
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLate-stage modification of peptides and proteins at cysteine with diaryliodonium salts†
Stephen A.
Byrne
 ab,
Max J.
Bedding
ab,
Leo
Corcilius
ab,
Daniel J.
Ford
ab,
Yichen
Zhong
c,
Charlotte
Franck
abc,
Mark
Larance
cd,
Joel P.
Mackay
bc and
Richard J.
Payne
*ab
ab,
Max J.
Bedding
ab,
Leo
Corcilius
ab,
Daniel J.
Ford
ab,
Yichen
Zhong
c,
Charlotte
Franck
abc,
Mark
Larance
cd,
Joel P.
Mackay
bc and
Richard J.
Payne
*ab
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
bAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, Sydney, NSW 2006, Australia
cSchool of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia
dCharles Perkins Centre, The University of Sydney, NSW 2006, Australia
First published on 30th September 2021
Abstract
The modification of peptides and proteins has emerged as a powerful means to efficiently prepare high value bioconjugates for a range of applications in chemical biology and for the development of next-generation therapeutics. Herein, we report a novel method for the chemoselective late-stage modification of peptides and proteins at cysteine in aqueous buffer with suitably functionalised diaryliodonium salts, furnishing stable thioether-linked synthetic conjugates. The power of this new platform is showcased through the late-stage modification of the affibody zEGFR and the histone protein H2A.
Introduction
Peptides and proteins occupy a central role as the functional biomolecules that drive most processes in living systems. Following ribosomal synthesis, additional molecular complexity is added to peptides and proteins through post-translational modifications (PTMs) that serve to expand and diversify the functionality possible from the 20 proteinogenic amino acids.1,2 These PTMs range from the alkylations and acylations that drive gene expression, to the addition of large glycan and lipid chains – many of which underpin crucial protein–protein interactions and therefore endow additional functionality to their cognate proteins.2–4 Furthermore, a large number of therapeutics known as ‘biologics’,5–9 including antibody–drug conjugates (ADCs), rely on the functionalisation of peptides and proteins with distinct entities.10 The importance of protein modifications in biology and the pharmaceutical/biotechnological industry has led to the search for new methods that can be used for efficient access to modified peptides and proteins in homogeneous form (either with PTMs or unnatural ‘designer’ modifications, e.g. PEGylation)11 from fully unprotected peptides and proteins.To date, a number of so called ‘bioconjugation’ methods have been developed that enable site-selective functionalisation of peptides and proteins.12 Necessitating high degrees of chemo- and regio-selectivity in order to generate homogeneous protein conjugates, the development of such methods has focussed on two overarching approaches: (1) the incorporation of non-proteinogenic amino acids or engineered peptide sequences which take advantage of unique chemical (bioorthogonal)13 or chemoenzymatic reactivity,14 and (2) the utilisation of selective chemical reactions at proteinogenic amino acids.15 While both approaches have been successfully executed for the synthesis of peptide and protein conjugates, targeting native amino acids for late-stage modification provides a more operationally simple approach. However, this remains a challenging endeavour given that the reaction must be selective in the presence of a plethora of functionality that decorate the side chains of proteins, and reaction conditions must be mild and biocompatible (i.e. aqueous solvent, pH 6–8, temperatures ≤ 37 °C).
Cysteine (Cys) represents an attractive amino acid for the development of functionalisation chemistry due to the unique nucleophilicity of its thiol side chain. This provides the potential for chemoselectivity,16 while the relatively low natural abundance of Cys in proteins (ca. 1.8%)17 provides an avenue for enhanced regioselectivity.18 Maleimide reagents are frequently employed to covalently modify Cys residues both in academic and industrial settings. Notably this chemistry is employed for the functionalisation of monoclonal antibodies with cytotoxic payloads (e.g. brentuximab vedotin, also known as Adcetris®).10 However, the resulting conjugates can suffer from poor stability due to retro-Michael reactivity following conjugation.19
This drawback has prompted the exploration of alternative chemistry that could be used to generate stable Cys conjugates chemoselectively. While several elegant Cys conjugation methods have been reported recently, including a number of novel Cys arylation methodologies (Scheme 1a and b),20 these approaches still suffer from modest chemoselectivity,21 the need for highly specific peptide sequences,22 or potential problems with biocompatibility owing to the use of transition metal reagents.23,24
 | ||
| Scheme 1 Previously reported (a) nucleophilic aromatic substitution (SnAr) and (b) oxidative addition of transition metal complexes for Cys arylation on peptides and proteins; R = functional cargo; X = halide or pseudohalide; (c) proposed use of diaryliodonium salts for direct arylation of Cys to generate thioether-linked peptide and protein conjugates described in this work. | ||
Hypervalent iodine chemistry has recently emerged as an attractive means to modify peptides and proteins.25–30 A report from Kirkwood et al. in 1947 that diphenyliodonium chloride reacts with Cys in boiling water to produce phenylcysteine31 provided an encouraging proof of principle that this class of reagents could be used for direct functionalisation of peptides and proteins through the thiol side chain of Cys residues. Furthermore the air- and moisture-stability of hypervalent iodine reagents,32 coupled with their simple access from cheap and readily available aryl iodide and aryl boronate precursors33 led us to propose their application in the development of a platform for the chemoselective arylation of peptides and proteins at Cys (Scheme 1c). The proposed mechanism of arylation involves initial nucleophilic attack of the electrophilic iodine(III) centre by the thiolate of Cys to afford a 3-centre 4-electron hypervalent bond, followed by reductive elimination to release an aryl iodide and generate the target thioether linkage (Scheme 1c).34 Herein, we report the development of diaryliodonium salts as reagents for the late-stage modification of peptides and proteins at Cys. We demonstrate the power of the methodology via the chemoselective functionalisation of the mucin 1 variable number tandem repeat (MUC1 VNTR), an affibody to the epidermal growth factor receptor Ac-Cys-ZEGFR:1907 (zEGFR),35 and the protein histone 2A (H2A).36
Results and discussion
The development of the direct Cys arylation chemistry began with the synthesis of a suitable diaryliodonium salt (1) that could be used to optimise the proposed chemistry on a model peptide system (Scheme 2). To this end, we chose to adopt a one-pot procedure reported by Olofsson et al.37 that uses readily available aryl iodide and aryl boronic acid precursors to synthesise the bis(4-trifluoromethylphenyl)iodonium tetrafluoroborate salt 1 (Scheme 2, see ESI† for synthetic details). With the bis(4-trifluoromethylphenyl)iodonium reagent 1 in hand we next investigated conditions for the proposed direct arylation chemistry on an unprotected model peptide bearing a Cys residue, namely H-CSPGYS-NH2 (2) that was prepared by Fmoc-strategy solid-phase peptide synthesis (SPPS) (Scheme 2a, see ESI† for synthetic details). Several reaction conditions and buffers were systematically screened on this model system (see ESI†). Optimal reaction conditions that emerged involved the use of aqueous 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer at pH 8.0, tris(2-carboxyethyl)phosphine (TCEP) to prevent cystine formation, with the addition of 10 vol% acetonitrile (MeCN) as co-solvent, which was necessary to dissolve the diaryliodonium salt 1 (Scheme 2a). Importantly, the reactions did not need to be conducted under an inert atmosphere and proceeded to 90% conversion in 90 min when performed open to air [as judged by analytical ultra-performance liquid-chromatography (UPLC) analysis] (Scheme 2b). The target arylation product 3 was isolated in 60% yield following high-performance liquid-chromatography (HPLC) purification and was characterised by nuclear magnetic resonance (NMR) spectroscopy and high-resolution electrospray ionisation mass-spectrometry (HR-ESI-MS) (Scheme 2c, see ESI† for details). Importantly, the chemoselectivity for the side chain of Cys was confirmed by a heteronuclear multiple bond correlation (HMBC) experiment revealing a correlation between the aromatic ipso-carbon and the methylene protons of Cys (Scheme 2c, see ESI†). Following the success of this initial model system, we explored the effect of aryl substituents on reactivity with Cys (see ESI† for details). Consistent with the previously established reactivity reported for this class of reagents,34,38 we observed a decrease in the rate of arylation and overall efficiency of the reaction when moving from an electron withdrawing aryl system to more electron rich systems (e.g. p-methoxyphenyl, see ESI†).39 | ||
| Scheme 2 (a) Arylation of model peptide 2 at Cys with 1 to generate aryl conjugate 3; (b) monitoring of arylation reaction by analytical UPLC; (c) HR-ESI-MS of conjugate 3 and confirmation of Cys chemoselectivity through HMBC. | ||
Having demonstrated the direct arylation of a simple model system, we next sought to synthesise a diaryliodonium salt equipped with an alkyne handle that would enable late-stage Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC)40,41 chemistry on peptide and protein substrates, taking advantage of the variety of azides that can be obtained commercially or through simple synthetic procedures. Attempts to prepare the tetrafluoroborate analogue of compound 4 (Scheme 3a) via the same procedure used to prepare 1 provided the product in extremely low yield and purity. Therefore, compound 4 was synthesised via an alternative route with a modified version of a protocol reported by DiMagno et al. (see ESI† for synthetic details).42
 | ||
| Scheme 3 (a) Arylation of peptide MUC1 VNTR 5 with diaryliodonium salt 4 to afford 6 and subsequent functionalisation by CuAAC in one-pot to furnish 7, 8, 9, and 10; (b) characterisation of MUC1 VNTR conjugates 7 (PEG10), 8 (GlcNAc), 9 (biotin), and 10 (TAMRA) by ESI-MS and analytical UPLC. | ||
With compound 4 in hand we aimed to apply our methodology to a larger peptide substrate containing 11 of the 20 proteinogenic amino acids, namely, MUC1 VNTR (5).43 The peptide was synthesised by Fmoc-strategy SPPS with a Cys residue installed internally (in place of a serine) to eliminate steric bias at position 11 (Scheme 3a, see ESI† for synthetic details). Pleasingly, the arylation of 5 with diaryliodonium salt 4 under the optimised conditions proceeded to 91% conversion within 120 min (as indicated by UPLC-MS analysis), and the product MUC1-alkyne (6) peptide conjugate could be isolated in 52% yield following HPLC purification (see ESI† for synthetic details). Importantly, the thioether linkage in 6 was stable for 24 h in plasma, and phosphate buffered saline containing 100 mM glutathione, dithiothreitol or TCEP (see ESI† for details). Diaryliodonium reagent 4 was itself stable to storage in solid form at room temperature for 18 months.
Alkyne-containing peptide 6 was subsequently reacted with azides bearing a PEG10, N-acetyl glucosamine (GlcNAc), biotin or fluorescent TAMRA functionality under CuAAC conditions40,41 to afford peptide conjugates 7–10 in good yields over the two-step process (see ESI† for details). However, arylation of peptide 5 with 4 and CuAAC could also be performed in a one-pot manner without any purification of the MUC1-alkyne intermediate 6 (Scheme 3a). This one-pot arylation-CuAAC chemistry on 5 was used to generate four peptide conjugates bearing PEG10 (7), GlcNAc (8), biotin (9) and TAMRA (10) functionalities. Each of the reactions proceeded smoothly to generate the conjugates with 91% conversion over the two steps (as judged by UPLC-MS analysis). These conjugates were then purified by HPLC to afford 7–10 in 28–57% isolated yield (Scheme 3b). We note that the modest yields in some cases were a result of poor recoveries from HPLC rather than inefficient reactions. The peptide conjugates were characterised by electrospray ionisation mass-spectrometry (ESI-MS) and analytical UPLC.
Having demonstrated the efficiency of the arylation chemistry on the MUC1 VNTR peptide, we next envisaged applying our novel protein conjugation platform to a more challenging system, namely the affibody zEGFR (11) equipped with an N-terminal Cys residue (Scheme 4, see ESI† for synthetic details). Pleasingly, arylation with diaryliodonium salt 4 proceeded to 95% conversion in 240 min (as judged by UPLC-MS analysis). In this case TCEP was necessary to prevent the formation of unreactive cystine during the reaction. The protein conjugate 12 was isolated in 45% yield following HPLC purification and characterised by ESI-MS, analytical UPLC, and matrix-assisted laser desorption ionisation-time of flight mass-spectrometry (MALDI-ToF MS) (Scheme 4). Importantly, the Cys-selectivity of the arylation reaction was confirmed by LC-MS/MS analysis of 12 following trypsin digestion (see ESI† for details). As with the MUC1 VNTR, we found that the arylation chemistry and CuAAC could be performed in one-pot without purification of intermediate 12. Specifically, arylation followed by CuAAC with azido-PEG3-biotin in the same pot furnished the affibody conjugate 13 (94% conversion over the two steps as judged by UPLC-MS), which was purified by HPLC and characterised by ESI-MS, analytical UPLC, and MALDI-ToF MS (Scheme 4, see ESI† for synthetic details). Circular dichroism (CD) spectroscopy of functionalised zEGFR affibodies 11–13 showed that the characteristic α-helical secondary structure of the proteins was maintained following conjugation (see ESI†).
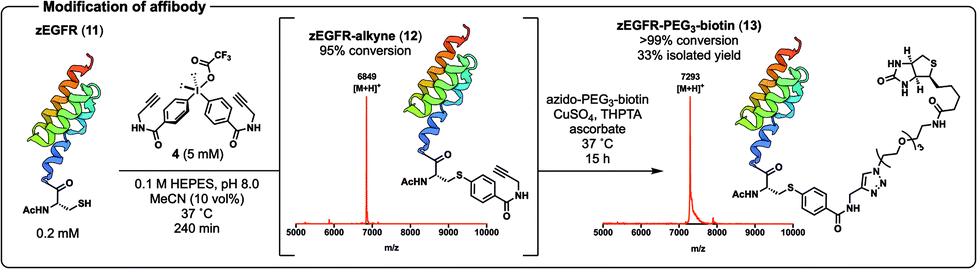 | ||
| Scheme 4 Arylation of zEGFR affibody 11 with 4 to afford the intermediate 12, which was converted to biotin conjugate 13 in one-pot using the CuAAC reaction. Structure of zEGFR from PDB (ID: 2KZI). | ||
Having demonstrated that alkyne-derived diaryliodonium salts can be used for the efficient functionalisation of peptides and proteins via CuAAC chemistry, we next sought to generate a ketone-containing diaryliodonium salt to access single-site-functionalised proteins primed for diversification with an alternative reaction manifold, namely the oxime-ligation.44 To this end, we synthesised diaryliodonium salt 14 (ref. 45) (see ESI†) and moved to assess the efficiency of oxime ligation chemistry on a more complex protein system, namely the histone H2A, one of the four proteins found in the octameric complex that make nucleosomes (Scheme 5a).46 The 129 residue and 14 kDa protein was expressed as the T120C mutant 15 by site-directed mutagenesis in Escherichia coli and purified by HPLC (see ESI† for details).36 It should be noted that the aqueous buffer used in these reactions was changed from HEPES to phosphate due to side-product formation when arylation of 15 was performed in HEPES (see ESI† for details). Gratifyingly, treatment of H2A 15 with diaryliodonium salt 14 in phosphate buffer led to the generation of the desired arylated conjugate 16 in just 60 min with 98% conversion. Purification by HPLC afforded 16 in 54% isolated yield (Scheme 5a, see ESI† for ESI-MS and analytical HPLC data). We also confirmed the Cys-selectivity of the arylation reaction by LC-MS/MS analysis of trypsin-digested 16 (see ESI† for details). The ketone-containing protein conjugate 16 was successfully functionalised with a variety of hydroxylamines using 5-methoxyanthranilic acid (5MA) as a nucleophilic catalyst47,48 (Scheme 5a). The resulting histone protein conjugates, functionalised with biotin (17), TAMRA (18), and the cell-penetrating peptide ‘penetratin’ (19),49 were each generated cleanly following oxime ligation in 1–20 h (83–93% conversions). Each of the constructs was purified by HPLC to afford the oxime conjugates 17–19 in 82–83% isolated yields (Scheme 5b, see ESI† for characterisation data).
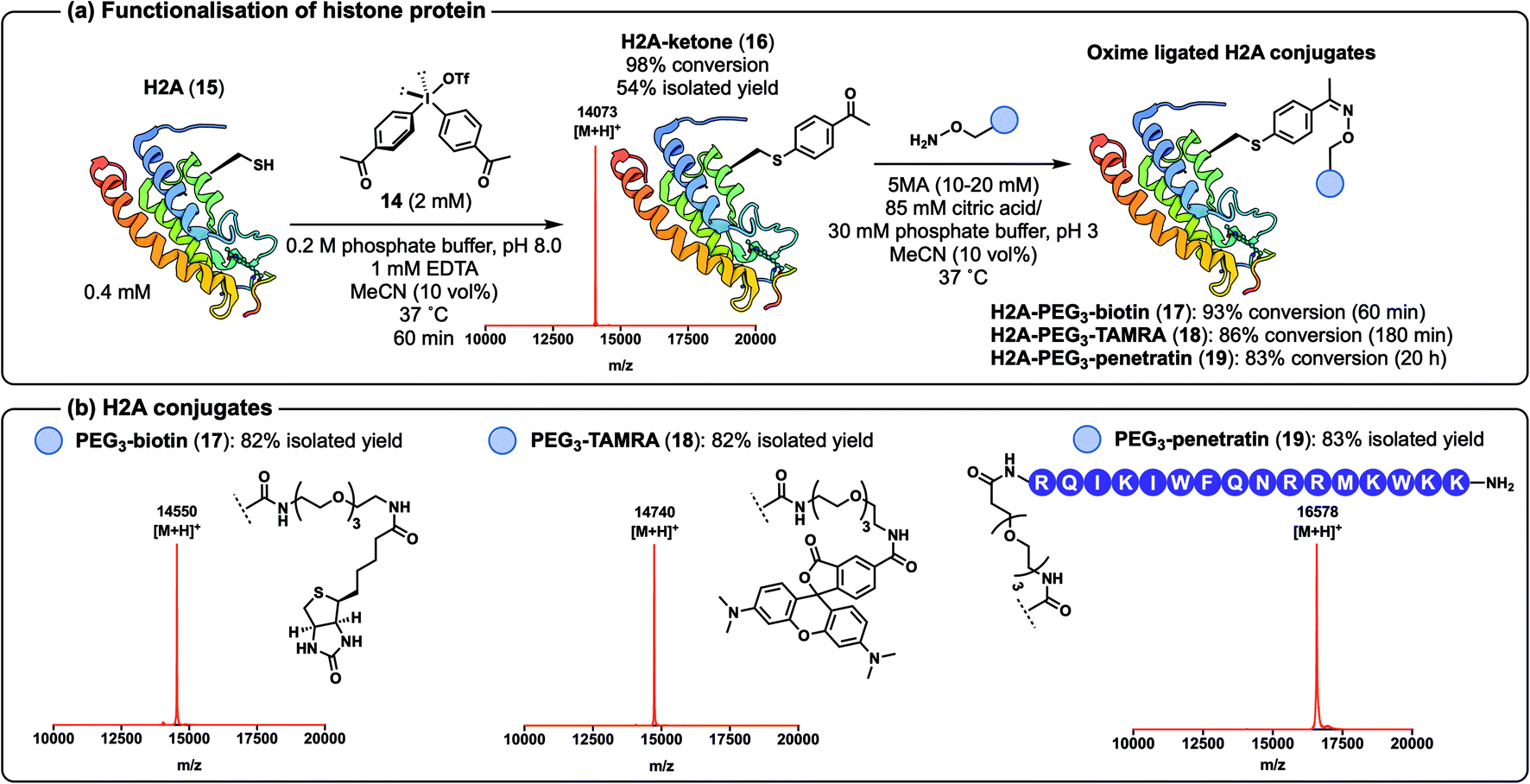 | ||
| Scheme 5 (a) Arylation of histone protein 15 to afford 16 for subsequent oxime-ligation reactions to furnish 17, 18, and 19; (b) characterisation of the oxime ligated H2A conjugates 17, 18, and 19 by MALDI-ToF MS. Structure of H2A from PDB (ID: 4QYL). | ||
Having thus far used our arylation chemistry to install a reactive handle for subsequent functionalisation, we next sought to employ the method to directly functionalise proteins (Scheme 6). PEGylation of proteins is known to improve their efficacy and half-life, and we therefore selected this system to test direct functionalisation chemistry.11 Towards this end, we first synthesised diaryliodonium salts equipped with mPEG4 (20) and mPEG8 (21) chains (Scheme 6, see ESI† for synthetic details). With the PEGylated diaryliodonium salts in hand we next investigated their efficiency in the late-stage modification of the affibody 11 (Scheme 6). Arylation of the affibody 11 with 20 proceeded smoothly to 90% conversion in 180 min in phosphate buffer. The PEGylated protein was subsequently purified by HPLC to afford 22 in 32% yield (Scheme 6, see ESI† for details). Likewise, we were delighted to find that treatment of 11 with 21 in phosphate buffer delivered the protein conjugate 23 with 90% conversion in 120 min and could be isolated by HPLC in 21% yield (Scheme 6, see ESI† for details). The functionalised affibodies were characterised by ESI-MS, MALDI-ToF MS, analytical UPLC, and CD spectroscopy (see ESI† for data). The Cys-selectivity of the arylation reactions was verified by LC-MS/MS analysis of the trypsin digested proteins (see ESI† for details).
 | ||
| Scheme 6 Arylation of affibody 11 with PEGylated diaryliodonium salts 20 and 21 to yield PEGylated proteins 22 and 23, respectively; MALDI-ToF MS spectra of purified 22 and 23. Structure of zEGFR from PDB (ID: 2KZI). | ||
Having validated our direct arylation chemistry on the affibody 11 we next moved to the histone protein 15. Gratifyingly, arylation of 15 with 20 progressed to 94% conversion in 180 min and purification by HPLC provided 24 in 53% isolated yield (Scheme 7, see ESI† for ESI-MS and analytical UPLC data). Likewise, we were pleased to find that treatment of the protein 15 with the diaryliodonium salt 21 led to 99% conversion to 25 in 120 min as judged by MALDI-ToF MS analysis, and subsequent HPLC purification provided the PEGylated protein 25 in 77% isolated yield (Scheme 7, see ESI† for ESI-MS and analytical UPLC data). The Cys-selectivity of each of the arylation reactions were unequivocally demonstrated by tryptic digestion and LC-MS/MS analysis of the proteolysed fragments (see ESI†).
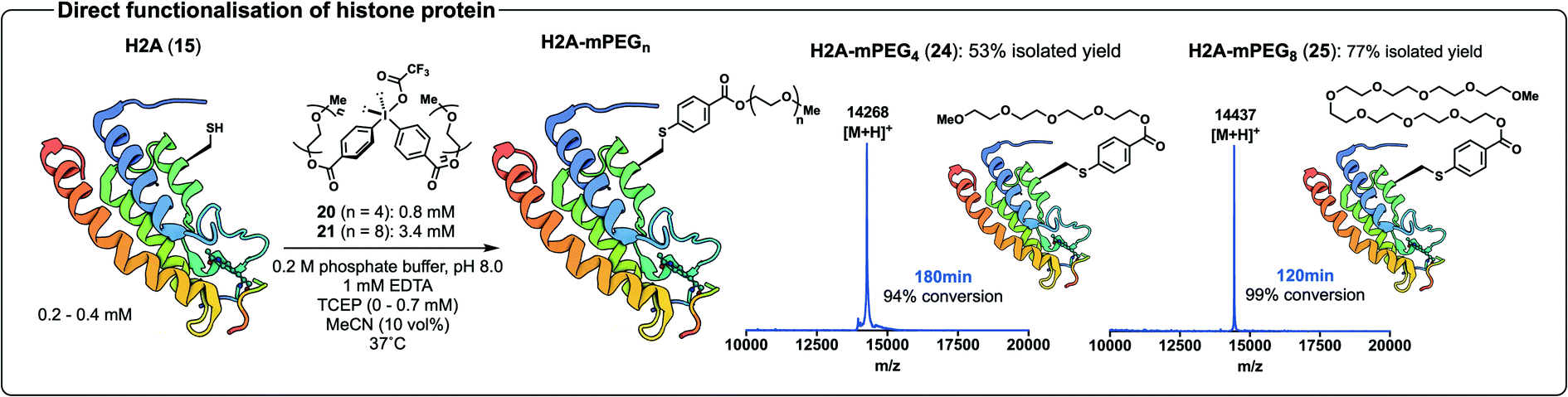 | ||
| Scheme 7 Arylation of histone protein 15 with 20 and 21 to yield protein conjugates 24 and 25, respectively; accompanied by MALDI-ToF MS spectra of crude 24 and 25 before HPLC purification. Structure of H2A from PDB (ID: 4QYL). | ||
Conclusions
In summary, we have shown that diaryliodonium salts are both mild and robust Cys selective reagents for the late-stage synthesis of a variety of stable peptide and protein conjugates. The power of this chemistry was highlighted through the installation of a variety of different functionalities in aqueous buffer that is compatible with proteins. The chemoselectivity of the methodology was also exemplified through the functionalisation of the affibody zEGFR and protein H2A that possessed each of the 20 proteinogenic amino acids. Given the overall simplicity and efficiency of the chemistry, we anticipate that the Cys functionalisation methodology reported here will find widespread application in the synthesis of high value peptide and protein conjugates in the future.Data availability
All experimental procedures, analytical, and spectroscopic data are provided in the ESI.† The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028382, username: E-mail: reviewer_pxd028382@ebi.ac.uk, password: 3UCckTu3.Author contributions
SB and RJP designed the project, SB and LC synthesised the diaryliodonium salts, SB synthesised the arylated model peptide, SB synthesised the MUC1 VNTR conjugates, SB synthesised the zEGFR conjugates, SB and DF synthesised the H2A conjugates, MJB synthesised azido-PEG3-biotin and azido-PEG3-TAMRA, MJB synthesised zEGFR, LC conducted the NMR analysis on the arylated model peptide, LC and DJF synthesised the alkoxyamines, YZ and JPM produced the H2A protein, CF conducted the CD spectroscopy, ML conducted LC-MS/MS experiments and data analysis, SB and RJP prepared the manuscript with assistance from all authors. RJP obtained funding for the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded through the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). We would like to thank Dr Nicholas Proschogo (The University of Sydney) for assistance with mass spectrometry and Dr Ian Luck (Sydney Analytical, The University of Sydney) for assistance with NMR spectroscopy. This work was supported by The University of Sydney Dean's International Postgraduate Research Scholarship (S. A. B.) and the Australian Government Research Training Program scholarship (M. J. B. and Y. Z.). Protein structures were created with http://BioRender.com.Notes and references
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Protein posttranslational modifications: The chemistry of proteome diversifications, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS PubMed.
- A. C. Conibear, Deciphering protein post-translational modifications using chemical biology tools, Nat. Rev. Chem., 2020, 4, 674–695 CrossRef CAS.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Native chemical ligation in protein synthesis and semi-synthesis, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- R. E. Thompson and T. W. Muir, Chemoenzymatic semisynthesis of proteins, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS PubMed.
- D. J. Drucker, Advances in oral peptide therapeutics, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS PubMed.
- C. Krejsa, M. Rogge and W. Sadee, Protein therapeutics: New applications for pharmacogenetics, Nat. Rev. Drug Discovery, 2006, 5, 507–521 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Synthetic therapeutic peptides: science and market, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Protein therapeutics: A summary and pharmacological classification, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Peptide therapeutics: Current status and future directions, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Strategies and challenges for the next generation of antibody–drug conjugates, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- J. Milton Harris and R. B. Chess, Effect of pegylation on pharmaceuticals, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Contemporary approaches to site-selective protein modification, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, From mechanism to mouse: A tale of two bioorthogonal reactions, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- M. Rashidian, J. K. Dozier and M. D. Distefano, Enzymatic labeling of proteins: Techniques and approaches, Bioconjugate Chem., 2013, 24, 1277–1294 CrossRef CAS PubMed.
- J. N. Degruyter, L. R. Malins and P. S. Baran, Residue-specific peptide modification: A chemist's guide, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed.
- P. Ochtrop and C. P. R. Hackenberger, Recent advances of thiol-selective bioconjugation reactions, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- A. Moura, M. A. Savageau and R. Alves, Relative amino acid composition signatures of organisms and environments, PLoS One, 2013, 8, e77319 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chemical modification of proteins at cysteine: Opportunities in chemistry and biology, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- P. A. Szijj, C. Bahou, V. Chudasama and V. Minireview, Addressing the retro-Michael instability of maleimide bioconjugates, Drug Discovery Today: Technol., 2018, 30, 27–34 CrossRef PubMed.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Arylation chemistry for bioconjugation, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS PubMed.
- D. A. Shannon, R. Banerjee, E. R. Webster, D. W. Bak, C. Wang and E. Weerapana, Investigating the proteome reactivity and selectivity of aryl halides, J. Am. Chem. Soc., 2014, 136, 3330–3333 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, π-Clamp-mediated cysteine conjugation, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, Organometallic gold(III) reagents for cysteine arylation, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Organometallic palladium reagents for cysteine bioconjugation, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- M. B. Hansen, F. Hubálek, T. Skrydstrup and T. Hoeg-Jensen, Chemo- and regioselective ethynylation of tryptophan-containing peptides and proteins, Chem.–Eur. J., 2016, 22, 1572–1576 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, A protein functionalization platform based on selective reactions at methionine residues, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, “Doubly orthogonal” labeling of peptides and proteins, Chem, 2019, 5, 2243–2263 CAS.
- K. Rahimidashaghoul, I. Klimánková, M. Hubálek, M. Korecký, M. Chvojka, D. Pokorný, V. Matoušek, L. Fojtík, D. Kavan, Z. Kukačka, P. Novák and P. Beier, Reductant-induced free radical fluoroalkylation of nitrogen heterocycles and innate aromatic amino acid residues in peptides and proteins, Chem.–Eur. J., 2019, 25, 15779–15785 CrossRef CAS PubMed.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Ethynylation of cysteine residues: From peptides to proteins in vitro and in living cells, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- G. Espuña, D. Andreu, J. Barluenga, X. Pérez, A. Planas, G. Arsequell and G. Valencia, Iodination of Proteins by IPy2BF4, a New Tool in Protein Chemistry, Biochemistry, 2006, 45, 5957–5963 CrossRef PubMed.
- R. B. Sandin, R. G. Christiansen, R. K. Brown and S. Kirkwood, The reaction of iodonium salts with thiol compounds, J. Am. Chem. Soc., 1947, 69, 1550 CrossRef CAS.
- A. Yoshimura and V. V. Zhdankin, Advances in synthetic applications of hypervalent iodine compounds, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- E. Merritt and B. Olofsson, Diaryliodonium salts: A journey from obscurity to fame, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
- H. Pinto De Magalhães, H. P. Lüthi and A. Togni, Reductive eliminations from λ 3-iodanes: Understanding selectivity and the crucial role of the hypervalent bond, Org. Lett., 2012, 14, 3830–3833 CrossRef PubMed.
- M. Friedman, A. Orlova, E. Johansson, T. L. J. Eriksson, I. Höidén-Guthenberg, V. Tolmachev, F. Y. Nilsson and S. Ståhl, Directed evolution to low nanomolar affinity of a tumor-targeting epidermal growth factor receptor-binding affibody molecule, J. Mol. Biol., 2008, 376, 1388–1402 CrossRef CAS PubMed.
- Y. Zhong, B. P. Paudel, D. P. Ryan, J. K. K. Low, C. Franck, K. Patel, M. J. Bedward, M. Torrado, R. J. Payne, A. M. van Oijen and J. P. Mackay, CHD4 slides nucleosomes by decoupling entry- and exit-side DNA translocation, Nat. Commun., 2020, 11, 1519 CrossRef CAS PubMed.
- M. Bielawski, D. Aili and B. Olofsson, Regiospecific one-pot synthesis of diaryliodonium tetrafluoroborates from arylboronic acids and aryl iodides, J. Org. Chem., 2008, 73, 4602–4607 CrossRef CAS PubMed.
- J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Arylation with unsymmetrical diaryliodonium salts: A chemoselectivity study, Chem.–Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed.
- A. M. Embaby, S. Schoffelen, C. Kofoed, M. Meldal and F. Diness, Rational Tuning of Fluorobenzene Probes for Cysteine-Selective Protein Modification, Angew. Chem., Int. Ed., 2018, 57, 8022–8026 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Analysis and optimization of copper-catalyzed azide–alkyne cycloaddition for bioconjugation, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click chemistry: Diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- L. Qin, B. Hu, K. D. Neumann, E. J. Linstad, K. McCauley, J. Veness, J. J. Kempinger and S. G. DiMagno, A mild and general one-pot synthesis of densely functionalized diaryliodonium salts, Eur. J. Org. Chem., 2015, 2015, 5919–5924 CrossRef CAS PubMed.
- D. W. Kufe, Mucins in cancer: Function, prognosis and therapy, Nat. Rev. Cancer, 2009, 9, 874–885 CrossRef CAS PubMed.
- D. K. Kömel and E. T. Kool, Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- K. B. Pal, J. Lee, M. Das and X. W. Liu, Palladium(II)-catalyzed stereoselective synthesis of C-glycosides from glycals with diaryliodonium salts, Org. Biomol. Chem., 2020, 18, 2242–2251 RSC.
- R. D. Kornberg, Chromatin structure: A repeating unit of histones and DNA, Science, 1974, 184, 868–871 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Nucleophilic Catalysis of Oxime Ligation, Angew. Chem., Int. Ed., 2006, 118, 7743–7746 CrossRef.
- P. Crisalli and E. T. Kool, Water-soluble organocatalysts for hydrazone and oxime formation, J. Org. Chem., 2013, 78, 1184–1189 CrossRef CAS PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, The third helix of the Antennapedia homeodomain translocates through biological membranes, J. Biol. Chem., 1994, 269, 10444–10450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03127a |
| This journal is © The Royal Society of Chemistry 2021 |