
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelation-assisted C–C bond activation of biphenylene by gold(I) halides†
Hélène
Beucher‡
 a,
Johannes
Schörgenhumer‡
a,
Estíbaliz
Merino
bc and
Cristina
Nevado
*a
a,
Johannes
Schörgenhumer‡
a,
Estíbaliz
Merino
bc and
Cristina
Nevado
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, Zurich, CH 8057, Switzerland. E-mail: cristina.nevado@chem.uzh.ch
bUniversidad de Alcalá, Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Andrés M. del Río (IQAR), Facultad de Farmacia, Alcalá de Henares, 28805, Madrid, Spain
cInstituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Ctra. de Colmenar Viejo, Km. 9.100, 28034, Madrid, Spain
First published on 28th October 2021
Abstract
A chelation-assisted oxidative addition of gold(I) into the C–C bond of biphenylene is reported here. The presence of a coordinating group (pyridine, phosphine) in the biphenylene unit enabled the use of readily available gold(I) halide precursors providing a new, straightforward entry towards cyclometalated (N^C^C)- and (P^C)-gold(III) complexes. Our study, combining spectroscopic and crystallographic data with DFT calculations, showcases the importance of neighboring, weakly coordinating groups towards the successful activation of strained C–C bonds by gold.
Activation of C–C bonds by transition metals is challenging given their inertness and ubiquitous presence alongside competing C–H bonds.1 Both the intrinsic steric hindrance as well as the highly directional character of the p orbitals involved in the σC–C bond impose a high kinetic barrier for this type of processes.2,3 Biphenylene, a stable antiaromatic system featuring two benzene rings connected via a four-membered cycle, has found widespread application in the study of C–C bond activation. Since the seminal report from Eisch et al. on the oxidative addition of a nickel(0) complex into the C–C bond of biphenylene,4 several other late transition metals have been successfully applied in this context.5 Interestingly, despite the general reluctance of gold(I) to undergo oxidative addition,6 its oxidative insertion into the C–C bond of biphenylene was demonstrated in two consecutive reports by the groups of Toste7a and Bourissou,7b respectively. The high energy barrier associated with the oxidation of gold could be overcome by the utilization of gold(I) precursors bearing ligands that exhibit either a strongly electron-donating character (e.g. IPr = [1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene])7a or small bite angles (e.g. DPCb = diphosphino-carborane).7b,8 In line with these two approaches, more sophisticated bidentate (N^C)- and (P^N)-ligated gold(I) complexes have also been shown to aid the activation of biphenylene at ambient temperature (Scheme 1a).7c,d
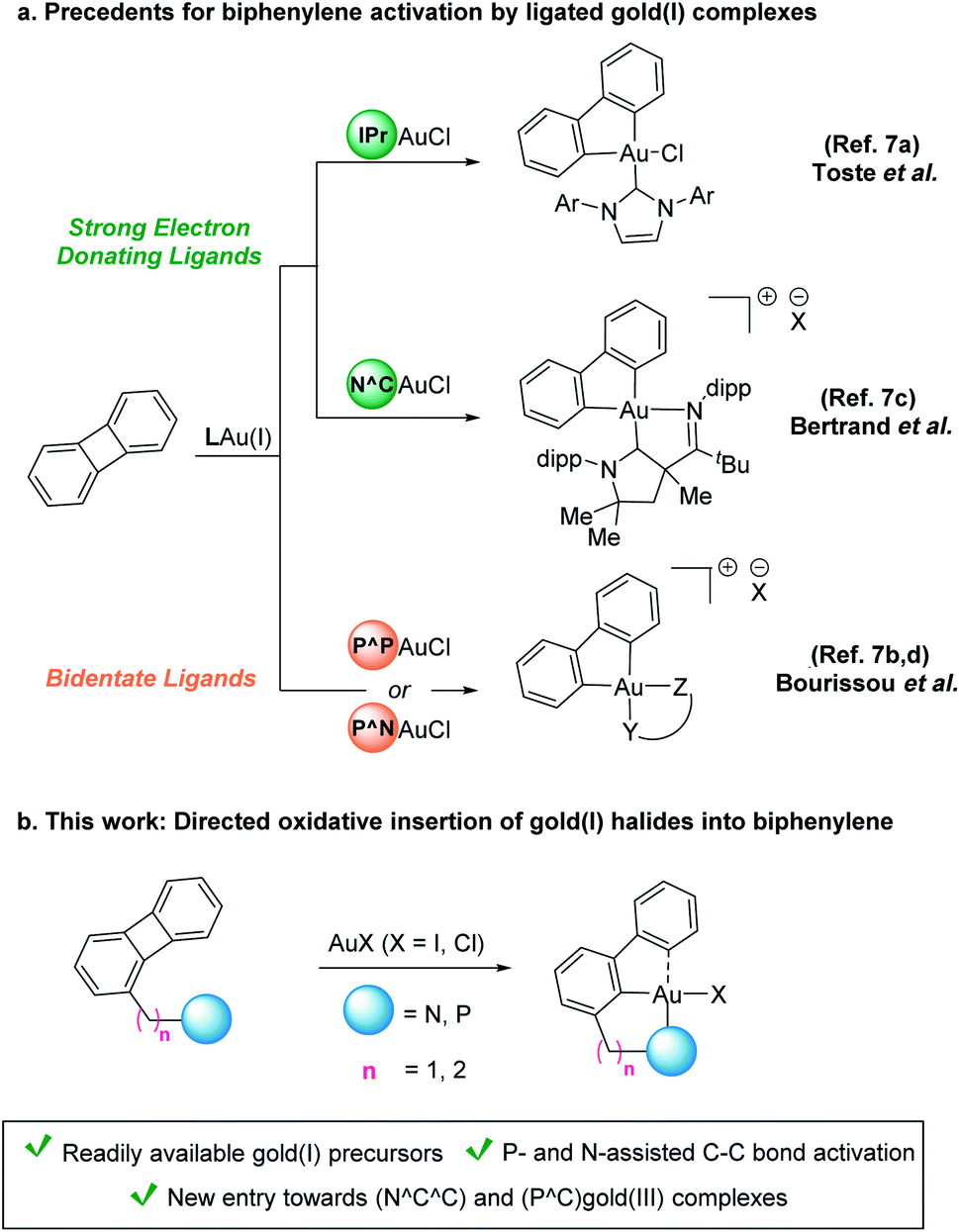 | ||
| Scheme 1 (a) Previous reports on oxidative addition of ligated gold(I) precursors onto biphenylene. (b) This work: pyridine- and phosphine-directed C–C bond activation of biphenylene by commercially available gold(I) halides. | ||
In this context, we hypothesized that the oxidative insertion of gold(I) into the C–C bond of biphenylene could be facilitated by the presence of a neighboring chelating group.9 This approach would not only circumvent the need for gold(I) precursors featuring strong σ-donor or highly tailored bidentate ligands but also offer a de novo entry towards interesting, less explored ligand templates. However, recent work by Breher and co-workers showcased the difficulty of achieving such a transformation.10
Herein, we report the oxidative insertion of readily available gold(I) halide precursors into the C–C bond of biphenylene. The appendage of both pyridine and phosphine donors in close proximity to the σC–C bond bridging the two aromatic rings provides additional stabilization to the metal center and results in a de novo entry to cyclometalated (N^C^C)- and (P^C)gold(III) complexes (Scheme 1b).
Our study commenced with the preparation of 5-chloro-1-pyridino-biphenylene system 2via Pd-catalyzed Suzuki cross coupling reaction between 2-bromo-3-methylpyridine and 2-(5-chlorobiphenylen-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 1 (Scheme 2).11 To our delight, the reaction of 2 with gold(I) iodide in toluene at 130 °C furnished complex κ3-(N^C^C)Au(III)–I 3 in 60% yield.12,13 Complex 3 was isolated as yellow plate-type crystals from the reaction mixture and its molecular structure was unambiguously assigned by NMR spectroscopy, high-resolution mass spectrometry (HR-MS) and crystallographic analysis. Complex 3 exhibits the expected square-planar geometry around the metal center, with a Au–I bond length of 2.6558(3) Å.14 The choice of a neutral weakly bound gold(I)-iodide precursor is key for a successful reaction outcome: similar reactions in the presence of [(NHC)AuCl + AgSbF6] failed to deliver the desired biscyclometalation adducts, as reported by Breher et al. in ref. 10. The oxidative insertion of gold(I) iodide into the four-membered ring of pyridino-substituted biphenylene provides a novel and synthetically efficient entry to κ3-(N^C^C)gold(III) halides. These species have recently found widespread application as precursors for the characterization of highly labile, catalytically relevant gold(III) intermediates,15a–d as well as for the preparation of highly efficient emitters in OLEDs.15e–g Previous synthetic routes towards these attractive biscyclometalated gold(III) systems involved microwave-assisted double C–H functionalization reactions that typically proceed with low to moderate yields.15a
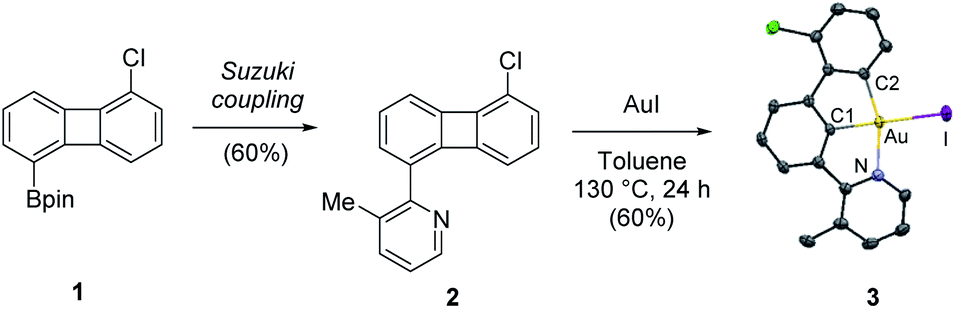 | ||
| Scheme 2 Synthesis of complex 3via oxidative addition of Au(I) into the C–C bond of pyridine-substituted biphenylene. X-ray structures of complex 3 with atoms drawn using 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. Additional selected bond distances [Å]: N–Au = 2.126(2), C1–Au = 1.973(2), C2–Au = 2.025(2), Au–I = 2.6558(3) and bond angles [deg]: N–Au–I = 99.25(6), N–Au–C1 = 79.82(9), C1–Au–C2 = 81.2(1), C2–Au–I = 99.73(8). For experimental details, see ESI.† | ||
Encouraged by the successful results obtained with the pyridine-substituted biphenylene and considering the prominent use of phosphines in gold chemistry,6,16 we wondered whether the same reactivity would be observed for a P-containing system. To this end, both adamantyl- and tert-butyl-substituted phosphines were appended in C1 position of the biphenylene motif. Starting from 5-chlorobiphenylene-1-carbaldehyde 4, phosphine-substituted biphenylenes 5a and 5b could be accessed in 3 steps (aldehyde reduction to the corresponding alcohol, Appel reaction and nucleophilic displacement of the corresponding benzylic halide) in 64 and 57% overall yields, respectively.13 The reactions of 5a and 5b with commercially available gold(I) halides (Me2SAuCl and AuI) furnished the corresponding mononuclear complexes 7a–b and 8a–b, respectively (Scheme 3).13 All these complexes were fully characterized and the structures of 7a, 7b and 8a were unambiguously characterized by X-ray diffraction analysis.13 Interestingly, the nature of the halide has a clear effect on the chemical shift of the phosphine ligand so that a Δδ of ca. 5 ppm can be observed in the 31P NMR spectra of 7a–b (Au–Cl) compared to 8a–b (Au–I), the latter being the more deshielded. The Au–X bond length is also impacted, with a longer Au–I distance (2.5608(1) Å for 8a) compared to that measured in the Au–Cl analogue (2.2941(7) Å for 7a) (Δd = 0.27 Å).13
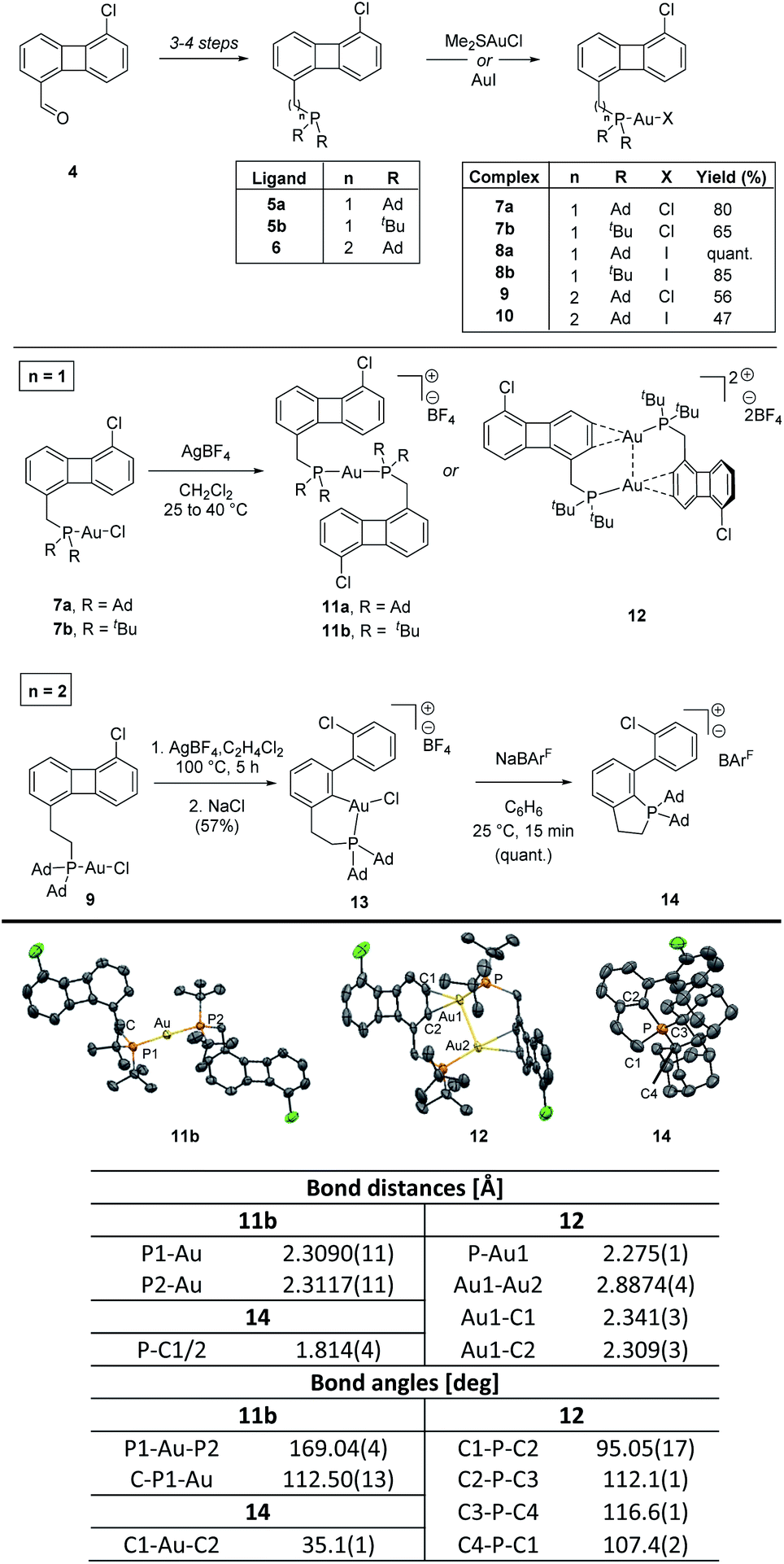 | ||
| Scheme 3 Synthesis and reactivity of complexes 7a–b, 8a–b, 9 and 10. X-ray structure of complexes 11b, 12 and 14 with atoms drawn using 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. For experimental details and X-ray structures see ESI.† | ||
Despite numerous attempts to promote the C–C activation in these complexes,10,13 all reactions resulted in the formation of highly stable cationic species 11a–b and 12, which could be easily isolated from the reaction media. In the case of cationic mononuclear-gold(I) complexes 11, a ligand scrambling reaction in which the chloride ligand is replaced by a phosphine in the absence of a scavenger, a process previously described for gold(I) species, can be used to justify the reaction outcome.17 The formation of dinuclear gold complex 12 can be ascribed to the combination of a strong aurophilic interaction between the two gold centers (Au–Au = 2.8874(4) Å) and the stabilizing η2-coordination of the metal center to the aromatic ring of biphenylene. Similar η2-coordinated gold(I) complexes have been reported but, to the best of our knowledge, only as mononuclear species.18
Taking into consideration the observed geometry of complexes 7a–b in the solid state,13 the facile formation of stable cationic species 11 and 12 and the lack of reactivity of the gold(I) iodides 8a–b, we hypothesized that the free rotation around the C–P bond was probably restricted, placing the gold(I) center away from the biphenylene system and thus preventing the desired oxidative insertion reaction. To overcome this problem, we set out to elongate the arm bearing the phosphine unit with an additional methylene group, introduced via a Wittig reaction from compound 4 to yield ligand 6, prepared in 4 steps in 27% overall yield. Coordination with Me2SAuCl and AuI resulted in gold(I) complexes 9 and 10, respectively (Scheme 3). The structure of 9 was unambiguously assigned by X-ray diffraction analysis and a similar environment around the metal center to that determined for complex 7a was observed for this complex.13
With complexes 9 and 10 in hand, we explored their reactivity towards C–C activation of the four-membered ring of biphenylene.19 After chloride abstraction and upon heating at 100 °C for 5 hours, ring opening of the biphenylene system was observed for complex 9. Interestingly, formation of mono-cyclometalated adduct 13 was exclusively observed (the structure of 13 was confirmed by 1H, 13C, 31P, 19F, 11B and 2D NMR spectroscopy and HR-MS).13 The solvent appears to play a major role in this process, as performing the reaction in non-chlorinated solvents resulted in stable cationic complexes similar to 11.13,20,21 The presence of adventitious water is likely responsible for the formation of the monocyclometalated (P^C)gold(III) complex 13 as when the reaction was carried out in C2H4Cl2 previously treated with D2O, the corresponding deuterated adduct 13-d could be detected in the reaction media. These results showcase the difficulties associated with the biscyclometalation for P-based complexes as well as the labile nature of the expected biscyclometalated adducts. Interestingly though, these processes can be seen as a de novo entry towards relatively underexplored (P^C)gold(III) species.22
The C–C activation was further confirmed by X-ray diffraction analysis of the phosphonium salt 14, which arise from the reductive elimination at the gold(III) center in 13 upon exchange of the BF4− counter-anion with the weakly coordinating sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF).13,23 The phosphorus atom is four-coordinate, with weak bonding observed to the distant counter-anion and a distorted tetrahedral geometry (C1–P–C2 = 95.05(17), C2–P–C3 = 112.1(1), C3–P–C4 = 116.6(1), C4–P–C1 = 107.4(2) deg). These results represent the third example in which the C(sp2)–P bond reductive elimination at gold(III) has been reported.24
Further, it is important to note that, in contrast to the reactivity observed for the pyridine-substituted biphenylene, neither P-coordinated gold(I) iodo complexes 8a, 8b nor 10 reacted to give cyclometalated products despite prolonged heating, which highlights the need for highly reactive cationized gold(I) species to undergo oxidative addition when phosphine ligands are flanking the C–C bond.13
To get a deeper understanding on the observed differences in reactivity for the N- vs. P-based directing groups, ground- and transition-state structures for the oxidative insertion of gold(I) halides in C1-substituted biphenylenes were computed by DFT calculations. The reactions of Py-substituted 2 with AuI to give 3 (I) and those of P-substituted 7a (II) and 9 (III) featuring the cationization of the gold(I) species were chosen as models for comparative purposes with the experimental conditions (Fig. 1 and S1–S10 in the ESI†).25–27 The computed activation energies for the three processes are in good agreement with the experimental data. The pyridine-substituted biphenylene I exhibits the lowest activation barrier for the oxidative insertion process (ΔG‡ = 34.4 kcal mol−1). The reaction on the phosphine-substituted derivatives II and III proved to be, after cationization of the corresponding gold(I) halide complexes (II-BF4, III-BF4) higher in energy (ΔG‡ = 39.6 and 46.3 kcal mol−1 respectively), although the obtained values do not rule out the feasibility of the C–C activation process. The transition state between I and I′ exhibits several interesting geometrical features: (a) the biphenylene is significantly bent, (b) the cleavage of the C–C bond is well advanced (dC–C = 1.898 Å in TSIvs. dC–C = 1.504 Å in I), and (c) the two C and the I atoms form a Y-shape around gold with minimal coordination from the pyridine (dN–Au = 2.742 Å in TSIvs. dN–Au = 2.093 Å in I and 2.157 Å in I′, respectively). The transition-state structures found for the P-based ligands (TSII and TSIII) also show an elongation of the C–C bond and display a bent biphenylene. However, much shorter P–Au distances (dP–Au = 2.330 Å for TSII and 2.314 Å for TSIII) can be observed compared to the pyridine-based system, as expected due to the steric and electronic differences between these two coordinating groups. Analogously, longer C–Au distances were also found for the P-based systems (dC1–Au = 2.152 Å for TSIvs. 2.235 Å and 2.204 Å for TSII and TSIII; dC2–Au = 2.143 Å for TSIvs. 2.219 Å and 2.162 Å for TSII and TSIII), with a larger deviation of square planarity for Au in TSIII compared to TSII.28,29 These results suggest that, provided the appropriate distance to the C–C bond is in place, the strong coordination of phosphorous to the gold(I) center does not prevent the C–C activation of biphenylene but other reactions (i.e. formation of diphosphine gold(I) cationic species, protodemetalation) can outcompete the expected biscyclometalation process. In contrast, a weaker donor such as pyridine offers a suitable balance bringing the gold in close proximity to the C–C bond and enables both the oxidative cleavage as well as the formation of the double metalation product.
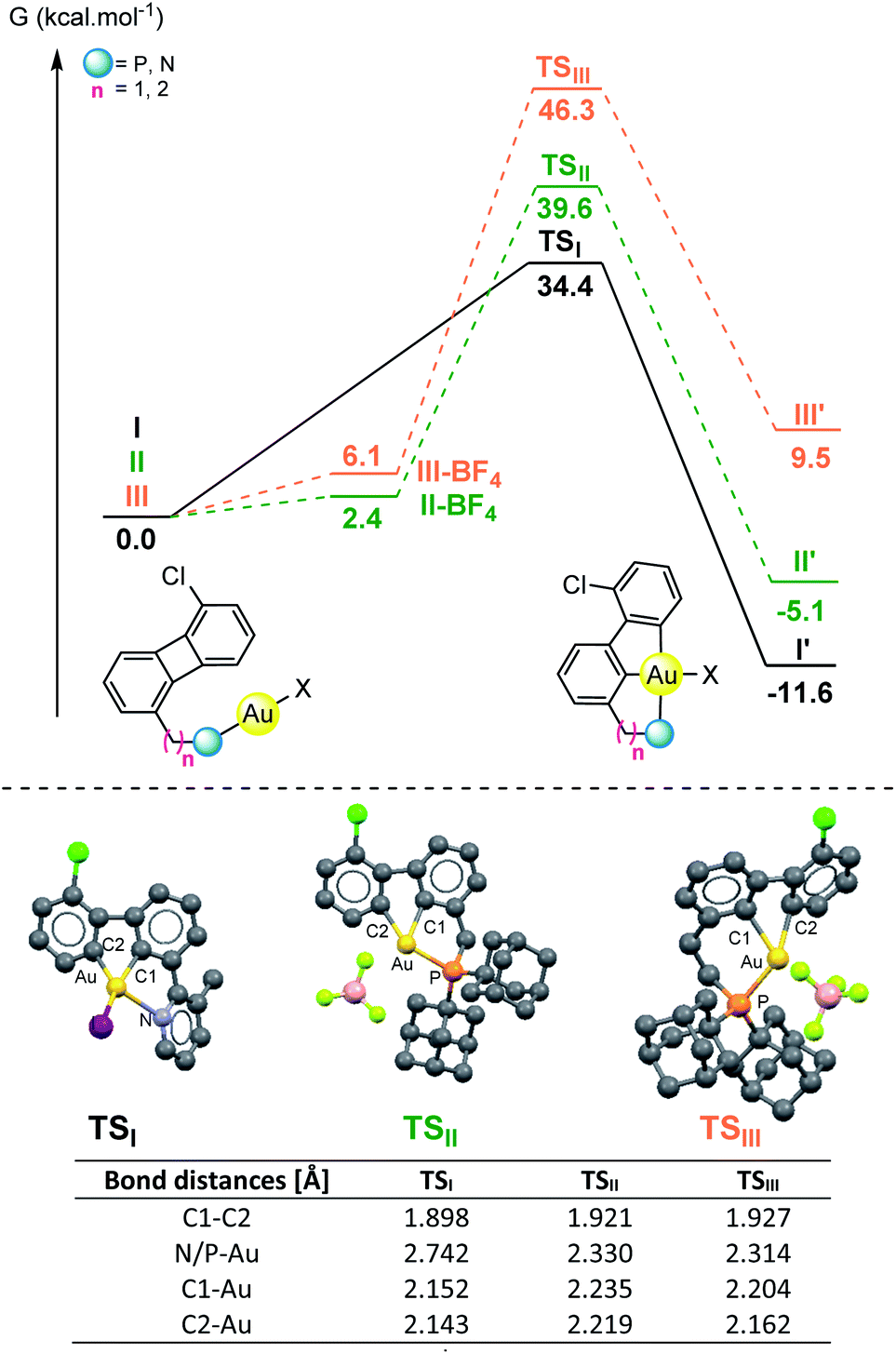 | ||
| Fig. 1 Energy profile (ΔG and ΔG‡ in kcal mol−1), optimized structures, transition states computed at the IEFPCM (toluene/1,2-dichloroethane)-B3PW91/DEF2QZVPP(Au,I)/6-31++G(d,p)(other atoms) level of theory for the C–C activation of biphenylene with gold(I) iodide from I and gold(I) cationic from II and III. Computed structures of the transition states (TSI, TSII and TSIII) and table summarizing relevant distances. | ||
Conclusions
In conclusion, biphenylene derivatives featuring both pyridine and phosphine coordinating groups at C2 position have been synthesized enabling the facile activation of the neighboring σC–C bond via oxidative cyclometalation from readily available gold(I) halide precursors. The reactions provide a new entry towards cyclometalated (N^C^C)- and (P^C)-gold(III) species. Aided by DFT calculations, we demonstrate the importance of the neighboring chelating group, as its coordination properties strongly influence the outcome of the reaction towards the formation of bis- or monocyclometalated complexes.Data availability
Deposition Numbers 2065053 (for 3), 2065060 (for 7a), 2065062 (for 7b), 2065061 (for 8a), 2065065 (for 9), 2065063 (for 11b), 2065064 (for 12) and 2065066 (for 14) contain the supplementary crystallographic data for this paper.Author contributions
Conceptualisation, supervision and writing – C. N.; investigation and methodology – H. B., J. S., E. M., C. N.; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Thomas Fox for his support in the NMR measurements and Prof. Anthony Linden and Dr Olivier Blacque for the X-ray structure determination of compounds 3, 7a, 7b, 8a, 9, 11b, 12 and 14. We are grateful for financial support by the Swiss National Science Foundation (R'Equip grant no. 206021_164018) for an X-ray diffractometer used in this work and the Comunidad de Madrid Research Talent Attraction Program (2018-T1/IND-10054) to E. M. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation.Notes and references
-
(a)
M. Murakami and N. Ishida, in Cleavage of Carbon-Carbon Single Bonds by Transition Metals, ed. M. Murakami and N. Chatani, Wiley-Hoboken, 2015 CrossRef
; (b) P. H. Chen, B. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340 CrossRef CAS PubMed
-
(a) C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Müller, T. Satoh and W. D. Jones, J. Mol. Catal. A: Chem., 2002, 189, 157 CrossRef CAS
- E. M. Siegbahn and M. R. A. Blomberg, J. Am. Chem. Soc., 1992, 114, 10548 CrossRef
- J. J. Eisch, A. M. Plotrotrowski, K. I. Han, C. Krüger and Y. H. Tsay, Organometallics, 1985, 4, 224 CrossRef CAS
- For selected references, see:
(a) C. Perthuisot and W. D. Jones, J. Am. Chem. Soc., 1994, 116, 3647 CrossRef CAS
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed
-
(a) C.-Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449 CrossRef CAS PubMed
- M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed
- For precedents on directed activation of biphenylene, see:
(a) Y. Koga, M. Kamo, Y. Yamada, T. Matsumoto and K. Matsubara, Eur. J. Inorg. Chem., 2011, 2869 CrossRef CAS
- W. Feuerstein and F. Breher, Dalton Trans., 2021, 50, 9754 RSC
- S.-L. Wang, M.-L. Pan, W.-S. Su and Y.-T. Wu, Angew. Chem., Int. Ed., 2017, 56, 14694 CrossRef CAS PubMed
- The introduction of a Me-substituent in meta position of the pyridine ring aimed to improve the solubility of the ligand. The corresponding pyridine-coordinated gold(I) halide intermediate was observed in solution but could not be isolated due to its labile character.
- See ESI† for further details.
- The analogous reaction with AuCl did not furnish the desired cyclometalated adduct, in line with the work reported in ref. 10, and with previous reports showcasing the high energy demand associated to the oxidative addition for AuCl and AuBr species, see: J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed
-
(a) R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287 CrossRef CAS PubMed
-
(a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed
-
(a) G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Betrand, J. Organomet. Chem., 2008, 693, 1674 CrossRef CAS PubMed
-
(a) E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455 CrossRef PubMed
- Even at elevated temperatures, 10 did not show any reactivity, in line with our observations for complexes 8a–b (Section 3 in the ESI†).
- When the reaction was carried out in C2D2Cl4, adduct 13 was also obtained according to HR-MS analysis.
- The influence of the solvent on the activation of biphenylene with cationic gold(I) complexes has been computed and the results can be found in Table S3 in the ESI.†.
- For previous reports on the synthesis of (P^C)gold(III) complexes, see:
(a) M. A. Bennett, K. Hoskins, W. R. Kneen, R. S. Nyholm, P. B. Hitchcock, R. Mason, G. B. Robertson and A. D. C. Towl, J. Am. Chem. Soc., 1971, 93, 4591 CrossRef CAS
- BF4 was also exchanged for other anions, such as triflate, tetraphenylborane and bistriflimide, but no C–P bond reductive elimination was observed in these cases.
-
(a) H. Kawai, W. J. Wolf, A. G. DiPasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587 CrossRef CAS PubMed
- The oxidative insertion of gold(I) complexes (iodide, chloride and cationic) in C1-substituted biphenylenes 2, 7 and 9 has been computed and the results can be found in the ESI.†.
- For previous DFT calculations on the oxidative addition of other metals to biphenylene, see:
(a) Z. Gu, G. B. Boursalian, V. Gandon, R. Padilla, H. Shen, T. V. Timofeeva, P. Tongwa, K. P. C. Vollhardt and A. A. Yakovenko, Angew. Chem., Int. Ed., 2011, 50, 9413 CrossRef CAS PubMed
- The involvement of a Wheland intermediate in the mechanism was also considered, yet no stable structures were found for this pathway.
- For a previous comparison between pyridine and phosphine coordination on the reactivity of gold(III) complexes, see: J. Serra, P. Font, E. D. Sosa Carrizo, S. Mallet Ladeira, S. Massou, T. Parella, K. Miqueu, A. Amgoune, X. Ribas and D. Bourissou, Chem. Sci., 2018, 9, 3832 RSC
- The NBOs for the transition state structures have been calculated and can be found in the ESI (Fig. S12–S14†).
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2065053, 2065060–2065066. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03814a |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |