogt, Organometallics, 2014, 33, 2798–2805 CrossRef CAS.
- W. Goertz, W. Keim, D. Vogt, U. Englert, M. D. K. Boele, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1998, 2981–2988 RSC.
- J. Zamora-Moreno, F. Murillo, M. A. Muñoz-Hernández, M. Grellier, S. Pan, S. Jalife, G. Merino, S. Sabo-Etienne and V. Montiel-Palma, Organometallics, 2018, 37, 3581–3587 CrossRef CAS.
- L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, A. Guy Orpen and T. F. Koetzle, J. Chem. Soc., Dalton Trans., 1991, 1789–1798 RSC.
- Y. Zhang, J. C. Lewis, R. G. Bergman, J. A. Ellman and E. Oldfield, Organometallics, 2006, 25, 3515–3519 CrossRef CAS.
- D. Schmitz, M. Kalter, A. C. Dunbar, M. Vöst, A. Fischer, K. Batke, G. Eickerling, K. Ruhland, J. Ebad-Allah, C. Kuntscher and W. Scherer, Eur. J. Inorg. Chem., 2020, 79–83 CrossRef CAS.
- S. Scholer, M. H. Wahl, N. I. Wurster, A. Puls, C. Hattig and G. Dyker, Chem. Commun., 2014, 50, 5909–5911 RSC.
- M. P. Mitoraj, M. G. Babashkina, K. Robeyns, F. Sagan, D. W. Szczepanik, Y. V. Seredina, Y. Garcia and D. A. Safin, Organometallics, 2019, 38, 1973–1981 CrossRef CAS.
- J. C. Lewis, J. Wu, R. G. Bergman and J. A. Ellman, Organometallics, 2005, 24, 5737–5746 CrossRef CAS.
- A. G. Jarvis, P. E. Sehnal, S. E. Bajwa, A. C. Whitwood, X. Zhang, M. S. Cheung, Z. Lin and I. J. S. Fairlamb, Chem.–Eur. J., 2013, 19, 6034–6043 CrossRef CAS PubMed.
- W. Yao, O. Eisenstein and R. H. Crabtree, Inorg. Chim. Acta, 1997, 254, 105–111 CrossRef CAS.
- T. S. Thakur and G. R. Desiraju, J. Mol. Struct.: THEOCHEM, 2007, 810, 143–154 CrossRef CAS.
- For [2-H]+ and [2-iPr]+ the lowest energy conformation was that seen in the crystal structures, while for [2-Me]+ one lower energy conformation and for [2-OMe]+ two additional conformations were located that were more stable than that derived from the crystal structures. See ESI† for full details.
- D. P. Fairlie and B. Bosnich, Organometallics, 1988, 7, 936–945 CrossRef CAS.
- A. D. Wilson, A. J. M. Miller, D. L. DuBois, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2010, 49, 3918–3926 CrossRef CAS PubMed.
- R. J. Pawley, G. L. Moxham, R. Dallanegra, A. B. Chaplin, S. K. Brayshaw, A. S. Weller and M. C. Willis, Organometallics, 2010, 29, 1717–1728 CrossRef CAS.
- M. A. Esteruelas, M. Oliván and A. Vélez, Inorg. Chem., 2013, 52, 5339–5349 CrossRef CAS.
- P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- These resonances are most likely the inner lines of a tightly-coupled AB doublet.
- Both 1H NMR spectra for 4-Me and 4-iPr were collected with a long aquisition delay time (10 s) to ensure reliable intregration for the hydride signals.
- A. C. Albeniz, G. Schulte and R. H. Crabtree, Organometallics, 1992, 11, 242–249 CrossRef CAS.
- A. Y. Verat, M. Pink, H. Fan, J. Tomaszewski and K. G. Caulton, Organometallics, 2008, 27, 166–168 CrossRef CAS.
- L. J. Sewell, A. B. Chaplin, J. A. B. Abdalla and A. S. Weller, Dalton Trans., 2010, 39, 7437–7439 RSC.
- N. Phillips, L. Treasure, N. H. Rees, R. Tirfoin, J. E. McGrady and S. Aldridge, Eur. J. Inorg. Chem., 2014, 4877–4885 CrossRef CAS.
- A. C. Cooper, J. C. Huffman and K. G. Caulton, Organometallics, 1997, 16, 1974–1978 CrossRef CAS.
- D. X. Hu, P. Grice and S. V. Ley, J. Org. Chem., 2012, 77, 5198–5202 CrossRef CAS.
- Selected examples of iPr cyclometallation: (a) M. A. Esteruelas, A. M. López, E. Oñate and E. Royo, Organometallics, 2005, 24, 5780–5783 CrossRef CAS; (b) L. J. L. Häller, M. J. Page, S. Erhardt, S. A. Macgregor, M. F. Mahon, M. A. Naser, A. Vélez and M. K. Whittlesey, J. Am. Chem. Soc., 2010, 132, 18408–18416 CrossRef PubMed; (c) N. S. Townsend, A. B. Chaplin, M. A. Naser, A. L. Thompson, N. H. Rees, S. A. Macgregor and A. S. Weller, Chem.–Eur. J., 2010, 16, 8376–8389 CrossRef CAS PubMed; (d) M. Moret, S. F. Keller, J. C. Slootweg and P. Chen, Inorg. Chem., 2009, 48, 6972 CrossRef CAS PubMed; (e) See ref. 14.
- R. C. Knighton, J. Emerson-King, J. P. Rourke, C. A. Ohlin and A. B. Chaplin, Chem.–Eur. J., 2018, 24, 4927–4938 CrossRef CAS PubMed.
- A. C. Cooper, E. Clot, J. C. Huffman, W. E. Streib, F. Maseras, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1999, 121, 97–106 CrossRef CAS.
- L. J. L. Häller, E. Mas-Marzá, M. K. Cybulski, R. A. Sanguramath, S. A. Macgregor, M. F. Mahon, C. Raynaud, C. A. Russell and M. K. Whittlesey, Dalton Trans., 2017, 46, 2861–2873 RSC.
- The maximum level of deuteration measured is 2-iPr-D14, which suggests that only two iPr groups undergo H/D exchange, likely a consequence of conformational constraints.
- N. M. West, S. Reinartz, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 2006, 128, 2059–2066 CrossRef CAS PubMed.
- C. Cheng, B. G. Kim, D. Guironnet, M. Brookhart, C. Guan, D. Y. Wang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 6672–6683 CrossRef CAS PubMed.
- A. Johnson, C. G. Royle, C. N. Brodie, A. J. Martínez-Martínez, S. B. Duckett and A. S. Weller, Inorg. Chem., 2021 DOI:10.1021/acs.inorgchem.0c03687.
- N. M. Scott, V. Pons, E. D. Stevens, D. M. Heinekey and S. P. Nolan, Angew. Chem., Int. Ed., 2005, 44, 2512–2515 CrossRef CAS.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- U. Fekl and K. I. Goldberg, J. Am. Chem. Soc., 2002, 124, 6804–6805 CrossRef CAS PubMed.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed.
- K. Burgess, W. A. Van der Donk, S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, J. Am. Chem. Soc., 1992, 114, 9350–9359 CrossRef CAS.
- J. M. Brown and G. C. Lloyd-Jones, J. Am. Chem. Soc., 1994, 116, 866–878 CrossRef CAS.
- I. A. I. Mkhalid, R. B. Coapes, S. N. Edes, D. N. Coventry, F. E. S. Souza, R. L. Thomas, J. J. Hall, S.-W. Bi, Z. Lin and T. B. Marder, Dalton Trans., 2008, 1055–1064 RSC.
- A. Johnson, A. J. Martínez-Martínez, S. A. Macgregor and A. S. Weller, Dalton Trans., 2019, 48, 9776–9781 RSC.
- M. L. H. Green and G. Parkin, in The Chemical Bond III: 100 years old and getting stronger, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2017, pp. 79–139 Search PubMed.
- M. Dietz, A. Johnson, A. Martínez-Martínez and A. S. Weller, Inorg. Chim. Acta, 2019, 491, 9–13 CrossRef CAS.
- A. Cassen, Y. Gloaguen, L. Vendier, C. Duhayon, A. Poblador-Bahamonde, C. Raynaud, E. Clot, G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2014, 53, 7569–7573 CrossRef CAS PubMed.
- A. B. Chaplin, A. I. Poblador-Bahamonde, H. A. Sparkes, J. A. K. Howard, S. A. Macgregor and A. S. Weller, Chem. Commun., 2009, 244–246, 10.1039/B816739G.
- H. C. Johnson, R. Torry-Harris, L. Ortega, R. Theron, J. S. McIndoe and A. S. Weller, Catal. Sci. Technol., 2014, 4, 3486–3494 RSC.
- M. K. Cybulski, N. A. Beattie, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Chem.–Eur. J., 2020, 26, 11141–11145 CrossRef CAS PubMed.
- M. Baranac-Stojanović, RSC Adv., 2014, 4, 308–321 RSC.
- D. Ajami and J. Rebek, Nat. Chem., 2009, 1, 87–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2064124–2064130. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01430g |
| This journal is © The Royal Society of Chemistry 2021 |