
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelative 3D cryo X-ray imaging reveals intracellular location and effect of designed antifibrotic protein–nanomaterial hybrids†
J.
Groen
 a,
A.
Palanca
bc,
A.
Aires
d,
J. J.
Conesa
ae,
D.
Maestro
b,
S.
Rehbein
f,
M.
Harkiolaki
g,
A. V.
Villar
bh,
A. L.
Cortajarena
*di and
E.
Pereiro
*a
a,
A.
Palanca
bc,
A.
Aires
d,
J. J.
Conesa
ae,
D.
Maestro
b,
S.
Rehbein
f,
M.
Harkiolaki
g,
A. V.
Villar
bh,
A. L.
Cortajarena
*di and
E.
Pereiro
*a
aMISTRAL Beamline, Experiments Division, ALBA Synchrotron Light Source, Cerdanyola del Valles, 08290 Barcelona, Spain. E-mail: epereiro@cells.es
bInstituto de Biomedicina y Biotecnologia de Cantabria (IBBTEC), University of Cantabria, CSIC, 39011 Santander, Spain
cDepartment of Anatomy and Cell Biology, University of Cantabria, 39011 Santander, Spain
dCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, 20014, Donostia San Sebastian, Spain. E-mail: alcortajarena@cicbiomagune.es
eNational Center for Biotechnology CSIC (CNB-CSIC), Department of Macromolecular Structures, Cantoblanco, 28049 Madrid, Spain
fHelmholtz-Zentrum Berlin für Materialien und Energie, Bessy II, D-12489 Berlin, Germany
gBeamline B24, Diamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
hDepartment of Physiology and Pharmacology, University of Cantabria, Avd. Herrera Oria s/n, Santander, Spain
iIkerbasque, Basque Foundation for Science, 48009 Bilbao, Spain
First published on 19th October 2021
Abstract
Revealing the intracellular location of novel therapeutic agents is paramount for the understanding of their effect at the cell ultrastructure level. Here, we apply a novel correlative cryo 3D imaging approach to determine the intracellular fate of a designed protein–nanomaterial hybrid with antifibrotic properties that shows great promise in mitigating myocardial fibrosis. Cryo 3D structured illumination microscopy (cryo-3D-SIM) pinpoints the location and cryo soft X-ray tomography (cryo-SXT) reveals the ultrastructural environment and subcellular localization of this nanomaterial with spatial correlation accuracy down to 70 nm in whole cells. This novel high resolution 3D cryo correlative approach unambiguously locates the nanomaterial after overnight treatment within multivesicular bodies which have been associated with endosomal trafficking events by confocal microscopy. Moreover, this approach allows assessing the cellular response towards the treatment by evaluating the morphological changes induced. This is especially relevant for the future usage of nanoformulations in clinical practices. This correlative super-resolution and X-ray imaging strategy joins high specificity, by the use of fluorescence, with high spatial resolution at 30 nm (half pitch) provided by cryo-SXT in whole cells, without the need of staining or fixation, and can be of particular benefit to locate specific molecules in the native cellular environment in bio-nanomedicine.
Introduction
The intracellular localization of specific therapeutic agents is a mandatory step towards understanding their mechanisms of action and effects at the cellular level. To this aim, a correlative imaging approach, which delivers information on the cellular ultrastructure and the molecular localization in whole cells under close-to-native conditions, is compulsory. An unambiguous molecular localization will allow, firstly, an understanding of the possible routes used to enter the cell, secondly, how the cell deals with a specific agent, and finally what are the effects induced by the treatment on the ultrastructural morphology, which is a direct reflection of the cellular homeostasis.1 Here we use a novel 3D cryo correlative light and X-ray tomography (CLXT) imaging approach, the protocol of which is described elsewhere,2 towards the investigation of a promising antifibrotic treatment for myocardial fibrosis as a model system. CLXT utilizes the gold standard for sample preparation of cryo-preserved cells, and combines 3D structured illumination microscopy (cryo-3D-SIM) to locate the therapeutic agent through its fluorescence signal, followed by soft X-ray tomography (cryo-SXT) to visualize this specific fluorescence signal in the cell ultrastructural context pinpointing its fate in the whole 3D cellular environment. This novel methodology is not only high-throughput, allowing imaging of cell populations under different conditions and time points, but it also benefits from the broad range of existing fluorescent dyes and proteins, opening its use to a wide range of biological studies.This work focuses on the particular example of cardiac fibrosis, a health condition that negatively influences the progression of many heart diseases and affects millions of people worldwide.3 Regardless of the etiology of the cardiac injury, cardiac fibrosis promotes the pathological increase of collagen deposition leading to heightened myocardial stiffness and dampened cardiac function, which are common features in patients with advanced cardiac failure.4 There is no preventive or curative treatment for cardiac fibrosis, due to the elusive mechanism of fibrosis and the lack of specific targets.5 Although a number of therapeutic agents prescribed for specific cardiovascular disorders have been shown to exert beneficial effects on myocardial fibrosis, in most cases an invasive surgery to save the patients live is needed.3 The transforming growth factor β (TGF-β) is a cytokine that triggers the activation of myocardial fibroblasts, regulating collagen production and deposition in the extracellular matrix.6 Together with the heat shock protein 90 (Hsp90), they participate in the pro-fibrotic signaling cascade promoting cardiac fibrosis.7 Transforming growth factor β (TGF-β) is a cytokine that triggers fibroblast activation, regulating collagen production and deposition in the extracellular matrix. Heat shock protein 90 (Hsp90) participates in the pro-fibrotic signaling cascade triggered by TGF-β promoting cardiac fibrosis.7 TGF-β is an essential cytokine excreted by many cell-types and involved in many important functional processes such as regulation of immune reaction, control of differentiation, regulation of proliferation and participation in inflammation among others.8 Due to these pleiotropic effects, in vivo TGF-β targeting to reduce fibrosis is always accompanied by a large number of side effects. As a result, a possible strategy to reduce fibrosis while avoiding side effects is the inhibition of a key factor involved in its profibrotic signaling cascade, such as Hsp90, which has been shown to play an essential role in TGF-β-mediated collagen production.9
The development of an engineered Hsp90 inhibiting module (CTPR390) based on a tetratricopeptide repeat (TPR) domain has shown great potential inhibiting Hsp90.10–12 In particular, previous studies have demonstrated its effectiveness to reduce myocardial pro-fibrotic events in vitro and in vivo.12 Further studies focused on re-engineering the Hsp90 inhibitory domain by its fusion to a related TPR domain that enables the stabilization of a metallic nanocluster, in this case a gold nanocluster (AuNC).13 This engineered protein–nanomaterial hybrid (Prot-NM) also showed efficient therapeutic activity by reducing the myocardial fibrosis and heart hypertrophy in an animal model of cardiac remodeling.14 In addition, the AuNC has shown to exhibit fluorescence (λexc = 370/λem = 445 nm),13 which can be used for localization, and it has shown to have a positive effect on the internalization of the Prot-NM.14 Herein, we have further investigated its in vitro localization and effect by looking at the cellular response at the ultrastructural level, testing both primary cardiac mouse fibroblast and the NIH-3T3 immortalized embryonic fibroblasts. We aim at unravelling the cellular effects of this promising therapeutic agent, and expect to directly correlate those effects with the Hsp90 inhibition. For this aim, we treated the fibroblasts with two protein hybrid systems, with and without the engineered Hsp90 inhibitory domain (TPR-Hsp90-AuNC and TPR-AuNC, respectively). Then, the treated fibroblasts were cryopreserved and imaged using CLXT.2 Furthermore, these results were complemented by confocal microscopy.
Cryo-3D-SIM at B24 beamline (Diamond Synchrotron, UK)15 was used to locate the Prot-NM in the cellular environment. Due to a sub-optimal laser excitation wavelength (405 nm) for the AuNC, resulting in low fluorescence emission, the Prot-NM was tagged with the fluorophore Alexa-488 (see S1†). Furthermore, to position the fluorescence signal in the context of the full cellular structure, cryo-SXT16,17 in the water window energy range was subsequently used18–26 at the Mistral beamline (Alba Synchrotron, Spain).27,28 The data collected with cryo-SXT was in addition used to obtain valuable information about specific organelle morphology and number which are an indication of cellular homeostasis.1 Indeed, the mitochondrial homeostasis in healthy cells is maintained through dynamic and tightly regulated fission and fusion events. Therefore, a change in the mitochondrial morphology and number gives insights into the overall health of the cell.29
The combination of cryo-3D-SIM and cryo-SXT is a novel and powerful correlative imaging approach15,30 which enables linking cellular function and structure with high spatial resolution under close-to-native conditions. Both techniques are high throughput and easy to use (e.g. same sample support and no intensive sample preparation needed) although 3D data correlation requires an appropriate fiducialisation strategy.2 Note that a full workflow under cryogenic conditions maximizes efficiency by maintaining sample integrity and quality between the microscopy modalities.
Herein, we investigated the intracellular fate of these two Prot-NM, with and without the Hsp90 binding domain, accurately locating them within specific cellular organelles while describing related clear morphological effects which have been found to be cell type-dependent, concluding that primary mouse fibroblasts are more resilient to treatment with Prot-NM than NIH-3T3 cells.
Results
Cryo-3D-SIM confirms the presence of the protein–nanomaterial hybrid in treated cells
CLXT was used to image four groups of both primary cardiac mouse fibroblasts and NIH-3T3 cells: the control group, TGF-β activated group, TGF-β activated and TPR-Hsp90-AuNC treated group (TPR-Hsp90-AuNC group), and finally TGF-β activated and TPR-AuNC treated group lacking the Hsp90 inhibitory domain (TPR-AuNC group). Comparing previous studies, it became apparent that the presence of the metal cluster on the Prot-NM has a positive effect on the internalization and the final therapeutic effect of the Hsp90 inhibitory activity.12–14 Considering this, only hybrid formulations with AuNCs were selected for evaluation.In the case of the TPR-Hsp90-AuNC group, the Alexa-488 specific signal was intense and well-defined in specific intracellular locations, confirming that the observed fluorescence signal corresponded indeed to the internalized TPR-Hsp90-AuNC as no equivalent signal was found in the other groups. Nonetheless, some low intensity auto-fluorescence signal was observed in the control and the TGF-β groups at both excitation wavelengths (405 nm and 488 nm) (see S1, S2 and S3†). In the case of the TPR-AuNC group a low but distinct signal was found (S2 and S3†) indicating the presence of TPR-AuNC at significantly lower concentrations than TPR-Hsp90-AuNC in the cell. The comparison of the maximum fluorescence signal value collected between the treatment groups showed a slight increase of the TPR-AuNC signal compared to the autofluorescence signal of the control (2-fold) and TGF-β (1.5-fold), and a 6-fold increase in value for the TPR-Hsp90-AuNC compared to the TPR-AuNC (see S3†). After cryo-3D-SIM imaging, cryo-SXT was performed on the same cells at the Mistral beamline (Fig. 1).
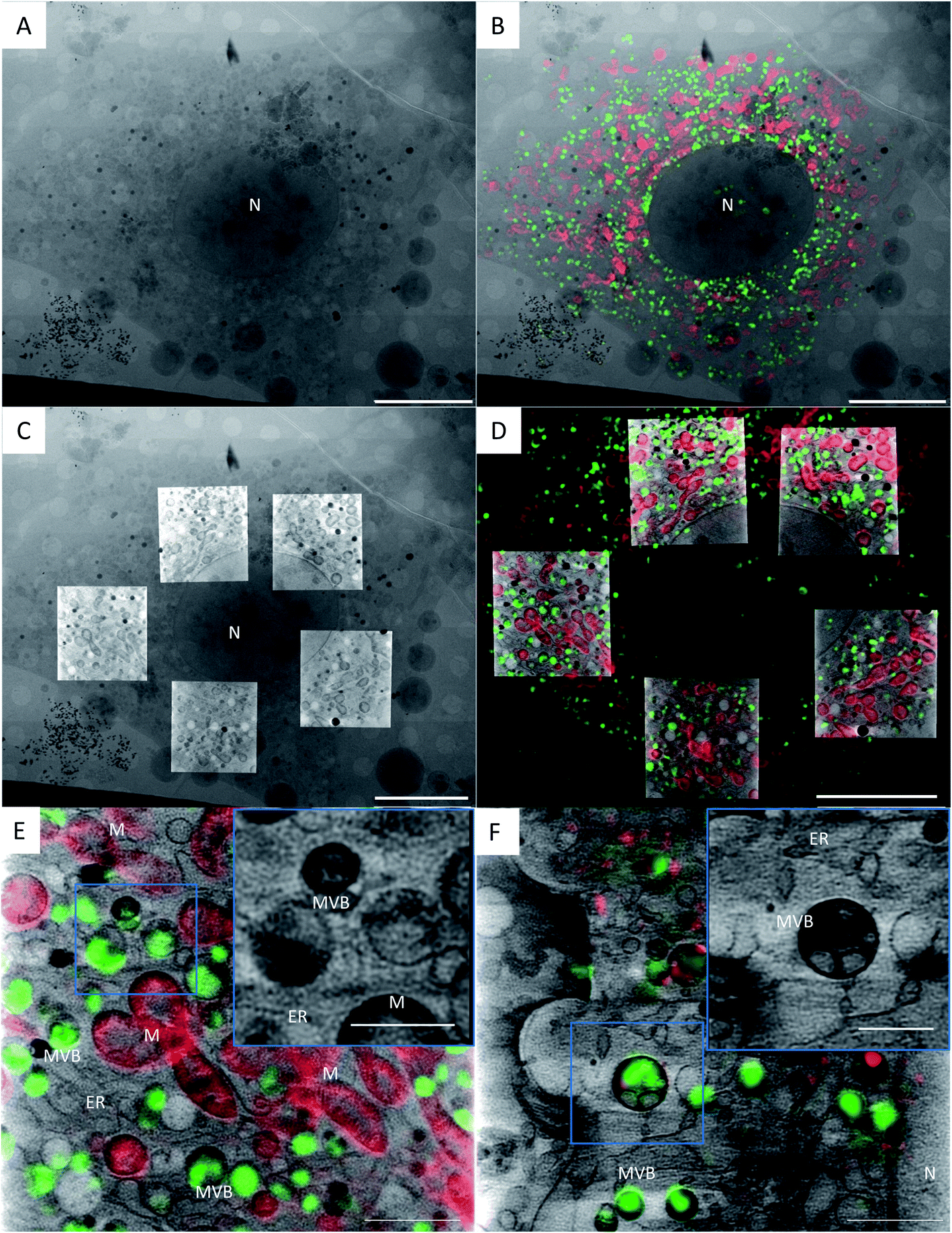 | ||
| Fig. 1 Correlation of a TPR-Hsp90-AuNC treated NIH-3T3 cell using cryo-3D-SIM showing the mitochondria in red (cmxros mitotracker) and the TPR-Hsp90-AuNC in green (alexa-488), and cryo-SXT showing the reconstructed linear absorption coefficient. (a) X-ray projection mosaic; (b) cryo-SIM signal projection overlaid on X-ray mosaic; (c) single slice of the tomographic reconstructions of 5 areas overlaid on the X-ray mosaic; (d) single slice of the tomographic reconstruction overlaid with a single slice of the cryo-3D-SIM, see S5† for the correlation accuracy map. Scale bar: (a–d) 10 μm; bottom row: single slice from a single tomographic reconstruction showing the reconstructed absorbance from SXT overlaid with the cryo-SIM signal from TPR-Hsp90-AuNC (green) coming from MVBs and from the mitochondria (red) in NIH-3T3 (E) and cardiac primary mice fibroblasts (F). Scale bar: 2 μm; inset 1 μm. N = nucleus, M = mitochondria, MVB = multivesicular bodies, ER = endoplasmic reticulum. | ||
Cryo-SXT provides ultrastructural information of the cell and places the fluorescence signal unambiguously within multivesicular bodies
Fig. 1 presents the correlated data of a TPR-Hsp90-AuNC treated NIH-3T3 cell. Fig. 1A shows the 2D X-ray projection mosaic (i.e. tile-scanned projections covering a grid square) of the full cell and Fig. 1B its overlay with the 2D SIM data projection. The red fluorescence signal corresponds to the mitotracker Red CMXRos (ThermoFisher) and the green to the Alexa488-labelled TPR-Hsp90-AuNC. For this cell, five areas within the cytoplasm surrounding the nucleus were chosen for tomography collection, and a single slice of the resulting combined reconstructions is shown in Fig. 1C, superimposed on top of the X-ray 2D mosaic. In Fig. 1D a single slice from the correlated 3D datasets is shown (see S4† for the correlation accuracy map and for a video of the whole 3D stack). No laser damage was noticed at the achievable resolution of cryo-SXT (30 nm half pitch according to the FSCe/o criterion, see S5†)19,31–33 after cryo-3D-SIM. Correlating both 3D datasets, the TPR-Hsp90-AuNC signal was unambiguously located in multivesicular bodies (MVBs) which were homogeneous in size (0.5–1 μm in diameter) and distributed throughout the cytoplasm (Fig. 1B, D and 2). The calculated correlation accuracy map shows an accuracy down to 70 nm although this will vary within the volume region and depends directly on the fiducialisation strategy chosen14 as shown in S4.† These MVBs showed vesicle-like compartments with different morphologies and higher linear absorption coefficient in the case of primary mouse fibroblasts, which might indicate a different mechanism of processing this TPR-Hsp90-AuNC accumulation by the cell (see Fig. 1 bottom and S6† for additional information on MVB morphologies in both cell types). Note that cryo-SXT directly reconstructs the linear absorption coefficient value of the elemental composition within each voxel, hence being quantitative and therefore allowing direct comparison.24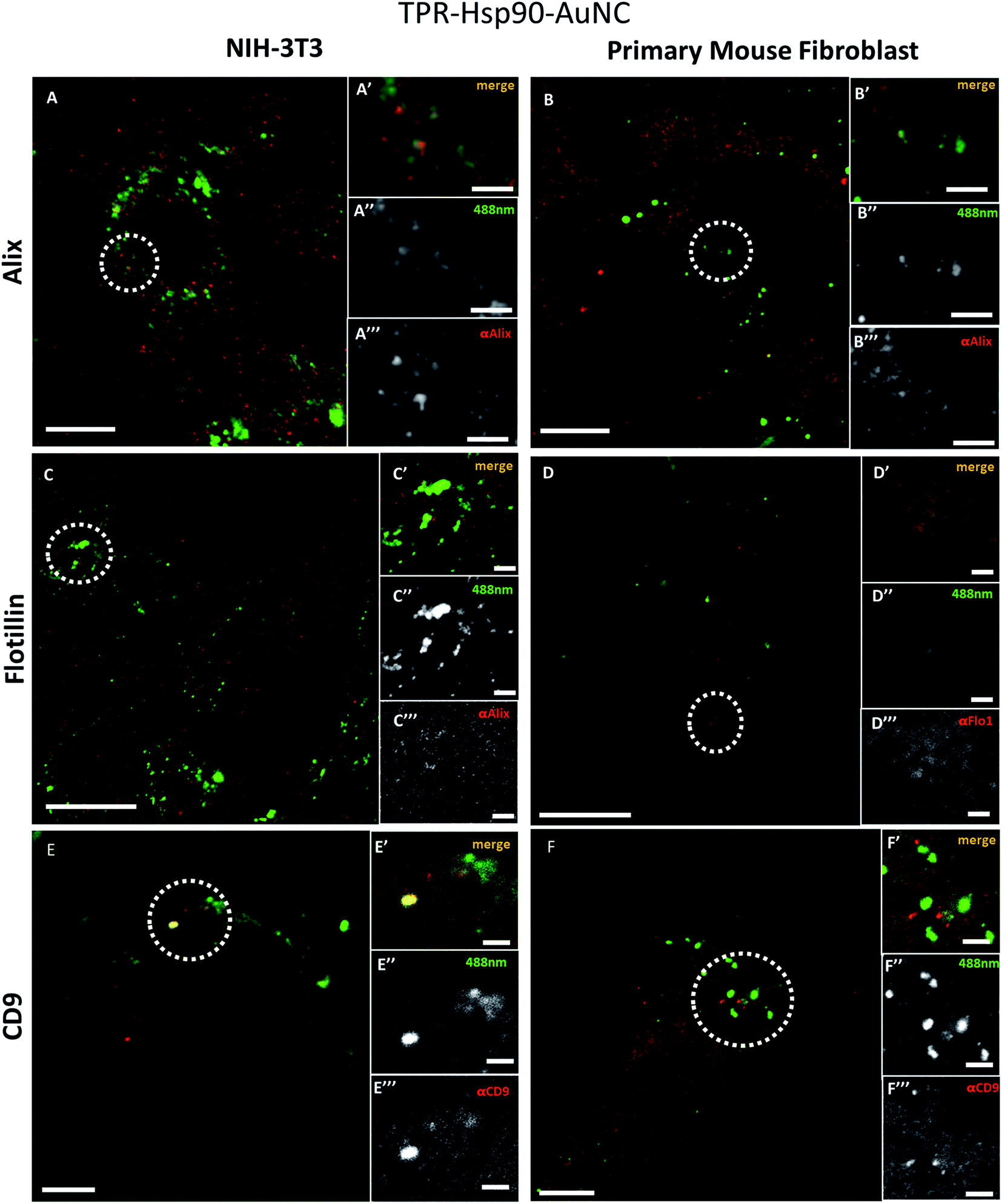 | ||
| Fig. 2 TPR-Hsp90-AuNC co-localization study on NIH-3T3 and primary mice fibroblasts, respectively, of anti-alix (a and b), anti-flotillin (c and d) and anti-cd9 (e and f) using confocal microscopy. The inset shows the merged (′), green (alexa488, ′′) and red (from the respective antibody, ′′′) signals. Scale bars: (a; b; e; and f) 5 μm; (c and d) 3 μm; insets for (a; b; c and d), 1 μm; insets for (e and f) 2 μm. | ||
In order to evaluate the number and size of MVBs observed upon the different treatments, we segmented the volumes using Amira software34 (see Fig. 4). Even though no direct comparison can be made between cell types due to the larger size of the primary cells compared to NIH-3T3, the appearance and tendencies observed in the two can be compared. When evaluating the TGF-β-treated groups, both cell types seem very similar to the control group, having an almost identical average and slightly decreased median MVB volume, the latter being more evident in NIH-3T3 than in primary fibroblasts. This result suggests that these MVBs are part of a normal cell function and not related or affected by stress-induction. In the case of cells treated with TPR-Hsp90-AuNC, a large increase in MVB number was found with respect to the control group (85% increase for NIH-3T3 and 74% for primary mouse fibroblasts, see Fig. 4). When focusing on the primary fibroblasts a non-significant decreased volume was observed. Together with the increase in MVB number, these results suggest the recent formation of new vesicles upon the treatments. Regarding the NIH-3T3 cells, the vesicles present an increase in volume, although non-significant, suggesting that these vesicles were formed early after incorporation of the therapeutic agent and matured by the time of vitrification. Finally, the samples treated with TPR-AuNC showed a slight increase in the number of MVBs compared to the untreated control (29% for NIH-3T3 and 11% for primary fibroblasts) and TGF-β treated cells, although not significant as overall numbers are very low. However, in both cell types this group presented the smallest average and median volume for the MVBs (t ≤ 0.05 for NIH-3T3 and t ≤ 0.01 for primary fibroblasts). Ultimately, with the exception of the TPR-Hsp90-AuNC samples, the MVBs evaluated in both cell types were very similar when the same treatment is compared.
Confocal microscopy reveals the nature of the organelle containing the therapeutic agent
To determine which type of vesicles contains the TPR-Hsp90-AuNCs and understand their role, different biomarkers were tested to evaluate their spatial association with the Alexa488 signal. The selected targets were: Alix, Flotillin-1, CD9 and LC3-II, all associated with different types of cellular vesicles as explained below. All assays were performed on cells fixed with paraformaldehyde (PFA) and imaged by confocal microscopy.Fig. 3 shows the comparison of the co-localization of Alix (Fig. 2A and B), Flotillin-1 (Fig. 2C and D), and CD-9 (Fig. 2E and F) with TPR-Hsp90-AuNC in NIH-3T3 and primary fibroblasts. Both anti-Alix and anti-Flotillin-1 are markers for MVB biogenesis while anti-CD9 (anti-tetraspanin CD9) is associated with adhesion, motility, membrane fusion, signal trafficking vesicles and cargo transfer from cellular vesicles.35,36 In NIH-3T3 cells a spatial co-localization (at the achievable confocal resolution) of TPR-Hsp90-AuNC with anti-Alix and anti-CD9 was observed. In addition, a low anti-Flotillin-1 signal was detected and there was no spatial association with the therapeutic molecule. In the case of primary mouse fibroblasts, some spatial association was observed with anti-Alix, although the signal was lower compared to the NIH-3T3 cells. Similar to NIH-3T3, some signal was detected from anti-Flotillin-1, although it appeared diffused. Finally, some foci were observed from anti-CD9, but there was no association with the TPR-Hsp90-AuNC. With the above observations, the two cell types appear to show the same tendency with the exception of the CD9 biomarker that showed co-localization with TPR-Hsp90-AuNC in NIH-3T3 and not in primary fibroblasts.
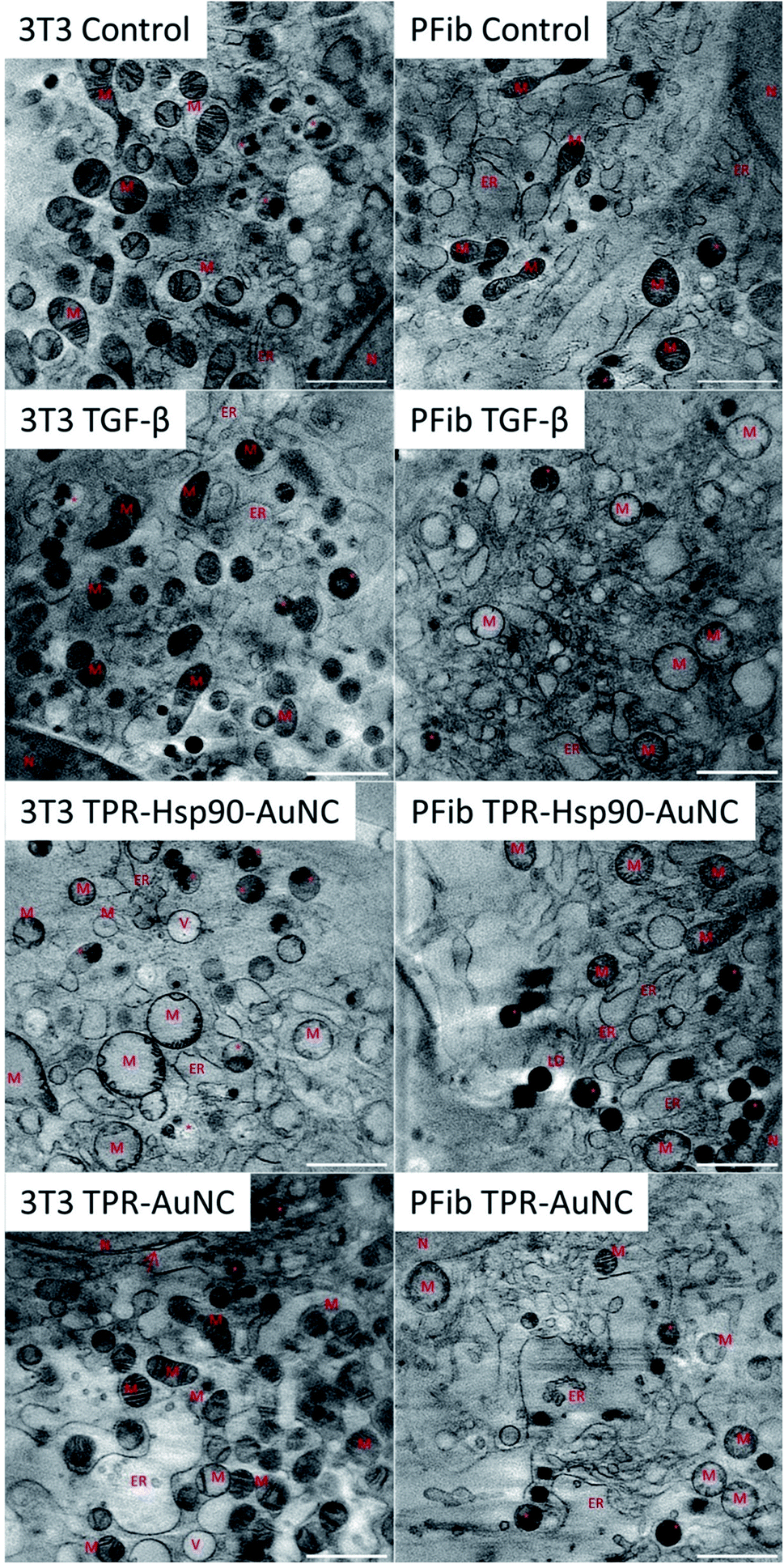 | ||
| Fig. 3 Snapshot of a representative tomographic reconstruction for each sample showing the reconstructed absorbance value to compare the morphology of the cell after treatment. * = MVB; M = mitochondria; N = nucleus, ER = endoplasmic reticulum, V = vacuole, LD = lipid droplet, arrow = nuclear double membrane. Scale bar: 2 μm. | ||
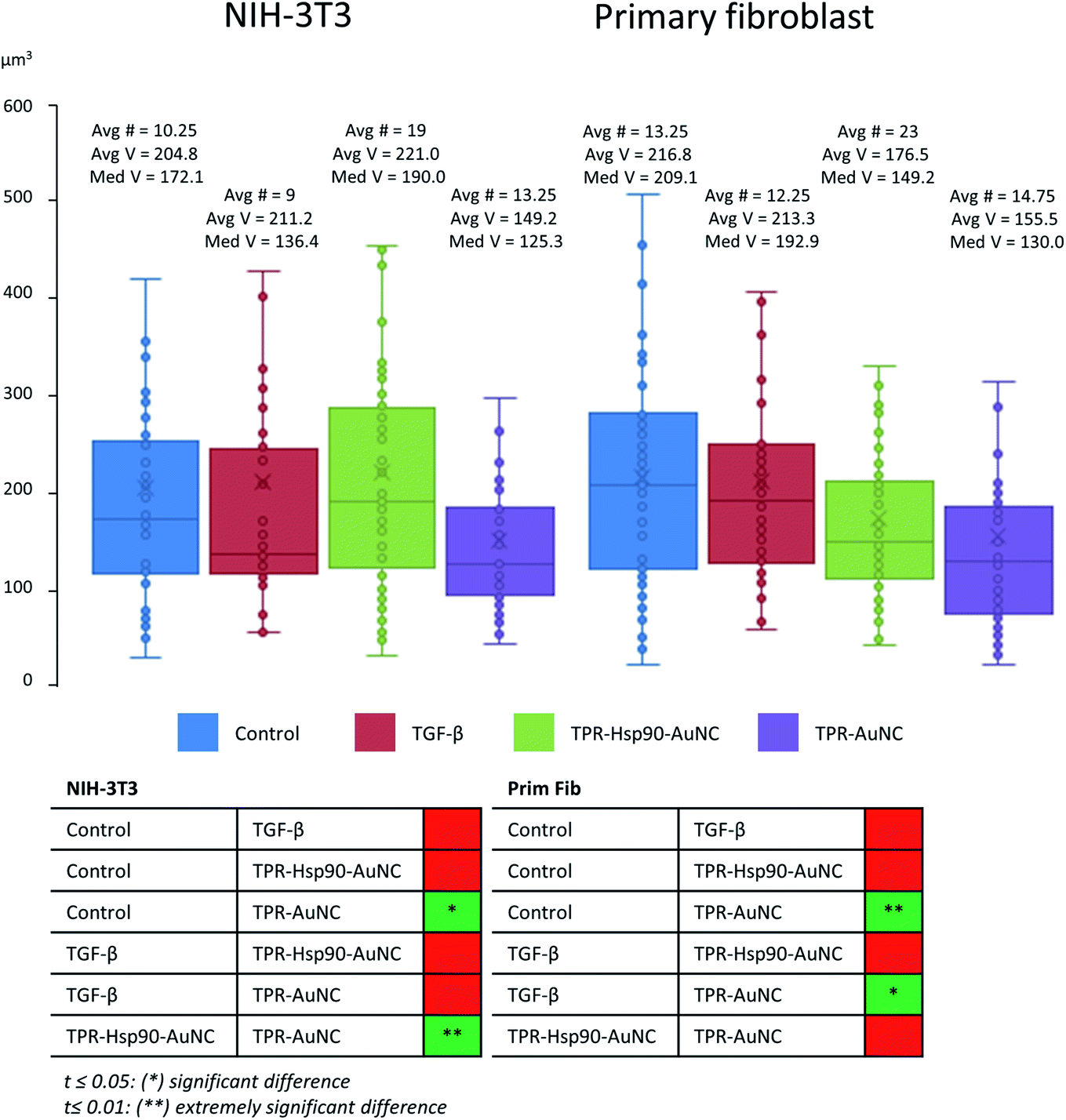 | ||
| Fig. 4 Comparison of MVB volumes of both NIH-3T3 and primary fibroblast cells measured after segmentation using Amira.34 Avg # represents the average number of segmented MVBs in a tomogram, Avg V stands for average volume and Med V stands for median volume (both in μm3). the measured data were tested for variance and the corresponding t-test was used to determine statistically relevant differences between groups, the results of which are shown in the table below. | ||
These biomarkers were also tested upon TPR-AuNC treatment (S7†). The TPR-AuNC group showed the same associations with anti-Alix for both cell types and a co-localization with anti-CD9 in NIH-3T3, however none in primary fibroblasts similar to TPR-Hsp90-AuNC. Moreover, and in contrast to TPR-Hsp90-AuNC, a clear spatial association between Flotillin-1 and TPR-AuNC was observed, especially in primary fibroblasts. Few foci were also observed in NIH-3T3 cells.
The last biomarker, anti-LC3-II, was chosen to test for autophagy and degradation via autophagosomes (S8†). A small number of foci were detected in both cell types for both treatments; however, only in NIH-3T3 cells treated with TPR-AuNC an association was observed. Overall, these results ruled out the conjecture that TPR-Hsp90-AuNC-containing MVBs were involved in the autophagosome pathway. The possibility of an enhancement of programmed cell-death apoptotic events caused by stress upon treatment was also tested using the activated caspase-3 (cleaved caspase-3 Asp175).37 No increments were seen for TPR-AuNC, TPR-Hsp90-AuNC, or TGF-β treated cells, which indicates that TGF-β did not induce apoptosis at the concentration used in this study. Moreover, the control cells were also labelled and imaged and showed no foci with any of the tested biomarkers (S9†).
Cryo-SXT enables a statistical analysis of the observed morphological differences upon treatment
In addition to elucidating the intracellular fate of Prot-NM, cryo-SXT allowed evaluating possible changes in the cellular ultrastructure upon treatment (Fig. 3). Of special interest was the mitochondrial morphology as their appearance is directly related to the cellular health, as already mentioned. In order to obtain statistical information about morphological changes of the different cell groups and to understand the effect of the treatment, the mitochondria were also segmented using Amira software.34 This segmentation allowed evaluating the number, volume and shape dependence of these organelles for each treatment with respect to the control cells.In the case of the NIH-3T3 cells, the TGF-β group presented an increased number of mitochondria (78% more compared to the control) with smaller volume and increased elongation (see Fig. 3 and 5). This indicates that the cells are both increasing their mitochondria surface area and are producing more mitochondria for upcoming changes. Previous work showed that treatment with TGF-β leads to differentiation or activation of the cell to increase collagen production,38 both being highly energy consuming processes. This confirms that the TGF-β treatment promotes the desired cellular pro-fibrotic events and that the changed mitochondrial morphology is a natural reaction to the activation with TGF-β. The TPR-Hsp90-AuNC group showed a smaller number of mitochondria (44% less compared to the control) with increased volume. This type of mitochondrial swelling is usually associated with cellular stress.39 In the case of TPR-AuNC, a similar effect on the cells to that of the TGF-β group was expected. Indeed, similar mitochondrial volume but with a slight increase in the number of mitochondria (20%) was observed. However, the mitochondrial elongation present in the TGF-β group was not observed.
 | ||
| Fig. 5 Comparison of mitochondrial volumes of both NIH-3T3 and primary mouse fibroblast cells, measured after segmentation using Amira.34 Avg # represents the average number of segmented mitochondria in a tomogram, Avg V stands for average volume and Med V stands for median volume (both in μm3). The measured data were tested for variance and the corresponding t-test was used to determine statistically relevant differences, the results of which are shown in the table below. | ||
When analyzing the cardiac primary mouse fibroblast, significant differences compared to the NIH-3T3 cells were observed (see Fig. 3 and 5). First, the number of mitochondria was similar in all four groups (see Fig. 5), although a decrease of 20% compared to the control is found in the TPR-Hsp90-AuNC group, following the same tendency as that for NIH-3T3. Second, the mitochondrial volume was also similar for all the groups except the TGF-β one which showed a significantly higher mean volume compared to the control. This can be explained by the increased energy demand, similar to the observed effect in NIH-3T3 cells. In terms of sphericity, there were significant differences between most of the groups. In contrast to what was seen in the NIH-3T3 cells, the control group had the most diverse shapes consistent with the expected continuous fission and fusion of mitochondria.29 For both TGF-β and the TPR-AuNC groups, mitochondria appeared rather spherical, while the TPR-Hsp90-AuNC group showed slightly greater variety of shapes, but still leaning towards spherical mitochondria. Mitochondria morphological changes due to fission and fusion are beneficial under conditions of high-energy demand40 as in the case of TGF-β induced collagen production in fibroblasts. Note that apart from the control, all groups have been ‘activated’ with TGF-β and therefore higher mitochondrial activity demand was expected.
Even though no direct comparison of the numbers between cell types can be done, as primary mouse fibroblasts are larger than NIH-3T3 cells, tendencies and the mitochondria appearance can be compared. Primary mouse fibroblasts seemed overall more stable, as no large divergence from the control was measured, although a significant reaction towards the activation with TGF-β was observed. In contrast, each treatment in NIH-3T3 cells causes a unique and seemingly extreme reaction. A significant reduction in mitochondrial size combined with increased number suggests mitochondrial fission events, which play a critical role in the attenuation of mitochondrial damage.41 In addition, the presence of many small, rounded mitochondria points to a stationary phase of the cell with low activity.42 These findings already suggest that the NIH-3T3 cells experience more difficulties when dealing with a sudden change in cellular homeostasis, as in the case of TGF-β activation. Furthermore, the mitochondrial swelling observed in the TPR-Hsp90-AuNC groups, which is a clear indicator of oxidative stress,43 supports this hypothesis. These results demonstrate that primary mouse fibroblasts are more resilient to an altered cellular homeostasis which is in this case induced by both TGF-β and the nanomaterial treatment compared to the immortalized embryonic NIH-3T3 fibroblasts.
It is worth mentioning that no structural changes have been observed in the cell nuclei, lipid droplets or vacuole numbers whereas in the case of the ER, some opening was observed in the groups treated with Prot-NM for both cell types with respect to their control cells. This type of ER enlargement is normal in cardiac fibroblasts in which fibrosis events have been triggered, as is also stimulated with the TGF-β treatment. This effect was already shown previously12 and was here confirmed. However, in the NIH-3T3 TPR-AuNC group this effect appeared even larger than for the TGF-β group (Fig. 3).
Finally, in order to confirm the findings of previous studies12 that this Prot-NM can indeed inhibit collagen production, NIH-3T3 cells were incubated for 48 h applying the same treatment groups and imaged. Note that, to this aim, tomograms were collected in extracellular areas between cells, where collagen bundles are expected. As was previously described for a similar Hsp90 inhibitory molecule (CTPR390-488),12 some collagen fibers were found in the TPR-Hsp90-AuNC group. However, these fibers seemed less structured and in lower quantity than for the TGF-β group, which presented clearly defined collagen bundles as expected (see S10†). Therefore, a complete inhibition of extracellular collagen production in NIH-3T3 cells was not achieved upon treatment, but a substantial reduction was indeed visible.
Discussion
Alix is an accessory protein of the endosomal sorting complex required for transport and has been related to the biogenesis of trafficking vesicles, such as extracellular vesicles.44 The cellular trafficking-related protein Flotillin-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 has been related to endocytosis and endosomal trafficking events. In addition, lipid raft-dependent endocytosis can be mediated by Flotillin-1 that would be recruited to the surface forming pre-endocytic clusters.45 TPR-Hsp90-AuNC did not show co-localization with Flotillin-1, while TPR-AuNC did. The lack of association for the first could suggest that the protein hybrid does not enter via endocytosis. The ability of TPR-Hsp90-AuNC to bind to extracellular Hsp9012 (may allow it to enter the cell bound to this chaperone through a different route than TPR-AuNC. This mechanism would also explain the lower intracellular concentration of TPR-AuNC as an active transport via Hsp90 naturally results in higher intracellular concentrations compared to a passive, endocytosis-dependent incorporation. After uptake of the nanomaterials, trafficking vesicles presenting Alix, which showed association with both Prot-NM, could then be the main transport route. The difference concerning the association with CD9 for both nanomaterials, observed only in NIH-3T3 cells, can be related to exocytosis events and may suggest a different cell-dependent way of dealing with the Prot-NM. This hypothesis is in line with our observation of an increased absorbance of the MVBs in primary fibroblasts compared to NIH-3T3 cells (Fig. 1 bottom and S6†) by cryo-SXT. Overall, these results suggest that the MVBs are mainly related to endosomal trafficking events, however the origin of the endosomal trafficking seems to differ depending, not only on cell type, but also on the presence of the Hsp90 binding module.
45 has been related to endocytosis and endosomal trafficking events. In addition, lipid raft-dependent endocytosis can be mediated by Flotillin-1 that would be recruited to the surface forming pre-endocytic clusters.45 TPR-Hsp90-AuNC did not show co-localization with Flotillin-1, while TPR-AuNC did. The lack of association for the first could suggest that the protein hybrid does not enter via endocytosis. The ability of TPR-Hsp90-AuNC to bind to extracellular Hsp9012 (may allow it to enter the cell bound to this chaperone through a different route than TPR-AuNC. This mechanism would also explain the lower intracellular concentration of TPR-AuNC as an active transport via Hsp90 naturally results in higher intracellular concentrations compared to a passive, endocytosis-dependent incorporation. After uptake of the nanomaterials, trafficking vesicles presenting Alix, which showed association with both Prot-NM, could then be the main transport route. The difference concerning the association with CD9 for both nanomaterials, observed only in NIH-3T3 cells, can be related to exocytosis events and may suggest a different cell-dependent way of dealing with the Prot-NM. This hypothesis is in line with our observation of an increased absorbance of the MVBs in primary fibroblasts compared to NIH-3T3 cells (Fig. 1 bottom and S6†) by cryo-SXT. Overall, these results suggest that the MVBs are mainly related to endosomal trafficking events, however the origin of the endosomal trafficking seems to differ depending, not only on cell type, but also on the presence of the Hsp90 binding module.
Cryo-SXT allowed us to draw conclusions regarding the reaction of the cell towards the treatment. The immortalized NIH-3T3 fibroblasts showed clear signs of stress in the form of mitochondrial swelling, likely due to the TPR-Hsp90-AuNC treatment. This swelling event was not observed for TPR-AuNC, which suggests a direct link between the molecular changes promoted by the binding of the hybrid functional protein to the C-terminal end of Hsp90,14 although the lower uptake could also explain this lack of visible swelling. Other studies have already reported that stress, especially oxidative stress, induces mitochondrial swelling, which would support our first hypothesis.43 Whether the stress is induced only by the Hsp90 inhibition46,47 or also because of the presence of the AuNC48,49 is therefore still to be answered.
Both cell types were affected by a low intracellular TPR-AuNC concentration, as shown by the morphological differences in mitochondria shape and number, as well as in the extra ER enlargement, especially in NIH-3T3 cells. This type of ER enlargement is normal in cardiac fibroblasts in which fibrosis events have been triggered as shown previously12 and confirmed here. Nonetheless, the fact that TPR-AuNC treatment has a noticeable effect on the cells, while missing the Hsp90 inhibitory domain suggests that the observed effect for the NIH-3T3 TPR-Hsp90-AuNC group is probably not entirely due to the Hsp90 inhibition. These effects could stem from the AuNC, the metallic component of the Prot-NM as was suggested before. Note that the increased Hsp90 production is not the only effect induced by the TGF-β activation. Another possibility could therefore be related to a different signaling cascade induced by the TGF-β. This hypothesis is supported by our observation in the TGF-β group, which showed a drastic morphological change compared to the control group. Ultimately, more research will be needed to investigate these observations and to determine which aspect of the treatment is causing the morphological changes observed. Finally, it is worth mentioning that these results reinforce the need of evaluating systematically the cellular structure upon application of specific treatments with adequate imaging techniques to be able to assess the cellular response.
Primary mouse fibroblasts, on the other hand, do not exhibit these symptoms of stress after overnight TPR-Hsp90-AuNC treatment, indicating that the cells had time to recover or adapt to the new conditions. The fact that the number of mitochondria is similar in every group, is in line with other work that related organelle size to cellular homeostasis,1 arguing that every cell type has an optimum number of organelles and a deviation from this number suggests a change in gene expression. While previous studies12–14 already highlighted the lack of negative effects, we now confirm that at the ultrastructural level TPR-Hsp90-AuNC is a viable option to inhibit the Hsp90 protein in primary mouse fibroblasts while also ensuring its ATPase activity and thereby keeping the cell in homeostasis. Despite the side effects in NIH-3T3 cells, we confirm here that Hsp90 inhibition at the C-terminal end reduces collagen overproduction in both cell types.
Methods
TPR preparation
The TPR variants used in this work have been previously described.11,14 The His-tagged proteins have been purified by affinity chromatography following standard protocols. C41 (DE3) cells transfected with the pProEx-HTA plasmid encoding the desired TPR gene, were grown in an orbital shaker at 37°C to an optical density of 0.6–0.8. Then the protein expression was induced with 1 mM isopropyl β-d-thiogalactoside (IPTG) and the cells were left for 16 hours at 20°C. The cells were centrifuged and lysed by sonication in 50 mM Tris, 500 mM NaCl, pH 8.0. Proteins were purified from the supernatant using a 5 ml HisTrap Q column (GE Healthcare). Then, the his-tag was removed by TEV protease cleavage and purified by gel filtration chromatography (HiLoad 16/60 Superdex 200 column).AuNC formation
AuNCs were synthesized and stabilized in the protein scaffolds using a protocol based on previously reported procedures.13,14 A protein at 20 μM was incubated with HAuCl4 (30 eq. with respect to protein) for 1 hour at 20 °C. Afterwards, a reduction step was applied by adding 100 eq. with respect to the Au concentration of sodium ascorbate and left to incubate at 50 °C for 72 h. Finally, the Prot-NM was purified by gel filtration (Sephadex G-25 column).Alexa labeling
Alexa Fluor 488 NHS ester was used to label the TPR-Hsp90-AuNC and TPR-AuNC proteins through their primary amines at a 1:2 stoichiometry to avoid over-labeling and potential disruption of the protein recognition activity.
Primary mouse fibroblast isolation
Live animal studies were approved by the University of Cantabria (Spain) Institutional Laboratory Animal Care and Use Committee in compliance with the Guide for the Care and Use of Laboratory Animals (ILAR, 1985) and were conducted in accordance with the “European Directive for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes” (European Communities Council Directive 86/606/EEC).Adult cardiac fibroblasts from mice C57BL6 strain (wildtype) were isolated directly from enzymatically digested hearts (collagenase, trypsin and DNase) followed by centrifugation and filtration. They were maintained in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% FBS and 100 U/ml penicillin–streptomycin. The cells (2 × 105) were plated in 35 mm cell culture dishes incubated at 37 °C in 5% CO2. For experimental procedures, low passage cells (p2) were seeded onto 60 mm cell culture dishes.
Sample preparation for CLXT
The sample preparation protocol is based on a previously published procedure2,50 and involves cryogenic liquids. Both NIH-3T3 and primary myocardial mouse fibroblasts were grown in complete culture medium (DMEM with 10% FBS and 1% Penicillin/Streptomycin) at 37 °C and 5% CO2 until a confluence of 70–80% was reached. At this point the cells are ready for subculture, or for sample preparation. As the sample support, gold quantifoil R 2/2 holey carbon-film microscopy grids (Au-G200F1) were used. Before cell deposition, grids were placed in P60 Petri-dishes, already separated by the type of treatment, and UV-sterilized. Spreading the grids at this stage minimizes grid handling and possible damaging. Cells are washed in PBS and seeded on top of the grids at a concentration of around 2–3 × 105 cells per ml (3 ml total in the case of a P60 Petri dish). After allowing the cells to settle and get accustomed to the surface for around 24 hours, they were treated. Both the NIH-3T3 and primary fibroblasts were divided into 4 groups: control, TGF-β activated, TGF-β activated and treated with TPR-Hsp90-AuNC, and finally TGF β activated and treated with TPR-AuNC lacking the Hsp90 inhibitory domain. Cells were activated with 1 ng ml−1 TGF-β final concentration and for the TPR-Hsp90-AuNC, as well as TPR-AuNC groups, the activated cells were treated with their respective Prot-NM at a final concentration of 0.8 μM. Note that the concentration of TGF-β to activate the cells was increased compared to the previous study (by 3 times).12 This increase provided a more consistent response of the cell population without affecting the gene-expression on a single cell basis (data not shown). After overnight incubation, Mitotracker Red CMXRos (ThermoFisher) was added to the grids, following manufacturer recommendations. After the incubation time of the fluorescent dye passed, a mix of 100 nm gold fiducial markers (BBI Solutions; 10× concentrated) and 200 nm Tetraspecks (ThermoFisher; 5× diluted) was prepared and 3 μl thereof was deposited on the grid followed by blotting of 2 s using a Whatman #1 filterpaper and vitrification through plunge freezing in liquid ethane precooled by liquid nitrogen in a Leica EM-CPC. Care has to be taken as grids have to be kept at cryogenic temperatures from this point on at all times. Pre-screening of the vitrified grids was done using a Linkam CMS196 cryo-stage on a Zeiss Axio Scope fluorescence microscope to determine the integrity of the grid as well as pinpointing cells of interest. A selection of grids is made at this point for correlative workflow.Cryo-3D-SIM
The selected grids, at least two per cell and condition, were sent to B24 at the Diamond Synchrotron (UK). The grids were imaged with a home-built cryo-3D-SIM51 to collect the green signal coming from the Alexa488 on the TPR proteins (λexc 488/λem 525, 40 mW, 100 ms), and red signal coming from mitotracker Red CMXRos (λexc 561/λem 605, 40 mW, 50–100 ms depending on staining efficiency) using a 100× 0.9 NA objective lens achieving a lateral resolution of roughly 210 nm full width at half maximum (FWHM) at an emission wavelength of 488 nm.15 For each sample-type three to six cells, preferably from different grids, were imaged and the collected data were processed on site.Cryo soft X-ray tomography
The grids were then sent back to Mistral at ALBA (Spain) for data collection using soft X-ray microscopy.27,28,50 The grids were loaded into the transmission X-ray microscope (TXM) chamber and the cells imaged prior with the cryo-3D-SIM were identified using the on-line visible light fluorescence microscope inside the TXM vacuum vessel with a long distance 20× objective. Once the areas were found again, tilt series were collected at a photon energy of 520 eV in areas showing interesting fluorescence signals in the cryo-3D-SIM data. In addition, other cells were imaged for morphological data evaluation, comparison and statistics. All data were collected using a Ni Fresnel zone plate lens with an outermost zone width of 25 nm (courtesy of S. Rebhein, HZB-Bessy II), giving an effective voxel size of 10 nm3. Depending on the sample and the position of the cell on the grid, images were collected covering a maximum angular range of −65° to +65° (or less) with a constant step size of 1°. While for the lower tilt angles an exposure time of 1–2 seconds was used, at higher tilt angles the exposure time was increased up to 4 seconds, to increase the signal-to-noise ratio while taking care to not oversaturate the CCD camera. The data were then pre-processed by normalization with the collected flatfield signal (flux impinging the sample) and the corresponding machine current, followed by deconvolution of the measured PSF of the TXM at the corresponding energy.31Fluorescence and immunofluorescence assays
The immunofluorescence assays of Hsp90 were performed using the aforementioned treatments. Then samples were fixed (4% paraformaldehyde), permeabilized (20–30 min 0.05%Triton-X in PBS 1×) and incubated overnight at 4°C with different primary antibodies anti-Alix (1:100, sc53540), anti-Flotillin-1 (1:100, sc74566), antiCD9 (1:100, donation from Sanchez-Madrid), anti-LC3-II (Cell Signaling #2775) and anti-caspase-2 (ab182657). The samples were washed with 0.05% Tween 20 in PBS, incubated for 30 min in the specific secondary antibody conjugated with Alexa-568 (ThermoFisher), washed in PBS and mounted with the Vectashield antifade mounting medium (Vector).
Confocal images were obtained in the microscopy facility (IDIVAL, Santander, Spain) with a confocal microscope NIKON A1R spectral (405 nm, 488 nm, 514 nm, 561 nm, 638 nm) and Fiji52 was used for data visualization.
Data processing
The tilt-series were aligned using the 100 nm Au fiducials and the IMOD software.53 The reconstruction was performed using the SIRT algorithm54 with tomo3d with long object compensation55 for a fast reconstruction, which is for visualization purposes only, or using the ART algorithm56 with tomoJ57 for linear absorption coefficient recovery. The tomographic reconstruction of each of the cells (several tomograms per cell collected) imaged before using the cryo-3D-SIM were then post-processed using Fiji52 to ensure they are aligned to each other by flattening and cropping in the z-direction to adjust the height, based on the carbon quantifoil support layer. The correlation was done using the ICY58 plugin in ecCLEM.59 First, the cryo-SXT reconstructions were 2D transformed to the X-ray mosaic to place them in the context of the whole cell as each tomogram covers a volume of 10 μm3 of the cell. To prevent excessively large files, cropping of the X-ray mosaic to the area containing the reconstructions is performed. The transformed files could then be combined into one for subsequent 3D correlation. Because of the chromatic shift of the 3D-SIM, the fluorescent channels were first aligned to each other using the 200 nm Tetraspecks. After this, the now aligned fluorescent channels were roughly transformed in 2D to the X-ray mosaic using large features, like, for example, the holes in the quantifoil film or using large organelles, like mitochondria. After this the fluorescent data were transformed in 3D using more fine features such as Tetraspecks or recognizable organelle shapes.Statistical analysis of organelles
The tomographic reconstructions were segmented using Amira software.34 Measurements of the segmentation were performed using the Amira module Label Analysis. For the volume, the native measure is used but for the sphericity a custom measure had to be created using the formula:with V being the volume of the label and A being the surface area. For each condition four tomographic reconstructions belonging to four different cells were segmented and measured. The statistical analysis was performed using Excel 2016s Data Analysis add-in. First, the datasets were tested for equal or unequal variance, followed by the corresponding two-tailed t-test using a cut-off value of 0.05 to test for significance between the groups.
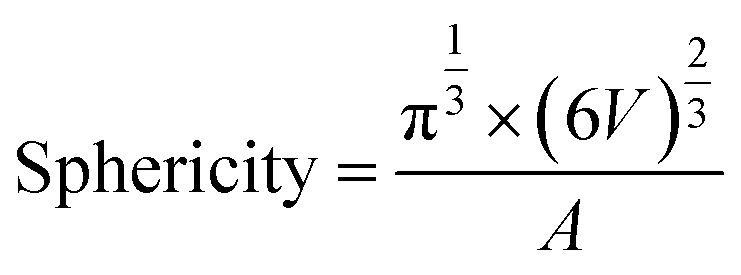
Conclusions
Herein, CLXT has allowed locating unambiguously and in the 3D cellular environment, a designed therapeutic agent inhibiting Hsp90 after being internalized by two different types of fibroblasts. Furthermore, this novel methodology enables evaluating the effects of the different treatments at the cellular level which is essential for a future practical usage at the clinical level. Cryo-3D-SIM provides excellent specificity using a wide range of common dyes and fluorescent proteins and adequate 3D resolution for correlative purposes, although higher sensitivity at the single molecule level and resolution would be desirable. Cryo-SXT in turn reveals the environmental context and the ultrastructural details, which allow identifying the designed protein–nanomaterial hybrid (Prot-NM) containing organelles and describing the morphological differences accompanying the different treatments. In both cases, the TPR-AuNC with and without the Hsp90 binding module were found in specific multivesicular bodies (MVBs), although in higher numbers in the first case. These MVBs likely play a functional role in cell homeostasis. Furthermore, we found substantial differences between NIH-3T3 and primary mouse fibroblast cells, which led us to conclude that the immortalized version of a cell type may not always be a good functional alternative when studying molecular processes as deviations from primary cells can occur.This detailed study, using correlatively cryo-3D-SIM and cryo-SXT to obtain specific intracellular location and ultrastructural information, highlights the relevance of a mechanistic understanding of the molecular process triggered by novel engineered Prot-NM with potential therapeutic applications. Here, a protein engineering-based biological drug (TPR-Hsp90-AuNC) with a demonstrated effect as an antifibrotic molecule has been evaluated. The results of this study reveal the effect of the Hsp90 inhibitory domain and its correlation with its antifibrotic function and mechanism. But at the same time, they show that the presence of a small metallic nanocluster of 4 to 12 atoms, in addition to its function as a stabilizing and tagging module, possibly has an effect on the cellular morphology, which still needs to be investigated further for a complete understanding. It is important to note that, considering the modularity of the engineered system, the inhibitory domain is interchangeable. This opens up possibilities for a wide variety of treatments that could benefit from the design of alternative Prot-NM. As has been shown here, CLXT is able to track the fate of the specific therapeutic agent with high correlation accuracy, as well as monitor with statistical significance the morphological changes due to the treatment, allowing shedding light on the cellular processes triggered. The methodology utilized is applicable to other disease models and systems and will always provide excellent specificity and spatial resolution. These latest results put the focus on the need to study the effects of nanomaterials at the cellular level under close-to-native conditions in order to be able to further propel nanomaterial-based drugs and nanomedicine into the clinic.
Data availability
The data of the main correlation figure was deposited at the BioImage Archive (https:// www.ebi.ac.uk/biostudies/BioImages) and EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/). The accession numbers for the data deposited at EMPIAR is EMPIAR-10839, and the accession numbers for the data deposited at the BioImage Archive is S-BSST728.Author contributions
J. G. performed all the experiments, with the exception of the confocal microscopy part, which was done by A. P. and A. V. V. Primary fibroblasts were isolated and shipped to Alba by D. M. Prot-NM was prepared and sent to Alba by A. A. Access to the SIM was provided by M. H. and the 25 nm Fresnel zone plate lens was built by S. R. Assistance with the X-ray microscope data collection was provided by E. P., and J. J. C. assisted and guided the data processing and correlation process. E. P. and A. L. C. directed the research and together with J. G. wrote the paper. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the help of R. Oliete (Alba) for his help with the sample preparation and C. Okolo for the cryo-3D-SIM data collected at B24 at Diamond Synchrotron during experimental session BI25162. Cryo-SXT data were collected at Mistral beamline at Alba Synchrotron during experimental sessions 2019093739 and 2020034355. The authors wish to thank F. Madrazo for his time and expertise at the IDIVAL image facility. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 754397. This work was partially supported by the European Research Council ERC-CoG-648071-ProNANO, ERC-PoC-841063-NIMM; Spanish State Research Agency, Spain; (BIO2015-72124-EXP), (MDM-2017-0720), (PID2019-111649RB-I00) and (RTI2018-095214-B-I00); the Basque Government (Elkartek KK-2017/00008; RIS3-2019222005); and the Next Generation and Innovation Valdecilla (PREVAL19/05), (IDIVAL INNVAL17/22), (INNVAL20/34), (NVAL18/11) and (NextVal 18/14). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720 (CIC biomaGUNE).Notes and references
- L. W. Cole, The Evolution of Per-cell Organelle Number, Front. Cell Dev. Biol., 2016, 4, 85 CrossRef PubMed.
- C. A. Okolo, I. Kounatidis, J. Groen, K. L. Nahas, S. Balint and T. M. Fish, et al., Sample preparation strategies for efficient correlation of 3D SIM and soft X-ray tomography data at cryogenic temperatures, Nat. Protoc., 2021, 16(6), 2851–2885 CrossRef CAS PubMed.
- K. E. Porter and N. A. Turner, Cardiac fibroblasts: At the heart of myocardial remodeling, Pharmacol. Ther., 2009, 123(2), 255–278 CrossRef CAS PubMed.
- S. Hinderer and K. Schenke-Layland, Cardiac fibrosis – A short review of causes and therapeutic strategies, Adv. Drug Delivery Rev., 2019, 146, 77–82 CrossRef CAS PubMed.
- M. Liu, B. López de Juan Abad and K. Cheng, Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies, Adv. Drug Delivery Rev., 2021, 173, 504–519 CrossRef CAS PubMed.
- T. Doetschman, J. v. Barnett, R. B. Runyan, T. D. Camenisch, R. L. Heimark and H. L. Granzier, et al., Transforming growth factor beta signaling in adult cardiovascular diseases and repair, Cell Tissue Res., 2012, 347(1), 203–223 CrossRef CAS PubMed.
- R. Datta, T. Bansal, S. Rana, K. Datta, S. Chattopadhyay and M. Chawla-Sarkar, et al., Hsp90/Cdc37 assembly modulates TGFβ receptor-II to act as a profibrotic regulator of TGFβ signaling during cardiac hypertrophy, Cell. Signalling, 2015, 27(12), 2410–2424 CrossRef CAS PubMed.
- M. Morikawa, R. Derynck and K. Miyazono, TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology, Cold Spring Harbor Perspect. Biol., 2016, 8(5), a021873 CrossRef PubMed.
- R. García, D. Merino, J. M. Gómez, J. F. Nistal, M. A. Hurlé and A. L. Cortajarena, et al., Extracellular heat shock protein 90 binding to TGFβ receptor I participates in TGFβ-mediated collagen production in myocardial fibroblasts, Cell. Signalling, 2016, 28(10), 1563–1579 CrossRef PubMed.
- A. L. Cortajarena, T. Kajander, W. Pan, M. J. Cocco and L. Regan, Protein design to understand peptide ligand recognition by tetratricopeptide repeat proteins, Protein Eng., Des. Sel., 2004, 17(4), 399–409 CrossRef CAS.
- A. L. Cortajarena, F. Yi and L. Regan, Designed TPR Modules as Novel Anticancer Agents, ACS Chem. Biol., 2008, 3(3), 161–166 CrossRef CAS PubMed.
- R. A. Cáceres, T. Chavez, D. Maestro, A. R. Palanca, P. Bolado and F. Madrazo, et al., Reduction of cardiac TGFβ-mediated profibrotic events by inhibition of Hsp90 with engineered protein, J. Mol. Cell. Cardiol., 2018, 123, 75–87 CrossRef PubMed.
- A. Aires, I. Llarena, M. Moller, J. Castro-Smirnov, J. Cabanillas-Gonzalez and A. L. Cortajarena, A Simple Approach to Design Proteins for the Sustainable Synthesis of Metal Nanoclusters, Angew. Chem., Int. Ed., 2019, 58(19), 6214–6219 CrossRef CAS PubMed.
- A. Aires, D. Maestro, J. Ruiz del Rio, A. R. Palanca, E. Lopez-Martinez and I. Llarena, et al., Engineering multifunctional metal/protein hybrid nanomaterials as tools for therapeutic intervention and high-sensitivity detection, Chem. Sci., 2021, 12(7), 2480–2487 RSC.
- M. A. Phillips, M. Harkiolaki, D. M. Susano Pinto, R. M. Parton, A. Palanca and M. Garcia-Moreno, et al., CryoSIM: super-resolution 3D structured illumination cryogenic fluorescence microscopy for correlated ultrastructural imaging, Optica, 2020, 7(7), 802 CrossRef CAS PubMed.
- D. Weiß, G. Schneider, B. Niemann, P. Guttmann, D. Rudolph and G. Schmahl, Computed tomography of cryogenic biological specimens based on X-ray microscopic images, Ultramicroscopy, 2000, 84(3–4), 185–197 CrossRef.
- G. Schneider, P. Guttmann, S. Heim, S. Rehbein, F. Mueller and K. Nagashima, et al., Three-dimensional cellular ultrastructure resolved by X-ray microscopy, Nat. Methods, 2010, 7(12), 985–987 CrossRef CAS PubMed.
- J. L. Carrascosa, F. J. Chichón, E. Pereiro, M. J. Rodríguez, J. J. Fernández and M. Esteban, et al., Cryo-X-ray tomography of vaccinia virus membranes and inner compartments, J. Struct. Biol., 2009, 168(2), 234–239 CrossRef CAS PubMed.
- F. J. Chichón, M. J. Rodríguez, E. Pereiro, M. Chiappi, B. Perdiguero and P. Guttmann, et al., Cryo X-ray nano-tomography of vaccinia virus infected cells, J. Struct. Biol., 2012, 177(2), 202–211 CrossRef PubMed.
- C. Hagen, P. Guttmann, B. Klupp, S. Werner, S. Rehbein and T. C. Mettenleiter, et al., Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells, J. Struct. Biol., 2012, 177(2), 193–201 CrossRef PubMed.
- E. M. H. Duke, M. Razi, A. Weston, P. Guttmann, S. Werner and K. Henzler, et al., Imaging endosomes and autophagosomes in whole mammalian cells using correlative cryo-fluorescence and cryo-soft X-ray microscopy (cryo-CLXM), Ultramicroscopy, 2014, 143, 77–87 CrossRef CAS PubMed.
- M. Chiappi, J. J. Conesa, E. Pereiro, C. O. S. Sorzano, M. J. Rodríguez and K. Henzler, et al., Cryo-soft X-ray tomography as a quantitative three-dimensional tool to model nanoparticle:cell interaction, J. Nanobiotechnol., 2016, 14(1), 15 CrossRef PubMed.
- A. J. Pérez-Berná, M. J. Rodríguez, F. J. Chichón, M. F. Friesland, A. Sorrentino and J. L. Carrascosa, et al., Structural Changes In Cells Imaged by Soft X-ray Cryo-Tomography During Hepatitis C Virus Infection, ACS Nano, 2016, 10(7), 6597–6611 CrossRef PubMed.
- J. Groen, J. J. Conesa, R. Valcárcel and E. Pereiro, The cellular landscape by cryo soft X-ray tomography, Biophys. Rev., 2019, 11(4), 611–619 CrossRef PubMed.
- B. Kepsutlu, V. Wycisk, K. Achazi, S. Kapishnikov, A. J. Pérez-Berná and P. Guttmann, et al., Cells Undergo Major Changes in the Quantity of Cytoplasmic Organelles after Uptake of Gold Nanoparticles with Biologically Relevant Surface Coatings, ACS Nano, 2020, 14(2), 2248–2264 CrossRef CAS PubMed.
- V. Weinhardt, J.-H. Chen, A. A. Ekman, J. Guo, S. G. Remesh and M. Hammel, et al., Switchable resolution in soft x-ray tomography of single cells, PLoS One, 2020, 15(1), e0227601 CrossRef CAS PubMed.
- E. Pereiro, J. Nicolás, S. Ferrer and M. R. Howells, A soft X-ray beamline for transmission X-ray microscopy at ALBA, J. Synchrotron Radiat., 2009, 16(4), 505–512 CrossRef CAS PubMed.
- A. Sorrentino, J. Nicolás, R. Valcárcel, F. J. Chichón, M. Rosanes and J. Avila, et al., MISTRAL: a transmission soft X-ray microscopy beamline for cryo nano-tomography of biological samples and magnetic domains imaging, J. Synchrotron Radiat., 2015, 22(4), 1112–1117 CrossRef CAS PubMed.
- R. J. Youle and A. M. van der Bliek, Mitochondrial Fission, Fusion, and Stress, Science, 2012, 337(6098), 1062–1065 CrossRef CAS PubMed.
- I. Kounatidis, M. L. Stanifer, M. A. Phillips, P. Paul-Gilloteaux, X. Heiligenstein and H. Wang, et al., 3D Correlative Cryo-Structured Illumination Fluorescence and Soft X-ray Microscopy Elucidates Reovirus Intracellular Release Pathway, Cell, 2020, 182(2), 515–530 CrossRef CAS PubMed.
- J. Otón, E. Pereiro, A. J. Pérez-Berná, L. Millach, C. O. S. Sorzano and R. Marabini, et al., Characterization of transfer function, resolution and depth of field of a soft X-ray microscope applied to tomography enhancement by Wiener deconvolution, Biomed. Opt. Express, 2016, 7(12), 5092 CrossRef PubMed.
- G. Cardone, K. Grünewald and A. C. Steven, A resolution criterion for electron tomography based on cross-validation, J. Struct. Biol., 2005, 151(2), 117–129 CrossRef PubMed.
- P. Reineck, A. N. Abraham, A. Poddar, R. Shukla, H. Abe and T. Ohshima, et al., Multimodal Imaging and Soft X-Ray Tomography of Fluorescent Nanodiamonds in Cancer Cells, Biotechnol. J., 2021, 16(3), 2000289 CrossRef CAS PubMed.
- ThermoFisher Scientific, Amira for Life & Biomedical Sciences, Mar, 8, 2021, Retrieved from: https://www.thermofisher.com/es/es/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software/amira-life-sciences-biomedical.html Search PubMed.
- Z. Andreu and M. Yáñez-Mó, Tetraspanins in Extracellular Vesicle Formation and Function, Front. Immunol., 2014, 5, 442 Search PubMed.
- M. F. Santos, G. Rappa, J. Karbanová, C. Vanier, C. Morimoto and D. Corbeil, et al., Anti-human CD 9 antibody Fab fragment impairs the internalization of extracellular vesicles and the nuclear transfer of their cargo proteins, J. Cell. Mol. Med., 2019, 23(6), 4408–4421 CrossRef CAS PubMed.
- R. H. Chen and T. Y. Chang, Involvement of caspase family proteases in transforming growth factor-beta-induced apoptosis, Cell Growth Differ., 1997, 8(7), 821–827 CAS.
- V. v. Petrov, R. H. Fagard and P. J. Lijnen, Stimulation of Collagen Production by Transforming Growth Factor-β1 During Differentiation of Cardiac Fibroblasts to Myofibroblasts, Hypertension, 2002, 39(2), 258–263 CrossRef CAS PubMed.
- T. Farmer, N. Naslavsky and S. Caplan, Tying trafficking to fusion and fission at the mighty mitochondria, Traffic, 2018, 19(8), 569–577 CrossRef CAS PubMed.
- B. Westermann, Bioenergetic role of mitochondrial fusion and fission, Biochim. Biophys. Acta, Bioenerg., 2012, 1817(10), 1833–1838 CrossRef CAS PubMed.
- T. Tokuyama, A. Hirai, I. Shiiba, N. Ito, K. Matsuno and K. Takeda, et al., Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients, Biomolecules, 2020, 10(3), 450 CrossRef CAS PubMed.
- K. J. de Vos, V. J. Allan, A. J. Grierson and M. P. Sheetz, Mitochondrial Function and Actin Regulate Dynamin-Related Protein 1-Dependent Mitochondrial Fission, Curr. Biol., 2005, 15(7), 678–683 CrossRef CAS PubMed.
- R. Rossignol, R. Gilkerson, R. Aggeler, K. Yamagata, S. J. Remington and R. A. Capaldi, Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells, Cancer Res., 2004, 64(3), 985–993 CrossRef CAS PubMed.
- A. Iavello, V. S. L. Frech, C. Gai, M. C. Deregibus, P. J. Quesenberry and G. Camussi, Role of Alix in miRNA packaging during extracellular vesicle biogenesis, Int. J. Mol. Med., 2016, 37(4), 958–966 CrossRef CAS PubMed.
- G. P. Otto and B. J. Nichols, The roles of flotillin microdomains – endocytosis and beyond, J. Cell Sci., 2011, 124(23), 3933–3940 CrossRef CAS PubMed.
- A. Sable, K. M. Rai, A. Choudhary, V. K. Yadav, S. K. Agarwal and S. v Sawant, Inhibition of Heat Shock proteins HSP90 and HSP70 induce oxidative stress, suppressing cotton fiber development, Sci. Rep., 2018, 8(1), 3620 CrossRef PubMed.
- J. Li, A. Csibi, S. Yang, G. R. Hoffman, C. Li and E. Zhang, et al., Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(1), E21–E29 CrossRef CAS PubMed.
- C. Y. Tay, Y. Yu, M. I. Setyawati, J. Xie and D. T. Leong, Presentation matters: Identity of gold nanocluster capping agent governs intracellular uptake and cell metabolism, Nano Res., 2014, 7(6), 805–815 CrossRef CAS.
- P. Jawaid, M. U. Rehman, Q.-L. Zhao, M. Misawa, K. Ishikawa and M. Hori, et al., Small size gold nanoparticles enhance apoptosis-induced by cold atmospheric plasma via depletion of intracellular GSH and modification of oxidative stress, Cell Death Discovery, 2020, 6(1), 83 CrossRef CAS PubMed.
- J. Groen, A. Sorrentino, L. Aballe, R. Oliete, R. Valcárcel and C. Okolo, et al., A 3D Cartographic Description of the Cell by Cryo Soft X-ray Tomography, J. Visualized Exp., 2021, 2021(169), e62190 Search PubMed.
- N. Vyas, N. Perry, C. A. Okolo, I. Kounatidis, T. M. Fish and K. L. Nahas, et at., Cryo-Structured Illumination Microscopic Data Collection from Cryogenically Preserved Cells, J. Visualized Exp., 2021, 2021(171), e62274 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair and T. Pietzsch, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, Computer Visualization of Three-Dimensional Image Data Using IMOD, J. Struct. Biol., 1996, 116(1), 71–76 CrossRef CAS PubMed.
- P. Gilbert, Iterative methods for the three-dimensional reconstruction of an object from projections, J. Theor. Biol., 1972, 36(1), 105–117 CrossRef CAS PubMed.
- J. I. Agulleiro and J. J. Fernandez, Fast tomographic reconstruction on multicore computers, Bioinformatics, 2011, 27(4), 582–583 CrossRef CAS PubMed.
- R. Gordon, R. Bender and G. T. Herman, Algebraic Reconstruction Techniques (ART) for three-dimensional electron microscopy and X-ray photography, J. Theor. Biol., 1970, 29(3), 471–481 CrossRef CAS PubMed.
- C. MessaoudiI, T. Boudier, C. Sorzano and S. Marco, TomoJ: tomography software for three-dimensional reconstruction in transmission electron microscopy, BMC Bioinf., 2007, 8(1), 288 CrossRef PubMed.
- F. de Chaumont, S. Dallongeville, N. Chenouard, N. Hervé, S. Pop and T. Provoost, et al., Icy: an open bioimage informatics platform for extended reproducible research, Nat. Methods, 2012, 9(7), 690–696 CrossRef CAS PubMed.
- P. Paul-Gilloteaux, X. Heiligenstein, M. Belle, M.-C. Domart, B. Larijani and L. Collinson, et al., eC-CLEM: flexible multidimensional registration software for correlative microscopies, Nat. Methods, 2017, 14(2), 102–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04183e |
| This journal is © The Royal Society of Chemistry 2021 |