
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent formation route to a new class of oxygen-based 1,3-dipoles and the modular synthesis of furans†
Huseyin
Erguven‡
b,
Cuihan
Zhou‡
a and
Bruce A.
Arndtsen
 *a
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A0B8, Canada. E-mail: bruce.arndtsen@mcgill.ca
bDepartment of Chemistry and Chemical Biology, Rutgers University, 123 Bevier Road, Piscataway, NJ 08854, USA
First published on 27th October 2021
Abstract
A new class of phosphorus-containing 1,3-dipoles can be generated by the multicomponent reaction of aldehydes, acid chlorides and the phosphonite PhP(catechyl). These 1,3-dipoles are formally cyclic tautomers of simple Wittig-type ylides, where the angle strain and moderate nucleophilicity in the catechyl-phosphonite favor their cyclization and also direct 1,3-dipolar cycloaddition to afford single regioisomers of substituted products. Coupling the generation of the dipoles with 1,3-dipolar cycloaddition offers a unique, modular route to furans from combinations of available aldehydes, acid chlorides and alkynes with independent control of all four substituents.
Introduction
1,3-Dipolar cycloaddition reactions offer a powerful approach to construct five-membered ring products.1 A prominent application of these systems is in the synthesis of nitrogen-containing heterocycles, such as the prototypical click reaction between substituted azides and alkynes to form triazoles.2 Less nitrogen-rich heterocycles can also be assembled with other 1,3-dipoles, such as pyrroles (from münchnones), pyrazoles (from sydnones), and related systems.3,4 This breadth is driven in part by the availability of stabilized nitrogen-based 1,3-dipoles of use in cycloaddition, many of which were developed in the pioneering work of Huisgen.1 In contrast, the use of oxygen-containing 1,3-dipoles to form oxygen-heterocycles, while viable, is much less prevalent. Heterocycles such as furans are important in pharmaceutically relevant structures, natural products, electronic materials, and as building blocks in organic synthesis.5–8 Unfortunately, 1,3-dipoles of utility in furan synthesis are often less stable and more challenging to form in a general fashion than their nitrogen analogues. For example, oxygen variants of münchnones are known (Fig. 1a),9 but are labile, have limited structural diversity, and importantly, are often formed from substituted diazo precursors that must themselves be synthesized. Carbonyl ylides can also allow access to partially reduced furan derivatives, but they are unstable intermediates available from synthetic diazo precursors or activated epoxide ring opening.10 Furans are instead typically generated by more classical cyclization methods, such as the Paal–Knorr synthesis, substitutions on pre-formed furans, or more recently developed metal catalyzed cyclization or coupling reactions.11,12 While all these methods are effective, they often require the initial generation of one or more substrates incorporating the correct substituents, which adds steps to the synthesis, creates waste and can limit their ease of generalization.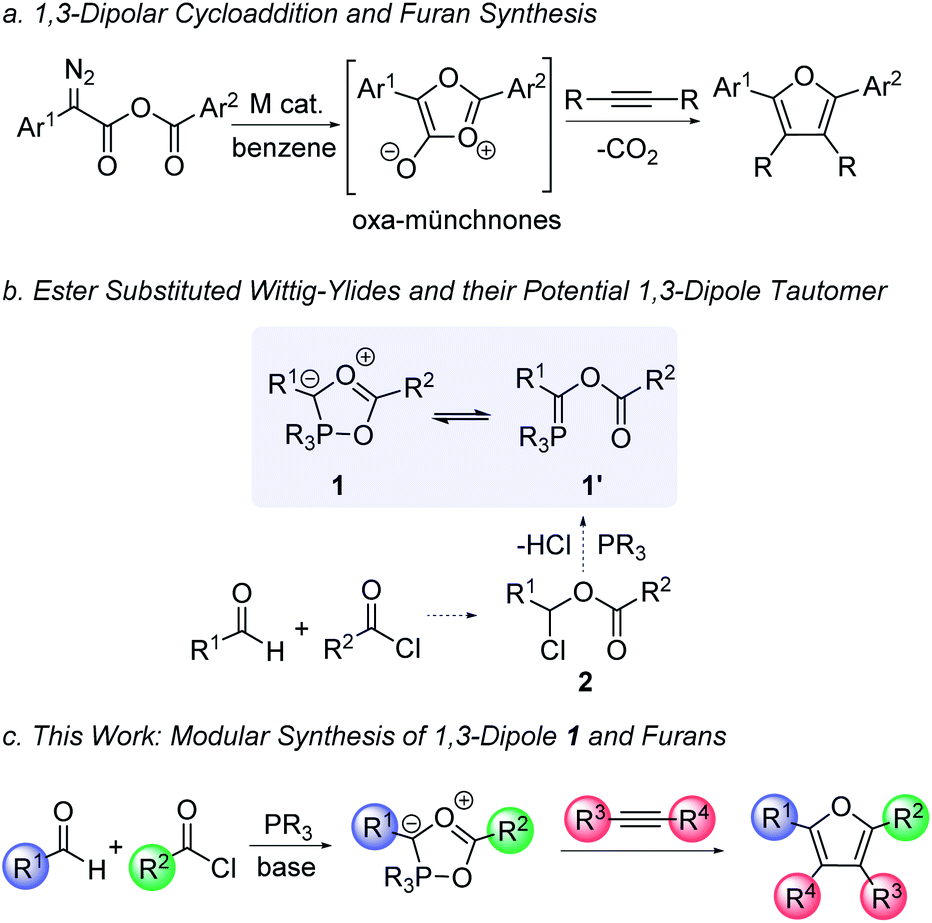 | ||
| Fig. 1 Cycloaddition approaches to furan derivatives and design of a new phosphorus-based 1,3-dipolar cycloaddition reagent. | ||
In considering methods to efficiently synthesize furans, we questioned if this might be enabled by forming a new 1,3-dipole: the phosphorus-containing 1 (Fig. 1b). Several phosphine-mediated syntheses of furans have recently been described, including systems employing intramolecular Wittig-reactions with alkyne, acylating agent and/or aldehyde substrates, although these typically require the use of α,β-unsaturated esters or pre-synthesized cyclization substrates.131 could offer a 1,3-dipolar cycloaddition route to these products with the ability to tune all four substituents and incorporate various alkyne or alkene dipolarophiles. 1 is formally a cyclized variant of a Wittig-type ylide 1′, and while appearing complex, could be accessible by the reaction of phosphines, acid chlorides and aldehydes (via acyl oxonium salts 2). We have recently reported the nitrogen version of these 1,3-dipoles prepared with imines for use in cycloaddition.14,15 However, the formation of the oxygen-based dipole 1 presents several challenges. First, aldehydes are weak nucleophiles and do not readily react with acyl chlorides:16 a critical first step in this transformation. Indeed, aldehydes are better known for their electrophilic reactivity and could instead undergo a direct reaction with the phosphorus reagent.17 Even more intrinsic to this chemistry, the generation of this 1,3-dipole requires the unknown cyclization of 1′ to 1, which would presumably be disfavored with the weakly chelating ester substituent. Nevertheless, previous studies have shown that the angle strain at the phosphorus center can be effective in favoring cyclization to generate a five-coordinate phosphorus,14c which led us to question if such a system might also allow the generation of this oxygen-based 1,3-dipolar cycloaddition reagent (1). We describe here our studies towards such a system. These studies demonstrate that the coupling of aldehydes, acylating agents and the correctly substituted phosphine can afford a multicomponent synthesis of a new class of 1,3-dipoles (Fig. 1c). Combining the formation of 1 with in situ 1,3-dipolar cycloaddition opens a modular synthesis of polysubstituted furans from three available reagents: aldehydes, acyl chlorides and alkynes.
Results and discussion
Our initial studies toward this system involved the reaction of benzoyl halide, p-tolualdehyde, and (catechyl)PPh followed by the NEtiPr2 base. In order to test for the formation of 1a, the electron deficient alkyne dimethylacetylenedicarboxylate (DMAD) was added in a subsequent step as a 1,3-dipolar cycloaddition substrate. Attempts at this reaction with benzoyl chloride led to no observable furan product under various conditions (e.g.Table 1, entry 1, see ESI Table S1† for full details). One possibility for the lack of reactivity may be slow coupling of the aldehyde with the acyl chloride, which typically requires the use of Lewis acid catalysts to activate the acyl chloride.18 However, the analogous reaction with added AlCl3 (entry 2) or more electrophilic benzoyl bromide or benzoyl iodide (entry 3) led to similar results, as did the generation of these acyl halides in situ (entries 4 and 5). It is known that aldehydes themselves can react with phosphites or phosphonites (i.e. the Abramov reaction),17 which could also inhibit the synthetic sequence. To block this possibility, the aldehyde was first combined with an in situ generated benzoyl iodide, followed by the addition of PhP(catechyl) and then DMAD and a base. We were pleased to find that furan 3a could now be observed in low yield (entry 6). The use of pre-formed benzoyl bromide or iodide further improves the yield of furan, with the latter now generating furan in 70% yield (entries 7 and 8).|
|
||||
|---|---|---|---|---|
| Entry | X | MX′ | PR3 | % 3ac |
| a 0.12 mmol acyl halide, 0.1 mmol aldehyde, 0.12 mmol PR3, 1 ml MeCN, and then 0.15 mmol MX′ for 12 h, followed by dimethyl acetylene dicarboxylate (21 mg, 0.15 mmol) and NEtiPr2 (19 mg, 0.15 mmol) for 1 h. b Acyl halide, MX′, and aldehyde in 1 ml CD3CN for 12 h, followed by the addition of PR3. c NMR yields vs. an internal standard. d In 1,2-dichloroethane. e 0.10 mmol AgOTf. f 2 h reaction with alkyne. g Yield after 1 h. | ||||
| 1 | Cl | — | PhP(catechyl) | — |
| 2 | Cl | AlCl3 | PhP(catechyl) | — |
| 3 | Br or I | — | PhP(catechyl) | — |
| 4 | Cl | TMSBr | PhP(catechyl) | — |
| 5 | Cl | NaI | PhP(catechyl) | — |
| 6b | Cl | NaI | PhP(catechyl) | 32 |
| 7b | Br | — | PhP(catechyl) | 45 |
| 8b | I | — | PhP(catechyl) | 70 |
| 9d | Cl | AgOTf | PhP(catechyl) | 80 |
| 10d,e | Cl | AgOTf | PhP(catechyl) | 87 |
| 11d | Cl | AgOTf | PPh3 | — |
| 12d | Cl | AgOTf | PCy3 | — |
| 13d | Cl | AgOTf | P(OCH2CF3)3 | — |
| 14d | Cl | AgOTf | (PhO)P(catechyl) | 8 |
| 15d,e,f | Cl | AgOTf | P(OPh)3 | 55 (48)g |
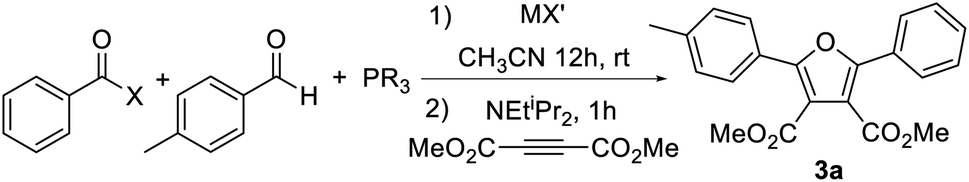
While these results were encouraging, the stepwise addition of reagents adds a layer of complexity to the overall synthesis. In addition, the procedure was found to be sensitive to substrates and reaction times, and change in the acyl halide or aldehyde led to drastically different results. We therefore probed alternative ways to access the 1,3-dipole. One option is to generate a more potent acylating agent that might react with the aldehyde more readily than the phosphonite. After examining various conditions (see Table S1†), it was found that this can be accomplished by the simple addition of AgOTf. Thus, the one pot reaction of benzoyl chloride, aldehyde and PhP(catechyl) in the presence of AgOTf, which presumably forms an electrophilic acyl triflate intermediate,19 can allow the formation of furan 3a in high yield upon DMAD and base addition (entry 9). Performing the reaction with equimolar AgOTf, which prevents the formation of excess acyl triflate, further increases the reaction efficiency (entry 10).20 As expected, the specific phosphorus reagent plays a key role in the reaction. For example, more electron rich phosphines such as PPh3 or PCy3 also react with aldehyde, acyl chloride and AgOTf. However, upon addition of a base and alkyne, these do not lead to cycloaddition to form furans, presumably reflecting the poor ability of these more electron rich phosphines to cyclize to a 1,3-dipolar form (entries 11 and 12). Moving to more electron poor phosphites such as P(OPh)3 or P(OCH2CF3)3 leads to diminished furan yields relative to PhP(catechyl) as well, arising in this case from a sluggish initial reaction of these phosphorus reagents with the aldehyde/acylating agent (entries 13 and 14), or slower subsequent cycloaddition reactivity (entry 15). Together, these results are consistent with the role of the moderate nucleophilicity of PhP(catechyl) in allowing the reaction with an in situ generated oxonium salt, together with that of the angle strain of the catechyl unit in favoring the formation of a five-coordinate phosphorus needed to access a 1,3-dipole (vide infra).21
Combining these optimized conditions with the availability of acyl chlorides, aldehydes and alkynes provides a straightforward pathway to synthesize a range of furan products (Table 2). For example, various electron poor dipolarophiles can be used in cycloaddition with 1 as a method to modulate the 3,4-substituents on the furan core. Of note, unsymmetrical dipolarophiles (3c–g) lead to a single product, where the electron withdrawing unit is directed away from the former aldehyde carbon. The latter is consistent with the localization of negative charge on this carbon in the 1,3-dipole, which, together with the large PR3 unit, directs cycloaddition regioselectivity.21 More electron rich alkynes, such as 1-hexyne or phenyl acetylene, do not react with 1. However, tethering the alkyne to the aldehyde can allow this cycloaddition as well to form the polycyclic furan 3h in moderate yield. A variety of aromatic aldehydes can be employed in the reaction, including those with either electron donating (3i–3m) or withdrawing groups (3p–3s), as well as those with meta and ortho-substitution (3j, 3k, 3q–3s). The aromatic substituents can be replaced with heteroaromatic units, such as from 3- or 2-thiophenyl aldehyde (3n, 3o). The acid chloride displays similar structural flexibility in proceeding with electron donating (3t–3u), electron withdrawing (3v–3y), ortho- (3u) or even very large mesityl (3g) substituents, as well as with extended conjugation (3z). Thiophenyl acid chloride can also be employed (3aa), and, when used with thiophenyl aldehyde, offers access to the alternating, π-conjugated 3bb. Alternatively, the use of cyano-substituted substrates generates 3v, a precursor to derivatives of the antimicrobial furamidine.22 Nucleophilic heterocycles such as pyridines or pyrroles are not compatible with the electrophilic reaction conditions, nor are simple alkyl substituents on the acid chloride or aldehyde, presumably due to their α-deprotonation. However, the tertiary alkyl adamantyl acid chloride (3cc) is a viable substrate. The reaction can also be carried out on the 1 mmol scale with similar yields (e.g.3a). Together, this offers a modular approach to access an array of substituted furans with the independent tuning of each substituent.
| a 0.24 mmol of acyl chloride, 0.2 mmol of aldehyde and PhP(catechyl) (52 mg, 0.24 mmol) in 1 ml of 1,2-dichloroethane, followed by AgOTf (62 mg, 0.24 mmol) for 12 h, then alkyne (0.30 mmol) and NEtiPr2 (39 mg, 0.30 mmol), 1 h. Isolated yields. b NEtiPr2 (65 mg, 0.50 mmol) at 80°C for 12 h. c NEtiPr2 (65 mg, 0.50 mmol), 100°C for 72 h. d Stirred for only 1 h after AgOTf addition. e Acyl chloride and AgOTf pre-mixed for 2 h before adding aldehyde and PhP(catechyl). f NMR yield vs. the internal standard of hexamethyl benzene. g Reaction on the 1.0 mmol scale. |
|---|

|
We next turned our attention to probing the structure of the phosphorus-based cycloaddition reagent formed in this system. Insight into the first step in the reaction can be obtained by monitoring the coupling of benzoyl chloride, p-tolualdehyde and PhP(catechyl) in the presence of AgOTf by 1H NMR analysis, which shows the formation of a new product (4a) in near quantitative yield. This compound can be precipitated from the mixture by addition of pentane, and has been characterized to be the cationic phosphonium salt shown (Fig. 2a). Of note, 1H NMR analysis shows the expected resonances for aromatic residues and the α-methylene hydrogen coupled to phosphorus (δ 6.82 ppm, 2JP–H = 4.5 Hz), as well as the expected phosphorus signal in 31P NMR (δ 24.3 ppm).14,23 The addition of the EtNiPr2 base at −35°C to 4a without a dipolarophile present led to the formation of a complex mixture of unidentifiable products, including Ph(catechyl)P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, suggesting that the putative 1,3-dipole generated here is unstable. However, the stability of this intermediate can be enhanced by incorporating a sterically encumbered mesityl substituent on the acid chloride together with an electron deficient 4-fluorobenzaldehyde. The generation of this product by deprotonation with the LiHMDS base at room temperature leads to the formation of a bright red product 1b in 93% yield (Fig. 2b). Of note, the in situ31P NMR spectra show an upfield shift of the phosphorus in 1b (δ 8.3 ppm) from the phosphonium salt 4b (δ 23.5 ppm). This is consistent with the generation of a 5-coordinate phosphorus, although it is not as shielded as previously noted for a phospha-münchnone (δ ca. −17 ppm).14a,c In addition, 13C NMR data reveal that the carbonyl unit (δ 170.3 ppm) is close to the typical region for esters, although with coupling to phosphorus (JC–P = 4.7 Hz). Together, these data suggest that the interaction between phosphorus and the ester oxygen is weak, and does not significantly perturb the carbonyl unit. Nevertheless, the ability of 1b to react as a 1,3-dipole can be seen in its reaction with 1,1-chlorocyanoethylene, which leads to the formation of what appear to be two isomeric compounds, as determined by in situ1H, 19F and 31P NMR data.24 One of these can be isolated by chromatography and has been characterized as the bicyclic adduct shown in Fig. 2b. Of note, 31P NMR analysis of 5b shows a signal at −11.3 ppm, consistent with a five-coordinate phosphorus. The two diastereotopic methylene protons of the former alkene at δ 3.57 and 3.45 ppm are upfield from chlorocyanoethylene, as expected for cycloaddition, and show now strong coupling to phosphorus (JH–P = 67.3 Hz and 54.3 Hz), while 13C NMR analysis shows a strong upfield shift of the former carbonyl unit to δ 109.8 ppm. NOE spectra also show the phosphorus phenyl substituent oriented near the p-fluorophenyl group, and away from the catechyl in the rigid bicyclic structure. All other NMR and HRMS data are consistent with the structure shown. Heating the 1,3-dipolar cycloaddition adduct 5b in the presence of a base leads to furan in 62% overall yield, demonstrating the intermediacy of 1,3-dipole 1b, and the cycloadduct 5b, in furan synthesis.
O, suggesting that the putative 1,3-dipole generated here is unstable. However, the stability of this intermediate can be enhanced by incorporating a sterically encumbered mesityl substituent on the acid chloride together with an electron deficient 4-fluorobenzaldehyde. The generation of this product by deprotonation with the LiHMDS base at room temperature leads to the formation of a bright red product 1b in 93% yield (Fig. 2b). Of note, the in situ31P NMR spectra show an upfield shift of the phosphorus in 1b (δ 8.3 ppm) from the phosphonium salt 4b (δ 23.5 ppm). This is consistent with the generation of a 5-coordinate phosphorus, although it is not as shielded as previously noted for a phospha-münchnone (δ ca. −17 ppm).14a,c In addition, 13C NMR data reveal that the carbonyl unit (δ 170.3 ppm) is close to the typical region for esters, although with coupling to phosphorus (JC–P = 4.7 Hz). Together, these data suggest that the interaction between phosphorus and the ester oxygen is weak, and does not significantly perturb the carbonyl unit. Nevertheless, the ability of 1b to react as a 1,3-dipole can be seen in its reaction with 1,1-chlorocyanoethylene, which leads to the formation of what appear to be two isomeric compounds, as determined by in situ1H, 19F and 31P NMR data.24 One of these can be isolated by chromatography and has been characterized as the bicyclic adduct shown in Fig. 2b. Of note, 31P NMR analysis of 5b shows a signal at −11.3 ppm, consistent with a five-coordinate phosphorus. The two diastereotopic methylene protons of the former alkene at δ 3.57 and 3.45 ppm are upfield from chlorocyanoethylene, as expected for cycloaddition, and show now strong coupling to phosphorus (JH–P = 67.3 Hz and 54.3 Hz), while 13C NMR analysis shows a strong upfield shift of the former carbonyl unit to δ 109.8 ppm. NOE spectra also show the phosphorus phenyl substituent oriented near the p-fluorophenyl group, and away from the catechyl in the rigid bicyclic structure. All other NMR and HRMS data are consistent with the structure shown. Heating the 1,3-dipolar cycloaddition adduct 5b in the presence of a base leads to furan in 62% overall yield, demonstrating the intermediacy of 1,3-dipole 1b, and the cycloadduct 5b, in furan synthesis.
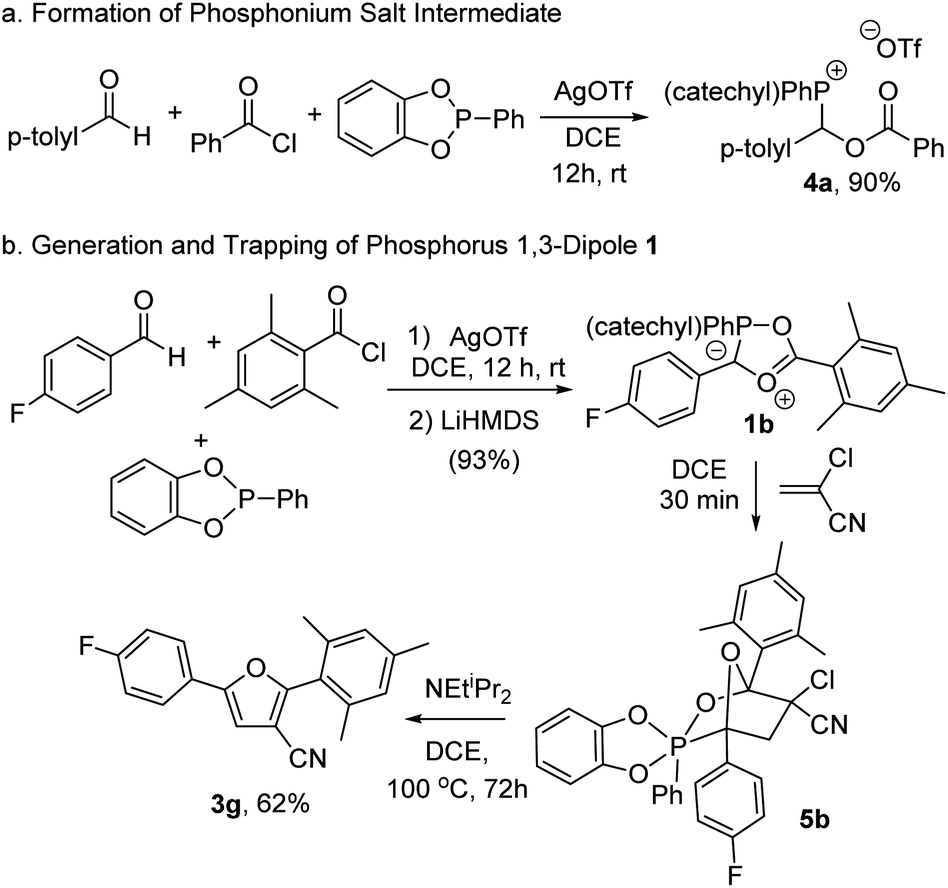 | ||
| Fig. 2 Characterization of the phosphorus-containing 1,3-dipole 1. | ||
Finally, the ability to generate the 1,3-dipole 1 in one pot from available reagents makes it straightforward to access new products. As an illustration, performing the reaction with available terephthaloyl chloride can allow the modular assembly of the novel oligomeric 6 in a one pot reaction incorporating an alternating and π-conjugated arene and furan heterocycles (Fig. 3a). Alternatively, other heterocycles can be generated by cycloaddition of new dipolarophiles. The latter can be seen with nitriles. The nitrile-tethered aldehyde 7 can be formed by O-alkylation of salicylaldehyde. The reaction of this aldehyde with benzoyl chloride and PhP(catechyl) to generate the 1,3-dipole leads to rapid cycloaddition to form the fused-ring oxazole 8 in two steps from available reagents (Fig. 3b). This represents what is to our knowledge a unique example of nitrile 1,3-dipolar cycloaddition to form oxazoles, and suggests that these 1,3-dipoles could prove to be of general utility in heterocycle formation.
 | ||
| Fig. 3 Synthesis of oligomeric furans and oxazoles via 1,3-dipolar cycloaddition with 1. | ||
Conclusions
In conclusion, we have described a modular method to construct furans from aldehydes, acyl chlorides and alkynes. This transformation proceeds via the formation of a new class of 1,3-dipoles, the phosphorus-containing 1. The latter is generated in a one pot reaction of aldehydes, acid chlorides and PhP(catechyl), and undergoes rapid and regioselective 1,3-dipolar cycloaddition with alkynes to assemble an array of substituted furans with control over each substituent. Considering the utility of 1,3-dipolar cycloaddition reactions, we anticipate that the ability to readily form these oxygen-based 1,3-dipoles should offer efficient access to a range of oxygen-containing heterocyclic products.Data availability
All experimental data are available in the ESI.†Author contribution
H.E. and C.Z. performed the research and data analysis described in the paper and conceived the ideas with B.A.A. B.A.A., H.E. and C.Z. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), McGill University (James McGill Research Fund), and the Fonds de recherche du Québec – Nature et Technologies (FQRNT) supported Centre for Green Chemistry and Catalysis for funding this research.Notes and references
- (a) M. Breugst and H. U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed; (b) A. Padwa,W. H. Pearson, Synthetic Applications of 1,3-dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Inc., 2002 CrossRef.
- (a) R. Ramapanicker, P. Chauhan, Click Chemistry: Mechanistic and Synthetic Perspectives,in Click Reactions in Organic Synthesis, ed. S. Chansekaran, Wiley-VCH, Weinheim, Germany, 2016, pp. 1–24 Search PubMed; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (c) S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC; (d) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (e) T. Cañeque, S. Müller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- For reviews: (a) H. L. Gingrich, J. S. Baum, MesoionicOxazolesin Oxazoles, The Chemistry of Heterocyclic Compounds, vol. 45, ed. I. J. Turchi, Wiley, New York, 1986, pp. 731–922 Search PubMed; (b) B. A. Arndtsen, Chem.–Eur. J., 2009, 15, 302–313 CrossRef CAS PubMed; (c) G. W. Gribble, Mesoionic Ring Systems, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products, ed. A. Padwa, W. H. Pearson, John Wiley & Sons, Inc, 2003, pp. 681–753 Search PubMed.
- For recent examples: (a) H. Erguven, D. C. Leitch, E. N. Keyzer and B. A. Arndtsen, Angew. Chem., Int. Ed., 2017, 56, 6078–6082 CrossRef CAS PubMed; (b) P. A. Champagne and H. N. Houk, J. Org. Chem., 2017, 82, 10980–10988 CrossRef CAS PubMed; (c) E. Yen-Pon, P. A. Champagne, L. Plougastel, S. Gabillet, P. Thuéry, M. Johnson, G. Muller, G. pieters, F. Taran, K. N. Houk and D. Audisio, J. Am. Chem. Soc., 2019, 141, 1435–1440 CrossRef CAS PubMed; (d) B. D. A. Shennan, P. W. Smith, Y. Ogura and D. J. Dixon, Chem. Sci., 2020, 11, 10354–10360 RSC; (e) M. A. Allen, R. A. Ivanovich and A. M. Beauchemin, Angew. Chem., Int. Ed., 2020, 59, 23188–23197 CrossRef CAS PubMed; (f) R. Guo, J. Jiang, C. Hu, L. L. Liu, P. Cui, M. Zhao, Z. Ke, C.-H. Tung and L. Kong, Chem. Sci., 2020, 11, 7053–7059 RSC; (g) D. Campeau, A. Pommainville and F. Gagosz, J. Am. Chem. Soc., 2021, 143, 9601–9611 CrossRef CAS PubMed; (h) T. Ezawa, Y. Sohtome, D. Hashizume, M. Adachi, M. Akakabe, H. Koshino and M. Sodeoka, J. Am. Chem. Soc., 2021, 143, 9094–9104 CrossRef CAS PubMed.
- (a) J. B. Sperry and D. L. Wright, Curr. Opin. Drug Discovery Dev., 2005, 8, 723–740 CAS; (b) Y.-J. Wu, Prog. Heterocycl.Chem., 2012, 24, 1–53 CrossRef.
- (a) A. Boto andL. Alvarez, Furan and Its Derivatives, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar andS. K. Chattopadhyay, 2011, pp. 97–152 Search PubMed; (b) B. A. Keay, J. M. Hopkins, P. W. Dibble, Furans and their Benzo Derivatives: Applications, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven andR. J. K. Taylor, Elsevier, Oxford, 2008, pp. 571–623 Search PubMed; (c) A. F. Pozharskii, A. T. Soldatenkov, and A. R. Katritzky, Heterocycles in Life and Society, ed. A.F. Pozharskii, A.T. Soldatenkov, A.R. Katritzky, 2011 CrossRef; (d) M. Saleem, H. J. Kim, M. S. Ali and Y. S. Lee, Nat. Prod. Rep., 2005, 22, 696–716 RSC.
- (a) H. Tsuji and E. Nakamura, Acc. Chem. Res., 2017, 50, 396–406 CrossRef CAS PubMed; (b) U. H. F. Bunz, Angew. Chem., Int. Ed., 2010, 49, 5037–5040 CrossRef CAS PubMed; (c) C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS PubMed; (d) J. T. Lin, P.-C. Chen, Y.-S. Yen, Y.-C. Hsu, H.-H. Chou and M.-C. P. Yeh, Org. Lett., 2009, 11, 97–100 CrossRef CAS PubMed.
- (a) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef CAS; (b) M. Pedro, T. Tomas, J. I. Delso and M. Rosa, Curr. Org. Chem., 2007, 11, 1076–1091 CrossRef; (c) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS PubMed.
- (a) M. Hamaguchi and T. Nagai, J. Chem. Soc., Chem. Commun., 1985, 4, 190–191 RSC; (b) M. Hamaguchi and T. Nagai, J. Chem. Soc., Chem. Commun., 1985, 19, 1319–1321 RSC; (c) M. Hamaguchi, N. Tomida, Y. Iyama and T. Oshima, J. Org. Chem., 2006, 71, 5162–5170 CrossRef CAS PubMed.
- (a) A. Padwa, Acc. Chem. Res., 1991, 24, 22–28 CrossRef CAS; (b) M. P. Doyle, D. C. Forbes, M. N. Protopopova, S. A. Stanley, M. M. Vasbinder and K. R. Xavier, J. Org. Chem., 1997, 62, 7210–7215 CrossRef CAS PubMed; (c) D. M. Hodgson, A. H. Labande, S. Muthusamy, Cycloadditions of Carbonyl Ylides Derived from Diazocarbonyl Compounds, in Organic Reactions, John Wiley & Sons, Inc., 2013, pp. 133–496 Search PubMed; (d) R. Huisgen, Angew. Chem., Int. Ed., 1977, 16, 572–585 CrossRef; (e) W. Chen, X. Fu, L. Lin, X. Yuan, W. Luo, J. Feng, X. Liu and X. Feng, Chem. Commun., 2014, 50, 11480–11483 RSC.
- For reviews: (a) D. M. X. Donnelly, M. J. Meegan, Furans and their Benzo Derivatives: (iii) Synthesis and Applications,in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees,Pergamon, Oxford, 1984, pp. 657–712 Search PubMed; (b) B. A. Keay, Chem. Soc. Rev., 1999, 28, 209–215 RSC; (c) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955–2020 CrossRef CAS; (d) R. C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850–852 CrossRef CAS PubMed; (e) H. N. C. Wong, X.-L. Hou, K.-S. Yeung, H. Huang, Five-Membered Heterocycles: Furan,inModern Heterocyclic Chemistry, 2011, pp.533-592 Search PubMed; (f) W. J. Moran and A. Rodríguez, Org. Prep. Proced. Int., 2012, 44, 103–130 CrossRef CAS; (g) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- For selected recent examples: (a) A. W. Sromek, M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 10500–10501 CrossRef CAS PubMed; (b) H. Ila, O. Baron, A. J. Wagner and P. Knochel, Chem. Commun., 2006, 583–593 RSC; (c) J. Barluenga, L. Riesgo, R. Vicente, L. A. López and M. Tomás, J. Am. Chem. Soc., 2008, 130, 13528–13529 CrossRef CAS PubMed; (d) L. Melzig, C. B. Rauhut and P. Knochel, Chem. Commun., 2009, 3536–3538 RSC; (e) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766–5769 CrossRef CAS PubMed; (f) Y. Yang, J. Yao and Y. Zhang, Org. Lett., 2013, 15, 3206–3209 CrossRef CAS PubMed; (g) J. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2015, 54, 11107–11111 CrossRef CAS PubMed; (h) A. Dey, M. A. Ali, S. Jana and A. Hajra, J. Org. Chem., 2017, 82, 4812–4818 CrossRef CAS PubMed; (i) V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS PubMed; (j) H. Jin and A. Furstner, Angew. Chem., Int. Ed., 2020, 59, 13618–13622 CrossRef CAS PubMed; (k) F.-L. Haut, C. Habiger, L. A. Wein, M. Podewitz and T. Magauer, J. Am. Chem. Soc., 2021, 143, 1216–1223 CrossRef CAS PubMed.
- (a) S. F. Kirsch, Org. Biomol. Chem., 2006, 4, 2076–2080 RSC; (b) T.-T. Kao, S. E. Syu, Y.-W. Jhang and W. Lin, Org. Lett., 2010, 12, 3066–3069 CrossRef CAS PubMed; (c) K.-W. Chen, S.-E. Syu, Y.-J. Jang and W. Lin, Org. Biomol. Chem., 2011, 9, 2098–2106 RSC; (d) Y. Wang, Y.-C. Luo, X.-Q. Hu and P.-F. Xu, Org. Lett., 2011, 13, 5346–5349 CrossRef CAS PubMed; (e) J. hu, Y. Wei and X. Tong, Org. Lett., 2011, 13, 3068–3071 CrossRef CAS PubMed; (f) J. Wang, R. Zhou, Z.-R. He and Z. He, Eur. J. Org. Chem., 2012, 6033–6042 CrossRef CAS; (g) W. Zou, Z.-R. He and Z. He, Chin. J. Org. Chem., 2015, 35, 1729–1745 CrossRef; (h) K. Tateishi, Y. Matsumoto and A. Saito, Eur. J. Org. Chem., 2019, 5603–5609 CrossRef CAS.
- (a) D. J. St. Cyr and B. A. Arndtsen, J. Am. Chem. Soc., 2007, 129, 12366–12367 CrossRef PubMed; (b) E. H. Krenske, K. N. Houk, B. A. Arndtsen and D. J. St. Cyr, J. Am. Chem. Soc., 2008, 130, 10052–10053 CrossRef CAS PubMed; (c) D. J. St.-Cyr, M. S. T. Morin, F. Belanger-Gariepy, B. A. Arndtsen, E. H. Krenske and K. N. Houk, J. Org. Chem., 2010, 75, 4261–4273 CrossRef CAS PubMed; (d) S. Aly, M. Romashko and B. A. Arndtsen, J. Org. Chem., 2015, 80, 2709–2714 CrossRef CAS PubMed; (e) T. Parvin and L. H. Choudhury, Tetrahedron, 2011, 67, 8213–8228 CrossRef PubMed.
- For applications: (a) M. S. T. Morin and B. A. Arndtsen, Org. Lett., 2014, 16, 1056–1059 CrossRef CAS PubMed; (b) L. V. Kayser, M. Vollmer, M. Welnhofer, H. Krikcziokat, K. Meerholz and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 10516–10521 CrossRef CAS PubMed; (c) L. V. Kayser, E. M. Hartigan and B. A. Arndtsen, ACS Sustainable Chem. Eng., 2016, 4, 6263–6267 CrossRef CAS; (d) A. Moquin, R. Hanna, T. Liang, H. Erguven, E. R. Gran, B. A. Arndtsen, D. Maysinger and A. Kakkar, Chem. Commun., 2019, 55, 9829–9832 RSC.
- R. Appel and H. Mayr, J. Am. Chem. Soc., 2001, 133, 8240–8251 CrossRef PubMed.
- (a) R. Engel, Phosphorus Addition atsp2 Carbon, inOrganic Reactions, 2004, pp. 175–248 Search PubMed; (b) R. Engel,J. L. Cohen, Synthesis of Carbon–Phosphorus Bonds, CRC Press, Boca Raton, 2nd edn, 2003 CrossRef.
- (a) R. Adams and E. H. Vollweiler, J. Am. Chem. Soc., 1918, 40, 1732–1746 CrossRef CAS; (b) R. Adams and E. H. Vollweiler, J. Am. Chem. Soc., 1921, 43, 651–659 CrossRef; (c) T. S. Chou and P. Knochel, J. Org. Chem., 1990, 55, 4791–4793 CrossRef CAS; (d) W. Su and J. Can, J. Chem. Res., 2005, 88–90 CrossRef CAS.
- F. Effenberger, J. K. Eberhard and A. H. Maier, J. Am. Chem. Soc., 1996, 118, 12572–12579 CrossRef CAS.
- The reaction of aldehyde, acid chloride, silver triflate and phosphonite is rapid (<1 h), but generates a 10–20% unidentified impurity that slowly converts to 4 over 12 h.
- M. S. T. Morin, D. J. St-Cyr, B. A. Arndtsen, E. H. Krenske and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17349–17358 CrossRef CAS PubMed.
- (a) B. P. Das and D. W. Boykin, J. Med. Chem., 1977, 20, 531–536 CrossRef CAS PubMed; (b) D. W. Boykin, J. Braz. Chem. Soc., 2002, 13, 763–771 CrossRef CAS; (c) B. Sauer, T. S. Skinner-Adams, A. Bouchut, M. J. Chua, C. Pierrot, F. Erdmann, D. Robaa, M. Shmidt, J. Khalife, K. T. Andrews and W. Sippl, Eur. J. Med. Chem., 2017, 127, 22–40 CrossRef CAS PubMed.
- J. C. Tebby, Handbook of Phosphorus-31 Nuclear Magnetic Resonance Data, 1990, CRCPress, 2017 Search PubMed.
- See ESI† for in situ1H, 19F, and 31P NMR data.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04088j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |