
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of benthol A and its challenging stereochemical assignment: opening up a new window for skeletal diversity of super-carbon-chain compounds†
Zhong-Ping
Jiang‡
a,
Shi-Hao
Sun‡
a,
Yi
Yu
b,
Attila
Mándi
 d,
Jiao-Yang
Luo
e,
Mei-Hua
Yang
e,
Tibor
Kurtán
*d,
Wen-Hua
Chen
*c,
Li
Shen
*b and
Jun
Wu
*a
d,
Jiao-Yang
Luo
e,
Mei-Hua
Yang
e,
Tibor
Kurtán
*d,
Wen-Hua
Chen
*c,
Li
Shen
*b and
Jun
Wu
*a
aSchool of Pharmaceutical Sciences, Southern Medical University, 1838 Guangzhou Avenue North, Guangzhou 510515, P. R. China. E-mail: wwujun2003@yahoo.com
bMarine Drugs Research Center, College of Pharmacy, Jinan University, 601 Huangpu Avenue West, Guangzhou 510632, P. R. China. E-mail: shenli6052@sina.com
cSchool of Biotechnology and Health Sciences, Wuyi University, Jiangmen, Guangdong Province 529020, P. R. China. E-mail: whchen@wyu.edu.cn
dDepartment of Organic Chemistry, University of Debrecen, PO Box 400, 4002 Debrecen, Hungary. E-mail: kurtan.tibor@science.unideb.hu
eKey Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100193, P. R. China
First published on 30th June 2021
Abstract
Super-carbon-chain compounds (SCCCs) are marine organic molecules featuring long polyol carbon chains with numerous stereocenters. Polyol–polyene compounds (PPCs) and ladder-frame polyethers (LFPs) are two major families. It is highly challenging to establish the absolute configurations of SCCCs. In this century, few new SCCC families have been reported. Benthol A, an aberrant SCCC, was obtained from a South China Sea benthic dinoflagellate that should belong to a new taxon. Its planar structure and absolute configuration, containing thirty-five carbon stereocenters, were unambiguously established by a combination of extensive NMR spectroscopic investigations, periodate degradation of the 1,2-diol groups, ozonolysis of the carbon–carbon double bonds, J-based configurational analysis, NOE interactions, modified Mosher's MTPA ester method, and DFT-NMR 13C chemical-shift calculations aided by DP4+ statistical analysis. Benthol A displayed potent antimalarial activity against Plasmodium falciparum 3D7 parasites. This new molecule combines extraordinary structural features, particularly eight scattered ether rings on a C72 backbone chain, which places it within a new SCCC family between PPCs and LFPs, herein termed polyol–polyether compounds. This suggestion was strongly supported by principal component analysis. The discovery of benthol A does not only provide new insights into the untapped biosynthetic potential of marine dinoflagellates, but also opens up a new window for skeletal diversity of SCCCs.
Introduction
Marine dinoflagellates produce massive and diverse super-carbon-chain compounds (SCCCs), the backbones of which are long and flexible carbon chains that are highly oxygen-functionalized.1,2 Representative examples are palytoxin and maitotoxin with molecular weights of 2680 and 3422 daltons, respectively. To date, maitotoxin is believed to have the longest carbon chain in nature, except for biopolymers.1–4 SCCCs exhibit remarkable bioactivities, such as activation effects of ion channels, and antifungal, antitumor, and anti-osteoclastic activities.1–8 However, the highly time-consuming stereochemical determination of SCCCs retards their potential applications in biomedicine.There is no doubt that fragment stereodivergent synthesis combined with NMR spectroscopic data comparison is an accurate and reliable approach to the stereochemical assignment of SCCCs.1–8 However, it normally takes decades to determine the configuration of a complex SCCC. For example, symbiodinolide, a SCCC containing a 62-membered lactone and 61 carbon stereocenters, was first identified in 2007 from the symbiotic dinoflagellate, Symbiodinium sp.9 Since then, chemists have constantly striven to resolve its absolute configuration through fragment stereodivergent synthesis. Nevertheless, the absolute configurations of the last two fragments on its backbone, viz., C53–C61 and C75–C79, remain to be determined.2,10–12
With the aid of modern molecular biological, algaculture engineering, chemical, spectroscopic, and computational techniques, the configurational determination of complex SCCCs based on natural chemical entity has become feasible, though it still takes years. For instance, gibbosols A–C were obtained by our group in 2017 as a new class of SCCCs, i.e., polyol–polyol compounds, from the marine pelagic dinoflagellate, Amphidinium gibbosum. The planar structures and absolute configurations of these SCCCs, containing thirty-six or thirty-seven carbon stereocenters, were successfully established in 2020 by combined use of chemical, spectroscopic, and computational approaches based on natural products.13,14
During the ongoing projects in search of novel SCCCs from South China Sea dinoflagellates, we turned our attention to new taxa of these unicellular algae. Comparison of the 18S rDNA sequence of a benthic dinoflagellate (strain: MDRC-02, Fig. S1†) with those in NCBI nucleotide database using the BLAST algorithm revealed that the closest relative is an uncultured eukaryote clone (XM.18S_7; percent identity: 99.65%), whereas the closest dinoflagellate strain is Amphidinium operculatum (strain: TAK-0; percent identity: 80.95%). Phylogenetic tree inferred from the 18S rDNA sequence alignment in the NCBI databases suggested that the monophyly of the benthic strain MDRC-02 and the uncultured eukaryote clone XM.18S_7 formed a distinct clade, which is not closely related to any other clades of the dinoflagellate strains (Fig. S2†). Hence, the benthic dinoflagellate strain MDRC-02 should belong to a new taxon of dinoflagellate, or at least a new species of the genus Amphidinium. UPLC-MS-guided isolation led to the discovery of an unprecedented SCCC, named benthol A (1) (Fig. 1A), from the above benthic strain MDRC-02. Its chain backbone possesses eight scattered ether rings. Unquestionably, this raises the enormous opportunity to discover novel SCCCs from new taxa of dinoflagellates. Herein, we report the isolation, structural elucidation, and bioactivity of 1. The principal component analysis score plot of 1 and previously reported 187 SCCCs strongly supports that 1 belongs to a new SCCC family.
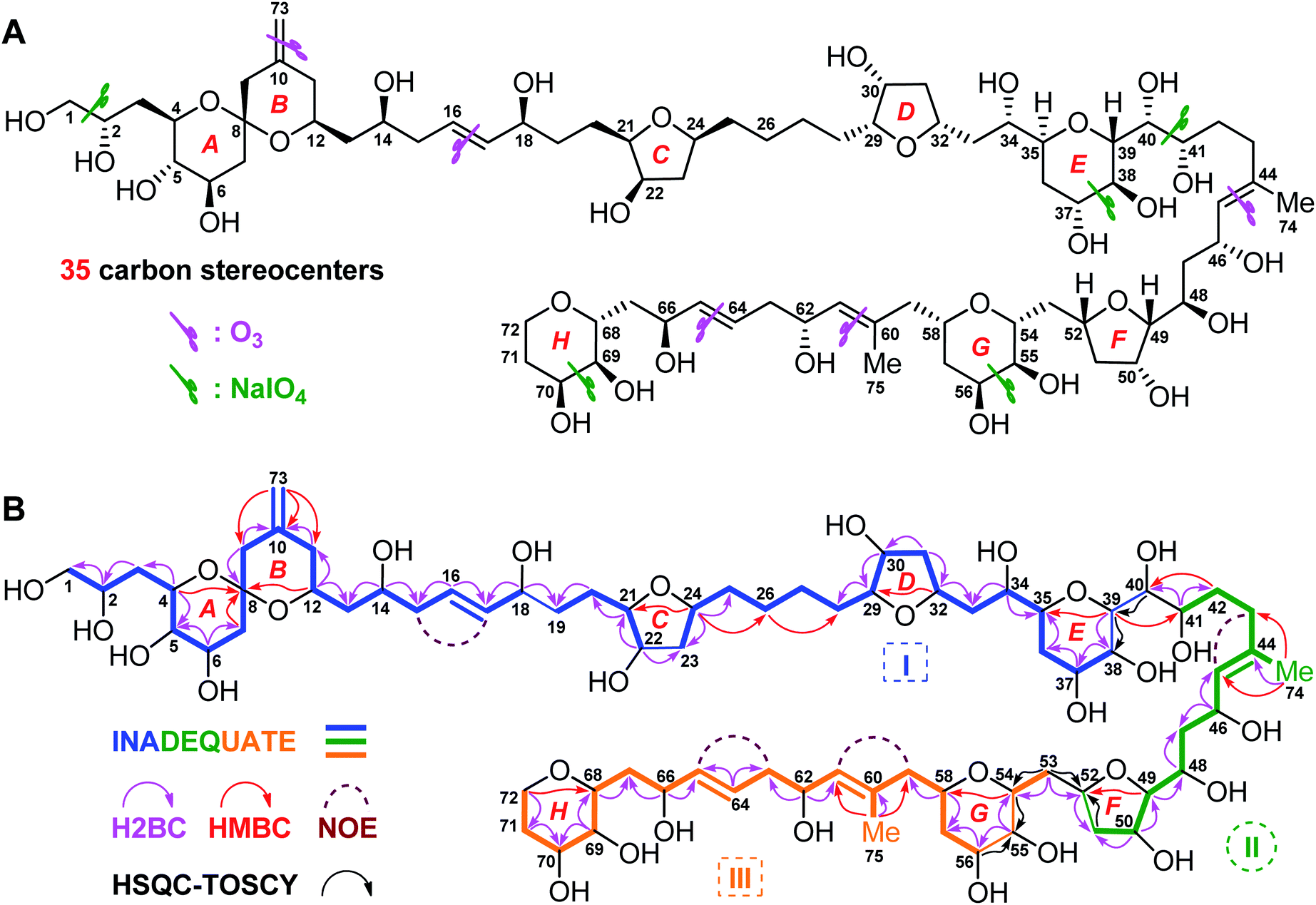 | ||
| Fig. 1 (A) Structure and absolute configuration of benthol A (1). (B) Key INADEQUATE, H2BC, HMBC, and HSQC-TOCSY correlations and diagnostic NOE interactions of benthol A (1). | ||
Results and discussion
Planar structure of benthol A
Benthol A (1) was obtained as a colorless solid. The molecular formula C75H126O30 with thirteen degrees of unsaturation was established by means of high resolution-electrospray ionization-mass spectrometry (HR-ESIMS) with a positive ion at m/z 1529.8234 ([M + Na]+, calcd 1529.8226) (Fig. S7–S8†). According to the 1H and 13C NMR spectroscopic data of 1 (Fig. S9–S22†), five degrees of unsaturation are due to five carbon–carbon double bonds. Thus, the molecule should contain eight rings.The 13C NMR spectroscopic data and the DEPT135 experiment of 1 (Fig. S23–S27†), combined with its 2D HSQC and sel-HSQC spectra (Fig. S28–S33†), revealed the presence of 75 carbon resonances for two methyl groups, 29 methylene groups (an olefinic, two oxygenated, and two overlapped signals at δC 41.73), 40 methine groups (six olefinic and 34 oxygenated, including two overlapped oxymethine signals at δC 73.22), and four quaternary carbons (three olefinic). After fed with NaH13CO3 in the culture medium of the dinoflagellate, 13C isotope enriched chemical entity of 1 (4.0 mg) was obtained for the 2D INADEQUATE experiment (Fig. S34–S36†).15,16 Consequently, three substructures, viz., I (from C1 to C40, and C73), II (from C41 to C52, and C74), and III (from C53 to C72, and C75), were determined by analysis of 2D INADEQUATE, HSQC, H2BC, HMBC, and HSQC-TOCSY correlations of 1 (Fig. S28–S41†). It is worth mentioning that, most carbon–carbon connections of the molecule were established by 2D INADEQUATE correlations (Fig. 1B).
For substructure I, the linear connections of C1 to C18, C19 to C22, and C23 to C38 were revealed by the corresponding 13C–13C correlations observed from the 2D INADEQUATE spectrum, and the relevant H2BC and HMBC crosspeaks. The linear connection of C18 and C19 and that of C22 and C23 were resolved by H2BC correlations between H18/C19 and H22/C23, respectively, whereas the branched connection of the C10![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C73 bond was assigned by the INADEQUATE correlation between C10/C73. The linear connection of C38 to C40 was deduced from HSQC-TOCSY correlations between C39/H38 and C40/H39. Ether bridges between C4/C8, C8/C12, C21/C24, C29/C32, and C35/C39 were determined by crucial HMBC crosspeaks between H4/C8, H12/C8, H24/C21, H32/C29, and H39/C35, respectively. Taken together, the above data gave substructure I, containing three tetrahydropyran (i.e., rings A, B, and E) and two tetrahydrofuran (i.e., rings C and D) moieties, among which rings A and B (from C4 to C12) constitute a 6,6-spiroketal motif (Fig. 1B).
C73 bond was assigned by the INADEQUATE correlation between C10/C73. The linear connection of C38 to C40 was deduced from HSQC-TOCSY correlations between C39/H38 and C40/H39. Ether bridges between C4/C8, C8/C12, C21/C24, C29/C32, and C35/C39 were determined by crucial HMBC crosspeaks between H4/C8, H12/C8, H24/C21, H32/C29, and H39/C35, respectively. Taken together, the above data gave substructure I, containing three tetrahydropyran (i.e., rings A, B, and E) and two tetrahydrofuran (i.e., rings C and D) moieties, among which rings A and B (from C4 to C12) constitute a 6,6-spiroketal motif (Fig. 1B).
For substructure II, the linear connection of C41 to C51 was established by the corresponding INADEQUATE and H2BC correlations. The linear connection of C51 and C52 was revealed by the H2BC crosspeak between H52/C51 and the HSQC-TOCSY correlation between C51/H52, whereas the branched connection of Me-74 and C44 was assigned by the INADEQUATE correlation between C74/C44, the H2BC crosspeak between H3-74/C44, and HMBC correlations between H3-74/C43 and H3-74/C45. An ether bridge between C49/C52 was determined by the crucial HMBC crosspeak between H49/C52. Taken together, the substructure II, containing a tetrahydrofuran moiety (i.e., ring F), was elucidated (Fig. 1B).
For substructure III, the linear connections of C53 and C54, and C56 to C72 were deduced by the corresponding INADEQUATE and H2BC correlations. The linear connection of C54 to C56 was resolved by H2BC correlations between H55/C54 and H55/C56, and HSQC-TOCSY crosspeaks between C54/H55 and C56/H55, whereas the branched connection of Me-75 and C60 was assigned by the INADEQUATE correlation between C75/C60, the H2BC crosspeak between H3-75/C60, and HMBC correlations between H3-75/C59 and H3-75/C61. Ether bridges between C54/C58 and C68/C72 were determined by crucial HMBC crosspeaks between H54/C58 and H2-72/C68, respectively. Taken together, the substructure III, containing two tetrahydropyran moieties (i.e., rings G and H), was assembled (Fig. 1B).
The presence of five tetrahydropyran and three tetrahydrofuran rings in 1, including a 6,6-spiroketal motif, was further confirmed by isotope shift experiments measured in CD3OD and CD3OH.17 Fifteen oxygenated carbons, viz., C8 (δ 99.76), C49 (δ 86.13), C29 (δ 84.69), C21 (δ 84.52), C24 (δ 79.06), C35 (δ 77.77), C52 (δ 76.54), C32 (δ 75.39), C68 (δ 75.01), C54 (δ 74.33), C4 (δ 73.54), C39 (δ 73.22), C58 (δ 70.96), C12 (δ 68.65), and C72 (δ 62.63), which exhibited no obvious deuterium-induced isotope shift, are involved in the ether linkages (Fig. S42–S46†). These results were in accordance with the aforementioned ether bridges assigned by the observed HMBC crosspeaks.
Finally, the H2BC correlation between H41/C40 and HMBC crosspeaks between H39/C41 and H2-42/C40 led to the connection of substructures I and II through the C40–C41 bond. Similarly, H2BC correlations between H2-53/C52 and the HSQC-TOCSY crosspeak between C53/H52 connected substructures II and III by the C52–C53 bond. NOE interactions between H2-15/H17, H2-43/H45, H2-59/H61, and H2-63/H65, and the large values of 3JH16,H17 (15.4 Hz) and 3JH64,H65 (15.4 Hz) concluded that the geometries of four carbon–carbon double bonds of 1, viz., C16C17, C44C45, C60C61, and C64C65, are all E-forms. Based on the above results, the planar structure of 1, containing an exomethylene moiety, two pendant methyl groups, eight ether rings, and twenty-two hydroxy groups as 1,2-diol, 1,3-diol, 1,4-diol, and 1,5-diol moieties, was completely elucidated (Fig. 1B).
Chemical degradation of benthol A
Owing to heavily overlapped 1H and 13C NMR signals of 1, particularly those of oxymethine groups crowded around 73.2 ppm, the determination of the relative configuration of 1 turned out to be a daunting task. To unambiguously establish the stereochemistry of the whole molecule, degradation reactions were applied to 1. NaIO4/NaBH4-mediated cleavage of the 1,2-diol groups of 1 with 13C natural abundance afforded two main products, viz., fragments 1A (from C2 to C40, and Me-73) and 1B (from C41 to C72, Me-74, and Me-75) (Fig. 2), whereas O3/NaBH4-mediated cleavage of the carbon–carbon double bonds of 1 with 13C-enriched abundance yielded five products, viz., fragments 1a (from C1 to C16), 1b (from C17 to C44, and Me-74), 1c (from C45 to C60, and Me-75), 1d (from C61 to C64), and 1e (from C65 to C72) (Fig. 2). The planar structures of these fragments were assigned by ESIMS, NMR spectroscopic data, and 1H–1H COSY, HSQC, HMBC, and NOESY spectra (Fig. S47–S144†). Most notably, 1b is a pair of epimers at C44.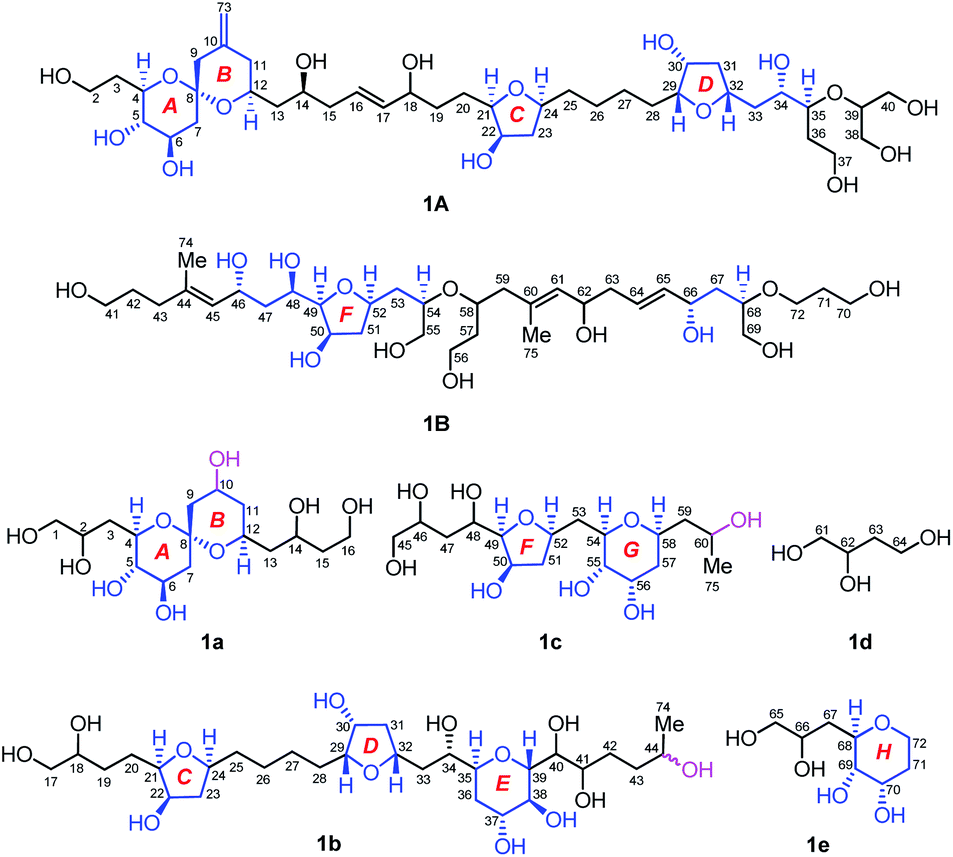 | ||
| Fig. 2 Structures of seven degradation fragments, viz., 1A, 1B, and 1a–1e. The relative configuration of each segment in blue color was assigned. Stereocenters at C10, C44, and C60 were generated as results of chemical degradation. Fragments 1a and 1c are optical isomers obtained from HPLC chiral separation, whereas 1b is a mixture of epimers at C44. | ||
Relative configuration of benthol A
To establish the relative configurations of the relevant segments in fragments 1A, 1B, 1a, 1b, 1c, and 1e (Fig. 2), extensive J-based configurational analysis (JBCA)18,19 and NOE interactions were employed. In fact, the relative configurations of four flexible segments, including C32–C35 in 1A and C46–C49, C52–C54, and C66–C68 in 1B, were assigned by JBCA referring to the typical criteria (Table S1†), whereas those of eight cyclic ether moieties (i.e., rings A–H) in the above fragments were determined by diagnostic NOE interactions. To sum up, the relative configurations of the C32–C35 segment in 1A, and the C46–C49, C52–C54, and C66–C68 segments in 1B were assigned as (anti/syn), (anti/anti), syn, and syn, respectively, by application of JBCA (Fig. S3†).The relative configuration of the 6,6-spiroketal moiety in 1a, including rings A (from C4 to C8) and B (from C8 to C12), were assigned by diagnostic NOE interactions and the distinctive values of 3JH,H (Fig. 3A). The chair conformation of ring A with the axial H4, H5, and H6 was revealed by NOE interactions between H4/H6, H5/H3a, H5/H3b, and H5/H7α, the large values of 3JH4,H5 (9.3 to 9.6 Hz), 3JH5,H6 (9.3 Hz), and 3JH6,H7α (9.0 Hz), and the intermediate value of 3JH6,H7β (5.2 Hz). Similarly, the chair conformation of ring B with the axial H12 and the equatorial H10 was supported by NOE interactions between H12/H2, H12/H4, and H12/H14, the large values of 3JH12,H11β (9.8 Hz) and 3JH12,H13β (9.9 Hz), and the intermediate values of 3JH10,H9β (5.4 Hz) and 3JH10,H11β (5.4 Hz). Most notably, the relative configuration of C8 in 1a was unequivocally assigned as S* based on NOE interactions between H7α/H9α, H7β/H9β, H4/H14, H6/H14, and H4/H12 (Fig. 3A). Finally, the relative configuration of the 6,6-spiroketal moiety in 1a was determined to be (4R*,5S*,6R*,8S*,12S*).
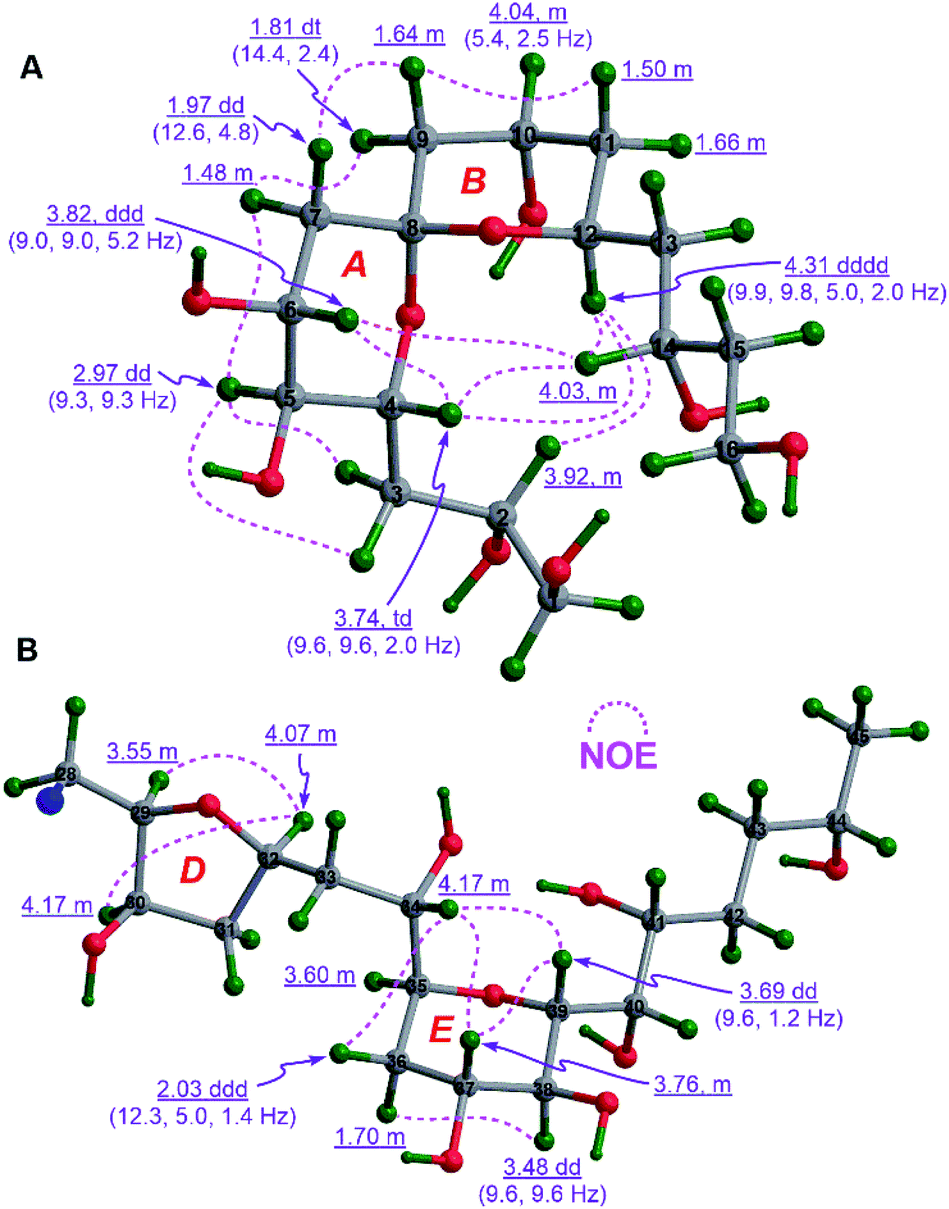 | ||
| Fig. 3 (A) The relative configuration of the 6,6-spiroketal moiety in 1a and (B) those of rings D and E in 1b assigned by diagnostic NOE interactions and 3JH,H values (the C17 to C27 moiety in 1b is omitted for clarity). | ||
NOE interactions between H29/H32 and H30/H32 of ring D in 1b revealed their cofacial relationship (Fig. 3B). The chair conformation of ring E in 1b with the axial H37, H38, and H39, and the equatorial H35 was corroborated by NOE interactions between H34/36β, H34/H37, H34/H39, H37/H39, and H36α/38, the large values of 3JH37,H38 (9.6 Hz) and 3J38,H39 (9.6 Hz), and the small value of 3JH35,H36β (1.4 Hz) (Fig. 3B). Combined with the aforementioned (anti/syn) relationship of the C32–C35 segment in 1A (Fig. S3A†), the relative configuration of the C29–C39 moiety in 1b was concluded to be (29R*,30R*,32R*,34S*,35S*,37R*,38S*,39R*). Additionally, the cofacial relationship of H21, H22, and H24 of ring C in 1b was deduced from NOE interactions between H21/H24 and H22/H24 (Fig. S4A†).
The cofacial relationship of H49, H50, and H52 in ring F of 1c was assigned by NOE interactions between H49/H52 and H50/H52, whereas the chair conformation of ring G in 1c with the axial H54, H55, and H58, and the equatorial H56 was supported by those between H54/H58 and H55/H57β, the large 3JH54,H55 (9.6 to 9.8 Hz), and the intermediate 3JH55,H56 (3.2 Hz) and 3JH56,H57β (5.8 Hz). Interestingly, the strong NOE interaction between H52/H54 revealed their same spatial orientation (Fig. 4A). Combined with the aforementioned (anti/anti) configuration of the C46–C49 segment (Fig. S3B†) and the syn relationship of the C52–C54 segment observed in 1B (Fig. S3C†), the relative configuration of 1c was thereby assigned as (46R*,48R*,49R*,50R*,52R*,54R*,55S*,56S*,58S*).
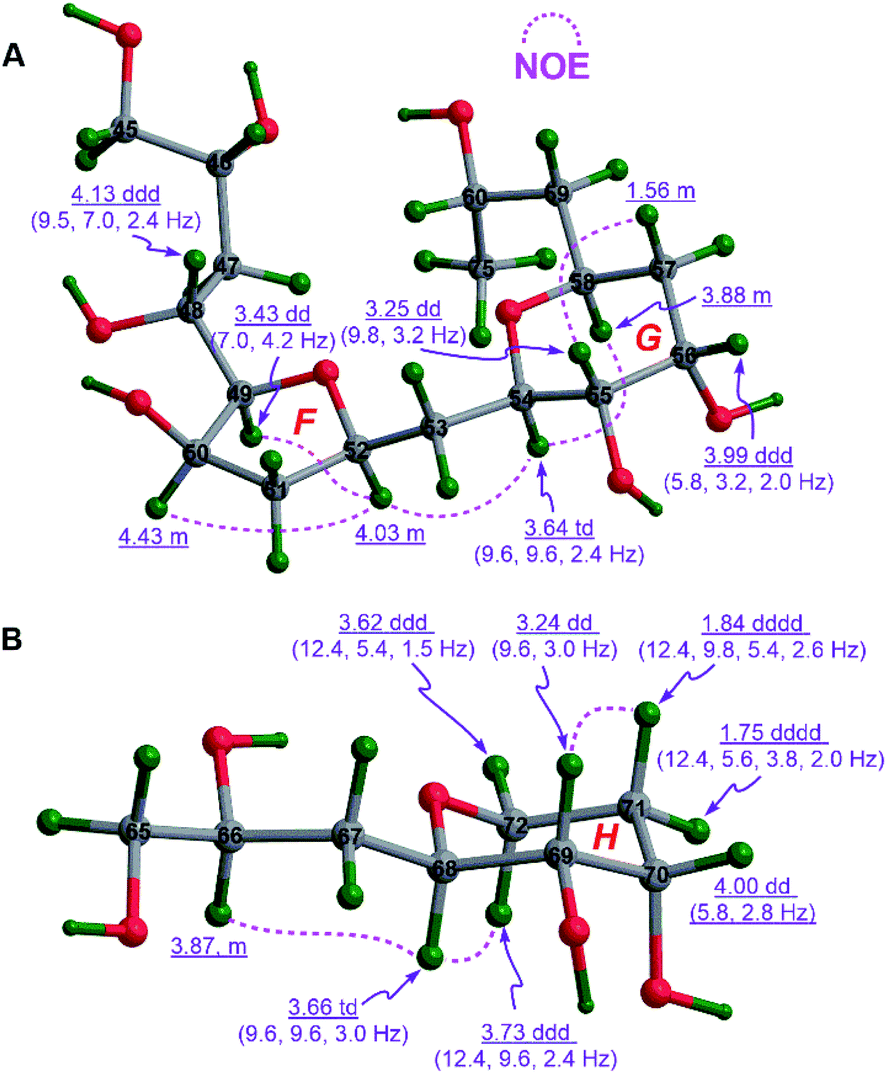 | ||
| Fig. 4 (A) The relative configurations of rings F and G in 1c and (B) that of ring H in 1e assigned by diagnostic NOE interactions and 3JH,H values. | ||
Similarly, the chair conformation of ring H in 1e with the axial H68 and H69, and the equatorial H70 was supported by NOE interactions between H68/H72α and H69/H71β, the large 3JH68,H69 (9.6 Hz), and the intermediate 3JH69,H70 (2.8 to 3.0 Hz) and 3JH70,H71β (5.4 to 5.8 Hz) (Fig. 4B).
Absolute configuration and bioactivities of benthol A
To determine the absolute configurations of 1A, 1B, 1a, 1b, 1c, and 1e, the modified Mosher's α-methoxy-α-trifluoromethyl-phenylacetic acid (MTPA) ester method was employed. The fragments 1A, 1B, 1b, and 1c were esterified with (R)- and (S)-MTPACl to produce the corresponding pairs of (S)- and (R)-MTPA esters (Fig. 5), viz., 1As/r (Fig. S145–S158†), 1Bs/r (Fig. S159–S172†), 1bs/r (Fig. S185–S200†), and 1cs/r (Fig. S201–S212†), respectively, whereas the fragments 1a and 1e reacted first with pivaloyl chloride to protect their terminal primary alcohol groups and then with (R)- and (S)-MTPACl to produce the corresponding pairs of (S)- and (R)-MTPA esters (Fig. 5), viz., 1as/r (Fig. S173–S184†) and 1es/r (Fig. S213–S222†), respectively.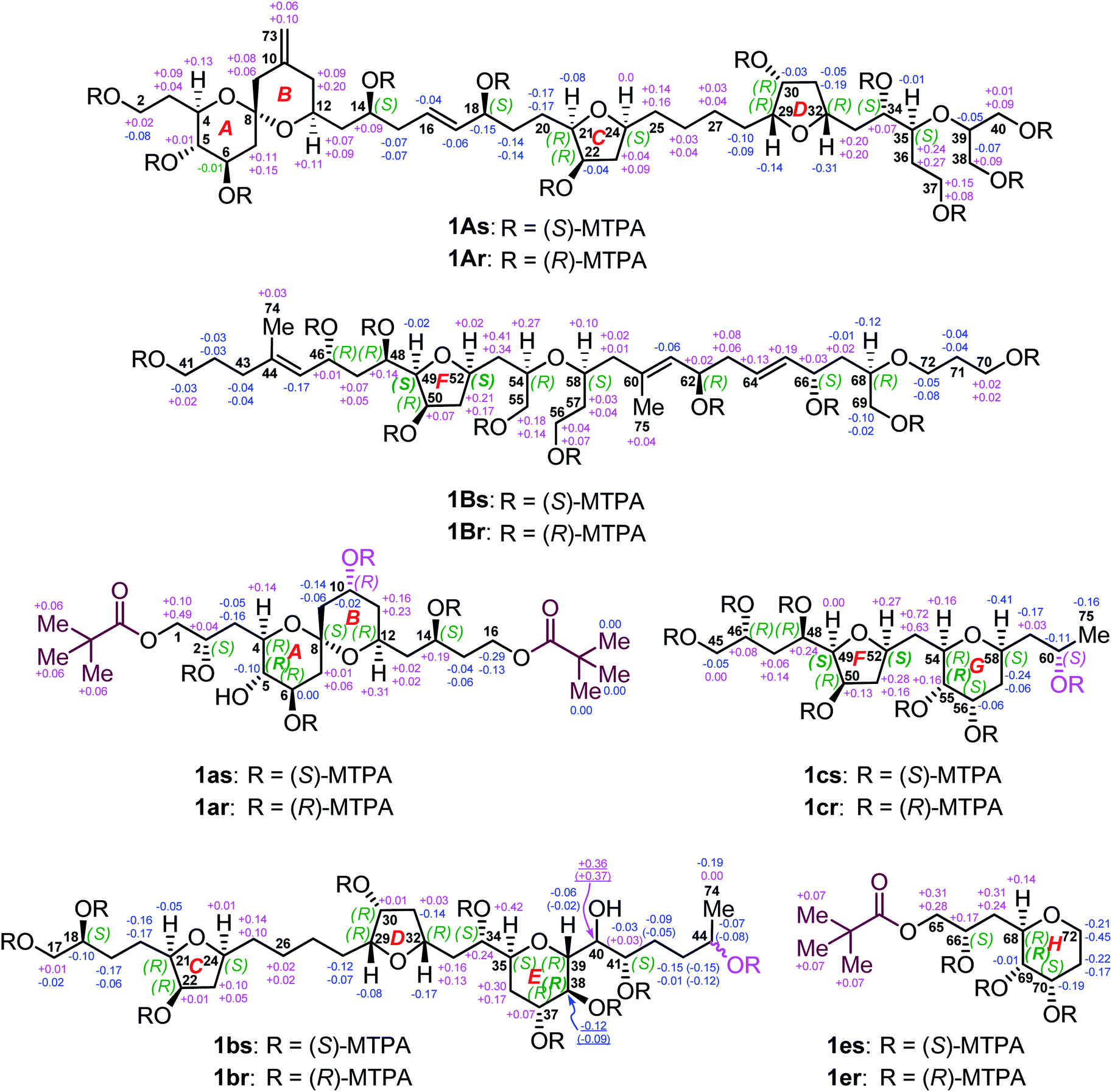 | ||
| Fig. 5 ΔδSR values obtained for fragments 1A, 1B, 1a, 1b, 1c, and 1e. Due to the different Cahn–Ingold–Prelog (CIP) priorities, the stereodescriptors at C49 and C52 of 1B, C5 of 1a, C38 of 1b, C49, C52, and C55 of 1c, and C69 of 1e, are opposite to those of 1Bs/r, 1as/r, 1bs/r, 1cs/r, and 1es/r, respectively. Owing to the epimeric C44 in 1bs/r, ΔδSR values of the C38–C44 segment and the Me-74 group are in pairs. However, 1as/r and 1cs/r are 10R and 60S isomers, respectively. | ||
Comparison of the ΔδSR values between 1as/r, the absolute configurations of C2, C6, C10, and C14 in 1a were assigned as S, R, R, and S, respectively, by typical Mosher ester analysis20 (Fig. 5). Based on the relative configuration of 1a (Fig. 3A), the absolute configurations of C4, C5, C8, and C12 in 1a were concluded to be R, S, S, and R, respectively (Fig. 5). Owing to the different CIP priority, the stereodescriptor at C5 in 1a is opposite to that of 1as/r. Similarly, the absolute configurations of C18, C22, and C41 in 1b were determined to be S, R, and S, respectively, by typical Mosher ester analysis.20 It is the same with 22R in 1A. Most notably, the above assigned 18S and 22R configurations were consistent with the ΔδSR sign distribution proposed for the acyclic 1,5-diol systems, as noted by Riguera,21–27viz., a type D syn-1,5-diol for the bis-MTPA esters of the C14–C18 diol moiety and a type B anti-1,5-diol for that of the C18–C22 diol moiety in 1A (Fig. 5). Based on the relative configuration of ring C in 1b (Fig. S4A†), the absolute configurations of C21 and C24 were concluded to be R and S, respectively. The absolute configurations of C34 and C37 in 1b were thereby assigned as S and R, respectively, by the sign distribution of ΔδSR values proposed for the 1,4-diol systems, i.e., a type A syn-1,4-diol in the bis-MTPA esters of the C34–C37 diol moiety (Fig. 5).21–27 Based on the aforementioned (29R*,30R*,32R*,34S*,35S*,37R*,38S*,39R*) configuration of the C29–C39 segment in 1b (Fig. 3B), the absolute configuration of this segment was successfully determined to be (29R,30R,32R,34S,35S,37R,38S,39R). Owing to the different CIP priority, the stereodescriptor at C38 in 1b is opposite to that of 1bs/r.
The absolute configurations of C46 and C48 in both 1B and 1c were identically assigned as R and R, respectively, by the sign distribution of ΔδSR values proposed for the 1,3-diol systems, i.e., a type A anti-1,3-diol for the bis-MTPA esters of the C46–C48 diol moiety in both 1B and 1c.21–27 The absolute configuration of C50 in 1B was established as R by typical Mosher ester analysis (Fig. 5).20 Based on the relative configuration of ring F in 1c (Fig. 4A), the absolute configurations of C49 and C52 were assigned as R and R, respectively. Due to the different CIP priorities, the stereodescriptors at C49 and C52 in 1c are opposite to that of 1cs/r. The absolute configurations of C55 and C56 in 1c were assigned as S and S, respectively, by the sign distribution of ΔδSR values proposed for the 1,2-diol systems, i.e., a type D anti-1,2-diol in the bis-MTPA esters of the C55–C56 diol moiety in 1c (Fig. 5).21–27 Owing to the different CIP priority, the stereodescriptor at C55 in 1c is opposite to that of 1cs/r. Based on the relative configuration of ring G in 1c (Fig. 4A), the absolute configurations of C54 and C58 were concluded to be R and S, respectively. The above results were consistent with the aforementioned relative configuration of 1c.
The absolute configurations of C62 and C66 in 1B were determined to be R and S, respectively, by the ΔδSR sign distribution proposed for the acyclic 1,5-diol systems, i.e., a type A anti-1,5-diol for the bis-MTPA esters of the C62–C66 diol moiety in 1B (Fig. 5).21–27 According to the syn relationship of the C66–C68 segment in 1B (Fig. S3D†), the absolute configuration of C68 was thereby assigned as R. Based on the relative configuration of ring H in 1e (Fig. 4B), the absolute configurations of C69 and C70 in 1e were determined to be S and S, respectively (Fig. 5). Most notably, the 62R configuration was reconfirmed based on the positive specific rotation ([α]25D = + 8 (c = 0.02, methanol)) of fragment 1d (Fig. 2),28,29 whereas the 66S configuration was further supported by the positive ΔδSR sign values of H2-65 and methyl protons of the terminal pivaloyl ester in 1e (Fig. 5). In addition, these results were corroborated by a type D anti-1,2-diol in the bis-MTPA esters of the C69–C70 diol moiety in 1e,21–27 though minor disturbance from the MTPA esters of C66 could be observed in the ΔδSR value of H69. Owing to the different CIP priority, the stereodescriptor at C69 in 1e is opposite to that of 1es/r (Fig. 5).
Thus far, the absolute configuration of C40 remained to be established. Due to its overlapped proton and carbon signals in 1b, JBCA could not be applied. To tackle this problem, DFT-NMR 13C chemical-shift calculations and DP4+ statistical analysis were employed.30,31 A model compound 1b′ representing the C34–C41 region of 1b was chosen for the calculation. The Merck Molecular Force Field (MMFF) conformational search was carried out on two possible C-40 epimers, viz., (34S,35S,37R,38S,39R,40R,41S)-1b′ and (34S,35S,37R,38S,39R,40S,41S)-1b′ (Fig. S4B†), producing 97 and 83 conformers, respectively, in a 21 kJ mol−1 energy window.32,33 The B3LYP/6-31+G(d,p) re-optimization of these conformers resulted in eight and twelve low-energy geometries with Boltzmann population above 1%. 13C NMR chemical-shift calculations were performed at the mPW1PW91/6-311+G(2d,p) level (Table S2†).34 The carbon chemical-shift differences and the corrected mean average error value35 showed a clear preference for the 40R epimer. The most characteristic chemical-shift difference could be observed at C41, adjacent to C40, with a Δδ value of 0.01 ppm for the 40R isomer versus that of 3.76 ppm for the 40S one (Table S3†). The DP4+ statistical analysis resulted in 99.93% confidence for the 40R epimer. Additionally, 3.41 Hz was computed for the 3JH39,H40 value of the 40R epimer, whereas 6.75 Hz was obtained for that of the 40S epimer. Evidently, the experimental 3JH39,H40 (1.2 Hz) further supported the absolute configuration of 40R in 1b.
Considering all the assignments of the absolute configurations for thirty-five carbon stereocenters in seven degradation products, viz., 1A, 1B, and 1a–1e, we may conclude that the absolute configuration of benthol A (1) is (2S,4R,5S,6R,8S,12S,14S,18S,21R,22R,24S,29R,30R,32R,34S,35S,37R,38S,39R,40R,41S,46R,48R,49R,50R,52R,54R,55S,56S,58S,62R,66S,68R,69S,70S) (Fig. 1A).
Nowadays, malaria is still one of the most deadly infectious diseases in the world, particularly in Africa and Asia. Due to rapid spread of resistant Plasmodium parasites, it is in urgent need to discover the next generation of potent antimalarial drugs with new chemical entities. Thus, benthol A (1) was assayed in vitro against the drug sensitive 3D7 strain of P. falciparum.36 As a consequence, it exhibited antiplasmodial activity with an EC50 value of 60.80 nM. Dihydroartemisinin was used as a positive control with an EC50 value of 4.12 nM. In addition, in vitro anti-HIV-1 assay37,38 revealed that 1 showed an inhibition rate of 57.1% at the concentration of 100.0 μM and 16.5% at the concentration of 20.0 μM, respectively. Efavirenz was used as a positive control with 90.2% inhibition at the concentration of 100.0 μM and 88.5% at the concentration of 20.0 μM, respectively.
Principal component analysis
To date, 187 SCCCs with molecular weights of greater than 1000 daltons have been isolated from marine dinoflagellates of eight genera, viz., Amphidinium, Gambierdiscus, Karenia, Karlodinium, Ostreopsis, Prorocentrum, Protoceratium, and Symbiodinium (Table S4†). Among them, polyol–polyene compounds (PPCs) and ladder-frame polyethers (LFPs), including 60 and 90 members, respectively, are two major families and occupy 79.8% of the total SCCCs.PPCs are mainly produced by dinoflagellates of the genera Amphidinium,39–63Karlodinium,64–69Ostreopsis,70,71 and Prorocentrum.72,73 These SCCCs are characterized by the presence of both a polyol and a polyene chain, connected by a central core containing two tetrahydropyran rings. Due to numerous stereocenters on flexible acyclic carbon chains of PPCs, only the absolute configurations of three molecules, i.e., AM3,61,63 KmTx2,65,67 and ostreol B,71 have hitherto been completely established. LFPs, featuring trans-fused polycyclic ether scaffolds, are generated by marine dinoflagellates of the genera Gambierdiscus,74–76Karenia,17,77–79 and Protoceratium.80,81 Marine toxins, such as ciguatoxins, brevetoxins, maitotoxin, and yessotoxins, are the representative examples.74–81 Owing to the universality of NOE interactions and ECD cotton effects for the stereochemical assignment of LFPs, the absolute configurations of these marine toxins have been determined.
Principal component analysis (PCA) is a widely used statistical technique for exploratory data analysis.82 It can reduce the dimensionality of complex data sets with the aim to achieve visualization loading in two- or three-dimensional plots.83 To categorize the SCCCs reported to date, PCA was applied to 188 SCCCs, including 1 and previously explored 187 SCCCs (Fig. 6). Eight parameters were considered, including molecular weight, and carbon numbers, ring numbers, hydroxy numbers, carbon–carbon double bond numbers, chiral carbon numbers, and spiroketal carbon numbers of the backbone chain, and the ratio of backbone carbons to ether rings. The model obtained by PCA showed a good fitting degree (R2X = 0.996) and a strong predictive ability [Q2(cum) = 0.882] (Fig. S5†). The PCA score plot revealed that PPCs and LFPs are the two largest independent groups. Except for PPCs, the other SCCCs from dinoflagellates of the genus Amphidinium are dispersively distributed. Most notably, 1 is distinctly separated from all the other SCCCs. It cannot be grouped with any other SCCCs from dinoflagellates of the genus Amphidinium (Fig. 6). The nearest neighbors of 1 are dinophysistoxin-4 (DTX-4, 131), DTX-5a (132), DTX-5b (133), DTX-5c (134), and prorocentroic acid (137) (Fig. 6 and S6†), all of which were obtained from dinoflagellates of the genus Prorocentrum. The above four DTX analogs are the derivatives of okadaic acid, containing three spiroketal moieties within the C38 backbone chain, whereas prorocentroic acid comprises a 6,8-dioxabicyclo[3.2.1]octane motif and two tetrahydropyran rings within the C60 backbone chain. The molecule of 1, however, contains eight scattered ether rings within the C72 backbone chain. Accordingly, 1 should be classified as the first member of a new SCCC family situated between PPCs and LFPs.
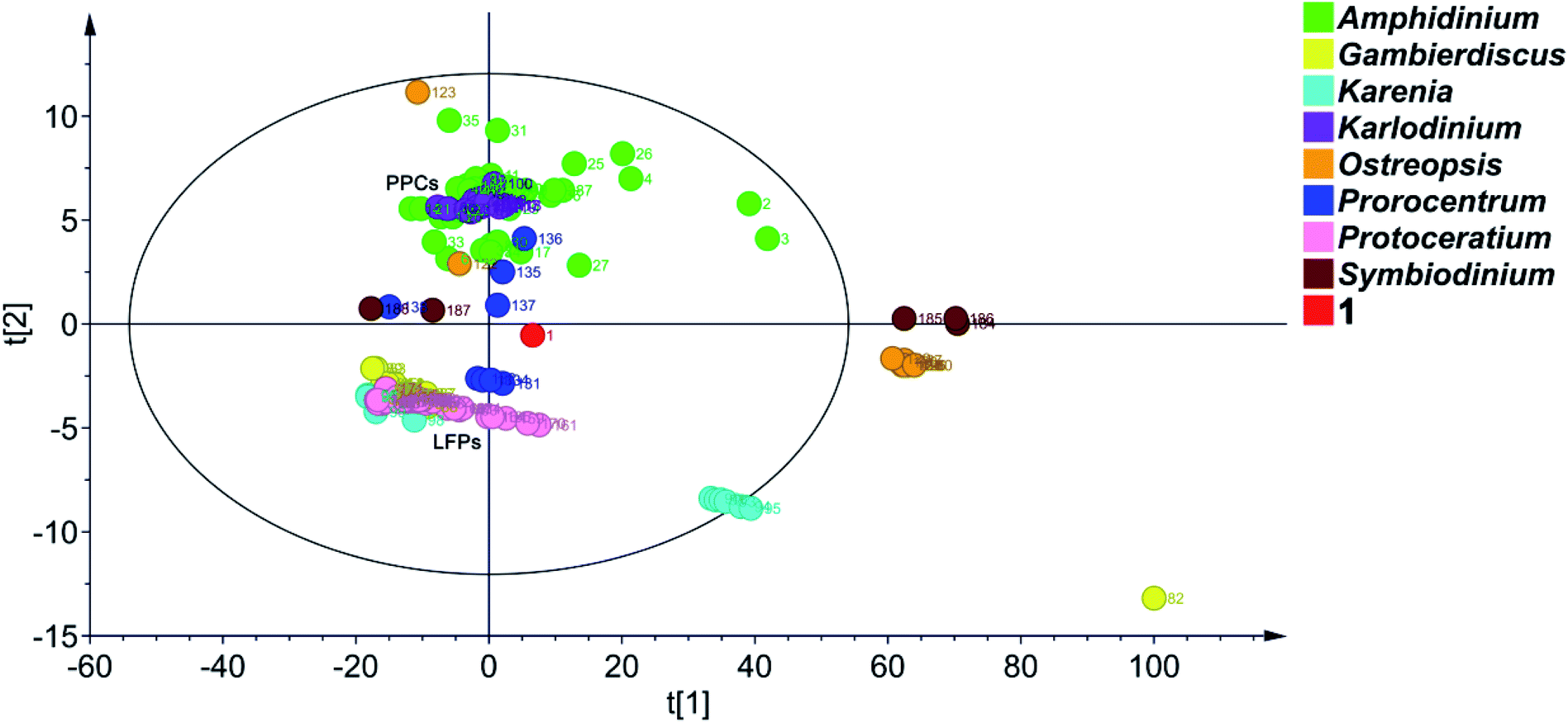 | ||
| Fig. 6 PCA score plot of 188 SCCCs. Benthol A is designated as the number 1 (The red dot) and previously reported 187 SCCCs as numbers 2 to 188 in the graph. R2X = 0.996, Q2(cum) = 0.882, ellipse: Hotelling's T2 (95%). | ||
Obviously, SCCCs from dinoflagellates of three genera, viz., Amphidinium, Ostreopsis, and Prorocentrum, are dispersively distributed, whereas those from the genera Karenia and Symbiodinium could be categorized into two main groups. SCCCs from the other three genera, viz., Karlodinium, Gambierdiscus, and Protoceratium, are concentrated. Maitotoxin (82), however, is isolated, mainly due to its huge molecular weight (Fig. 6). It may be concluded that dinoflagellates of the genera Amphidinium, Ostreopsis, and Prorocentrum harbor SCCCs with the richest structural diversity.
In fact, the first member of most SCCC families was reported in the last century. As aforementioned, symbiodinolide (184) was identified with a 62-membered lactone in 2007.9 However, its analog zooxanthellatoxin-A (186) was reported in 1995.12 The above two SCCCs are grouped with the inseparable isomeric mixture zooxanthellamides C1–C5 (185)84 to form a distinct SCCC family, termed super macrolides (Fig. 6).
It is worth mentioning that, amdigenols A (2) and D (3), containing three tetrahydropyran rings within the C98 backbone chain and four tetrahydropyran moieties within the C101 backbone chain, respectively, were obtained as dimeric AM analogs from the Okinawan dinoflagellate Amphidinium sp. in 2012 and 2020, respectively. The two SCCCs are grouped to form an independent SCCC family in the PCA score plot (Fig. 6).
It is reported that PPCs, such as AM3 and KmTx8, featuring both a hydrophilic polyol and a hydrophobic polyene chain, exhibited remarkable antifungal and antitumor activities.8,61,67 These pore-forming agents, taking a hairpin conformation in the lipid bilayer membrane, can specifically bind to membrane sterols and then perforate the membranes without altering the membrane integrity.63,67 It was demonstrated that AM3 directly interacts with the membrane cholesterol or ergosterol through the strict molecular recognition of the 3β-OH group,85 whereas KmTx8 was proposed to exhibit a cytotoxic mode-of-action against cancer cells through cholesterol and other membrane binding interactions.67 Structure–activity relationship investigations of AM3 analogs revealed that the proper length of the polyol chains in these PPCs plays an important role in their membrane-disrupting activities. When the length of the polyol chains exceeds 35 carbons, the membrane-disrupting activity of the AM3 analog decreases significantly.59 LFPs, such as ciguatoxins, brevetoxins, and maitotoxin, are activators of Na+ or Ca2+ channels in mammalian cell membranes.8,74–79 Although the target proteins and mode-of-action of maitotoxin remain unknown, its potent Ca2+ influx activity could be attributed to both hydrophilic and hydrophobic regions of this molecule. The trans-fused polycyclic ether feature of LFPs might be responsible for the toxicity of these marine toxins.8
The structure of benthol A (1), containing eight scattered ether rings on a C72 backbone chain, is totally different from those of PPCs and LFPs. It cannot be divided into two separated hydrophilic and hydrophobic regions. In fact, our preliminary bioassays revealed that benthol A did not show either obvious antifungal activity against the model fungal species Aspergillus niger or significant cytotoxicity against eight human cancer cell lines, viz., A375, AGS, A549, A2780, A2780/T, HCT-8, HCT-8/T, and MDA-MB-231, at the concentration of 100.0 μM. Evidently, the bioactivity profile of benthol A might be completely different from those of PPCs and LFPs. Most notably, in-depth mechanistic investigations of the potent antiplasmodial activity of benthol A are warranted.
Marine dinoflagellates are exclusively focused on the generation of polyketides. From the standpoint of biosynthesis, SCCCs are unique and huge polyketides.86 It is well known that small molecule polyketides, accounting for the majority of all the metabolites in bacteria, fungi, phytoplankton, plants, and invertebrates, are believed to be mainly synthesized by polyketide synthases. With the aid of modern techniques, the molecular bases of various polyketide synthases in bacteria and fungi have been thoroughly elucidated.87,88 However, large and complex genomes of dinoflagellates, and their limited genetic data have been hampering the molecular studies of SCCC biosynthesis. To date, the biosynthetic knowledge of SCCCs has been accessed only by means of biochemical labeling studies.80,89 The biosynthetic investigation of SCCCs through genome mining is still in the early stages.
Conclusions
In summary, an unprecedented SCCC, named benthol A, was isolated from the unexplored benthic dinoflagellate collected in the South China Sea. Its planar structure, containing a 6,6-spiroketal motif, three tetrahydrofuran rings, three tetrahydropyran moieties, and twenty-two hydroxy groups, was elucidated by means of extensive NMR spectroscopic investigations, particularly the 2D-INADEQUATE spectrum, acquired with 13C-enriched chemical entity. The absolute configurations of thirty-five carbon stereocenters were unambiguously established by a combination of periodate degradation of the 1,2-diol groups, ozonolysis of the carbon–carbon double bonds, JBCA, NOE interactions, modified Mosher's MTPA ester method, and DFT-NMR 13C chemical-shift calculations aided by DP4+ statistical analysis. Additionally, benthol A displayed potent antimalarial activity against Plasmodium falciparum 3D7 parasites. The fascinating structural features of benthol A place it as the first member of a new SCCC family located between PPCs and LFPs, herein termed polyol–polyether compounds. This suggestion was strongly supported by the PCA score plot of 188 SCCCs reported to date. To our knowledge, this is the first report of a SCCC containing eight scattered ether rings. The discovery of benthol A does not only provide new insights into underexplored chemical space of SCCCs, but also opens up a new window for skeletal diversity of SCCCs. This work demonstrates that new taxa of dinoflagellates harbor unique biosynthetic machineries for the production of novel SCCC families with signature features.Data availability
All experimental procedures, spectroscopic and computational data are provided as the ESI.Author contributions
Conceptualization, J. W. and L. S.; methodology, W. H. C., T. K., M. H. Y., J. W., and L. S.; investigation, Z. P. J., S. H. S., Y. Y., A. M., and J. Y. L.; writing – original draft, S. H. S., A. M., J. Y. L., W. H. C., T. K., J. W., and L. S.; writing – review & editing, W. H. C., J. W., and L. S.; funding acquisition, T. K., J. W., and L. S.; resources, M. H. Y., T. K., J. W., and L. S.; supervision, T. K., J. W., and L. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Guangdong Basic and Applied Basic Research Foundation, Funds for Distinguished Young Scholar, PR China (2020B1515020056) and the National Natural Science Foundation of China (U20A2001 and 31770377). The Hungarian authors were supported by the EU and co-financed by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. T. K. and A. M. thank the National Research, Development, and Innovation Office (NKFI K120181 and FK134653) for the financial support. The Governmental Information-Technology Development Agency (KIFÜ) is acknowledged for CPU time. The authors are grateful to Dr Zhi-Hui Xiao, Ai-Jun Sun, and Yun Zhang (Equipment Public Service Center, South China Sea Institute of Oceanology, Chinese Academy of Sciences) for recording 700 MHz NMR spectra and HR-ESIMS.Notes and references
- D. Uemura, Chem. Rec., 2006, 6, 235 CrossRef CAS.
- M. Kita and D. Uemura, Chem. Rec., 2010, 10, 48 CrossRef CAS.
- M. Murata, H. Naoki, T. Iwashita, S. Matsunaga, M. Sasaki, A. Yokoyama and T. Yasumoto, J. Am. Chem. Soc., 1993, 115, 2060 CrossRef CAS.
- T. Yasumoto and M. Murata, Chem. Rev., 1993, 93, 1897 CrossRef CAS.
- J. Kobayashi and M. Ishibashi, Chem. Rev., 1993, 93, 1753 CrossRef CAS.
- J. Kobayashi and T. Kubota, J. Nat. Prod., 2007, 70, 451 CrossRef CAS.
- S. A. Rasmussen, A. J. C. Andersen, N. G. Andersen, K. F. Nielsen, P. J. Hansen and T. O. Larsen, J. Nat. Prod., 2016, 79, 662 CrossRef CAS.
- T. Oishi, Bull. Chem. Soc. Jpn., 2020, 93, 1350 CrossRef CAS.
- M. Kita, N. Ohishi, K. Konishi, M. Kondo, T. Koyama, M. Kitamura, K. Yamada and D. Uemura, Tetrahedron, 2007, 63, 6241 CrossRef CAS.
- H. Takamura, T. Fujiwara, Y. Kawakubo, I. Kadota and D. Uemura, Chem.–Eur. J., 2016, 22, 1979 CrossRef CAS PubMed.
- H. Takamura, T. Fujiwara, Y. Kawakubo, I. Kadota and D. Uemura, Chem.–Eur. J., 2016, 22, 1984 CrossRef CAS.
- H. Nakamura, T. Asari and A. Murai, J. Am. Chem. Soc., 1995, 117, 550 CrossRef CAS.
- W. S. Li, R. J. Yan, Y. Yu, Z. Shi, A. Mándi, L. Shen, T. Kurtán and J. Wu, Angew. Chem. Int. Ed., 2020, 59, 13028 ( Angew. Chem. , 2020 , 132 , 13128 ) CrossRef CAS PubMed.
- W. S. Li, Z. Luo, Y. L. Zhu, Y. Yu, J. Wu and L. Shen, Mar. Drugs, 2020, 18, 590 CrossRef CAS PubMed.
- M. S. Lee, D. J. Repeta, K. Nakanishi and M. G. Zagorski, J. Am. Chem. Soc., 1986, 108, 7855 CrossRef CAS PubMed.
- F. M. D. Ismail, L. Nahar and S. D. Sarker, Phytochem. Anal., 2021, 32, 7 CrossRef CAS PubMed.
- Y. Hamamoto, K. Tachibana, P. T. Holland, F. Shi, V. Beuzenberg, Y. Itoh and M. Satake, J. Am. Chem. Soc., 2012, 134, 4963 CrossRef CAS PubMed.
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org. Chem., 1999, 64, 866 CrossRef CAS PubMed.
- G. Bifulco, P. Dambruoso, L. Gomez-Paloma and R. Riccio, Chem. Rev., 2007, 107, 3744 CrossRef CAS PubMed.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, J. Org. Chem., 2005, 70, 3778 CrossRef CAS PubMed.
- J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2012, 112, 4603 CrossRef CAS PubMed.
- K. Konno, T. Fujishima and Z. P. Liu, Chirality, 2002, 13, 72 CrossRef PubMed.
- H. Takamura, Y. Kadonaga, Y. Yamano, C. Han, Y. Aoyama, I. Kadota and D. Uemura, Tetrahedron Lett., 2009, 50, 863 CrossRef CAS.
- C. Han, Y. Yamano, M. Kita, H. Takamura and D. Uemura, Tetrahedron Lett., 2009, 50, 5280 CrossRef CAS.
- H. Takamura, Y. Kadonaga, Y. Yamano, C. Han, I. Kadota and D. Uemura, Tetrahedron, 2009, 65, 7449 CrossRef CAS.
- H. Takamura, Y. Kadonaga, I. Kadota and D. Uemura, Tetrahedron, 2010, 66, 7569 CrossRef CAS.
- E. Van der Eyckenl, H. D. Wilde, L. Deprez and M. Vandewalle, Tetrahedron Lett., 1987, 28, 4759 CrossRef.
- V. K. Tandon, A. M. Van Leusen and H. Wynberg, J. Org. Chem., 1983, 48, 2767 CrossRef CAS.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946 CrossRef CAS.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526 CrossRef CAS PubMed.
- W. S. Li, A. Mándi, J. J. Liu, L. Shen, T. Kurtán and J. Wu, J. Org. Chem., 2019, 84, 2596 CrossRef CAS PubMed.
- A. Mándi and T. Kurtán, Nat. Prod. Rep., 2019, 36, 889 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664 CrossRef CAS.
- S. Qiu, E. De Gussem, K. A. Tehrani, S. Sergeyev, P. Bultinck and W. Herrebout, J. Med. Chem., 2013, 56, 8903 CrossRef CAS PubMed.
- W. S. Wang, J. M. Yao, Z. Chen, Y. M. Sun, Y. Q. Shi, Y. F. Wei, H. J. Zhou, Y. F. Yu, S. Z. Li and L. P. Duan, Br. J. Pharmacol., 2020, 177, 5569 CrossRef CAS PubMed.
- J. L. Zhao, J. M. Feng, Z. Tan, J. M. Liu, J. Y. Zhao, R. D. Chen, K. B. Xie, D. W. Zhang, Y. Li, L. Y. Yu, X. G. Chen and J. G. Dai, J. Nat. Prod., 2017, 80, 1819 CrossRef CAS PubMed.
- J. L. Ren, X. P. Zou, W. S. Li, L. Shen and J. Wu, Mar. Drugs, 2018, 16, 434 CrossRef CAS PubMed.
- M. Satake, M. Murata, T. Yasumoto, T. Fujita and H. Naoki, J. Am. Chem. Soc., 1991, 113, 9859 CrossRef CAS.
- G. K. Paul, N. Matsumori, M. Murata and K. Tachibana, Tetrahedron Lett., 1995, 36, 6279 CrossRef CAS.
- G. K. Paul, N. Matsumori, K. Konoki, M. Murata and K. Tachibana, J. Mar. Biotechnol., 1997, 5, 124 CAS.
- Y. Doi, M. Ishibashi, H. Nakamichi, T. Kosaka, T. Ishikawa and J. Kobayashi, J. Org. Chem., 1997, 62, 3820 CrossRef CAS.
- T. Kubota, M. Tsuda, Y. Doi, A. Takahashi, H. Nakamichi, M. Ishibashi, E. Fukushi, J. Kawabata and J. Kobayashi, Tetrahedron, 1998, 54, 14455 CrossRef CAS.
- M. Murata, S. Matsuoka, N. Matsumori, G. K. Paul and K. Tachibana, J. Am. Chem. Soc., 1999, 121, 870 CrossRef CAS.
- T. Houdai, S. Matsuoka, M. Murata, M. Satake, S. Ota, Y. Oshima and L. L. Rhodes, Tetrahedron, 2001, 57, 5551 CrossRef CAS.
- X. Huang, D. Zhao, Y. Guo, H. Wu, L. Lin, Z. Wang, J. Ding and Y. Lin, Bioorg. Med. Chem. Lett., 2004, 14, 3117 CAS.
- X. Huang, D. Zhao, Y. Guo, H. Wu, E. Trivellone and G. Cimino, Tetrahedron Lett., 2004, 45, 5501 CrossRef CAS.
- N. Morsy, S. Matsuoka, T. Houdai, N. Matsumori, S. Adachi, M. Murata, T. Iwashita and T. Fujita, Tetrahedron, 2005, 61, 8606 CrossRef CAS.
- T. Kubota, A. Takahashi, M. Tsuda and J. Kobayashi, Mar. Drugs, 2005, 3, 113 CrossRef CAS.
- R. Echigoya, L. Rhodes, Y. Oshima and M. Satake, Harmful Algae, 2005, 4, 383 CrossRef CAS.
- K. Washida, T. Koyama, K. Yamada, M. Kita and D. Uemura, Tetrahedron Lett., 2006, 47, 2521 CrossRef CAS.
- N. Morsy, T. Houdai, S. Matsuoka, N. Matsumori, S. Adachi, T. Oishi, M. Murata, T. Iwashita and T. Fujita, Bioorg. Med. Chem., 2006, 14, 6548 CrossRef CAS.
- S. Huang, C. Kuo, Y. Lin, Y. Chen and C. Lu, Tetrahedron Lett., 2009, 50, 2512 CrossRef CAS.
- Y. Meng, R. M. Van Wagoner, I. Misner, C. Tomas and J. L. C. Wright, J. Nat. Prod., 2010, 73, 409 CrossRef CAS.
- N. Hanif, O. Ohno, M. Kitamura, K. Yamada and D. Uemura, J. Nat. Prod., 2010, 73, 1318 CrossRef CAS.
- T. Inuzuka, Y. Yamamoto, K. Yamada and D. Uemura, Tetrahedron Lett., 2012, 53, 239 CrossRef CAS.
- G. Nuzzo, A. Cutignano, A. Sardo and A. Fontana, J. Nat. Prod., 2014, 77, 1524 CrossRef CAS PubMed.
- T. Inuzuka, K. Yamada and D. Uemura, Tetrahedron Lett., 2014, 55, 6319 CrossRef CAS.
- M. Satake, K. Cornelio, S. Hanashima, R. Malabed, M. Murata, N. Matsumori, H. Zhang, F. Hayashi, S. Mori, J. S. Kim, C. H. Kim and J. S. Lee, J. Nat. Prod., 2017, 80, 2883 CrossRef CAS PubMed.
- A. Cutignano, G. Nuzzo, A. Sardo and A. Fontana, Mar. Drugs, 2017, 15, 157 CrossRef PubMed.
- Y. Wakamiya, M. Ebine, M. Murayama, H. Omizu, N. Matsumori, M. Murata and T. Oishi, Angew. Chem. Int. Ed., 2018, 57, 6060 ( Angew. Chem. , 2018 , 130 , 6168 ) CrossRef CAS.
- K. A. Martínez, C. Lauritano, D. Druka, G. Romano, T. Grohmann, M. Jaspars, J. Martín, C. Díaz, B. Cautain, M. de la Cruz, A. Ianora and F. Reyes, Mar. Drugs, 2019, 17, 385 CrossRef PubMed.
- Y. Wakamiya, M. Ebine, N. Matsumori and T. Oishi, J. Am. Chem. Soc., 2020, 142, 3472 CrossRef CAS.
- R. M. Van Wagoner, J. R. Deeds, M. Satake, A. A. Ribeiro, A. R. Place and J. L. C. Wright, Tetrahedron Lett., 2008, 49, 6457 CrossRef CAS.
- J. Peng, A. R. Place, W. Yoshida, C. Anklin and M. T. Hamann, J. Am. Chem. Soc., 2010, 132, 3277 CrossRef CAS.
- R. M. Van Wagoner, J. R. Deeds, A. O. Tatters, A. R. Place, C. R. Tomas and J. L. C. Wright, J. Nat. Prod., 2010, 73, 1360 CrossRef CAS.
- A. L. Waters, J. Oh, A. R. Place and M. T. Hamann, Angew. Chem. Int. Ed., 2015, 54, 15705 ( Angew. Chem. , 2015 , 127 , 15931 ) CrossRef CAS PubMed.
- P. Cai, S. He, C. Zhou, A. R. Place, S. Had, L. Ding, H. Chen, Y. Jiang, C. Guo, Y. Xu, J. Zhang and X. Yan, Harmful Algae, 2016, 58, 66 CrossRef CAS PubMed.
- S. A. Rasmussen, S. B. Binzer, C. Hoeck, S. Meier, L. S. de Medeiros, N. G. Andersen, A. Place, K. F. Nielsen, P. J. Hansen and T. O. Larsen, J. Nat. Prod., 2017, 80, 1287 CrossRef CAS PubMed.
- B. S. Hwang, E. Y. Yoon, H. S. Kim, W. Yih, J. Y. Park, H. J. Jeong and J. R. Rho, Bioorg. Med. Chem. Lett., 2013, 23, 3023 CrossRef CAS PubMed.
- B. S. Hwang, E. Y. Yoon, E. J. Jeong, J. Park, E. H. Kim and J. R. Rho, J. Org. Chem., 2018, 83, 194 CrossRef CAS PubMed.
- K. Sugahara, Y. Kitamura, M. Murata, M. Satake and K. Tachibana, J. Org. Chem., 2011, 76, 3131 CrossRef CAS PubMed.
- H. J. Domínguez, D. Cabrera-García, C. Cuadrado, A. Novelli, M. T. Fernández-Sánchez, J. J. Fernández and A. H. Daranas, Org. Lett., 2021, 23, 13 CrossRef PubMed.
- M. Murata, A. M. Legrand, Y. Ishibashi, M. Fukui and T. Yasumoto, J. Am. Chem. Soc., 1990, 112, 4380 CrossRef CAS.
- K. Yogi, N. Oshiro, Y. Inafuku, M. Hirama and T. Yasumoto, Anal. Chem., 2011, 83, 8886 CrossRef CAS PubMed.
- J. R. Chekan, T. R. Fallon and B. S. Moore, Curr. Opin. Chem. Biol., 2020, 59, 119 CrossRef CAS PubMed.
- Y. Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773 CrossRef CAS.
- K. Tanaka, Y. Itagaki, M. Satake, H. Naoki, T. Yasumoto, K. Nakanishi and N. Berova, J. Am. Chem. Soc., 2005, 127, 9561 CrossRef CAS PubMed.
- R. Suzuki, R. Irie, Y. Harntaweesup, K. Tachibana, P. T. Holland, D. T. Harwood, F. Shi, V. Beuzenberg, Y. Itoh, S. Pascal, P. J. B. Edwards and M. Satake, Org. Lett., 2014, 16, 5850 CrossRef CAS PubMed.
- M. Yamazaki, M. Izumikawa, K. Tachibana, M. Satake, Y. Itoh and M. Hashimoto, J. Org. Chem., 2012, 77, 4902 CrossRef CAS PubMed.
- P. Ciminiello, E. Fattorusso, M. Forino, R. Poletti and R. Viviani, Chem. Res. Toxicol., 2000, 13, 770 Search PubMed.
- G. Ivosev, L. Burton and R. Bonner, Anal. Chem., 2008, 80, 4933 CrossRef CAS PubMed.
- M. Yu, C. Zhou, D. Tian, H. M. Jia, Z. Q. Li, C. Yang, Y. M. Ba, H. K. Wu and Z. M. Zou, J. Pharm. Biomed. Anal., 2021, 198, 114004 CrossRef CAS PubMed.
- K. Onodera, H. Nakamura, Y. Oba, Y. Ohizumi and M. Ojika, J. Am. Chem. Soc., 2005, 127, 10406 CrossRef CAS PubMed.
- R. A. Espiritu, N. Matsumori, M. Tsuda and M. Murata, Biochemistry, 2014, 53, 3287 CrossRef CAS PubMed.
- R. M. Van Wagoner, M. Satake and J. L. C. Wright, Nat. Prod. Rep., 2014, 31, 1101 RSC.
- J. Clardy and C. Walsh, Nature, 2004, 432, 829 CrossRef CAS PubMed.
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524 CrossRef CAS.
- S. L. MacKinnon, A. D. Cembella, I. W. Burton, N. Lewis, P. LeBlanc and J. A. Walter, J. Org. Chem., 2006, 71, 8724 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental Fig. S1–S5, Tables S1–S4, and experimental procedures; supplemental Fig. S6–S221, including copies of HR-ESIMS for compound 1 and fragments 1A, 1B, 1d, 1As/r, 1Bs/r, 1as/r, 1bs/r, and 1cs/r; LR-ESIMS for fragments 1a–1c, 1e, and 1es/r; and 1D and 2D NMR spectra for compound 1 and fragments 1A, 1B, 1a–1e, 1As/r, 1Bs/r, 1as/r, 1bs/r, 1cs/r, and 1es/r. See DOI: 10.1039/d1sc02810c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |