
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmon-enabled N2 photofixation on partially reduced Ti3C2 MXene†
Binbin
Chang
a,
Yanzhen
Guo
c,
Donghai
Wu
c,
Li
Li
 *a,
Baocheng
Yang
*c and
Jianfang
Wang
*b
*a,
Baocheng
Yang
*c and
Jianfang
Wang
*b
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: lli@chem.ecnu.edu.cn
bDepartment of Physics, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China. E-mail: jfwang@phy.cuhk.edu.hk
cHenan Provincial Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou 450006, China. E-mail: baochengyang@infm.hhstu.edu.cn
First published on 20th July 2021
Abstract
Benefiting from the superior conductivity, rich surface chemistry and tunable bandgap, Ti3C2 MXene has become a frontier cocatalyst material for boosting the efficiency of semiconductor photocatalysts. It has been theoretically predicted to be an ideal material for N2 fixation. However, the realization of N2 photofixation with Ti3C2 as a host photocatalyst has so far remained experimentally challenging. Herein, we report on a sandwich-like plasmon- and an MXene-based photocatalyst made of Au nanospheres and layered Ti3C2, and demonstrate its efficient N2 photofixation in pure water under ambient conditions. The abundant low-valence Ti (Ti(4−x)+) sites in partially reduced Ti3C2 (r-Ti3C2) produced by surface engineering through H2 thermal reduction effectively capture and activate N2, while Au nanospheres offer plasmonic hot electrons to reduce the activated N2 into NH3. The Ti(4−x)+ active sites and plasmon-generated hot electrons work in tandem to endow r-Ti3C2/Au with remarkably enhanced N2 photofixation activity. Importantly, r-Ti3C2/Au exhibits ultrahigh selectivity without the occurrence of competing H2 evolution. This work opens up a promising route for the rational design of efficient MXene-based photocatalysts.
Introduction
Nitrogen is a requisite nutrient for all organisms on the Earth. Although N2 occupies ∼78 vol% of the atmosphere, its efficient utilization by organisms is greatly hindered because of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (945 kJ mol−1).1,2 Atmospheric N2 is continuously fixed into biologically usable forms of nitrogen, such as NH3 and NO3−. At the same time, the fixed forms of nitrogen are also continuously converted back to N2 in nature, constituting a giant nitrogen cycle (Fig. S1†).3 In this cycle, synthesized NH3 not only is used as an indispensable chemical feedstock but also can act as a potential hydrogen carrier owing to its high hydrogen density (17.6 wt%) and low liquefying pressure (∼8 atm).4,5 In addition, the obtained H2 and O2 can be utilized in fuel cells. As a result, the realization of such a nitrogen cycle will be meaningful in developing sustainable clean energy and relieving environmental pollution. The NH3 synthesis is a critical step in the cycle. Traditionally, NH3 is produced through the industrial Haber–Bosch process. However, this process requires high temperatures (>300 °C) and high pressures (>100 atm) with a massive energy consumption and a huge amount of CO2 emission.6,7 Because of the energy input and carbon footprint, it is highly desired to explore a promising artificial nitrogen fixation strategy under benign conditions for a sustainable, green and safe NH3 production.
N bond (945 kJ mol−1).1,2 Atmospheric N2 is continuously fixed into biologically usable forms of nitrogen, such as NH3 and NO3−. At the same time, the fixed forms of nitrogen are also continuously converted back to N2 in nature, constituting a giant nitrogen cycle (Fig. S1†).3 In this cycle, synthesized NH3 not only is used as an indispensable chemical feedstock but also can act as a potential hydrogen carrier owing to its high hydrogen density (17.6 wt%) and low liquefying pressure (∼8 atm).4,5 In addition, the obtained H2 and O2 can be utilized in fuel cells. As a result, the realization of such a nitrogen cycle will be meaningful in developing sustainable clean energy and relieving environmental pollution. The NH3 synthesis is a critical step in the cycle. Traditionally, NH3 is produced through the industrial Haber–Bosch process. However, this process requires high temperatures (>300 °C) and high pressures (>100 atm) with a massive energy consumption and a huge amount of CO2 emission.6,7 Because of the energy input and carbon footprint, it is highly desired to explore a promising artificial nitrogen fixation strategy under benign conditions for a sustainable, green and safe NH3 production.
Nitrogen photofixation offers an approach for achieving the energy-saving and environmentally friendly NH3 synthesis under ambient conditions with renewable solar energy as the driving force.8–10 In N2 photofixation, H2 is replaced by H2O as a reducing agent. N2 and 3H2O are converted to 2NH3 and 1.5O2.11–17 The key is to design an efficient photocatalyst. Two-dimensional (2D) photocatalysts exhibit unique merits in solar-to-chemical energy conversion.18–22 MXenes, a family of 2D layered transition metal carbides, nitrides or carbonitrides, have aroused much interest since the first report in 2011.23 Benefiting from the metallic conductivity, abundant surface terminal groups, large surface-to-volume ratio, excellent hydrophilic and ion transport properties, MXenes have been explored in diverse fields.24–26 Ti3C2, the first reported MXene, possesses several unique characteristics.24,25 (i) Its high conductivity enables excellent charge transfer kinetics, favoring the rapid migration and efficient separation of photogenerated electrons and holes; (ii) abundant exposed metal sites offer many active sites for catalysis; (iii) adjustable terminal groups (OH, O, F, etc.) bring a tunable bandgap and optical absorption, enabling facile regulation of the photocatalytic performance; (iv) excellent surface hydrophilicity improves the interfacial connection with other materials, facilitating the formation of heterostructures. Ti3C2 has therefore become a research hotspot in photocatalysis.27–30 Theoretical calculations have shown that Ti3C2 presents valid N2-philicity and the ability to chemisorb and activate N2, suggesting that Ti3C2 can be a promising material for N2 capture and reduction.31 Unfortunately, the extremely low Fermi level and metallic nature of Ti3C2 make it a superior electron acceptor, resulting in a poor separation efficiency of photogenerated electrons and holes.32,33 In addition, the narrow bandgap of Ti3C2 makes N2 photoreduction difficult.34 As a result, it has remained challenging to realize N2 photofixation solely with Ti3C2.
Plasmonic metal nanoparticles possess extraordinary optical properties that arise from localized surface plasmon resonance (LSPR) and offer a powerful means for boosting the photocatalytic activity.18,35 LSPR-enhanced photocatalysis mainly relies on the extension of light absorption to the long-wavelength range and the enhancement of the local electric field. Plasmonic hot electrons can drive reduction reactions and improve photocatalytic NH3 synthesis.18,36–38 The plasmon-intensified electric field can promote the photogeneration of charge carriers in semiconductors.39,40 As a result, combining plasmonic metal nanoparticles with 2D MXenes is expected to be a feasible strategy for constructing efficient photocatalysts for NH3 production.
Herein, we report on the construction of a Au nanosphere-embedded, partially reduced, and layered Ti3C2 (r-Ti3C2) photocatalyst with a unique sandwich-like architecture for efficient N2 photofixation under ambient conditions. r-Ti3C2 shows an expanded layer spacing and exposes many low-valence Ti sites (Ti(4−x)+) on the edge and basal planes, which serve as active sites for N2 activation. The unique sandwich-like, Au nanosphere-embedded r-Ti3C2 not only provides a large number of Ti(4−x)+ active sites but also brings a high contact area between Au nanospheres and r-Ti3C2, improving the probability of excited charge carriers to interact with the reaction solution. Moreover, the embedding of Au nanospheres hinders the self-stacking of the r-Ti3C2 layers, benefiting the exposure of the active sites and boosting the effective utilization of the active sites and charge carriers. The hot electrons photoexcited on the plasmonic Au nanospheres inject into r-Ti3C2 and thereby reduce Ti(4−x)+ site-activated N2 into NH3. The charge carrier recombination is largely suppressed because the electrons and holes are located in r-Ti3C2 and the Au nanospheres, respectively. The photocatalysts exhibit a superior activity for N2 photofixation in pure water at ambient temperature and pressure. Our strategy opens up new opportunities for designing MXene/(plasmonic metal) nanostructures to achieve efficient photo-driven N2 fixation.
Results and discussion
Materials synthesis and characterization
The Au nanosphere-embedded, partially reduced, and layered Ti3C2 with a unique sandwich-like architecture was prepared by a solvent-driven approach (Fig. 1a; see Experimental in the ESI†). First, the Al layers in layered Ti3AlC2 particles (Fig. S2†) were etched by HF to produce layered Ti3C2 with abundant O, OH and F terminations. During etching, some carbon atoms were replaced by oxygen atoms in the lattice of Ti3C2.41 Second, Ti3C2 was then thermally treated in a N2/H2 atmosphere to alter its surface chemistry and generate oxygen vacancies (OVs) through H2 reduction. Third, the Au nanospheres were driven by H2O and gradually interlaminated in layered r-Ti3C2, resulting in a unique sandwich-like structure.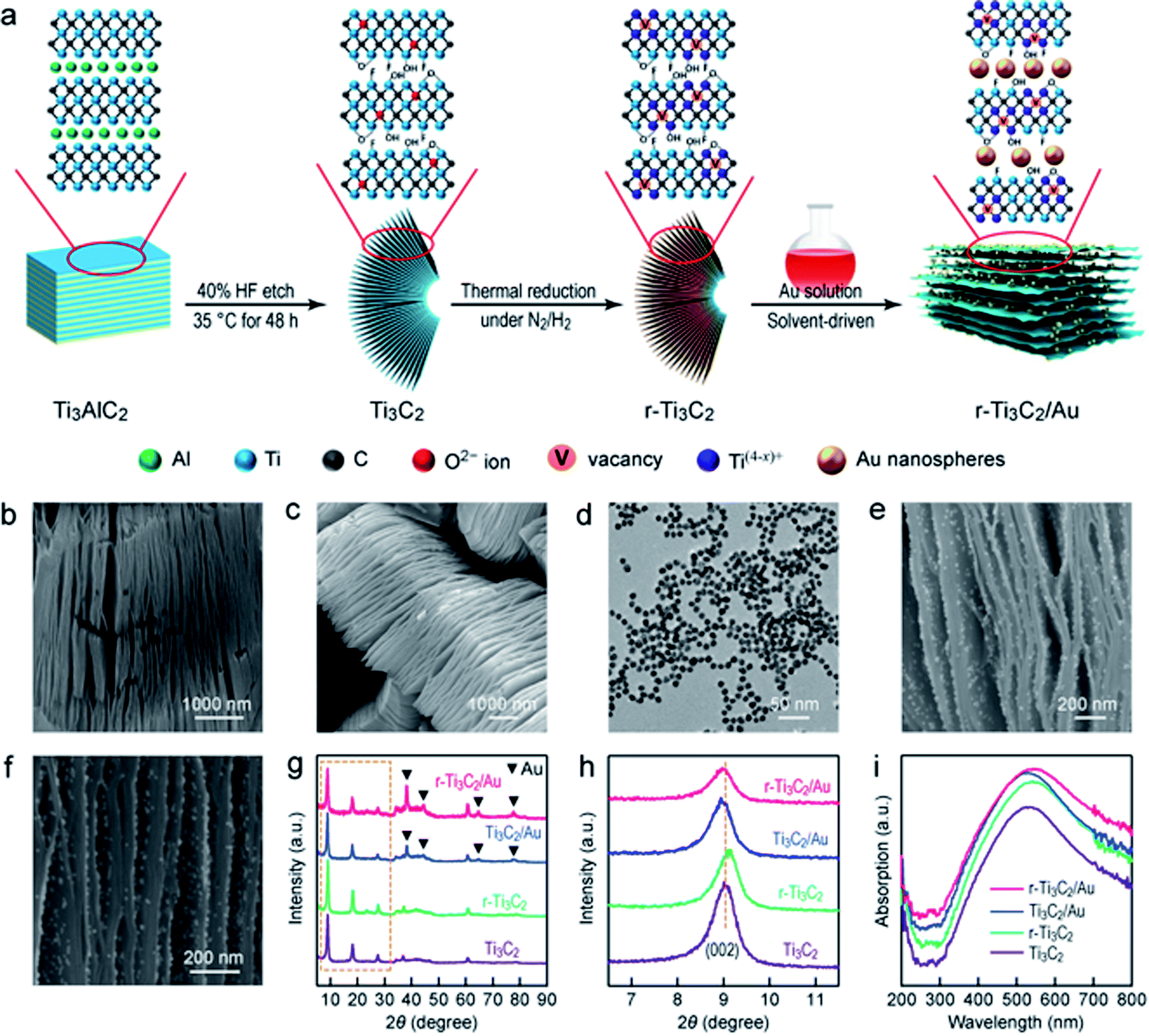 | ||
| Fig. 1 Synthesis of the sandwich-like r-Ti3C2/Au nanostructure. (a) Schematic illustrating the synthesis process. The Ti3C2 layer and Au nanosphere components are not drawn on the same size scale. (b) SEM image of Ti3C2. (c) SEM image of r-Ti3C2. (d) TEM image of the Au nanospheres. (e) SEM image of Ti3C2/Au. (f) SEM image of r-Ti3C2/Au. (g) XRD patterns of Ti3C2, r-Ti3C2, Ti3C2/Au and r-Ti3C2/Au. (h) XRD peaks of Ti3C2, r-Ti3C2, Ti3C2/Au and r-Ti3C2/Au for the (002) lattice planes. (i) Absorption spectra of Ti3C2, r-Ti3C2, Ti3C2/Au and r-Ti3C2/Au. | ||
Scanning electron microscopy (SEM) imaging (Fig. 1b) reveals the accordion-like multilayered structure of Ti3C2, suggesting the successful etching of the Al layers. The layered texture is well kept in r-Ti3C2 (Fig. 1c), indicating that the H2 treatment does not deteriorate the original layered structure. The Au nanospheres dispersed in aqueous solutions exhibit an extinction peak at 518 nm (Fig. S3†). Their sizes are uniform (Fig. 1d), as revealed by transmission electron microscopy (TEM), with an average diameter of 12.8 ± 0.7 nm (Fig. S4†). In the low-magnification SEM images of Ti3C2/Au and r-Ti3C2/Au (Fig. S5†), a unique sandwich-like architecture is clearly observed. The Au nanospheres are interlaminated between the MXene layers and uniformly distributed on the basal planes. High-magnification SEM imaging further shows clearly that the Au nanospheres are uniformly interlaminated in the MXene layers (Fig. 1e and f). The X-ray diffraction (XRD) of Ti3AlC2 (Fig. S6a†) reveals a strong and typical diffraction pattern of the pure Ti3AlC2 phase (JCPDS no. 52-0875). After HF treatment, the strong (002) peak exhibits a downshift from 9.55° to 9.05°, which corresponds to a c-lattice parameter (c-LP) increase from 18.53 Å to 19.48 Å, implying an interlayer spacing of 0.98 nm in Ti3C2. In addition, the disappearance of the intense peak at 2θ = 39.08° in Ti3AlC2 after HF etching verifies the removal of the Al layers.42 Taken together, these results confirm the successful synthesis of Ti3C2 MXene. After H2 reduction, r-Ti3C2 displays a similar XRD pattern to Ti3C2 (Fig. 1g), except an additional (006) diffraction peak at 2θ = 28.5° (Fig. S6b†), reflecting a better-organized multilayered structure with more opened Ti3C2 layers.43 The XRD patterns also reveal the coexistence of the cubic Au phase (JCPDS no. 01-1172) and Ti3C2 in Ti3C2/Au and r-Ti3C2/Au (Fig. 1g). Compared to Ti3C2, the (002) peak shows clear and different shifts among r-Ti3C2, Ti3C2/Au and r-Ti3C2/Au (Fig. 1h). A slight shift of the (002) peak to a higher angle of 9.12° for r-Ti3C2 corresponds to a c-LP decrease from 19.48 Å to 19.25 Å, signifying a reduced interlayer spacing of 0.96 nm in r-Ti3C2. The small c-LP change of r-Ti3C2 can be ascribed to the elimination of H2O molecules interlaminated between the Ti3C2 layers or surface reactions during H2 reduction.23,44 The (002) peak shifts down to 2θ = 8.95° for sandwich-like Ti3C2/Au and r-Ti3C2/Au, which corresponds to a c-LP of 19.68 Å, indicative of an enlarged interlayer spacing of 1.00 nm. The increased interlayer spacing is probably caused by the intercalation of the Au nanospheres between the Ti3C2 and r-Ti3C2 segments.45 Each segment contains multiple Ti3C2 layers, but its overall thickness is greatly reduced in comparison with the sample before the intercalation of the Au nanospheres. In the Raman spectra of Ti3AlC2 (Fig. S7†), four major peaks appearing at 145, 260, 410 and 605 cm−1 are the characteristic Raman bands of Ti3AlC2. They arise from the ω1, ω2, ω3 and ω4 Raman-active vibrational modes.46,47 Ti3C2 and r-Ti3C2 show similar Raman peaks, with a highly intensified peak at 153 cm−1, along with three weak peaks at 260, 428 and 610 cm−1. All the four peaks can be ascribed to the Raman-active vibrational modes of Ti–C,48,49 suggesting the successful etching of the Al atoms and the preservation of the Ti3C2 layers. In addition, two broad bands observed at 1350 and 1610 cm−1 for Ti3C2 and r-Ti3C2 correspond to the D- and G-bands of carbon, manifesting the existence of disordered carbon and ordered graphitic carbon, respectively. The larger peak intensities of the D- and G-bands in r-Ti3C2 reveal the existence of more disordered carbon after H2 reduction, suggesting the formation of more oxygen defects in the carbon layers of r-Ti3C2.50 Such oxygen defects can bring more active sites for N2 adsorption on the surface of r-Ti3C2.
Ti3C2 shows clear absorption in the spectral region of 300–800 nm with a maximum at ∼520 nm (Fig. 1i). r-Ti3C2 also exhibits a broad absorption band in the region of 300–800 nm, but the absorption maximum exhibits a slight redshift. This phenomenon is probably caused by the changes in the surface chemistry and terminal groups of r-Ti3C2, resulting in a changed bandgap.51 Despite the broad light absorption, very few charge carriers can be photogenerated and separated in Ti3C2 and r-Ti3C2.32–34 On the other hand, the efficient photothermal conversion of Ti3C2 MXene can facilitate surface catalytic reactions through the conversion of light to heat to activate the supported catalyst.52 Unlike common plasmonic Au/semiconductor hybrid photocatalysts,36,53 the absorption peak from the LSPR band of the Au nanospheres cannot be clearly observed in Ti3C2/Au or r-Ti3C2/Au. This result is likely caused by the spectral overlap between the LSPR of the Au nanospheres and the strong broad absorption of Ti3C2 MXene, which is reflected by the enhanced absorption in the region of 500–600 nm. The charge transport ability is another key factor in photocatalysis. Electrochemical impedance spectroscopy (EIS) measurements were conducted under white light illumination in a N2 atmosphere (Fig. S8†). A smaller semicircle in the obtained Nyquist plot implies a better charge transfer capability at the electrode–electrolyte interface. r-Ti3C2/Au exhibits the smallest arc radius, signifying a good charge transfer ability. The improved charge transfer ability can be attributed to the effective interfacial charge transfer in the sandwich-like r-Ti3C2/Au. Though the metallic Ti3C2 is difficult to photogenerate charge carriers, it can serve as an electron acceptor to capture the hot electrons generated by the plasmonic Au nanospheres owing to its superior electrical conductivity.32
Nitrogen photofixation
The photocatalytic N2 fixation experiments were performed in N2-saturated water under light illumination and ambient conditions in a quartz reactor (Fig. S9†). The produced ammonia amount was determined by Nessler's method, as shown by the calibration curve (Fig. S10†). Fig. 2a displays the time-dependent NH4+ concentrations over different photocatalysts under white light illumination. The Au nanospheres were found to be inactive for N2 photofixation under both white and visible light. NH4+ was hardly detected over Ti3C2. The NH4+ concentration reached 10.7 and 18.3 µmol L−1 in 6 h over r-Ti3C2 and Ti3C2/Au, respectively. The N2 photofixation activity of the r-Ti3C2/Au catalyst was greatly boosted to 216.8 µmol L−1 in 6 h. Under visible light (λ >400 nm), the produced NH4+ amounts of all catalysts decreased (Fig. 2b). The ammonia generation rates were normalized against the illumination time and the catalyst amount under both white and visible light (Fig. 2c). The NH4+ generation rate over r-Ti3C2/Au is 22.6 (12.4) µmol h−1 gcat−1 under white light (visible light) illumination, which is 5.8 (5.9) and 10.2 (10.3) times those of Ti3C2/Au and r-Ti3C2, respectively.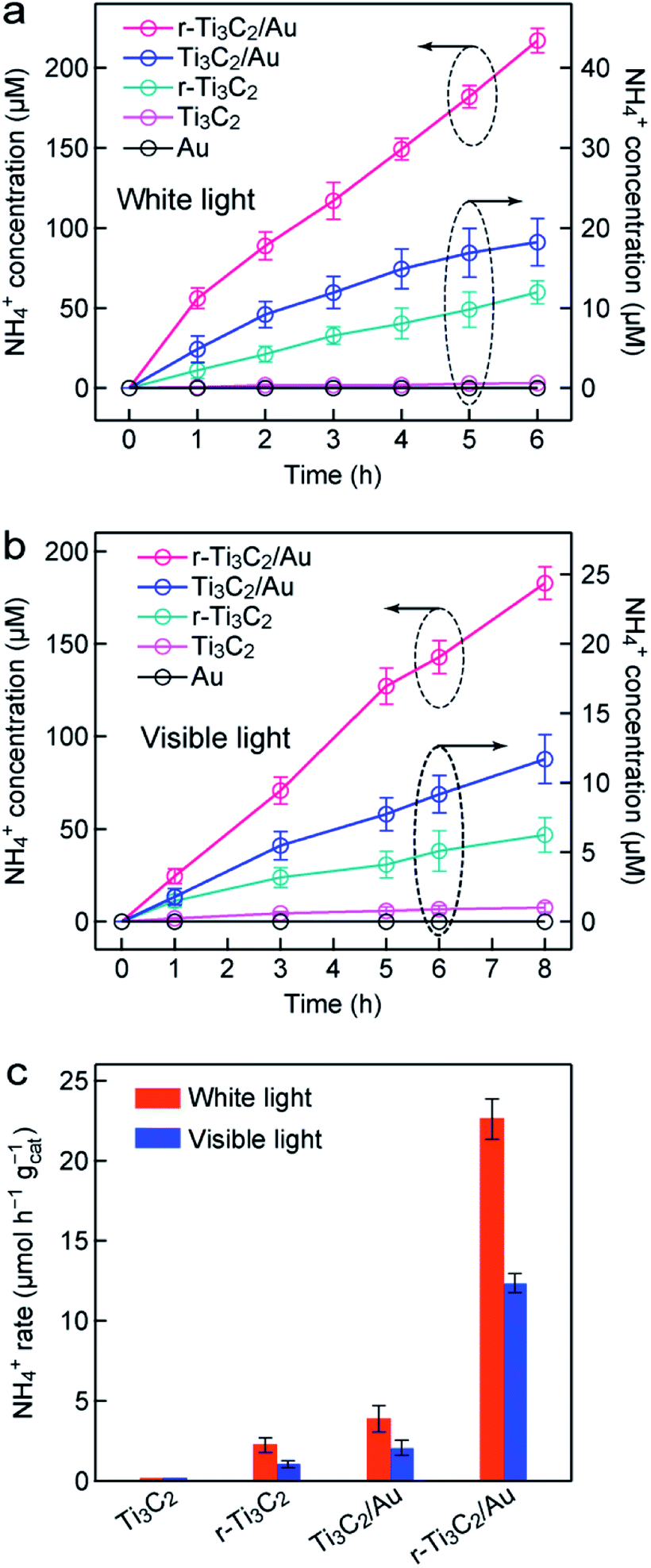 | ||
| Fig. 2 N2 photofixation over Au, Ti3C2, r-Ti3C2, Ti3C2/Au, and r-Ti3C2/Au. (a) Time courses of the ammonia concentrations measured under white light illumination. (b) Time courses of the ammonia concentrations measured under visible light illumination (λ >420 nm). (c) Ammonia production rates under white light and visible light. The rates for the Au nanospheres are not drawn because the Au nanospheres exhibit no activity for N2 photofixation. | ||
To verify the nitrogen and proton sources of the produced NH3, control experiments were carried out with r-Ti3C2/Au (Fig. S11†). NH4+ was not detected when N2 or H2O was replaced with Ar or aprotic acetonitrile, suggesting that the nitrogen and proton sources for the NH4+ formation are from N2 and H2O, respectively. In addition, NH4+ cannot be generated in dark, suggesting that light is an essential driving force for N2 photofixation. To further corroborate the origin of the produced NH3, an isotope labeling experiment was performed using 14N2 and 15N2 as the nitrogen sources. The obtained 14NH4Cl and 15NH4Cl were measured by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. 3a). The triplet and doublet peaks corresponding to 14NH4+ and 15NH4+ can be clearly observed in the 1H NMR spectra of the photocatalytic reaction solutions when 14N2 and 15N2 were used as the feed gas, respectively. This result verifies that the produced NH4+ indeed originated from N2 photofixation. Moreover, the evolution of O2 was also detected during the N2 photofixation process in a sealed reactor (Fig. S12a†). To assess whether O2 was produced during the N2 photofixation reaction catalyzed by r-Ti3C2/Au, the reactor was evacuated, bubbled with N2, and then sealed. Upon illumination for 1 h under white light, O2 was detected. The generated O2 should result from the oxidization of H2O by the hot holes in the Au nanospheres.54 Its amount is about three fourths that of NH4+ (Fig. S12b†), close to the stoichiometric ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 according to the reaction N2 + 3H2O → 2NH3 + 1.5O2, proving that NH3 is generated by coupling the activated N2 with the protons from H2O. Importantly, H2 was not detected during the N2 photofixation process (Fig. S12a†), suggesting the absence of the competing hydrogen evolution reaction. r-Ti3C2/Au is therefore a highly selective photocatalyst for N2 fixation.
:4 according to the reaction N2 + 3H2O → 2NH3 + 1.5O2, proving that NH3 is generated by coupling the activated N2 with the protons from H2O. Importantly, H2 was not detected during the N2 photofixation process (Fig. S12a†), suggesting the absence of the competing hydrogen evolution reaction. r-Ti3C2/Au is therefore a highly selective photocatalyst for N2 fixation.
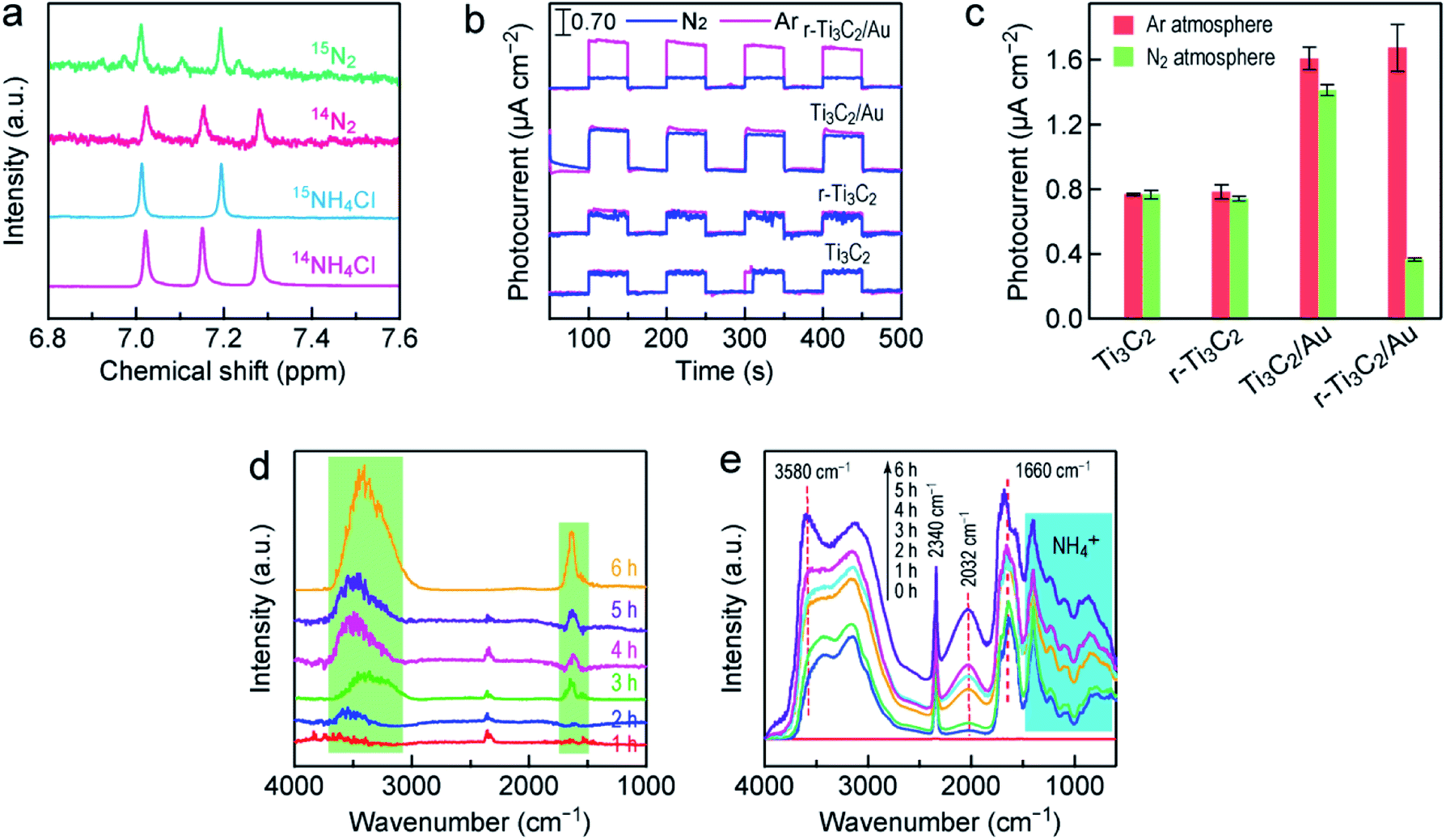 | ||
| Fig. 3 Understanding the photocatalytic N2 fixation process. (a) 1H NMR spectra of the solutions after the N2 fixation reaction over r-Ti3C2/Au in 15N2 and 14N2 atmospheres. The bottom two spectra were taken from the standard 15NH4Cl and 14NH4Cl solutions. (b) Photocurrent responses of the different catalysts recorded in Ar and N2 atmospheres, with the white light illumination switched on and off repeatedly. (c) Photocurrent densities of the different catalysts in Ar and N2 atmospheres under white light illumination. (d) In situ DRIFT spectra recorded as a function of time during the N2 photofixation reaction over r-Ti3C2/Au in the Ar atmosphere. (e) In situ DRIFT spectra recorded as a function of time during the N2 photofixation reaction over r-Ti3C2/Au in the N2 atmosphere. | ||
To reveal the mechanism of the photocatalytic N2 fixation, photocurrent tests were first performed under white light illumination in both N2 and Ar atmospheres (Fig. 3b). For Ti3C2, the photocurrent densities measured in Ar and N2 are nearly the same and reach ∼0.76 µA cm−2 (Fig. 3c). The photocurrent density of r-Ti3C2 is similar to that of Ti3C2, and the photocurrent in N2 shows a slight reduction. In the presence of the Au nanospheres, the photocurrents of Ti3C2/Au and r-Ti3C2/Au are enhanced in Ar because of the LSPR effect, and both reach ∼1.65 µA cm−2. There is a small decrease of ∼0.15 µA cm−2 in the photocurrent density of Ti3C2/Au when the Ar atmosphere changed to N2, which means a slight electron consumption for the reduction of N2 molecules. Remarkably, the photocurrent density of r-Ti3C2/Au in N2 is only one fifth of that in Ar, suggesting that the four fifth difference in photocurrent is consumed to reduce N2 molecules. As a result, r-Ti3C2/Au displays a remarkable N2 photofixation activity (Fig. 2). To look into the reaction process of adsorbed H2O and N2 and prove the activation and reduction of N2 on the surface of r-Ti3C2/Au, in situ diffuse-reflectance infrared Fourier transform spectroscopy (DRIFTS) was employed to monitor the N2 photofixation process. To record the DRIFT spectra, r-Ti3C2/Au was exposed to water vapor-saturated N2 under white light illumination, which allows for the investigation of the time-dependent change of the molecular species adsorbed on the catalyst. In the control experiment performed in an Ar atmosphere, two clear absorption peaks at 1660 and 3580 cm−1, corresponding to the characteristic bending modes of adsorbed H2O molecules,36 were observed. Their intensities were enhanced as the reaction time was prolonged (Fig. 3d). No absorption bands related to the N-containing species were detected, which further indicates that the nitrogen in NH3 truly originated from N2 molecules. The time-dependent DRIFT spectra recorded after the injection of N2 under white light reveal that several absorption peaks gradually appear as the illumination time was prolonged from 0 to 6 h. The signal at 2340 cm−1 can be ascribed to strongly chemisorbed N2 molecules.55 The absorption band at 2032 cm−1 is believed to arise from the Ti–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N complex formed through N2 binding to the Ti3+ sites.56 The absorption band at ∼3160 cm−1 corresponds to the adsorbed ammonia.9,37 Furthermore, the characteristic absorption band at 1410 cm−1 assigned to the NH4+ deformation vibration is intensified with increasing illumination time.37,57 These DRIFTS results provide strong evidence that N2 molecules can be adsorbed, activated and further reduced to form NH4+ under light illumination.
N complex formed through N2 binding to the Ti3+ sites.56 The absorption band at ∼3160 cm−1 corresponds to the adsorbed ammonia.9,37 Furthermore, the characteristic absorption band at 1410 cm−1 assigned to the NH4+ deformation vibration is intensified with increasing illumination time.37,57 These DRIFTS results provide strong evidence that N2 molecules can be adsorbed, activated and further reduced to form NH4+ under light illumination.
To further understand the photocatalytic N2 fixation performance of r-Ti3C2/Au, the effect of the Au nanosphere amount in r-Ti3C2/Au was systematically investigated. The amount of embedded Au nanospheres was determined by inductively coupled plasma mass spectrometry (ICP-MS; Fig. S13†). Fig. 4a shows the high-magnification SEM images of the Au/r-Ti3C2 samples that were embedded with the 12.8 nm-sized Au nanospheres at 0.49, 1.11, 1.78 and 2.28 wt% relative to the total amount of Au and r-Ti3C2, respectively. The unique sandwich-like architecture is observable for all samples. The Au nanospheres are uniformly distributed on the basal planes even when the loaded Au amount is increased to 1.78 wt%. As the embedded Au amount is increased to 2.28 wt%, aggregation occurs. The NH4+ concentrations catalyzed by the Au/r-Ti3C2 samples with different Au amounts increase with the illumination time under white light (Fig. 4b). When the embedded Au amount is increased from 0.49 to 1.78 wt%, the produced NH4+ amount steadily increases within the same reaction time. The r-Ti3C2/Au sample with the Au amount of 1.78 wt% gives the highest amount of the produced NH4+, whose concentration reaches 307.8 µmol L−1 in 6 h. However, when the embedded Au amount is further increased to 2.28 wt%, the generated NH4+ concentration is clearly reduced. Similarly, the normalized NH4+ production rate first increases with the Au amount, reaches the highest value of 31.8 µmol h−1 gcat−1 at the loaded Au amount of 1.78 wt%, and then decreases (Fig. 4c). To further explore the relationship between the N2 photofixation activity and the LSPR effect of the Au nanospheres, r-Ti3C2/Au with different Au nanosphere sizes and amounts were prepared. The uniform Au nanospheres with average sizes of 16.1 ± 0.8 (Fig. S14†) and 21.0 ± 0.8 nm (Fig. S15†) were synthesized. The unique sandwich-like structure with the uniform distribution of the Au nanospheres in the interlayers was obtained. Similarly, a high Au amount results in a reduction in the NH4+ production. Fig. 4d summarizes the relationship between the NH4+ generation rate and the loaded Au nanosphere size and amount. For the same Au size, the NH4+ production rates show a nearly volcano-shaped dependence on the embedded Au amount. An optimal N2 photofixation performance is achieved at a particular Au amount, which increases with the Au nanosphere diameter. The optimal Au amount for each size can be attributed to the enhanced LSPR effect with increasing amounts of Au nanospheres. The decrease in the N2 photofixation rate at a higher Au amount should be caused by the aggregation of the Au nanospheres and/or the blocking of the active sites on r-Ti3C2 for N2 adsorption by the excessive Au nanospheres. The N2 photofixation rates for the r-Ti3C2/Au samples containing the 20 nm-sized Au nanospheres are generally smaller than those of the samples containing the 13 nm- and 16 nm-sized Au nanospheres. This is probably because the number of Au nanospheres plays a more important role than the absorption cross-section for the LSPR effect in N2 photofixation. The optimal sample was found to be r-Ti3C2/Au containing the 16 nm-sized Au nanospheres at 2.45 wt%. This sample gives a NH4+ production rate of 33.8 µmol h−1 gcat−1.
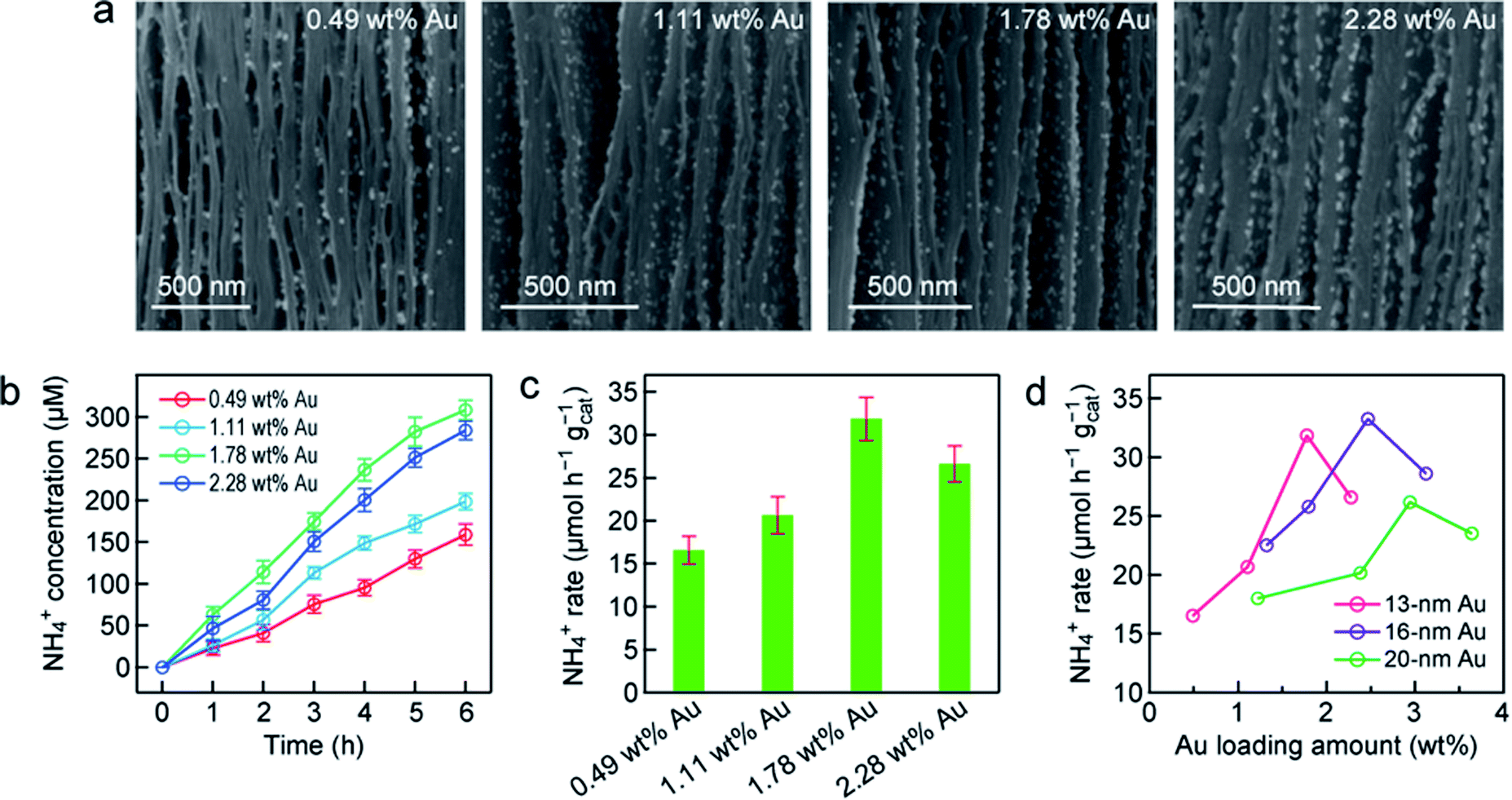 | ||
| Fig. 4 Nitrogen photofixation over different r-Ti3C2/Au samples. (a) SEM images of r-Ti3C2/Au containing the 13 nm-sized Au nanospheres at different amounts indicated in the images. (b) Time courses of the ammonia concentrations measured under white light for the samples shown in (a). (c) Ammonia production rates under white light for the samples shown in (a). (d) Photocatalytic ammonia production rates as a function of the loaded Au amount for r-Ti3C2/Au containing the differently sized Au nanospheres. | ||
Au nanospheres capped with different molecules were next employed to study their assembly with r-Ti3C2 and in turn their effect on N2 photofixation. Au nanospheres coated with cetyltrimethylammonium bromide (CTAB) were chosen. They exhibit good dispersibility and uniform sizes with an average diameter of 19.8 ± 0.7 nm (Fig. S16†). The CTAB-capped Au nanospheres were found to predominantly assemble onto the edges of the r-Ti3C2 layers (Fig. 5a). The resultant sample is therefore denoted r-Ti3C2/edge-Au. The CTAB-capped Au nanospheres are positively charged with a zeta potential of +34.2 mV, and the layered r-Ti3C2 sample is negatively charged with a zeta potential of −28.8 mV (Fig. S17†). The two components can therefore spontaneously assemble together through electrostatic attraction during solvent evaporation. The assembly results in an intimate contact between the Au nanospheres and the edges of r-Ti3C2. It also implies that the electrostatic attraction force is stronger than the solvent-driven force. In contrast, the citrate-capped Au nanospheres cannot be adsorbed onto the edges of r-Ti3C2 due to electrostatic repulsion. They are driven into the interlayers of r-Ti3C2 by the solvent, producing the sandwich-like nanostructure. The SEM images (Fig. 5b) of r-Ti3C2/edge-Au reveal that the Au nanospheres are distributed along the edges of the layered r-Ti3C2 without clear aggregation, even when the Au amount is increased from 1.25 to 3.75 wt%. The N2 photofixation activity of r-Ti3C2/edge-Au increases with increasing Au amounts (Fig. S18†). The photocatalytic N2 fixation performances of r-Ti3C2/Au with the Au nanospheres located at different positions are compared under the identical nanosphere diameter of ∼20 nm and the close amounts of Au (Fig. 5c). Considerable reductions in the NH4+ production rate are seen for the r-Ti3C2/edge-Au catalysts. The reductions are probably caused by the relatively large average distance from the edge-positioned Au nanospheres, which act as the electron sources, to the active sites on the layers of r-Ti3C2. The large distance increases the probability for electrons to get lost during their transport.
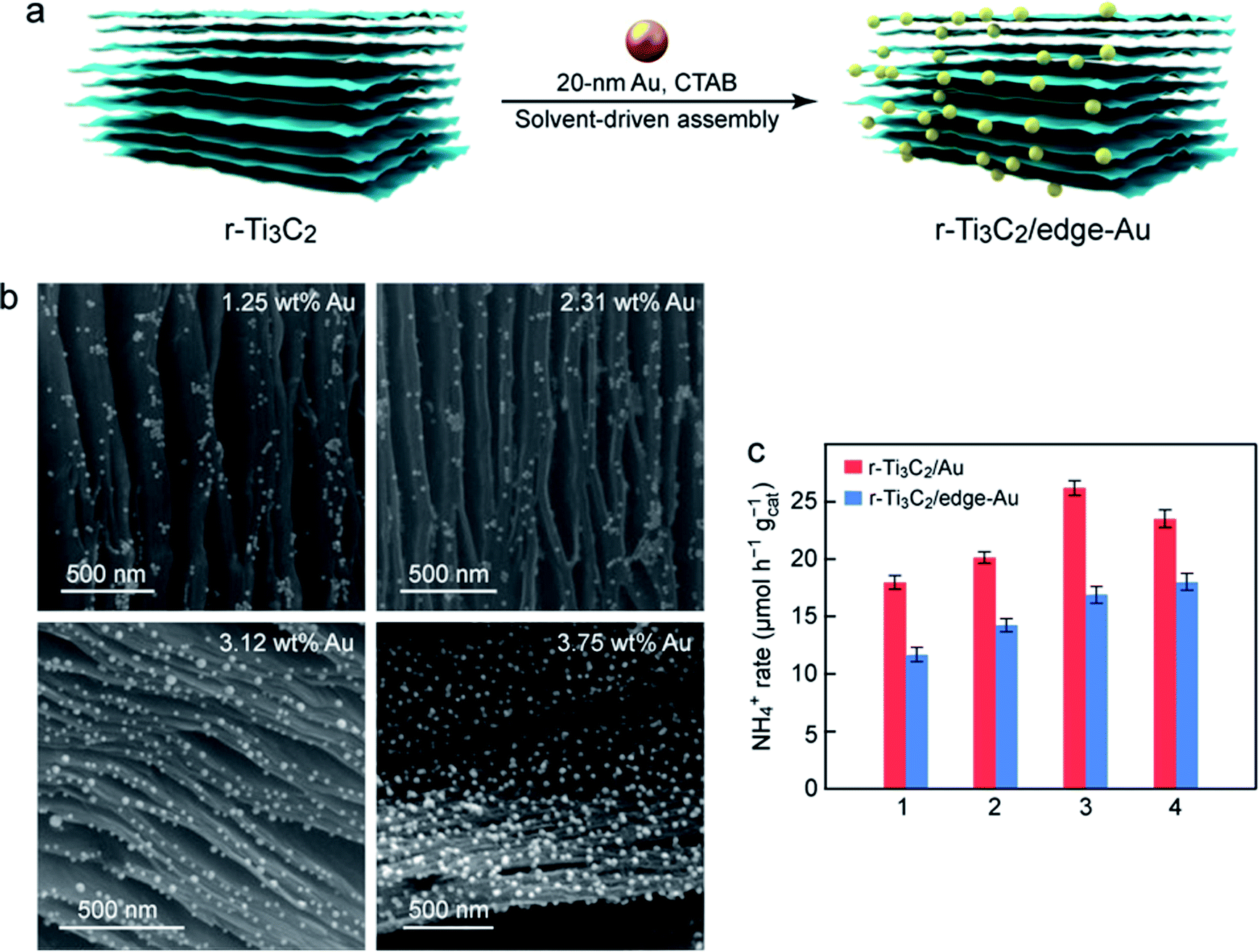 | ||
| Fig. 5 Characterization and N2 photofixation of r-Ti3C2/edge-Au. (a) Schematic illustrating the synthesis process of r-Ti3C2/edge-Au. (b) SEM images of r-Ti3C2/edge-Au with different loaded Au amounts. (c) Comparison of the ammonia production rates over the sandwich-like r-Ti3C2/Au and the r-Ti3C2/edge-Au catalysts under white light illumination. The sizes of the Au nanospheres in both catalysts are ∼20 nm. The loaded Au amounts in both catalysts are nearly the same in each group from 1 to 4. | ||
Active sites and N2 photofixation mechanism
The active sites of Ti3C2 MXene are vital for N2 adsorption and activation and crucial for the utilization of Ti3C2 MXene in N2 photofixation. X-ray photoelectron spectroscopy (XPS) was performed on Ti3C2 and r-Ti3C2 to examine the active sites (Fig. 6a and b). The peak at 454.6 eV for Ti3C2 and r-Ti3C2 can be assigned to the Ti–C bond. The peak appearing at 458.5 eV for both samples can be assigned to TiO2−x, which arises mainly from the Ti atoms surrounded by O2− ions in the lattice owing to the replacement of some carbon atoms by oxygen atoms during etching.37,50 The peaks at 455.3 (460.2), 456.1(461.2) and 457.1 eV (462.3 eV) come from 2p3/2 (2p1/2) of Ti2+, Ti3+ and Ti4+, respectively. Compared to Ti3C2, r-Ti3C2 exhibits an enhanced fraction of Ti3+ with a diminished fraction of Ti4+ (Table S1†), manifesting the partial reduction of Ti4+ to the low-valence states of Ti3+ or Ti2+ through H2 treatment. The low-valence state Ti3+ has been known to be active for N2 chemisorption. No clear changes were observed in the Ti 2p peaks for Ti3C2/Au and r-Ti3C2/Au, suggesting that the embedding of the Au nanospheres does not change the chemical state of Ti. The O 1s spectra of Ti3C2 and r-Ti3C2 can be fitted with two peaks at 529.4 and 531.2 eV (Fig. S19a†), which are, respectively, derived from Ti–O and Ti–OH. The decreased intensity ratio between Ti–OH and Ti–O for r-Ti3C2 is attributed to the H2 treatment. After the loading of the Au nanospheres, a new peak appears at ∼532.3 eV (Fig. S19b†), suggesting the strong affinity between the Au nanospheres and the Ti3C2 layers through the formation of Au–O–Ti.58 The peaks at 83.7 and 87.3 eV in Ti3C2/Au and r-Ti3C2/Au (Fig. S19c†) arise, respectively, from Au 4f5/2 and Au 4f7/2, further verifying the successful assembly of the Au nanospheres onto the Ti3C2/r-Ti3C2 layers. Low-temperature electron paramagnetic resonance (EPR) spectra were measured to further confirm the existence of Ti3+ and surface OVs (Fig. 6c). The EPR spectrum of Ti3C2 shows a weak signal at g = 1.998, which originated from the delocalized electrons of the O2− ions in the lattice.11,59 In comparison, r-Ti3C2 displays a stronger EPR peak with g = 2.004, demonstrating the formation of OVs caused by the H2 treatment and the existence of more Ti3+ in r-T3C2.57 Both XPS and EPR therefore reveal that r-Ti3C2 possesses a number of Ti3+ species and OVs in its framework, which can act as active sites for N2 chemisorption. N2 temperature-programmed desorption (TPD) further revealed the N2 adsorption ability of different samples. Only one peak at ∼100 °C was detected for Ti3C2 (Fig. 6d), which is caused by N2 physisorption. In addition to the physisorption peak, r-Ti3C2 presents a strong peak at a higher temperature of ∼250–300 °C, which should have originated from the chemisorption of N2 molecules.60 These results indicate that N2 chemisorption occurs at the Ti3+ sites on the r-Ti3C2 framework through the electron donation from the OV-induced Ti3+, as further discussed below.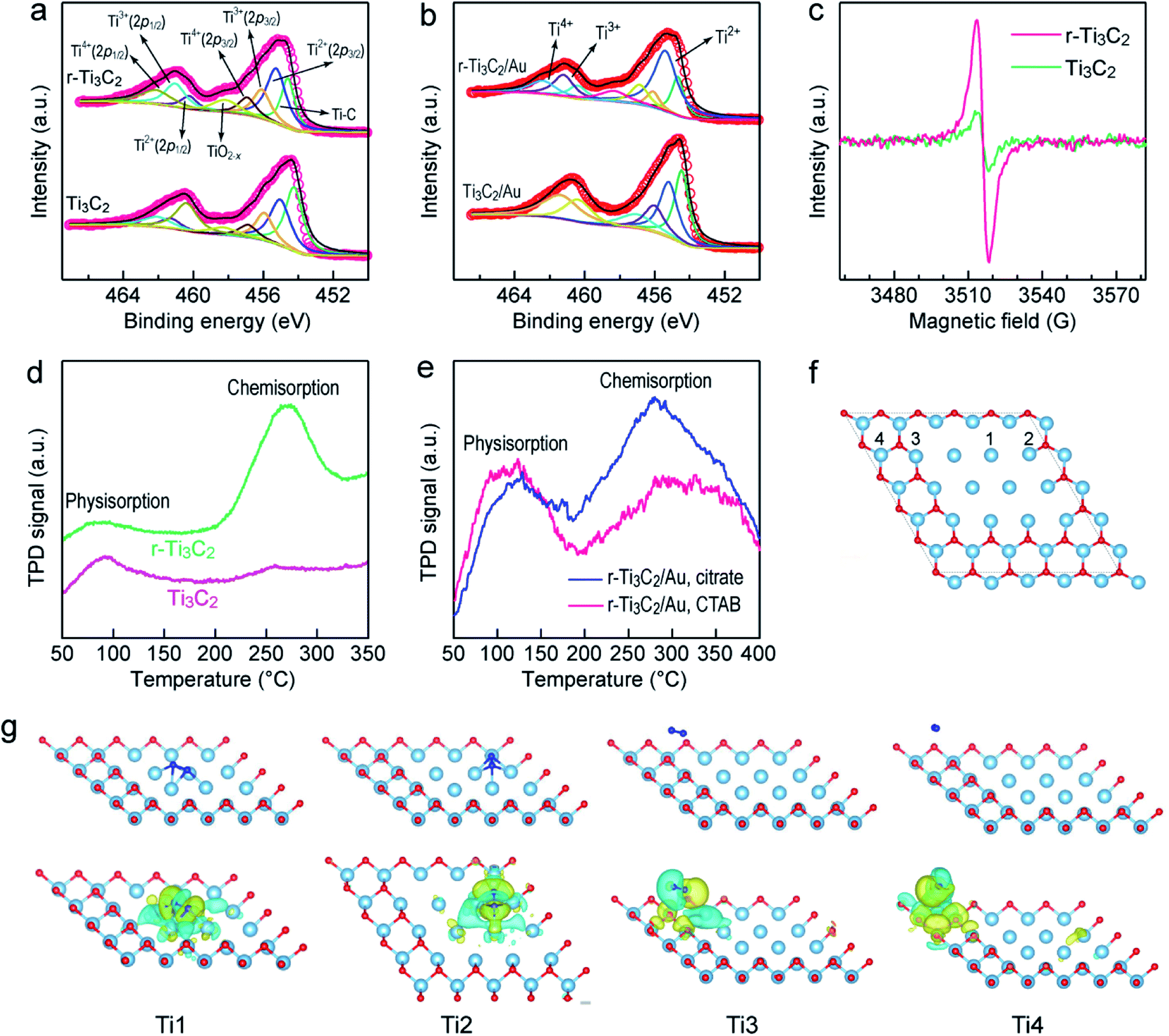 | ||
| Fig. 6 Adsorption and activation of N2 molecules. (a) Ti 2p XPS spectra of Ti3C2 and r-Ti3C2. (b) Ti 2p XPS spectra of Ti3C2/Au and r-Ti3C2/Au. (c) EPR spectra of Ti3C2 and r-Ti3C2. (d) N2 TPD profiles of Ti3C2 and r-Ti3C2. (e) N2 TPD profiles of r-Ti3C2/Au and r-Ti3C2/edge-Au. (f) Various Ti sites on r-Ti3C2. Only the top Ti layer terminated with a layer of O atoms is shown. (g) Adsorption configuration (top row) and charge density difference (bottom row) of a N2 molecule adsorbed at different Ti sites on the r-Ti3C2 surface. Light blue balls: Ti; red balls: O; and dark blue balls: N. Yellow cloud: electron enrichment and cyan cloud: electron depletion. | ||
Density functional theory (DFT) calculations were performed to investigate the chemisorption and activation of N2 molecules on different Ti sites of Ti3C2 MXene (Fig. 6f). Upon adsorption of a N2 molecule on the Ti1 site, the optimized N2 coordination configuration shows that the N2 molecule is chemisorbed on a Ti–Ti tripolymer center through a dinuclear end-on coordination mode, in which one N atom binds with the Ti–Ti tripolymer and the other binds with the Ti–Ti dimer (Fig. 6g). When a N2 molecule is adsorbed on the Ti2 site, the optimized coordination configuration is similar to that of N2 coordinated to the Ti1 site, but the adjacent two Ti1 sites are combined to form a Ti1–Ti2 tripolymer and a Ti1–Ti2 dimer (Fig. 6g). The Ti3 and Ti4 sites cannot adsorb N2 molecules (Fig. 6g), which is probably caused by the weak interaction due to the strongly electronegative nature of the surface O-terminal groups.61 To disclose the electron transfer between r-Ti3C2 and the adsorbed N2, the charge density difference was calculated (Fig. 6g, bottom row). A clear charge density difference is observed for the Ti1 and Ti2 sites, suggesting the occurrence of electron transfer from r-Ti3C2 to the N2 molecule. The electron-enriched isosurface on the adsorbed N2 molecule exhibits a π-orbital feature, indicating that the d-orbital electrons on the adjacent Ti atoms at the Ti1-site are transferred to the captured N2 molecule. As electrons are injected into the N2 molecule, the NN triple bond is considerably weakened with an elongated bond length. The triple bond lengths of the N2 molecules adsorbed at the Ti1 and Ti2 sites, respectively, increased to 1.351 and 1.345 Å, much longer than the value of 1.114 Å for a free N2 molecule (Fig. S20a and b†). In addition, a distinct reduction in the electron density between the two N atoms is also observed, implying that electrons are donated from the highest occupied σ-orbital of the N2 molecule to the adjacent Ti sites. Such phenomena of electron donation from N2 to the metal and back-donation from the metal to N2 also happen in M (transition metal)–N2 complexes and have been discovered for many metals.1,62,63 The strong activation of N2 molecules through electron back-donation from transition metals with available d-orbital electrons plays a crucial role in boosting the photocatalytic N2 fixation activity of the r-Ti3C2 catalyst.
In order to better understand the N2 chemisorption, we also performed DFT calculations to examine the adsorption energy (Ead) of N2 at different sites on r-Ti3C2 MXene (Fig. S20†). A comparison of the Ead values of a N2 molecule at the Ti1 site with different adsorption configurations reveals that the optimal N2 coordination configuration mentioned above gives the largest Ead value of −3.525 eV, meaning that N2 activation can be spontaneously realized when it is adsorbed at the Ti1 site. A comparison of the Ead values of N2 at different sites (Fig. S20†) shows that Ti1 is the strongest active site for N2 adsorption in r-Ti3C2 MXene. In addition, the adjacent Ti1 sites can help to build the optimal coordination configuration for N2 activation. The more the Ti1 sites are involved, the higher the Ead value is. Moreover, the Ti1 site does not bond with any O atoms. The bond length and adsorption energy of the N2 molecule at the Ti1 site obtained in our calculations for Ti3C2 MXene are very close to those obtained in previous works for Ti2C MXene.64,65
Solar utilization efficiency and stability of r-Ti3C2/Au
To evaluate the light utilization efficiency, the wavelength-dependent apparent quantum efficiencies (AQE) of r-T3C2/Au were determined by measuring the amount of produced ammonia in pure water under monochromatic light illumination (Fig. 7a). The AQE spectrum of r-Ti3C2/Au matches well with its absorption spectrum, suggesting that the ammonia evolution is photo-driven. Specifically, the AQE value for r-Ti3C2/Au reaches 0.697% at 520 nm owing to the synergistic effect of the strong plasmonic light harvesting capability of the Au nanospheres and the N2-activation capability of r-Ti3C2. Such an AQE value is higher than those obtained in many previous works at this wavelength (Tables S2 and S3†). In addition, we also examined the N2 photofixation activity of r-Ti3C2/Au under AM 1.5G solar light illumination in a sealed reactor filled with N2. The NH3 production rate was 21.26 µmol h−1 gcat−1, and O2 was also generated at a rate of 16.12 µmol h−1 gcat−1 in this sealed system (Fig. S21†). The molar ratio of the produced NH3 and O2 is close to the theoretical stoichiometric ratio. Accordingly, the solar-to-ammonia conversion efficiency (SACE) was calculated to be 0.013%. The stability of r-Ti3C2/Au was evaluated by performing successive rounds of reaction under white and visible light illumination (Fig. 7b). Almost ∼95% of the original ammonia generation activity was preserved after five successive cycles, indicative of the high stability of r-Ti3C2/Au. The unique sandwich-like structure with uniformly distributed Au nanospheres in the interlayers was well maintained in the used r-Ti3C2/Au catalyst (Fig. 7c). The slight decrease in the NH3 production rate during the cycling tests is believed to be caused by the slight aggregation of the loaded Au nanospheres (Fig. 7c). A negligible amount of the leached Au was detected in the fifth cycle reaction solution by ICP-AES. The stability of r-Ti3C2/Au was further confirmed by the nearly unchanged XRD spectrum (Fig. 7d). The photocatalytic stability can be ascribed to the fact that electrons are constantly produced and transferred from the Au nanospheres to Ti3C2 under light illumination, which can inhibit the oxidation of Ti3C2.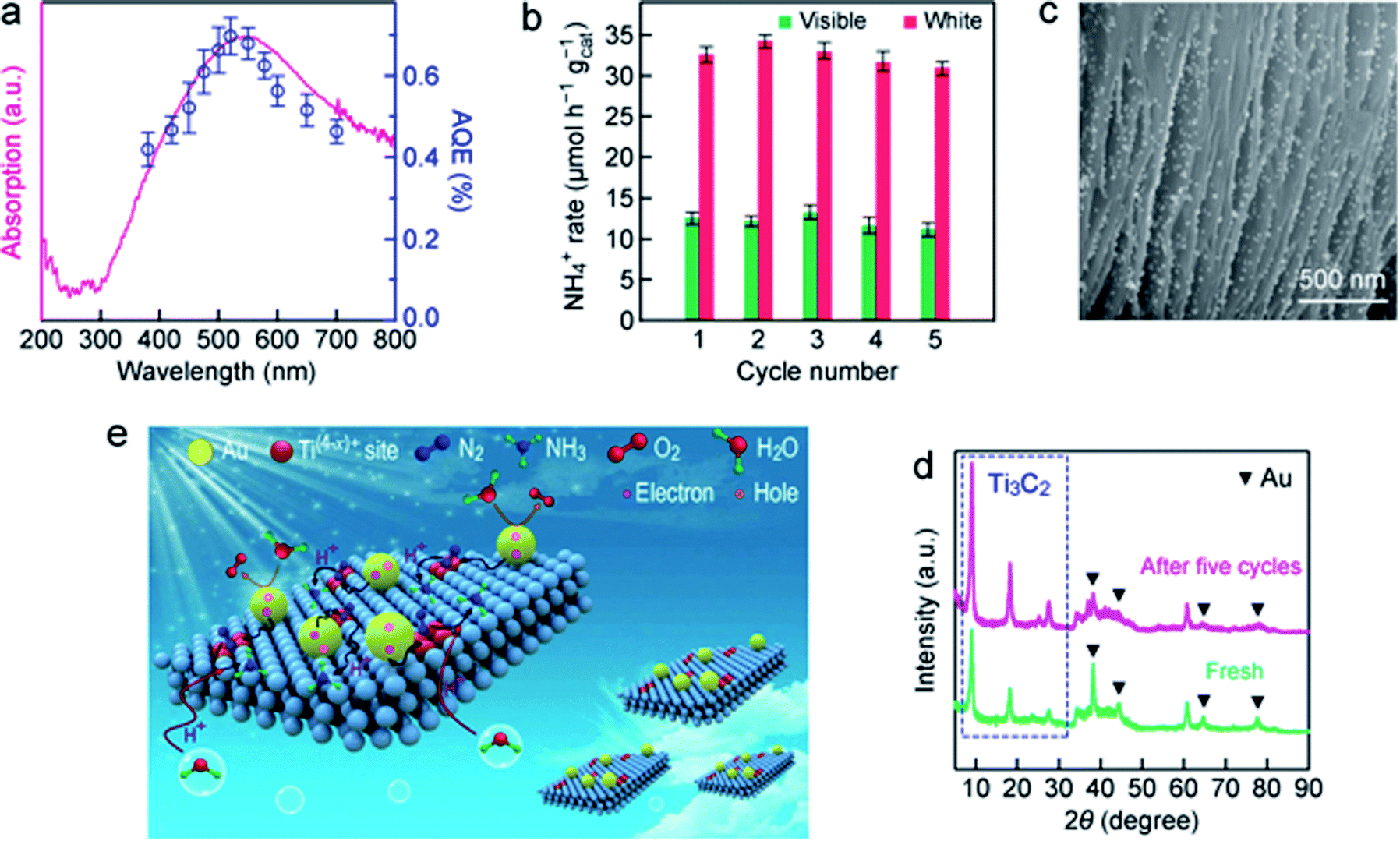 | ||
| Fig. 7 AQEs and recyclability of r-Ti3C2/Au for N2 photofixation. (a) AQE and absorption spectra. (b) Cyclic tests for N2 photofixation under white and visible light. (c) SEM image of r-Ti3C2/Au after the fifth cycle of test. (d) XRD patterns of the fresh sample and those after the fifth cycle of test. (e) Artistic illustration of the proposed mechanism for photocatalytic N2 fixation with r-Ti3C2/Au. | ||
Based on the above results, we propose a reaction mechanism for photocatalytic N2 fixation with r-Ti3C2/Au under ambient conditions (Fig. 7e). Benefiting from the good hydrophilicity, the r-Ti3C2/Au photocatalyst can be stably and uniformly dispersed in water. r-Ti3C2 possesses numerous low-valence Ti(4−x)+ sites that are associated with OVs and generated through H2 thermal reduction. They are active sites for capturing and activating N2 molecules. Under light illumination, photogenerated hot electrons from the Au nanospheres are injected into r-Ti3C2, which subsequently reduce the activated N2 at the Ti(4−x)+ sites. The hot holes remaining on the Au nanospheres are consumed through the oxidation of H2O to produce O2. The produced NH3 is accumulated in the aqueous reaction solution.
Conclusions
We have demonstrated the potential of Ti3C2 MXene to photocatalytically fix N2 in pure water under ambient conditions. Partially reduced layered Ti3C2 MXene is synthesized and integrated with Au nanospheres in a uniform sandwich-like structure through a controlled solvent-driven approach. r-Ti3C2 exposes abundant low-valence Ti sites, which act as active sites for capturing and activating N2 molecules. The embedded Au nanospheres donate plasmonic hot electrons to reduce the activated N2. Importantly, the unique sandwich-like architecture prevents the self-stacking of the Ti3C2 layers, which favors the exposure of the active sites for utilization. The abundant Ti(4−x)+ active sites and the LSPR effect work in tandem to endow r-Ti3C2/Au with a remarkably high N2 photofixation activity. The design in this work not only broadens the photocatalytic applications of MXene in N2 fixation as a host material but also opens up an avenue for the surface engineering of MXene with plasmonic nanoparticles to further explore the potential of MXene as a promising photocatalyst.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
B. B. C. performed the experiments and wrote the manuscript. Y. Z. G. assisted with the N2 photofixation experiments. D. H. W. carried out the density functional theory calculations. L. L., B. C. Y. and J. F. W. designed the research work. J. F. W. revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51872110), Shanghai Science and Technology Committee (STCSM, 18490740500), and the Research Grants Council of Hong Kong (GRF, 14305819).Notes and references
- H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547 RSC.
- C. J. M. Van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183 RSC.
- Y. M. Liu and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2794 CrossRef CAS PubMed.
- G. F. Chen, X. R. Cao, S. Q. Wu, X. Y. Zeng, L. X. Ding, M. Zhu and H. H. Wang, J. Am. Chem. Soc., 2017, 139, 9771 CrossRef CAS PubMed.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304 RSC.
- S. Licht, B. C. Cui, B. H. Wang, F. F. Li, J. Lau and S. Z. Liu, Science, 2014, 345, 637 CrossRef CAS PubMed.
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yan, Q. Jiang and X. B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- H. Li, C. L. Mao, H. Shang, Z. P. Yang, Z. H. Ai and L. Z. Zhang, Nanoscale, 2018, 10, 15429 RSC.
- H. Li, J. Shang, Z. H. Ai and L. Z. Zhang, J. Am. Chem. Soc., 2015, 137, 6393 CrossRef CAS PubMed.
- W. K. Wang, H. M. Zhang, S. B. Zhang, Y. Y. Liu, G. Z. Wang, C. H. Sun and H. J. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189 CrossRef CAS.
- C. M. Janet, S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, J. Phys. Chem. C, 2010, 114, 2622 CrossRef CAS.
- X. L. Xue, R. P. Chen, H. W. Chen, Y. Hu, Q. Q. Ding, Z. T. Liu, L. B. Ma, G. Y. Zhu, W. J. Zhang, Q. Yu, J. Liu, J. Ma and Z. Jin, Nano Lett., 2018, 18, 7372 CrossRef CAS PubMed.
- C. K. Yao, R. Wang, Z. S. Wang, H. Lei, X. P. Dong and C. Z. He, J. Mater. Chem. A, 2019, 7, 27547 RSC.
- Q. X. Liu, L. H. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102 RSC.
- Y. C. Hao, X. L. Dong, S. R. Zhai, H. C. Ma, X. Y. Wang and X. F. Zhang, Chem.–Eur. J., 2016, 22, 18722 CrossRef CAS PubMed.
- H. L. Jia, A. Du, H. Zhang, J. H. Yang, R. B. Jiang, J. F. Wang and C. Y. Zhang, J. Am. Chem. Soc., 2019, 141, 5083 CrossRef CAS PubMed.
- H. Diarmand-Khalilabad, A. Habibi-Yangjeh, D. Seifzadeh, S. Asadzadeh-Khaneghah and E. Vesali-Kermani, Ceram. Int., 2019, 45, 2542 CrossRef CAS.
- Y. F. Sun, S. Gao, F. C. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623 RSC.
- Y. F. Sun, S. Gao, F. C. Lei, C. Xiao and Y. Xie, Acc. Chem. Res., 2015, 48, 3 CrossRef CAS PubMed.
- Q. Su, Y. Li, R. Hu, F. Song, S. Y. Liu, C. P. Guo, S. M. Zhu, W. B. Liu and J. Pan, Adv. Sustainable Syst., 2020, 4, 2000130 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- X. B. Hui, X. L. Ge, R. Z. Zhao, Z. Q. Li and L. W. Yin, Adv. Funct. Mater., 2020, 30, 2005190 CrossRef CAS.
- Z. Li and Y. Wu, Small, 2019, 15, 1804736 CrossRef PubMed.
- J. H. Peng, X. Z. Chen, W. J. Ong, X. J. Zhao and N. Li, Chem, 2019, 5, 18 CAS.
- R. D. Tang, S. Xiong, D. X. Gong, Y. C. Deng, Y. C. Wang, L. Su, C. X. Ding, L. H. Yang and C. J. Liao, ACS Appl. Mater. Interfaces, 2020, 12, 56663 CrossRef CAS PubMed.
- X. Li, Y. Bai, X. Shi, N. Su, G. Z. Nie, R. M. Zhang, H. B. Nie and L. Q. Ye, Mater. Adv., 2021, 2, 1570 RSC.
- L. M. Azofra, N. Li, D. R. Macfarlane and C. H. Sun, Energy Environ. Sci., 2016, 9, 2545 RSC.
- K. L. Huang, C. H. Li, H. Z. Li, G. M. Ren, L. Wang, W. T. Wang and X. C. Meng, ACS Appl. Nano Mater., 2020, 3, 9581 CrossRef CAS.
- L. F. Hong, R. T. Guo, Y. Yuan, X. Y. Ji, Z. S. Li, Z. D. Lin and W. G. Pan, Mater. Today Energy, 2020, 18, 100521 CrossRef.
- J. Z. Qin, X. Hu, X. Y. Li, Z. F. Yin, B. J. Liu and K. H. Lam, Nano Energy, 2019, 61, 27 CrossRef CAS.
- J. H. Yang, Y. Z. Guo, W. Z. Lu, R. B. Jiang and J. F. Wang, Adv. Mater., 2018, 30, 1802227 CrossRef PubMed.
- J. H. Yang, Y. Z. Guo, R. B. Jiang, F. Qin, H. Zhang, W. Z. Lu, J. F. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497 CrossRef CAS PubMed.
- C. Y. Hu, X. Chen, J. B. Jin, Y. Han, S. M. Chen, H. X. Ju, J. Cai, Y. R. Qiu, C. Gao, C. M. Wang, Z. M. Qi, R. Long, L. Song, Z. Liu and Y. J. Xiong, J. Am. Chem. Soc., 2019, 141, 7807 CrossRef CAS PubMed.
- T. T. Hou, L. L. Chen, Y. Xin, W. K. Zhu, C. Y. Zhang, W. H. Zhang, S. Q. Liang and L. B. Wang, ACS Energy Lett., 2020, 5, 2444 CrossRef CAS.
- P. Zhang, T. Wang and J. L. Gong, Adv. Mater., 2015, 27, 5328 CrossRef CAS PubMed.
- J. H. Yang, H. Y. Bai, Y. Z. Guo, H. Zhang, R. B. Jiang, B. C. Yang, J. F. Wang and J. C. Yu, Angew. Chem., Int. Ed., 2021, 60, 927 CrossRef CAS PubMed.
- T. B. Limbu, B. Chitara, J. D. Orlando, M. Y. Garcia Cervantes, S. Kumari, Q. Li, Y. Tang and F. Yan, J. Mater. Chem. C, 2020, 8, 4722 RSC.
- Y. Y. Wen, T. E. Rufford, X. Z. Chen, N. Li, M. Q. Lyu, L. M. Dai and L. Z. Wang, Nano Energy, 2017, 38, 368 CrossRef CAS.
- M. Ghidiu, S. Kota, J. Halim, A. W. Sherwood, N. Nedfors, J. Rosen, V. N. Mochalin and M. W. Barsoum, Chem. Mater., 2017, 29, 1099 CrossRef CAS.
- C. J. Zhao, Q. Wang, H. Zhang, S. Passerini and X. Z. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 15661 CrossRef CAS PubMed.
- G. D. Zou, Z. W. Zhang, J. X. Guo, B. Z. Liu, Q. R. Zhang, C. Fernandez and Q. M. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 22280 CrossRef CAS PubMed.
- J. X. Low, L. Y. Zhang, T. Tong, B. J. Shen and J. G. Yu, J. Catal., 2018, 361, 255 CrossRef CAS.
- A. A. Khan, M. Tahir and A. Bafaqeer, Energy Fuels, 2020, 34, 9810 CrossRef CAS.
- R. B. Rakhi, B. Ahmed, M. N. Hedhili, D. H. Anjum and H. N. Alshareef, Chem. Mater., 2015, 27, 5314 CrossRef CAS.
- X. L. Li, X. W. Yin, M. K. Han, C. Q. Song, H. L. Xu, Z. X. Hou, L. T. Zhang and L. F. Cheng, J. Mater. Chem. C, 2017, 5, 4068 RSC.
- Y. Yoon, T. A. Le, A. P. Tiwari, I. Kim, M. W. Barsoum and H. Lee, Nanoscale, 2018, 10, 22429 RSC.
- H. Wang, Y. Wu, X. Z. Yuan, G. Zeng, J. Zhou, X. Wang and J. W. Chew, Adv. Mater., 2018, 30, 1704561 CrossRef PubMed.
- R. Y. Li, L. B. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752 CrossRef CAS PubMed.
- Y. Z. Guo, J. H. Yang, D. H. Wu, H. Y. Bai, Z. Yang, J. F. Wang and B. C. Yang, J. Mater. Chem. A, 2020, 8, 16218 RSC.
- S. Y. Moon, H. C. Song, E. H. Gwag, L. I. Nedrygailov, C. Lee, J. J. Kim, W. H. Doh and J. Y. Park, Nanoscale, 2018, 10, 22180 RSC.
- S. Y. Wang, X. Hai, X. Ding, K. Chang, Y. G. Xiang, X. G. Meng, Z. X. Yang, H. Chen and J. H. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- J. Liu, M. S. Kelley, W. Q. Wu, A. Banerjee, A. P. Douvalis, J. S. Wu, Y. B. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530 CrossRef CAS PubMed.
- Y. X. Zhao, Y. F. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- D. Liu, G. Zhang, Q. H. Ji, Y. Y. Zhang and J. H. Li, ACS Appl. Mater. Interfaces, 2019, 11, 25758 CrossRef CAS PubMed.
- C. D. Lv, Y. M. Qian, C. S. Yan, Y. Ding, Y. Y. Liu, G. Chen and G. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246 CrossRef CAS PubMed.
- X. M. Li, X. Sun, L. Zhang, S. M. Sun and W. Z. Wang, J. Mater. Chem. A, 2018, 6, 3005 RSC.
- L. R. Johnson, S. Sridhar, L. Zhang, K. D. Fredrickson, A. S. Raman, J. Jang, C. Leach, A. Padmanabhan, C. C. Price, N. C. Frey, A. Raizada, V. Rajaraman, S. Saiprasad, X. Tang and A. Vojvodic, ACS Catal., 2020, 10, 253 CrossRef CAS.
- Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2013, 257, 2551 CrossRef CAS.
- M. A. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef CAS PubMed.
- J. D. Gouveia, Á. Morales-García, F. Viñes, J. R. B. Gomes and F. Illas, ACS Catal., 2020, 10, 5049 CrossRef CAS.
- S. Li, G. Liu, Z. Liu, W. Hu and H. Deng, J. Mater. Chem. A, 2019, 7, 19950 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02772g |
| This journal is © The Royal Society of Chemistry 2021 |