
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon polarisation-assisted hydrogen-bonded ferroelectrics in liquid crystalline domains†
Guohao
Yuan
ab,
Yuko
Kimura
a,
Takayuki
Kobayashi
a,
Takashi
Takeda
 ab,
Norihisa
Hoshino
ab and
Tomoyuki
Akutagawa
*abc
ab,
Norihisa
Hoshino
ab and
Tomoyuki
Akutagawa
*abc
aGraduate School of Engineering, Tohoku University, Sendai 980-8579, Japan. E-mail: akutagawa@tohoku.ac.jp
bInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
cNational Institute for Material Science (NIMS), 1-2-1 Tsukuba, 305-0047, Japan
First published on 28th September 2021
Abstract
An alkylamide-substituted (−NHCOC10H21) hydrogen-bonded dibenzo[18]crown-6 derivative (1) was prepared to stabilise the ionic channel structure in a discotic hexagonal columnar (Colh) liquid crystal. The introduction of simple M+X− salts such as Na+PF6− and K+I− into the ionic channel of 1 enhanced the ionic conductivity of the Colh phase of the M+·(1)·X− salts, with the highest ionic conductivity reaching ∼10−6 S cm−1 for K+·(1)·I− and Na+·(1)·PF6− at 460 K, which was approximately 5 orders of magnitude higher than that of 1. The introduction of non-ferroelectric 1 into the ferroelectric N,N′,N′′-tri(tetradecyl)-1,3,5-benzenetricarboxamide (3BC) elicited a ferroelectric response from the mixed Colh phase of (3BC)x(1)1−x with x = 0.9 and 0.8. The further doping of M+X− into the ferroelectric Colh phase of (3BC)0.9(1)0.1 enhanced the ferroelectric polarisation assisted by ion displacement in the half-filled ionic channel for the vacant dibenzo[18]crown-6 of (3BC)0.9[(M+)0.5·(1)·(X−)0.5]0.1.
Introduction
Intermolecular hydrogen-bonding interactions have been used for the association and dissociation of each molecule in flexible and transformable molecular-assembly structures,1 which are used in the formation of biological structures.2 Among the various types of hydrogen-bonding interaction, amide-type intermolecular hydrogen-bonding interactions (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯N–H−) play an important role in the formation of biological molecular assembly structures such as α-helixes and β-sheets in polypeptides, collagen, and keratin.2,3 The bonding energy of amide-type hydrogen-bonding interactions is typically in the range of 5–10 kJ mol−1; therefore, the structural reconstruction of proteins in biological systems is effectively activated by both the association and dissociation processes of each molecule in flexible molecular assemblies.1–4 Such amide-type intermolecular hydrogen-bonding interactions are also useful for the construction of functional supramolecular assemblies.5,6 For instance, the one-dimensional (1D) O⋯N–H− hydrogen-bonding interaction has been effectively utilised for constructing low-dimensional molecular assembly structures.7–9 Moreover, a typical 1D molecular assembly has been reported in the discotic liquid crystal phase of a benzene derivative bearing multiple hydrophobic alkylamide (−CONHCnH2n+1) chains, where the effective intermolecular hydrogen-bonding interactions formed a 1D hydrogen-bonded π-stacking columnar assembly that further assembled into a discotic hexagonal columnar (Colh) liquid crystal phase.10 The 1D intermolecular O⋯N–H− hydrogen-bonding interactions can also form nanofibers and organogels,11–14 wherein the three-dimensional (3D) entanglement of each 1D fibrous molecular assembly generates micropores that capture the solvents in the organogel state. An excellent organogellation ability has been reported for cyclohexane-1,2-dialkylamide and benzene-1,3,5-trialkylamide derivatives.14–17 Thus, hydrogen-bonded alkylamide chains are one of the interesting functional units that can form flexible 1D supramolecular assemblies such as Colh liquid crystals, organogels, and nanofibers.
O⋯N–H−) play an important role in the formation of biological molecular assembly structures such as α-helixes and β-sheets in polypeptides, collagen, and keratin.2,3 The bonding energy of amide-type hydrogen-bonding interactions is typically in the range of 5–10 kJ mol−1; therefore, the structural reconstruction of proteins in biological systems is effectively activated by both the association and dissociation processes of each molecule in flexible molecular assemblies.1–4 Such amide-type intermolecular hydrogen-bonding interactions are also useful for the construction of functional supramolecular assemblies.5,6 For instance, the one-dimensional (1D) O⋯N–H− hydrogen-bonding interaction has been effectively utilised for constructing low-dimensional molecular assembly structures.7–9 Moreover, a typical 1D molecular assembly has been reported in the discotic liquid crystal phase of a benzene derivative bearing multiple hydrophobic alkylamide (−CONHCnH2n+1) chains, where the effective intermolecular hydrogen-bonding interactions formed a 1D hydrogen-bonded π-stacking columnar assembly that further assembled into a discotic hexagonal columnar (Colh) liquid crystal phase.10 The 1D intermolecular O⋯N–H− hydrogen-bonding interactions can also form nanofibers and organogels,11–14 wherein the three-dimensional (3D) entanglement of each 1D fibrous molecular assembly generates micropores that capture the solvents in the organogel state. An excellent organogellation ability has been reported for cyclohexane-1,2-dialkylamide and benzene-1,3,5-trialkylamide derivatives.14–17 Thus, hydrogen-bonded alkylamide chains are one of the interesting functional units that can form flexible 1D supramolecular assemblies such as Colh liquid crystals, organogels, and nanofibers.
The formation of organogels in CH2Cl2, N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), nitrobenzene, and benzonitrile has been reported for N,N′,N′′-tri(octadecyl)-1,3,5-benzenetricarboxamide,18,19 which forms π-stacking 1D molecular assemblies through intermolecular O⋯N–H− hydrogen-bonding interactions.20–25 The formation of a Colh liquid crystal phase has been reported for a symmetrical three-chain system of N,N′,N′′-trialkyl-1,3,5-benzenetricarboxamide.10 The introduction of three –CONHCnH2n+1 chains into the benzene core effectively generates intermolecular interactions to form the Colh phase. The π-stacking columns, aided by the intermolecular O⋯N–H− hydrogen-bonding interactions, have been characterized by the single-crystal X-ray diffraction analysis of N,N′,N′′-trimethyl-1,3,5-benzenetricarboxamide and N,N′,N′′-trimethoxyethyl-1,3,5-benzenetricarboxamide.26–28 For these hydrogen-bonded 1D supramolecular assemblies, the interesting physical response of ferroelectric switching has been reported in the Colh phase of N,N′,N′′-trialkyl-1,3,5-benzenetricarboxamide, which generated polarisation–electric field (P–E) hysteresis curves, typical of the ferroelectric ground state.29–32 The 1D intermolecular O⋯N–H− hydrogen-bonded interactions along the π-stacking direction can be inverted by applying an external electric field, which results in dipole inversion and ferroelectricity. The two different hydrogen-bonding orientations (O⋯N–H− and –N–H⋯O) along the π-stacking axis can transform into each other under the application of an outer electric field.33–37
Another type of interesting molecular system with a selective ion recognition ability has been extensively designed with crown ether derivatives for application in phase-transfer catalysis, ion separation, ion sensing, and ion transport in the solution phase.38 The chemical designs of the pore environment such as [12]crown-4, [15]crown-5, and [18]crown-6 are useful because of their high affinity to Li+, Na+, and K+ ions.39 In the solid state, a regular array of crown ether can form a 1D ionic channel by the regular overlapping of the central cavity. Simple ionic channel structures such as (Li+)x([15]crown-5), (Li+)x([18]crown-6), and (Na+)x([18]crown-6) can coexist with an electrically conducting π-stacking column of [Ni(dmit)2] molecules in the highly electrically conducting single-crystals of (Li+)x([15]crown-5)[Ni(dmit)2]2, (Li+)x([18]crown-6)[Ni(dmit)2]2, and (Na+)x([18]crown-6)[Ni(dmit)2]2, where the motional freedom of Li+ ions is coupled with the conduction electrons.40,41 In addition, crown ether fused liquid crystalline phthalocyanine derivatives have been prepared to fabricate ionic channels, where the cation binding 1D fibrous supramolecular assembly and π-stacking column have been reported.42–44 The molecular frameworks of crown ethers are interesting candidates for controlling the ionic motion in a molecular assembly, which has been applied to solid state ionics.45 Among the various types of molecular assembly, the lattice periodicity of liquid crystal phases such as nematic (N) and smectic (Sm) states is much lower than that of the single crystalline state, and the flexible molecular assembly structure enables the design of the dynamic motion of ions. Many ionic liquid crystalline materials have a relatively high ionic conductivity of up to 1.5 × 10−4 S cm−1 in the Sm phase at around 350 K.46–48 Interestingly, ion-conductive liquid crystalline materials based on the diaza[18]crown-6 derivative bearing decylalkoxy-p-cyanobiphenyl chains have been reported by Espinet et al.,49 and the introduction of K+I− into the crown ether imparted an ionic conductivity of 3.0 × 10−7 S cm−1 at 530 K, which is lower than that of a K+-free crown ether. The lower conductivity of the K+-doped crown ether is due to the effective K+-capturing ability of the diaza[18]crown-6 unit and the insufficient formation of a channel-type molecular assembly in the absence of a regular array of crown ethers. Therefore, the formation of a 1D regular array of crown ethers should be one of the key points to increase the ionic mobility in molecular assemblies. Another interesting liquid crystalline material with ion-transport capability has been reported with a triphenylene π-core bearing n-alkoxy-substituted crown ethers,50 where the ionic conductivity of the K+I−-doped molecular assembly was 20 times larger than that of the K+SCN−-doped assembly and 100 times larger than that of the K+BF4− assembly. Therefore, the counter-anion plays an important role in controlling the K+ conductivity in the 1D channel.51 The liquid crystalline ion-captured M+(crown ether) usually modifies the thermal phase transition behaviour significantly. The addition of an alkali metal salt into a mesogenic state changes it into a non-mesogenic state due to an increase in intermolecular interactions.52–55 Among the variety of inorganic anions, the soft I− ion can significantly stabilise the thermal liquid crystal phases.56,57
A combination of specific chemical structures between the hydrogen-bonded alkylamide chains and crown ether can realize an interesting 1D molecular assembly with an ionic channel, which enables the design of multi-functional molecular assemblies such as ion-conductive ferroelectrics. For instance, the ionic conductivity of organic materials is related to their mechanical, electrical, and optical properties.58 Although the presence of ionic conductivity usually disturbs the ferroelectric polarisation due to leakage current,59 ion-conductive ferroelectrics based on typical inorganic perovskites such as Bi4Ti3O12 have been reported,60–62 which showed a relatively high ionic conductivity of ∼10−2 S cm−1 at 923 K and a high remanent polarisation of ∼50 μC cm−2 along the a–c plane at 298 K. These ion-conductive ferroelectrics with a non-linear electric response have attracted much attention for potential application in transducers, actuators, sensors, etc.63 Although ion-conductive inorganic ferroelectrics have been developed, there is no report on organic molecular materials.
In this paper, we report the molecular design, synthesis, and physical characterization of a dibenzo[18]crown-6 derivative bearing four –NHCOC10H21 chains (1 in Scheme 1). The cavity size of 1 fits a K+ ion well, and the four –NHCOC10H21 chains formed intermolecular O⋯N–H− hydrogen-bonding interactions to generate a 1D columnar molecular assembly exhibiting both ferroelectricity and ionic conductivity. Although the formation of a 1D hydrogen-bonded columnar structure of 1 has been observed in the Colh liquid crystal phase, no ferroelectric response was observed in the P−E hysteresis curve in the Colh phase. It has been reported that the introduction of the nearest-neighbouring –CONHCnH2n+1 chains on the benzene π-core suppresses the ferroelectricity due to steric hindrance to the rotation of the hydrogen-bonded alkylamide chains. In addition, the mode of substitution of –NHCOCnH2n+1 chains on benzene, i.e., the direct bonding of the nitrogen atom with the benzene π-core, suppresses the ferroelectric response due to the steric hindrance to rotation. The ferroelectric response was unfortunately suppressed in the Colh phase of 1. Therefore, we first evaluated the ion-doping effect for 1 to control the ionic conduction behaviour of the M+·(1)·X− system, where M+ = Na+, K+, and Cs+ and X− = Br−, I−, PF6−, SCN−, and CH3COO−. To induce ferroelectricity, we fabricated mixed liquid crystals of 1 and N,N′,N′′-tris(tetradecyl)-1,3,5-benzenetricarboxamide (3BC in Scheme 1) wherein the 1D ionic channel of 1 and the 1D ferroelectric hydrogen-bonded chains of 3BC coexist as (3BC)x(1)1−x (x is the mixing ratio of 3BC). The molecular assembly structures and the dielectric and ferroelectric responses of (3BC)x(1)1−x were evaluated for the mixing ratios of x = 0.9, 0.8, and 0.7, and the ferroelectric properties of the ion-doped mixed systems, (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1, were examined to clarify the size effect of the alkali metal ion (M+) in the ionic channels. In the M+ doped mixed Colh phase of (3BC)x[M+y·(1)·X−y]1−x, y is the mixing ratio of M+X− per dibenzo[18]crown-6 unit in molecule 1. Therefore, the condition of y = 0.5 corresponds to a half site occupation inside the ionic channel of molecule 1, i.e. M+50·(1)100 for 100 dibenzo[18]crown-6 columns.
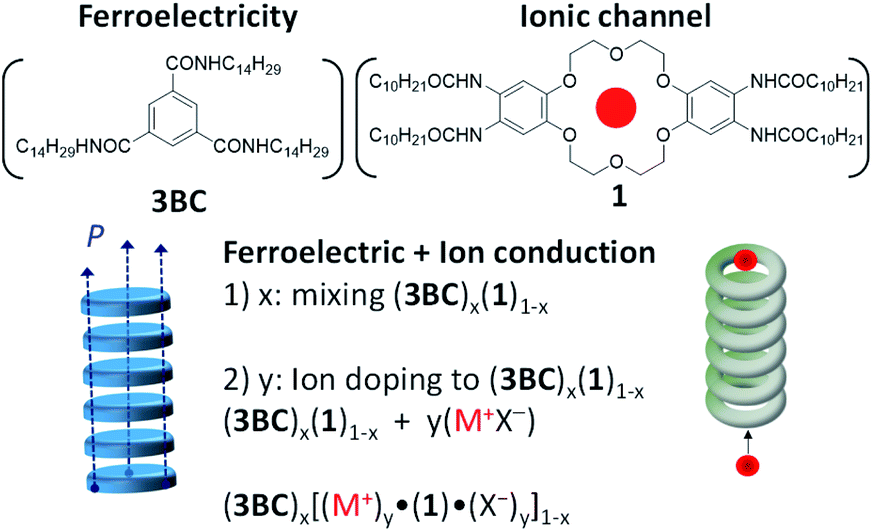 | ||
| Scheme 1 Molecular structures of the alkylamide (–NHCOC10H21)-fused dibenzo[18]crown-6 derivative (1) and ferroelectric N,N′,N′′-tris(tetradecyl)-1,3,5-benzenetricarboxamide (3BC). 3BC and 1 formed the ferroelectric and ionic channel type Colh phase. (1) Formation of the mixed Colh phase of (3BC)x(1)1−x, where x is the mixing ratio of 3BC. (2) Formation of the M+ doping mixed Colh phase of (3BC)x[M+y·(1)·X−y]1−x, where y is the mixing ratio of M+X− per dibenzo[18]crown-6 unit in molecule 1. | ||
Results & discussion
Molecular assemblies and phase transition behaviour of 1 and M+·(1)·X− salts
The Colh liquid crystal phase of 3BC was observed in the temperature range of 320–470 K (Fig. S1a†). The intermolecular amide-typeO⋯N–H− hydrogen-bonding interaction formed a π-stacking column of (3BC)∞ with a hexagonal columnar arrangement. In contrast, four hydrogen-bonded –NHCOC10H21 chains of molecule 1 formed the same Colh phase above 475 K, whose phase transition temperature to the Colh phase was 155 K higher than that of 3BC due to the high thermal stability of 1. Before phase transition into the Colh phase, two kinds of phase transition, S1–S2 and S2–Colh, were observed at 380 and 475 K with transition enthalpy changes (ΔH) of 10.1 and 14.1 kJ mol−1, respectively (Fig. S1a†). The Colh phase of 1 was significantly stable compared with that of 3BC due to the effective intermolecular hydrogen-bonding interaction between the four –NHCOC10H21 chains. The polarized optical microscope (POM) images under the cross-Nicole optical arrangement showed fluidic and birefringence behaviours for the Colh phases of both 1 and 3BC. A focal conic texture was observed in the POM image of 1 at 500 K (Fig. S1b†). The TG analysis of 1 revealed a high thermal stability up to 510 K (Fig. S2†), which corresponded to a relatively narrow thermal stability of the Colh phase of 1 from 475 to 510 K.
The formation of the hydrogen-bonded 1D molecular assembly structure was also confirmed by the formation of transparent organogels of 1 in CHCl3 or toluene with a concentration of ∼1 mM (Fig. S1c†). Molecule 1 was insoluble in CH3OH, C2H5OH, THF, CH3CN, hexane, etc., and the white powder precipitated from DMF and DMSO in the absence of an organogellation behaviour. The 1D hydrogen-bonded stack of dibenzo[18]crown-6 moieties formed an ionic channel through the O⋯N–H− hydrogen-bonding interaction (Fig. S3 and S4†), which further assembled into a 3D entangled structure with the solvent remaining in the micropores to form the organogel. The formation of the Colh phase of 1 was confirmed from the temperature-dependent PXRD profile (Fig. S5†). One sharp diffraction peak was observed at 2θ = 2.48° for the Colh phase at 493 K, which was assigned to d100 = 3.56 nm of the hexagonal columnar lattice (Fig. S5†). The d100 spacing is shorter than the maximum length of molecule 1 (∼4 nm), which is consistent with the inter-digitated molecular assembly structure of the lateral alkyl chains. The broad diffraction peak at 2θ = 20.3° could be assigned to the periodicity of d001 = 0.438 nm, corresponding to the melting state of the four alkyl chains and the average stacking distance of [18]crown-6 in the Colh phase.
The cavity size of [18]crown-6 in 1 fitted the K+ cation well, forming a stable K+-inclusion complex, K+·(1), which requires a counter-anion (X−) to compensate for the total charge of the K+·(1)·X− salt (Table S1†). The molecular structure of X− also affected the thermal phase transition behaviour of the Colh and isotropic liquid (I.L.) phases; therefore, we first evaluated the TG and DSC profiles of the five kinds of K+·(1)·X− salt with X− = Br−, I−, PF6−, SCN−, and CH3COO−. The structural modification of X− from symmetrical (Br−, I−, and PF6−) to linear (SCN− and CH3COO−) anions affects the formation of the Colh phase. Furthermore, the cation size affects the ion-inclusion ability of [18]crown-6 although the size-fitted K+·(1)·X− is the most tightly bound supramolecular assembly structure. From these points of view, we fabricated two different salts, Na+·(1)·PF6− and Cs+·(1)·(CO32−)0.5, where the Na+ and Cs+ cations of Na+·(1)·X− and Cs+·(1)·(X2−)0.5 salts were weakly bound in the cavity of [18]crown-6 in contrast to K+·(1)·X−. Here, a half amount of Cs+2CO32− was utilised to compensate for the occupation state in the ionic channel and total charge. Another important point is the mixing ratio of M+X− into the Colh phase of 1; different mixing ratios of (K+)y·(1)·(SCN−)y with y = 0.3 to 1.0 were used to evaluate the vacant ionic sites in the channel. The presence of vacant [18]crown-6 sites will enhance the ionic conductivity (σion) compared with that of a fully K+-occupied ionic channel.
Fig. 1a shows the DSC profiles of K+·(1)·SCN− (red) and K+·(1)·I− (blue). The phase transition from the solid to the Colh phase was observed from the DSC profiles and POM images (Fig. 1c and d), with both fluidic and birefringence behaviours confirmed in the Colh phase. Typical focal conic and/or spherulitic textures were observed in the Colh mesophase of the M+·(1)·X− salts. Although the S2–Colh phase transition temperature of 1 was relatively high at 473 K, an equimolar addition of the M+X− salt into the liquid crystalline 1 decreased the phase transition temperature from the solid to the Colh phase by approximately 50–100 K depending on the X− anions (Table S1†). It should be noted that the introduction of the M+X− salt into the Colh liquid crystalline 1 destabilised the solid phase and formed the Colh phase over a relatively wide temperature range.
 | ||
| Fig. 1 Phase transition behaviour of M+·(1)·X− salts and the formation of the Colh phase. (a) DSC profiles of K+·(1)·SCN− (red) and K+·(1)·I− (blue). (b) Temperature ranges of the solid phase (red) and Colh phase (blue). POM images of the Colh phase of (c) K+·(1)·I− at 483 K and (d) K+·(1)·SCN− at 473 K. | ||
The S–Colh phase transition temperatures of the K+·(1)·X− salts depended on the counter-anion X−. For instance, the phase transition temperatures to the Colh phase decreased in the order of Br− (T = 448 K), AcO− (T = 437 K), PF6− (T = 354 K), SCN− (T = 350 K), and I− (T = 328 K). In addition, the Colh–I.L. phase transitions were clearly observed at 478 K for K+·(1)·I− and at 483 K for K+·(1)·SCN− salts (Fig. 1a). The phase transition behaviours of S–Colh and Colh–I.L. were not affected by the addition of a hard anion such as Br−, but were effectively modulated by soft anions such as I−, PF6−, SCN−, and AcO−. The phase transition behaviours of the four anions were similar to each other and showed the lowering of the S–Colh phase transition temperature, which suggests the complete inclusion of K+ into the [18]crown-6 cavity and a mixed state of 1 and K+X− forming the K+·(1)·X− salt. When the K+ cation was replaced with a Na+ and/or Cs+ cation, the S–Colh phase transition temperatures of Na+·(1)·PF6− and Cs+·(1)·(CO32−)0.5 were 388 and 380 K, respectively, while their Colh–I.L. phase transitions occurred at 472 and 453 K. Therefore, almost uniform and complete Na+ and Cs+ capturing states in the absence of domain separation were confirmed in the Colh phases of both Na+·(1)·PF6− and Cs+·(1)·(CO32−)0.5.
The temperature-dependent PXRD profiles of the M+·(1)·X− salts were consistent with the formation of the Colh phase. The d-spacing of the hexagonal lattice with d100 = 3.9 nm for the M+·(1)·X− salts was consistent with that of 1 with d100 = 3.4–3.9 nm at 443 K (Fig. S3†). However, the crystalline domains of inorganic M+X− coexisted in the K+·(1)·Br−, K+·(1)·PF6−, and K+·(1)·I− salts due to the appearance of subtle sharp diffraction peaks at around 2θ = 20°, which indicates the domain separation of the liquid crystalline (M+)y·(1)·(X−)y and inorganic M+X− salts. In contrast, the uniform mixing of M+X− into the ionic channel of 1 was confirmed for the K+·(1)·SCN−, K+·(1)·AcO−, Na+·(1)·PF6−, and Cs+·(1)·(CO32−)0.5 salts (Fig. S6 and S7†) owing to the absence of sharp Bragg diffraction peaks for the crystalline domain of the inorganic dopant, M+X−.
Ionic conductivity of ion-capturing M+·(1)·X− salts
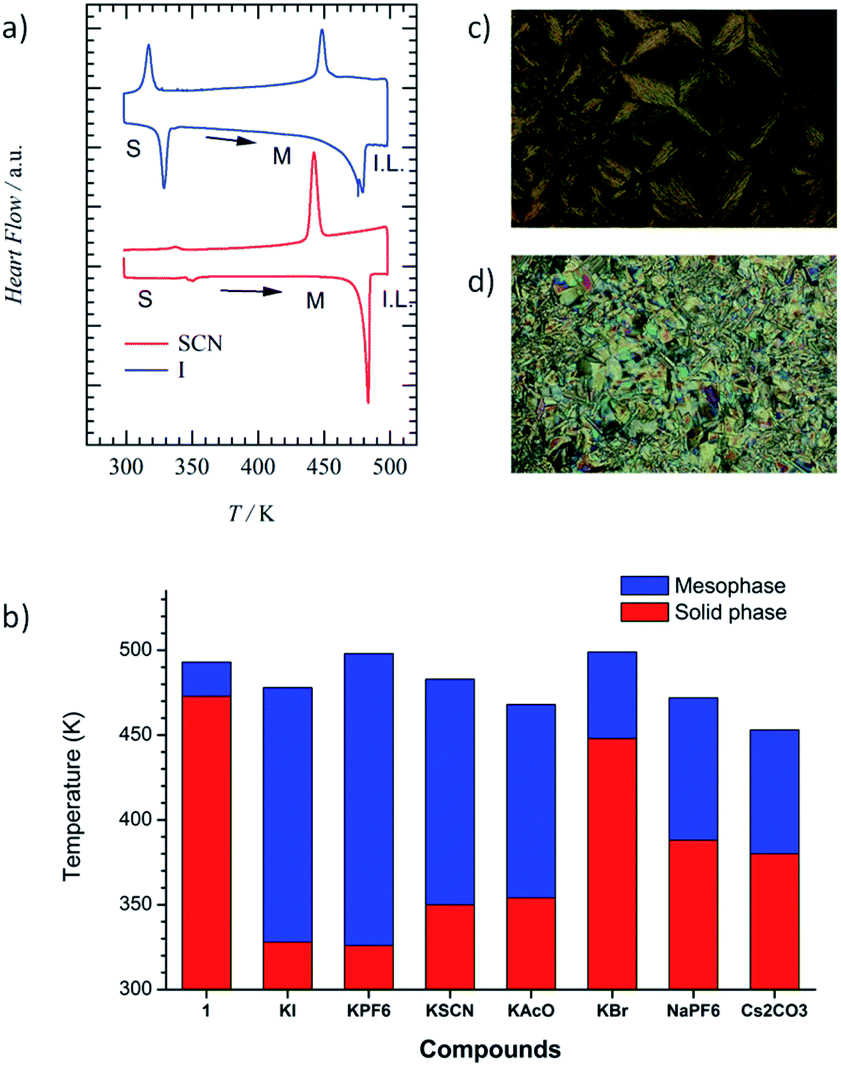
Fig. 2a and b show the anion (X−) and cation (M+)-dependent σion–T plots of the M+·(1)·X− salts, respectively. Table 1 summarizes σion at 460 K and activation energy (Ea). The temperature-dependent Cole–Cole (Z′–Z′′) plots of 1 and M+·(1)·X− salts showed ideal semicircle traces corresponding to the ionic conduction behaviours (Fig. S8†). The highest σion of ∼10−5 S cm−1 was observed for K+·(1)·I− and Na+·(1)·PF6− before phase transition in the I.L. state (Table S2†). Additionally, the σion of the Colh phase was ∼10−6 S cm−1 for most of the K+·(1)·X− salts, including X− = AcO−, I−, PF6−, and PF6−, which was approximately 6 times higher than that of M+-free 1. The significant increase in conductivity can be explained by the ionic transport pathway in the hydrogen-bonded array of [18]crown-6 in the M+·(1)·X− salts. The K+ conductivity (σK+) of the K+·(1)·X− salts depended on the counter-anion, X−, which decreased in the order of I− (σK+ = 1.53 × 10−5 S cm−1 at 486 K), PF6− (σK+ = 6.10 × 10−6 S cm−1 at 487 K), AcO− (σK+ = 1.73 × 10−6 S cm−1 at 469 K), SCN− (σK+ = 1.46 × 10−6 S cm−1 at 480 K), and Br− (σK+ = 2.30 × 10−8 S cm−1 at 490 K). The σK+ of the crystalline K+·(1)·Br− salt with domain separation was 3 orders of magnitude lower than that of the K+·(1)·I− salt, where the mixing state of the counter-anion, X−, affected the phase transition behaviour and σK+. The X− anions were bound to the positively charged K+(dibenzo[18]crown-6) unit through electrostatic interaction, where the size and affinity of X− anions to the cationic crown ether affected the σK+.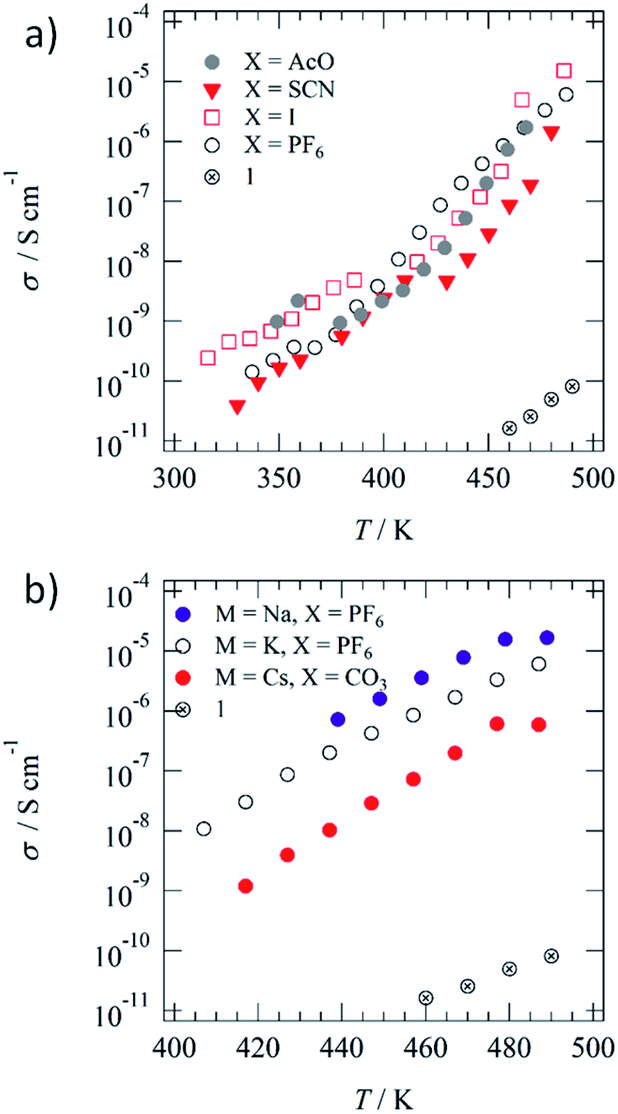 | ||
| Fig. 2 Temperature-dependent ionic conductivity of M+·(1)·X−. (a) Anion (X−)-dependent σK+–T plots of K+·(1)·X− (X− = AcO−, SCN−, I−, and PF6−) and K+-free 1. (b) Cation (X−)-dependent σion–T plots of Na+·(1)·PF6−, K+·(1)·PF6−, Cs+·(1)·(CO32−)0.5, and M+-free 1. | ||
| Compound | σ ion, S cm−1 at 460 K | E a, eV |
|---|---|---|
| 1 | 1.6 × 10−11 | 1.07 |
| K+·(1)·Br− | 9.7 × 10−9 | 0.44–0.63 |
| K+·(1)·AcO− | 7.3 × 10−7 | 0.13–2.15 |
| K+·(1)·I− | 4.6 × 10−6 | 0.54–2.17 |
| K+·(1)·PF6− | 9.2 × 10−6 | 0.37–1.26 |
| Na+·(1)·PF6− | 3.6 × 10−6 | 1.40 |
| Cs+·(1)·(CO32−)0.5 | 8.2 × 10−8 | 1.70 |
| K+·(1)·SCN− | 8.6 × 10−8 | 1.64 |
| (K+)0.8·(1)·(SCN−)0.8 | 7.0 × 10−7 | 0.82–1.90 |
| (K+)0.5·(1)·(SCN−)0.5 | 9.2 × 10−7 | 1.60 |
| (K+)0.3·(1)·(SCN−)0.3 | 9.5 × 10−8 | 1.59 |
Both the S–Colh and Colh–I.L. phase transition temperatures of the K+·(1)·X− salts decreased by the K+-inclusion array of [18]crown-6 units compared with that of M+-free 1, which indicates a decrease in the intermolecular interaction between the hydrogen-bonded K+·1 columns. The inclusion of K+ into the ionic channel increased the σK+, compared with that of the M+-free ionic channel of 1. The size-matching K+ ion in the cavity of dibenzo[18]crown-6 effectively stabilised the supramolecular structure of K+(dibenzo[18]crown-6), which was further connected by the four O⋯H–N− amide-type hydrogen-bonding interactions along the 1D molecular assembly of the ionic channel. The presence of X− anions between the thermally melting hydrogen-bonded 1D columns decreased the intermolecular van der Waals interaction between the alkyl chains in the Colh phase, which also decreased the Colh–I.L. phase transition temperature. In contrast, the addition of K+ into the ionic channel destabilised the 1D columnar assembly due to the electrostatic K+⋯K+ repulsive interaction, which decreased the Colh–I.L. phase transition temperature. The almost complete melting and mixing state of soft X− anions such as I− and PF6− decreased the intermolecular interaction of the crystal state, which destabilised the solid phase and lowered the S–Colh phase transition temperature.
Fig. 2b summarizes the cation (Na+, K+, and Cs+)-dependent σion–T plots of Na+·(1)·PF6−, K+·(1)·PF6−, and Cs+·(1)·(CO32−)0.5 salts. The Na+ conductivity (σNa+) of the Na+·(1)·PF6− salt (σNa+ = 1.7 × 10−5 S cm−1 at 487 K) was approximately 3 and 30 times larger than those of K+·(1)·PF6− (σK+ = 6.1 × 10−6 S cm−1 at 489 K) and Cs+·(1)·(CO32−)0.5 (σCs+ = 5.9 × 10−7 S cm−1 at 487 K), respectively. A much smaller cation effectively increases the σion due to the high carrier mobility. The size-matching K+ ion in the ionic channel was tightly bound in the cavity of [18]crown-6, while the slightly smaller Na+ in the Na+([18]crown-6) ionic channel had a much higher motional freedom than that of the K+([18]crown-6) array. Therefore, σNa+ was 3 times higher than σK+. In contrast, the ionic radius of the Cs+ cation is much larger than the pore size of [18]crown-6, which disturbed the Cs+ transport along the ionic channel and drastically lowered the σCs+. The occupancy state of the M+ cation in the ionic channel affected the σion values. For instance, the σion value of the fully M+-occupied ionic channel of an M+([18]crown-6) array should be lower than that of a partially occupied M+ vacant (M+)x([18]crown-6) array due to a decrease in the electrostatic M+⋯M+ repulsive interaction.
Herein, we evaluated the K+ occupation (y) effect on σK+ in the ionic channel of the (K+)y·(1)·(SCN−)y salts with different amounts of K+ cation (y = 0.0, 0.3, 0.5, 0.8, and 1.0). The σK+ values of (K+)y·(1)·(SCN−)y with y = 0.0, 0.3, 0.5, 0.8, and 1.0 at 470 K were 2.54 × 10−11, 2.24 × 10−7, 5.01 × 10−6, 9.21 × 10−7, and 1.88 × 10−7 S cm−1, respectively (Fig. S9 and Table S2†). The maximum σK+ of the (K+)y·(1)·(SCN−)y salts was obtained at y = 0.5 with a half-filled K+ ion in the ionic channel of (K+)0.5·(1)·(SCN−)0.5, which effectively reduced the Coulomb repulsive interaction between the nearest-neighbouring K+ ions. The σK+ value of (K+)0.5·(1)·(SCN−)0.5 at 470 K was approximately 25 and 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher than those of the fully K+-occupied (K+)·(1)·(SCN−) and M+-free 1, respectively. The presence of a carrier and vacant M+ ionic sites is essential for increasing the σion along the 1D ionic channel structure.
000 times higher than those of the fully K+-occupied (K+)·(1)·(SCN−) and M+-free 1, respectively. The presence of a carrier and vacant M+ ionic sites is essential for increasing the σion along the 1D ionic channel structure.
All the M+·(1)·X− salts showed similar σion values at temperatures before the phase transition to the I.L. state, where the σion values were dominated by the mass of the transport carrier. The σCs+ of the Cs+·(1)·(CO32−)0.5 salt at 447 K (2.9 × 10−8 S cm−1) was lower than the σNa+ of the Na+·(1)·PF6− salt at 449 K (1.6 × 10−6 S cm−1) and the σK+ of the K+·(1)·AcO− salt at 449 K (2.0 × 10−7 S cm−1) due to the complete dissociation of the M+⋯X− pair in the Colh phase. The σion in the Colh phase is usually dominated by the thermally activated carrier hopping process between the mobile M+ sites in the 1D ionic channel of the M+([18]crown-6) array, where semiconducting temperature dependence is observed for all the M+·(1)·X− salts. The σK+ of the K+·(1)·SCN− salt at 480 K (1.46 × 10−6 S cm−1) is similar to the σLi+ of a zwitterionic liquid crystal derivative of Li+-doped bis(trifluoromethylsulfonyl)imide and propylene carbonate in the SmA phase (10−4 to 10−5 S cm−1)64 and is larger than the σLi+ of a propylene carbonate-based columnar liquid crystalline material (10−6 to 10−8 S cm−1).65 The dielectric properties of ionic liquid have been theoretically discussed to predict the association–dissociation state,66,67 which can also apply to the ionic liquid crystalline phase. The M+ binding ability in the ionic liquid crystal of M+·(1)·X− salts influenced the magnitude of σion, where the diffusion of M+ was directly associated with the σion value.
Phase-separated Colh phase between ionic channel 1 and ferroelectric 3BC
Although molecule 1 formed the hydrogen-bonded Colh phase, ferroelectric P–E hysteresis was not observed over the measured temperature range. The rotation of the hydrogen-bonded –NHCOC10H21 chains was suppressed in the 1D array of (1)∞ due to a large steric hindrance compared with that of the –CONHCnH2n+1 chains. Therefore, we focused on the thermally stable Colh phase of ferroelectric 3BC (Scheme 1) to mix with the Colh phase of ionic channel (1)∞. Although the same kinds of Colh phase can be mixed together, the question whether the hydrogen-bonded columnar 1 and 3BC can be mixed together remains. The hydrogen-bonded infinite columns of (1)∞ and (3BC)∞ are thermally stable and form a homogeneous single column without the mixed state of [(3BC)x(1)1−x]∞, where the same molecules tend to stack together in the same 1D column. In contrast, there are two kinds of mixing state for each 1D column of (1)∞ and (3BC)∞. The first is a homogeneous random mixing state without domain separation, and the second is an inhomogeneous domain-separated mixing state.68,69 The mixing with and without domain separation can be distinguished by the DSC and PXRD profiles of the Colh phase. When the two different hydrogen-bonded columns of (1)∞ and (3BC)∞ can coexist in the same Colh phase, interestingly, both the ionic channel and ferroelectric chain coexist.The S–Colh and Colh–I.L. phase transition temperatures were 340 and 485 K, respectively, during the heating process, while the mixed liquid crystal of (3BC)0.9(1)0.1 (x = 0.9) exhibited double Colh–I.L. phase transition behaviours at around 474 and 480 K due to the phase-separated domains (Fig. 3a). Similarly, two S–Colh and Colh–I.L. phase transition temperatures of (3BC)0.8(1)0.2 (x = 0.8) were observed at 333 and 341 K and 478 and 483 K, respectively, during the heating process corresponding to the phase-separated states of the two kinds of domain. The POM image of the Colh phase of (3BC)0.9(1)0.1 at 450 K exhibited a focal conic texture similar to that of 3BC (Fig. S10†). When 1 was mixed into the Colh phase of 3BC above x > 0.3, the phase-separated state between the focal conic domain of 3BC and the homeotropic dark domain of 1 was observed in the POM images (Fig. S10†).
 | ||
| Fig. 3 Phase transition and mixed state of ionic channel (1)∞ and ferroelectric (3BC)∞ in the Colh phase. (a) DSC profiles of the mixed Colh liquid crystal of (3BC)x(1)1−x with x = 1, 0.9, and 0.8. (b) POM image of (3BC)0.9(1)0.1 at 450 K. (c) PXRD profiles of the Colh phases of (3BC)0.9(1)0.1 at 380 K, 3BC at 380 K, and 1 at 380 K. (d) Schematic of the phase separation state of the ferroelectric 3BC domain (blue columns) and ionic channel 1 domain (red columns). | ||
The PXRD profile of the Colh phase of (3BC)0.9(1)0.1 at 380 K clearly indicates the domain-separated state of ferroelectric 3BC and ionic channel 1 due to the appearance of two low-angle diffraction peaks with d100 spacings of 2.7 and 3.6 nm, respectively, which were relatively consistent with the d100 spacing of 3BC and 1 (Fig. 3c). The hydrogen-bonded columns of (3BC)∞ and (1)∞ were stabilised by the formation of a segregated stacking structure of the same molecules, which further assembled to form a single domain of each Colh phase. Therefore, the hydrogen-bonded columns of (3BC)∞ and (1)∞ coexisted in the domain-separated state (Fig. 3d), which was consistent with the two d100 spacings observed in the PXRD profile. The maximum molecular lengths of 1 and 3BC were approximately 4.0 and 4.5 nm, respectively, assuming the all-trans conformations of –NHCOC10H21 and –CONHC14H29 chains. The average inter-column distances (d100) of (1)∞ and (3BC)∞ in the Colh phases were shorter than those of the ideal molecular lengths, which suggests the presence of inter-digitated molecular assembly structures of alkylamide chains in the Colh phases.
Ferroelectricity of (3BC)x(1)1−x
Fig. 4a shows the temperature-dependent ferroelectric P–E hysteresis curves of the phase-separated Colh phases of mixed (3BC)0.9(1)0.1 together with that of 3BC. The addition of a small amount of a similar hydrogen-bonding Colh liquid crystal usually supressed the ferroelectric response.68,69 There was no ferroelectric response from the Colh phase of 1, while the introduction of 1 into the ferroelectric 3BC domain gave rise to P−E hysteresis curves. The remanent polarisation (Pr) of 3BC at 353 K (f = 1 Hz) was Pr = 1.1 μC cm−2 (Fig. S11†), while the Pr of mixed (3BC)0.9(1)0.1 at 353 K was approximately 56% lower (0.48 μC cm−2) than that of pure 3BC. | ||
| Fig. 4 Temperature-dependent P–E hysteresis curves of Colh phases for mixed crystals of (a) (3BC)0.9(1)0.1 and (b) plot of mixing ratio (x) vs. Pr of (1)x(3BC)1−x. | ||
The further introduction of 1 into 3BC in mixed (3BC)0.8(1)0.2 effectively lowered the Pr to 0.26 μC cm−2 at 373 K (Fig. S12†), which is 24% of the initial Pr of 3BC. The introduction of non-ferroelectric 1 into ferroelectric 3BC drastically suppressed the ferroelectric P–E responses due to the phase-separated domains and the reduction of inter-columnar ferroelectric interaction. Although the Pr was suppressed by the introduction of 1 into 3BC, the coercive electric field (Eth) of (3BC)x(1)1−x was the same as that of 3BC. Fig. 4b shows the mixing ratio (x) vs. Pr plots of mixed (3BC)x(1)1−x. A linear correlation was observed for a mixing ratio of x = 1 to 0.8. With the addition of 30% 1 ((3BC)0.7(1)0.3), the ferroelectric response completely disappeared, which was consistent with the extrapolation in the linear x–Pr plots.
Ferroelectricity of ion-doped (3BC)0.9[(M+)y·(1)·(X−)y]0.1
The doping effect of M+X− on the ferroelectric P–E response of the mixed Colh phase of (3BC)0.9(1)0.1 (x = 0.9) was evaluated to fabricate ion-conductive organic ferroelectrics. The motional freedom of the M+ cation in the ionic channel (1)∞ coexisting with the ferroelectric domain of 3BC affects the ferroelectric P–E response of the mixed Colh phase of (3BC)0.9(1)0.1.We introduced three kinds of cation, Na+, K+, and Cs+, into the ionic channel of (3BC)0.9(1)0.1 to form three ion-doped salts, (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1. The three kinds of cation (Na+, K+, and Cs+) have different dynamic behaviours according to the cation size; the mobility of the Na+ cation was higher than that of K+ and the larger Cs+ cation could not pass through the cavity of [18]crown-6. The occupancy states of Na+, K+, and Cs+ cations in the ionic channel were fixed as 50% probability (a half site in the ionic channel) to maintain enough mobile environments. The difference in the dynamic behaviours of the cations was evaluated from the P–E hysteresis responses of the mixed Colh phase. Fig. 5a–c show the temperature-dependent P–E hysteresis curves of the mixed Colh phase of (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1, respectively. The Pr of all the ion-doped salts increased with increasing temperature, and the maximum Pr values of (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1 salts were 1.2 μC cm−2 at 353 K, 1.1 μC cm−2 at 363 K, and 0.9 μC cm−2 at 353 K, respectively.
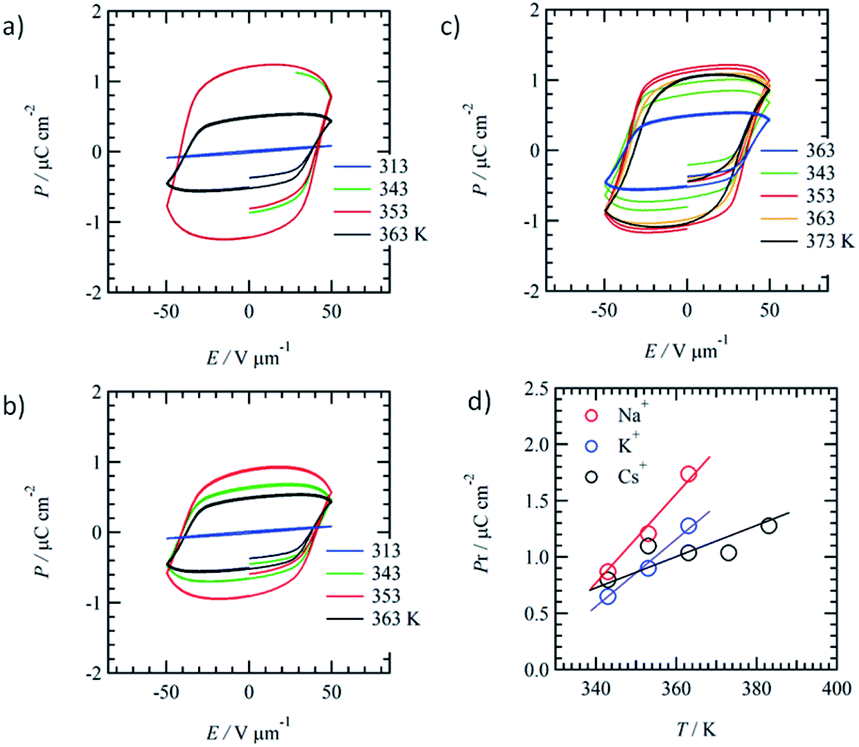 | ||
| Fig. 5 Temperature-dependent P–E hysteresis curves of ion-doped mixed crystals of (a) (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (b) (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (c) (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1. (d) Temperature-dependent Pr values of Na+, K+, and Cs+-doped (3BC)0.9(1)0.1. | ||
Fig. 5d shows the temperature-dependent Pr of the Na+, K+, and Cs+-doped (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1 salts. Although the Pr values of the three salts at 343 K were almost the same at ∼0.7 μC cm−2, the Pr values increased with increasing temperature in the order Na+ > K+ > Cs+. At 363 K, the Pr values of (3BC)0.9[(Na+)0.5·(1)·(PF6−)0.5]0.1, (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1, and (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1 were 1.74, 1.28, and 1.04 μC cm−2, which indicate the different dynamic behaviours of the cations at high temperatures.
The remanent polarisation (Pr) of 3BC at 353 K (f = 1 Hz) was 1.1 μC cm−2 (Fig. S11†), while the Pr of mixed (3BC)0.9(1)0.1 at 353 K was approximately 56% lower (0.48 μC cm−2) than that of pure 3BC. The further introduction of 1 into 3BC in mixed (3BC)0.8 (1)0.2 effectively lowered the Pr to 0.26 μC cm−2 at 373 K (Fig. S12†), which is 24% of the initial Pr of 3BC. The introduction of non-ferroelectric 1 into ferroelectric 3BC drastically suppressed the ferroelectric P–E responses due to the phase-separated domains and the reduction of inter-columnar ferroelectric interaction. Although the Pr was suppressed by the introduction of 1 into 3BC, the Eth of (3BC)0.9(1)0.1 was the same as that of 3BC.
The difference in the Pr values of the three salts was discussed from the viewpoint of a possible dynamic behaviour of the 50%-occupied Na+, K+, and Cs+ cations (a half occupied cation for all sites) in the ionic channel of (M+)0.5(1), which affected the polarisation of the (3BC)0.9[(M+)0.5·(1)·(X−)0.5]0.1 salts. Scheme 2 shows the possible role of the ionic transport channel of 1 in the mixed Colh phase of the (3BC)0.9[(M+)y·(1)·(X−)y]0.1 salts with M+ = Na+, K+, and Cs+. The ionic conduction along the electric field (E) can generate a local electric field (Eloc) along the columnar direction due to the generation of a concentration gradient of M+ in the ionic channels, where the 50%-occupied M+ cations in the ionic channels of (Na+)0.5([18]crown-6), (K+)0.5([18]crown-6), and (Cs+)0.5([18]crown-6) were responsible for the electric field E. However, the dynamic behaviours of Na+, K+, and Cs+ cations in the ionic channels were different. The Na+ cation, which is smaller than the cavity of [18]crown-6, can easily modulate the Na+ positions in the ionic channel and generate a local electric field (Eloc) and ionic polarisation (Pion) along the applied E direction, which results in a linear paraelectric P–E correlation and contributes to the overall Pr due to the effective application of the additional Eloc. Although the K+ mobility in the (K+)0.5([18]crown-6) ionic channel was lower than the Na+ mobility in the (Na+)0.5([18]crown-6) channel, the application of E of a sufficient magnitude at a high-temperature region generates a concentration gradient of K+ cations and Pion along the ionic channel. Therefore, compared with the Na+ salt, the K+ salt exhibited a subtle weaker temperature-dependent Pr for the (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1 salt (Fig. 5d).
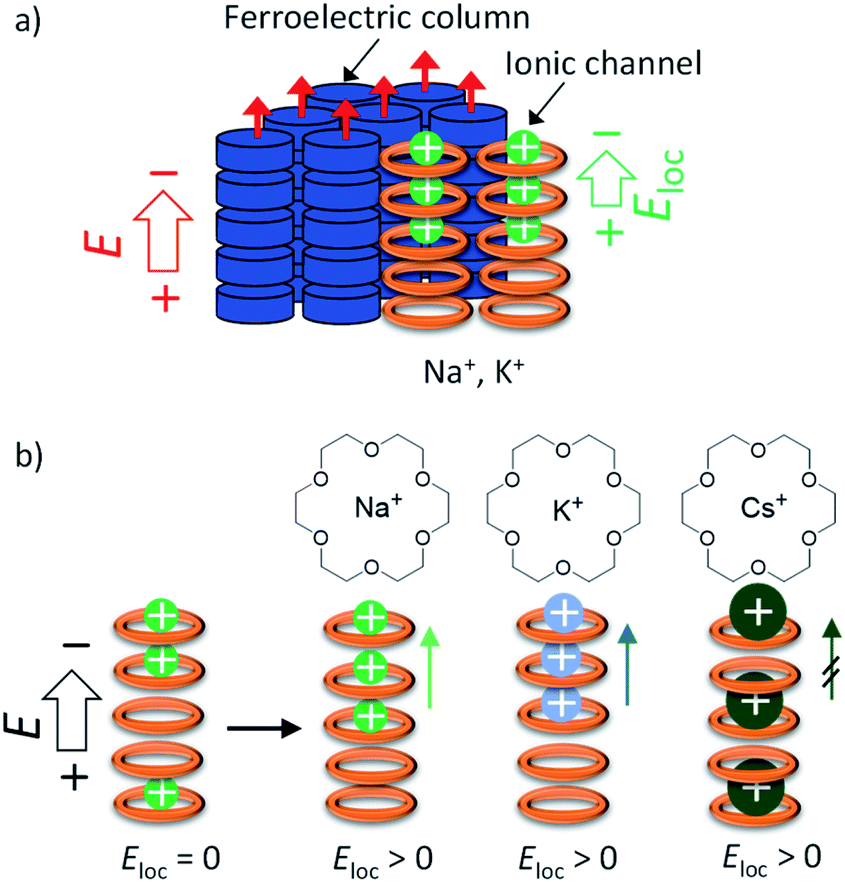 | ||
| Scheme 2 Coupling between the ferroelectricity of 3BC and M+-inclusion (M+)0.5(1) in the mixed Colh phase of (3BC)0.9[(M+)y·(1)·(X−)y]0.1 salts with M+ = Na+, K+, and Cs+. (a) Coexistence of the ferroelectric domain of 3BC (blue column) and ionic channel of (M+)y·(1)·(X−)y; M+X− = Na+PF6−, K+PF6−, and Cs+(CO32−)0.5. The ionic conduction along the electric field (E) generates a local electric field (Eloc) along the columnar direction. (b) 50%-occupied (M+)0.5([18]crown-6) ionic channels of (Na+)0.5([18]crown-6), (K+)0.5([18]crown-6), and (Cs+)0.5([18]crown-6) under electric field E. | ||
In contrast, the large Cs+ cation could not pass through the cavity of [18]crown-6, which resulted in a small contribution to the Pion term due to the subtle Cs+ displacement along the direction of the ionic channel. Therefore, the temperature-dependent Pr enhancement for the Cs+-doped (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1 salt was much smaller than those for the Na+ and K+-doped salts. The occupation states of Na+ and K+ cations under the condition of E ≠ 0 V relaxed from the biased state to the randomly occupied M+ state under the condition of E = 0 V, which also contributed to the P–E hysteresis curves with the polarisation enhancement factor of the ionic displacement effect.
Conclusions
An alkylamide-substituted (−NHCOC10H21) dibenzo[18]crown-6 derivative (1) was prepared to form anO⋯H–N− amide-type hydrogen-bonding-assisted 1D ionic channel by the overlapping of the central cavity of [18]crown-6. Both the organogellation and nanofiber formation of 1 clearly supported the 1D molecular assembly structures through the O⋯H–N− hydrogen-bonding interactions. An ionic conduction behaviour was observed for M+X−-doped 1, with domain-separated uniform mixing states confirmed. The highest σion of ∼10−6 S cm−1 was obtained for the Colh phases of K+·(1)·I− and Na+·(1)·PF6− salts before melting into the I.L. state, whose σion was approximately 6 orders of magnitude higher than that of M+-free 1. The significant σion enhancement by the M+X− doping of 1 can be explained by the ionic transport along the hydrogen-bonded [18]crown-6 array. The σion values of the (K+)y·(1)·(SCN−)y salts were tuned to control the K+ occupation probability, with the maximum σK+ obtained for (K+)0.5·(1)·(SCN−)0.5 wherein K+ ions occupied 50% of the possible sites in the ionic channel, which reduced the Coulomb K+⋯K+ repulsive interaction. Although the phase-separated domains of non-ferroelectric 1 and ferroelectric 3BC were observed in the mixed Colh phase of (3BC)x(1)1−x, the Colh phases at x = 0.9 and 0.8 showed ferroelectric P–E hysteresis responses. M+X− was doped into the ferroelectric Colh phases of (3BC)0.9(1)0.1 to modulate the ferroelectric polarisation assisted by the dynamic M+ displacements in the 50%-occupied ionic channel of (M+)0.5(1). The small Na+ cation effectively enhanced the polarisation through Na+ transport along the ionic channel to generate a concentration gradient of Na+ ion and ionic polarisation. A similar ionic polarisation effect was observed in the K+-doped ionic channel of the (3BC)0.9[(K+)0.5·(1)·(PF6−)0.5]0.1 salt. In contrast, the relatively low mobility of the large Cs+ cation in the (3BC)0.9[(Cs+)0.5·(1)·(CO32−)0.25]0.1 salt led to a subtle Cs+ ion displacement, contributing only slightly to the overall polarisation. The ionic motion in the ionic channels can assist the ionic polarisation behaviours of the ion-conductive liquid crystalline molecular ferroelectrics. Such new kinds of multi-functional organic material have the potential to control the polarisation state in flexible memory devices.
Data availability
Experimental section, TG charts, IR spectra, PXRD patterns, Z′–Z′′ plots, log(sK+)–T−1 plots, POM images, and P−E hysteresis curves.Author contributions
Y. G., Y. K., and T. K. conducted the synthetic experiments and analysed the data. T. T. performed the molecular design and preparations. N. H. carried out the dielectric measurements. T. A. prepared the manuscript and conceived the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (KAKENHI) (Grant Numbers: JP19H00886, JP20H05865, JP20K05442, and JP20H04655), Japan Science and Technology Agency, Core Research for Evolutional Science and Technology (Grant Number: JPMJCR18I4), and the “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” project supported by the Ministry of Education, Culture, Sports, Science and Technology.Notes and references
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed.
- Advances in Protein Chemistry, Peptide Solvation and H−Bonds, ed. R. L. Baldwin and D. Baker, Elsevier Academic Press, Amsterdam, vol. 72, 2006 Search PubMed.
- The Amide Linkage, A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley-Interscience, New Jersey, 2003.
- L. Stryer, Biochemistry, Freeman, New York, 1995 Search PubMed.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Outline of a Comprehensive Hydrogen bond Theory, Oxford Univ. Press, Oxford, 2009 Search PubMed.
- Supramolecular Assembly via Hydrogen Bonds I and II, ed. D. M. P. Mingos, Springer, Berlin, 2004 Search PubMed.
- M. L. Bushey, T. Q. Nguyen, W. Zhang, D. Horoszewski and C. Nuckolls, Angew. Chem., Int. Ed., 2004, 43, 5446–5453 CrossRef CAS PubMed.
- T. Akutagawa, Mater. Chem. Front., 2018, 2, 1064–1073 RSC.
- T. Akutagawa, Bull. Chem. Soc. Jpn., 2021, 94, 1400–1420 CrossRef CAS.
- Y. Matsunaga, N. Miyajima, Y. Nakayasu, S. Sakai and M. Yonenaga, Bull. Chem. Soc. Jpn., 1988, 61, 207–210 CrossRef CAS.
- R. G. Weiss and P. Terech, Molecular Gels, Springer, Dordrecht, 2006 Search PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- P. P. Bose, M. G. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198 RSC.
- K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed., 1996, 35, 1949–1951 CrossRef CAS.
- K. W. Tong, S. Dehn, J. E. Webb, K. Nakamura, F. Braet and P. Thordarson, Langmuir, 2009, 25, 8586–8592 CrossRef CAS PubMed.
- Y. Shishido, H. Anetai, T. Takeda, N. Hoshino, S.-i. Noro, T. Nakamura and T. Akutagawa, J. Phys. Chem. C, 2014, 118, 21204–21214 CrossRef CAS.
- B. Xing, C.-W. Yu, K.-H. Chow, P.-L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS PubMed.
- Y. Yasuda, E. Iishi, H. Inada and Y. Shirota, Chem. Lett., 1996, 25, 575–576 CrossRef.
- K. Hanabusa, C. Koto, M. Kimura, H. Shirai and A. Kakehi, Chem. Lett., 1997, 26, 429–430 CrossRef.
- A. R. Palmans, J. A. Vekemans, R. A. Hikmet, H. Fischer and E. W. Meijer, Adv. Mater., 1998, 10, 873–876 CrossRef CAS.
- L. Brunsveld, A. Schenning, M. Broeren, H. Janssen, J. Vekemans and E. W. Meijer, Chem. Lett., 2000, 29, 292–293 CrossRef.
- J. Roosma, T. Mes, P. Leclère, A. R. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 1120–1121 CrossRef CAS PubMed.
- P. J. Stals, M. M. Smulders, R. Martín-Rapún, A. R. Palmans and E. W. Meijer, Chem.–Eur. J., 2009, 15, 2071–2080 CrossRef CAS PubMed.
- S. Cantekin, D. W. Balkenende, M. M. Smulders, A. R. Palmans and E. W. Meijer, Nat. Chem., 2011, 3, 42–46 CrossRef CAS PubMed.
- C. M. Leenders, L. Albertazzi, T. Mes, M. M. Koenigs, A. R. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, Chem. Commun., 1999, 1945–1946 RSC.
- S. Ranganathan, K. Muraleedharan, C. C. Rao, M. Vairamani, I. L. Karle and R. D. Gilardi, Chem. Commun., 2001, 2544–2545 RSC.
- M. Kristiansen, P. Smith, H. Chanzy, C. Baerlocher, V. Gramlich, L. McCusker, T. Weber and P. Pattison, Cryst. Growth Des., 2009, 9, 2556–2558 CrossRef CAS.
- A. Sugita, K. Suzuki and S. Tasaka, Chem. Phys. Lett., 2004, 396, 131–135 CrossRef CAS.
- A. Sugita, K. Suzuki, A. Kubono and S. Tasaka, Jpn. J. Appl. Phys., 2008, 47, 1355–1358 CrossRef CAS.
- C. F. Fitié, W. C. Roelofs, M. Kemerink and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 6892–6893 CrossRef PubMed.
- C. F. Fitié, W. C. Roelofs, P. C. Magusin, M. Wübbenhorst, M. Kemerink and R. P. Sijbesma, J. Phys. Chem. B, 2012, 116, 3928–3937 CrossRef PubMed.
- T. Akutagawa, T. Takeda and N. Hoshino, Chem. Commun., 2021, 57, 8378–8401 RSC.
- H. Anetai, Y. Wada, T. Takeda, N. Hoshino, S. Yamamoto, M. Mitsuishi, T. Takenobu and T. Akutagawa, J. Phys. Chem. Lett., 2015, 6, 1813–1818 CrossRef CAS PubMed.
- H. Anetai, K. Sambe, T. Takeda, N. Hoshino and T. Akutagawa, Chem.–Eur. J., 2019, 25, 11233–11239 CAS.
- J. Wu, T. Takeda, N. Hoshino and T. Akutagawa, J. Phys. Chem. B, 2020, 124, 7067–7074 CrossRef CAS PubMed.
- H. Anetai, T. Takeda, N. Hoshino, H. Kobayashi, N. Saito, M. Shigeno, M. Yamaguchi and T. Akutagawa, J. Am. Chem. Soc., 2019, 141, 2391–2397 CrossRef CAS PubMed.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- G. W. Gokel, W. M. Lee and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- T. Akutagawa, T. Hasegawa, T. Nakamura, S. Takeda, T. Inabe, K. Sugiura, Y. Sakata and A. E. Underhill, Chem.–Eur. J., 2001, 7, 4902–4912 CrossRef CAS.
- T. Nakamura, T. Akutagawa, K. Honda, A. E. Underhill, A. T. Coomber and R. H. Friend, Nature, 1998, 394, 159–160 CrossRef CAS.
- C. F. van Nostrum, A. W. Bosman, G. H. Gelinck, P. G. Schouten, J. M. Warman, A. P. M. Kentgens, M. A. C. Devillers, A. Meijerink, S. J. Picken, U. Sohling, A. –J. Schouten and R. J. M. Nolte, Chem.–Eur. J., 1995, 1, 171–182 CrossRef CAS.
- C. F. van Nostrum, S. J. Picken and R. J. M. Nolte, Angew. Chem., Int. Ed., 1994, 33, 2173–2175 CrossRef.
- C. F. van Nostrum, S. J. Picken, A. –J. Schouten and R. J. M. Nolte, J. Am. Chem. Soc., 1995, 117, 9957–9965 CrossRef CAS.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
- B. Soberats, M. Yoshio, T. Ichikawa, H. Ohno and T. Kato, J. Mater. Chem. A, 2015, 3, 11232–11238 RSC.
- T. Ohtake, Y. Takamitsu, K. Ito-Akita, K. Kanie, M. Yoshizawa, T. Mukai, H. Ohno and T. Kato, Macromolecules, 2000, 33, 8109–8111 CrossRef CAS.
- V. Conejo-Rodríguez, C. Cuerva, R. Schmidt, M. Bardají and P. Espinet, J. Mater. Chem. C, 2019, 7, 663–672 RSC.
- P. Staffeld, M. Kaller, P. Ehni, M. Ebert, S. Laschat and F. Giesselmann, Crystals, 2019, 9, 74–93 CrossRef.
- Y. Inokuchi, O. V. Boyarkin, R. Kusaka, T. Haino, T. Ebata and T. R. Rizzo, J. Phys. Chem. A, 2012, 116, 4057–4068 CrossRef CAS PubMed.
- G. X. He, F. Wada, K. Kikukawa, S. Shinkai and T. Matsuda, J. Org. Chem., 1990, 55, 541–548 CrossRef CAS.
- V. Percec, W. D. Cho, G. Ungar and D. J. Yeardley, Chem.–Eur. J., 2002, 8, 2011–2025 CrossRef CAS.
- N. Steinke, W. Frey, A. Baro, S. Laschat, C. Drees, M. Nimtz, C. Hägele and F. Giesselmann, Chem.–Eur. J., 2006, 12, 1026–1035 CrossRef CAS PubMed.
- N. Steinke, M. Jahr, M. Lehmann, A. Baro, W. Frey, S. Tussetschläger, S. Sauer and S. Laschat, J. Mater. Chem., 2009, 19, 645–654 RSC.
- M. Kaller, S. Tussetschlager, P. Fischer, C. Deck, A. Baro, F. Giesselmann and S. Laschat, Chem.–Eur. J., 2009, 15, 9530–9542 CrossRef CAS PubMed.
- M. Kaller, C. Deck, A. Meister, G. Hause, A. Baro and S. Laschat, Chem.–Eur. J., 2010, 16, 6326–6337 CrossRef CAS PubMed.
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef PubMed.
- D. M. Smyth, Ferroelectrics, 1994, 151, 115–124 CrossRef CAS.
- D. V. Deyneko, D. A. Petrova, S. M. Aksenov, S. Y. Stefanovich, O. V. Baryshnikova, S. S. Fedotov, P. C. Burns, M. B. Kosmyna, A. N. Shekhovtsov and B. I. Lazoryak, CrystEngComm, 2019, 21, 1309–1319 RSC.
- H. Sun, H. J. Sohn, O. Yamamoto, Y. Takeda and N. Imanishi, Electro. Chem. Soc., 1999, 146, 1672–1676 CrossRef CAS.
- B. I. Lazoryak, O. V. Baryshnikova, S. Y. Stefanovich, A. P. Malakho, V. A. Morozov, A. A. Belik, I. A. Leonidov, O. N. Leonidova and G. V. Tendeloo, Chem. Mater., 2003, 15, 3003–3010 CrossRef CAS.
- C. Long, W. Ren, Y. Li, L. Liu, Y. Xia and H. Fan, J. Mater. Chem. C, 2019, 7, 8825–8835 RSC.
- Q. Jiang, M. N. Womersley, P. A. Thomas, J. P. Rourke, K. B. Hutton and R. C. C. Ward, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 094102 CrossRef.
- Y. P. Li, Y. H. R. Yang, Q. Zhao, W. C. Song, J. Han and X. H. Bu, Inorg. Chem., 2012, 51, 9642–9648 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Adv. Mater., 2009, 21, 1591–1594 CrossRef CAS.
- C. Held, L. F. Cameretti and G. Sadowski, Fluid Phase Equilib., 2008, 270, 87–96 CrossRef CAS.
- M. Bülow, X. Ji and C. Held, Fluid Phase Equilib., 2019, 492, 26–33 CrossRef.
- H. Anetai, K. Sambe, T. Takeda, N. Hoshino and T. Akutagawa, Chem.–Eur. J., 2019, 25, 11233–11239 CAS.
- J. Wu, T. Takeda, N. Hoshino and T. Akutagawa, J. Phys. Chem. B, 2020, 124, 7067–7074 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental; thermal stability and TG charts; vibrational IR spectra of molecule 1 and M+·(1)·X−; PXRD profiles of M+·(1)·X− salts; temperature-dependent Z′–Z′′ plots of M+·(1)·X− salts; POM images of (3BC)1−x(1)x. See DOI: 10.1039/d1sc03301h |
| This journal is © The Royal Society of Chemistry 2021 |