
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the primary and secondary coordination spheres on nitric oxide adsorption and reactivity in cobalt(II)–triazolate frameworks†
Julia
Oktawiec
 a,
Henry Z. H.
Jiang
a,
Ari B.
Turkiewicz
a and
Jeffrey R.
Long
*abc
a,
Henry Z. H.
Jiang
a,
Ari B.
Turkiewicz
a and
Jeffrey R.
Long
*abc
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: jrlong@berkeley.edu
bDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, California 94720, USA
cMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 19th October 2021
Abstract
Nitric oxide (NO) is an important signaling molecule in biological systems, and as such, the ability of porous materials to reversibly adsorb NO is of interest for potential medical applications. Although certain metal–organic frameworks are known to bind NO reversibly at coordinatively unsaturated metal sites, the influence of the metal coordination environment on NO adsorption has not been studied in detail. Here, we examine NO adsorption in the frameworks Co2Cl2(bbta) (H2bbta = 1H,5H-benzo(1,2-d:4,5-d′)bistriazole) and Co2(OH)2(bbta) using gas adsorption, infrared spectroscopy, powder X-ray diffraction, and magnetometry. At room temperature, NO adsorbs reversibly in Co2Cl2(bbta) without electron transfer, with low temperature data supporting spin-crossover of the NO-bound cobalt(II) centers of the material. In contrast, adsorption of low pressures of NO in Co2(OH)2(bbta) is accompanied by charge transfer from the cobalt(II) centers to form a cobalt(III)–NO− adduct, as supported by diffraction and infrared spectroscopy data. At higher pressures of NO, characterization data indicate additional uptake of the gas and disproportionation of the bound NO to form a cobalt(III)–nitro (NO2−) species and N2O gas, a transformation that appears to be facilitated by secondary sphere hydrogen bonding interactions between the bound NO2− and framework hydroxo groups. These results provide a rare example of reductive NO binding in a cobalt-based metal–organic framework, and they demonstrate that NO uptake can be tuned by changing the primary and secondary coordination environment of the framework metal centers.
Introduction
Nitric oxide (NO) is recognized as a key gasotransmitter with a number of important physiological roles, including as the endothelium-derived relaxing factor and as a neurotransmitter involved in memory formation and stroke damage.1,2 Given its influence on vasodilation, administration of NO gas and NO donor drugs are an important therapeutic strategy, and NO gas administration is under investigation as a potential supportive treatment for coronaviruses, in particular SARS-CoV-2.3–6 The reactivity of NO with proteins containing metal cofactors is critical to the therapeutic effects of this gas.7–10 For example, metal-containing biomolecules, including guanylate cyclase,11 cytochrome c oxidase,12 hemoglobin,13,14 and cobalamin,15–18 bind nitric oxide at the metal site, an event that can either enable or interfere with critical metabolic pathways. In such metalloproteins, nitric oxide binding is facilitated by the low reduction potentials of the transition metal-containing cofactors to bind NO via an electron-transfer mechanism, generating an oxidized metal and a reduced NO adduct.7,14,19–21 However, because of the deleterious reactions that can occur in the presence of excess nitric oxide, careful and controlled delivery is an active area of research.22Porous solid-state materials have generated considerable interest as NO-releasing therapeutics due to their tunability and high surface areas, and metal–organic frameworks in particular represent a key class of materials under investigation.22–24 Composed of metal ion or cluster nodes and multitopic organic linkers, metal–organic frameworks display high degrees of thermal and chemical stability in addition to considerable chemical and structural variability.25,26 Adsorption of NO has been reported in a number of frameworks, including Cu3(btc)2 (HKUST-1; btc3− = 1,3,5-benzenetricarboxylate),27 M2(dobdc) (M = Mg, Co, Ni, and Zn; dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate),28–32 and Zr6O4(OH)4(bdc)6 (UiO-66; bdc2− = 1,4-benzenedicarboxylate) and its derivatives.33 Many of these materials physisorb NO at coordinatively-unsaturated metal centers, with little evidence of charge transfer. However, reduction of nitric oxide upon adsorption has been characterized in copper(I)- and iron(II)-containing frameworks, such as Cu2Zn3Cl2(btdd) (CuI-MFU-4l; H2btdd = bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin),34 CuI-ZrTpmC* ({[Zr6O4(OH)4(C6H5COO)4]3(Cu(CH3CN)TpmC*)8}, where TpmC*8 = 1,1′,1′′-methanetriyltris-(3,5-dimethyl-1H-pyrazole-4-carboxylic acid)),35 Fe2(dobdc),28 MIL-88(Fe) derivatives (Fe3OX(L), where X = F, Cl, or OH and L2− = fumarate),36 Fe(bddpi)(DMF) (bddpi2− = 2,6-bis(1H-pyrazolyl)-pyromellitic diimide; DMF = N,N′-dimethylformamide),37 and [Zn3FeO(bdc)3]8 (Fe-MOF-5).38,39 In the case of Fe2(dobdc) and MIL-88(Fe), the NO unit is formally retained upon adsorption and is released in the presence of humidity.28,36 In contrast, upon binding at the copper(I) sites in Cu-MFU-4l and CuI-ZrTpmC* or the iron(II) sites in Fe-MOF-5, three equivalents of NO disproportionate to form N2O and a metal-bound NO2− species.34,35,38 Characteristic of most of these examples is that NO binding is strengthened exclusively via an electron transfer mechanism, in the absence of contributions from favorable non-covalent interactions.
Recently, we demonstrated that the framework Co2(OH)2(bbta) (H2bbta = 1H,5H-benzo(1,2-d:4,5-d′)bistriazole; Fig. 1c) reversibly binds O2via electron transfer to form a cobalt(III)–superoxo adduct that is stabilized by hydrogen bonding with the bridging hydroxo groups of the framework.40 In contrast, the related framework Co2Cl2(bbta) (Fig. 1a) exhibits much weaker uptake of O2 with no associated electron transfer. Here, we show that synergistic electron transfer and stabilizing hydrogen bonding interactions can also promote strong binding of NO in Co2(OH)2(bbta) (Fig. 1d–f), and, at higher pressures, disproportionation of NO to yield a cobalt(III)–nitro (NO2−) species. Additionally, the framework Co2Cl2(bbta) is shown to exhibit strong and reversible adsorption of NO at 298 K (Fig. 1e and f), with evidence for some low-spin cobalt(II) character at low temperatures (100 K), due to back-bonding from bound NO. The unique binding of NO in these frameworks is characterized by gas adsorption, infrared spectroscopy, powder X-ray diffraction, and magnetometry measurements.
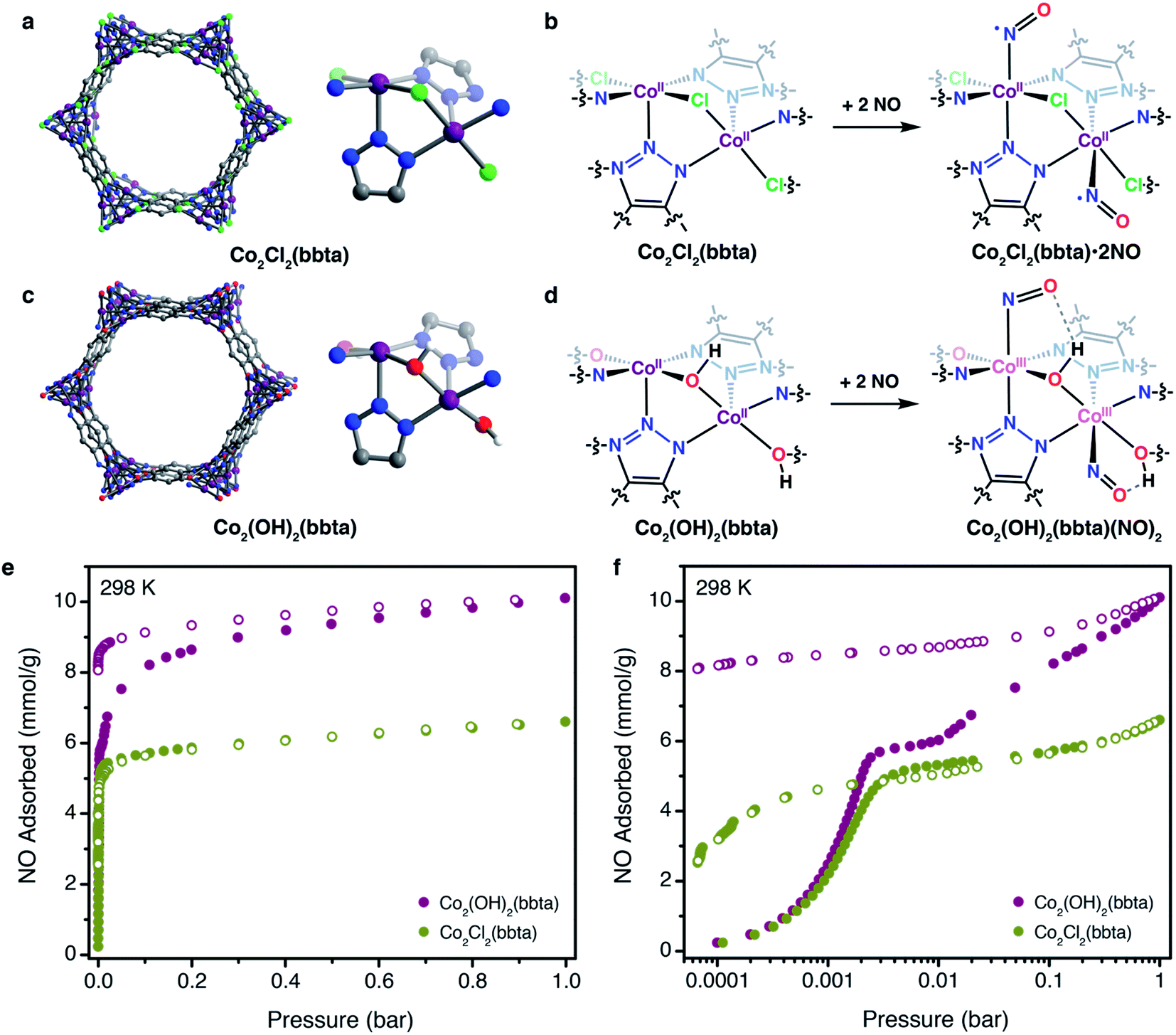 | ||
| Fig. 1 Structures of Co2Cl2(bbta) (a) and Co2(OH)2(bbta) (c), depicting the hexagonal framework pores and the local coordination environment of the cobalt centers in each material. Purple, red, blue, grey, and white spheres represent Co, O, N, C, and H atoms, respectively. Schemes illustrating NO binding to the cobalt(II) centers of Co2Cl2(bbta) (b) and Co2(OH)2(bbta) (d). In the latter material, the more electron-rich cobalt centers reduce NO upon binding, generating cobalt(III)–NO− adducts stabilized in part by a hydrogen bonding interactions with the framework bridging hydroxo groups, as indicated by spectroscopic and powder X-ray diffraction data. NO adsorption isotherms obtained for Co2Cl2(bbta) (green symbols) and Co2(OH)2(bbta) (purple symbols) at 298 K, with pressure shown on a linear (e) and logarithmic (f) scale. Closed and open circles represent adsorption and desorption points, respectively. | ||
Results and discussion
The frameworks Co2(OH)2(bbta) and Co2Cl2(bbta) have previously been studied as adsorbents for O2,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40,41 and CO2,42 as well as for the electrocatalytic oxidation of water to oxygen and photocatalytic reduction of CO2 to CO.43,44 These two isostructural frameworks contain hexagonal pores lined with chains of coordinatively unsaturated cobalt centers bridged by triazolate and chloride or hydroxide groups (Fig. 1a). The solvent molecules that bind these cobalt centers can be removed by heating under vacuum, allowing the resulting, five-coordinate cobalt ions to interact with adsorbates and show promising behavior for many applications. In the case of O2 binding, the strength of adsorption is dramatically enhanced when the framework bridging ligand is changed from chloride to hydroxide.40,41 This result is consistent with a more electron-rich ligand environment for the cobalt centers in Co2(OH)2(bbta) relative to those in Co2Cl2(bbta), which enables them to more readily reduce O2, although hydrogen-bond donation from the hydroxide ligand to the resulting superoxide also contributes significantly.45 Because NO, like O2, is capable of accepting an electron from a redox-active metal center,7 we sought to investigate how the electronic environment of the two frameworks may also give rise to distinct NO adsorption behavior.
40,41 and CO2,42 as well as for the electrocatalytic oxidation of water to oxygen and photocatalytic reduction of CO2 to CO.43,44 These two isostructural frameworks contain hexagonal pores lined with chains of coordinatively unsaturated cobalt centers bridged by triazolate and chloride or hydroxide groups (Fig. 1a). The solvent molecules that bind these cobalt centers can be removed by heating under vacuum, allowing the resulting, five-coordinate cobalt ions to interact with adsorbates and show promising behavior for many applications. In the case of O2 binding, the strength of adsorption is dramatically enhanced when the framework bridging ligand is changed from chloride to hydroxide.40,41 This result is consistent with a more electron-rich ligand environment for the cobalt centers in Co2(OH)2(bbta) relative to those in Co2Cl2(bbta), which enables them to more readily reduce O2, although hydrogen-bond donation from the hydroxide ligand to the resulting superoxide also contributes significantly.45 Because NO, like O2, is capable of accepting an electron from a redox-active metal center,7 we sought to investigate how the electronic environment of the two frameworks may also give rise to distinct NO adsorption behavior.
Notably, NO adsorption isotherms collected for both materials at 298 K exhibit an initial steep rise at low pressures, indicative of strong framework–gas interactions (Fig. 1e). At 0.1 bar, both materials adsorb approximately 92% of their theoretical capacity, assuming one molecule of NO per cobalt center (5.30 and 6.02 mmol g−1 for Co2Cl2(bbta) and Co2(OH)2(bbta), respectively; Fig. 1f). Both frameworks exhibit a much higher affinity for NO than O2,40 and the NO isotherm for Co2(OH)2(bbta) is only slightly steeper than that of Co2Cl2(bbta) at low pressures. While the uptake of both frameworks begins to plateau above 120 mbar, the uptake of Co2(OH)2(bbta) rises again to reach a capacity of 10.1 mmol g−1 (23.3 wt%) at 1 bar, one of the highest gravimetric capacities of any NO absorbent. This value can be contrasted to those previously observed, such as 6.5 mmol g−1 for Fe2(dobdc),28 ∼7.2 mmol g−1 Ni2(dobdc),23 and 7.1 mmol g−1 for UiO-66-NH2.33 Thus, its capacity corresponds to an unexpected 1.57 equiv. of NO per cobalt center, suggestive of additional reactivity beyond the binding of NO to the metal sites. In contrast, the NO capacity of Co2Cl2(bbta) is 6.6 mmol g−1 at 1 bar, which corresponds to 1.14 equiv. of NO gas per metal center. Interestingly, both frameworks also exhibit hysteresis upon desorption (Fig. 1f), although their recyclability of adsorption is drastically different: Co2Cl2(bbta) retains 97.4% of its adsorption capacity over three adsorption/desorption cycles with regeneration at 363 K, while the capacity of Co2(OH)2(bbta) is greatly reduced after one cycle, even with attempted regeneration at temperatures up to 423 K (see the ESI and Fig. S1–S3† for details).
Nitric oxide uptake in both frameworks was further characterized by powder X-ray diffraction, infrared spectroscopy, and SQUID magnetometry. The powder X-ray diffraction pattern for Co2Cl2(bbta) dosed with 2 mbar of NO gas at 298 K features changes in unit cell parameters and peak intensities relative to the desolvated framework, indicative of structural changes upon NO adsorption (Fig. S21 and S22†). Rietveld refinement yielded a structural model of NO-dosed Co2Cl2(bbta) (Co2Cl2(bbta)(NO)1.59), wherein NO is bound in a bent, end-on fashion with a Co–N–O angle of 136.8(6)°, a Co–NNO distance of 1.860(9) Å, and an N–O distance of 1.123(9) Å (Fig. 2a). The apparent compression of the N–O bond relative to that of free NO (1.154 Å) may be due to librational disorder. The unit cell volume of Co2Cl2(bbta) decreases only slightly by 98.3 Å3 (a 2.2% decrease) upon NO dosing, although the metal–ligand bonds increase slightly compared to those in the activated material. For example, two Co–Ntriazolate bond distances increase from 2.064(10) and 2.106(6) Å in Co2Cl2(bbta) to 2.094(5) and 2.135(9) Å in the NO-dosed framework, while the Co–Cl bond length increases from 2.377(3) to 2.3920(17) Å. Thus, the decrease in unit cell volume can be attributed primarily to a decrease in the Cl–Co–Cl bond angle from 179.59(14)° to 174.23(11)°.
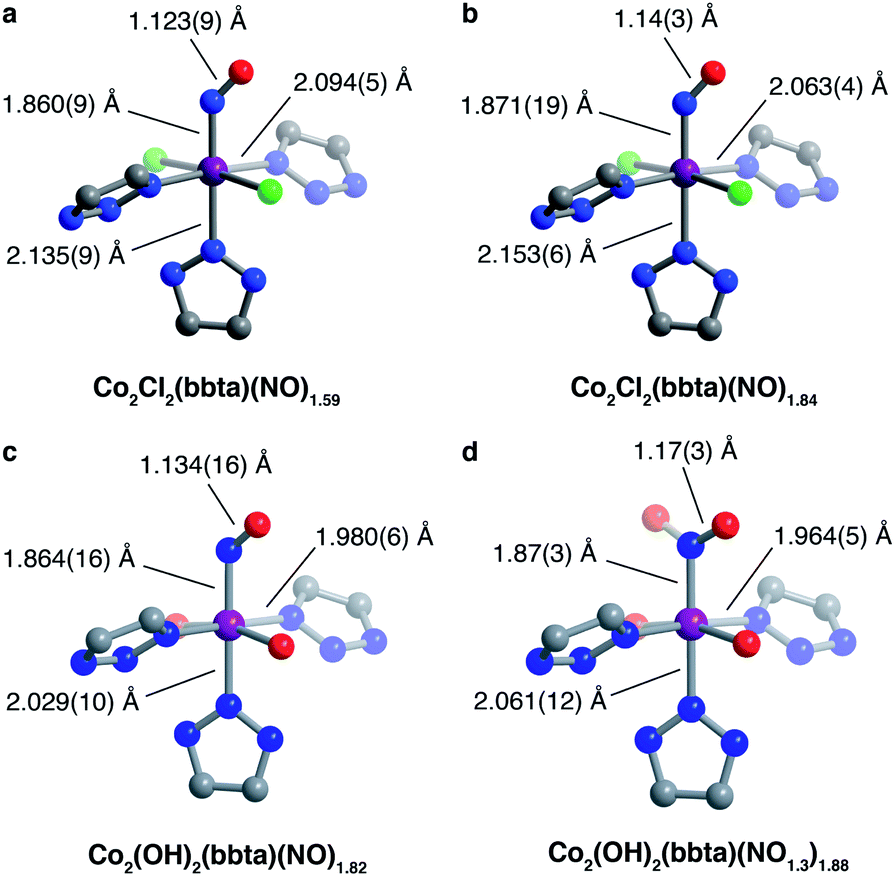 | ||
| Fig. 2 Structural models determined from analysis of powder X-ray diffraction data for Co2Cl2(bbta) dosed with 2 mbar NO gas at 298 K (a) and 100 K (b), as well as Co2(OH)2(bbta) dosed with 2 mbar (c) and 200 mbar NO gas (d) at 298 K. Purple, red, blue, green, and grey spheres represent Co, O, N, Cl, and C atoms respectively; H atoms are not shown as their positions could not be determined. In (a)–(c), the occupancy of the represented oxygen atom position is half of that of the NO-based nitrogen atom, and the position of a second oxygen atom with the same degree of occupancy (generated by symmetry) is not shown for clarity. For the structure of Co2(OH)2(bbta) dosed with 200 mbar NO, the occupancy of the NO oxygen atom increases to 56.1(4)% of the NO nitrogen atom occupancy, nearly 15% more than would be expected for the structure with bound NO. As a result, it is indicative of the presence of both bound NO and NO2− species. | ||
Interestingly, the structural changes upon NO binding do not appear to be related to changes in the cobalt oxidation or spin state, as the cobalt–ligand bond lengths of the framework are consistent with those of high-spin cobalt(II).46–48 However, upon slowly cooling to 100 K (Fig. 2b), the occupancy of the adsorbed NO species increases from 79.5(9)% to 91.8(12)%, concomitant with an elongation of the axial Co–Ntriazolate bond (by 0.018 Å, to 2.153(6) Å) and a contraction of the equatorial Co–Ntriazolate (by 0.031 Å, to 2.063(4) Å) and Co–Cl bonds (by 0.012 Å, to 2.380(3) Å). The changes in bond lengths suggest that the NO-bound cobalt centers may have partial low-spin character at low temperatures. Additionally, the elongation of the bond trans to the NO adduct is consistent with the strong trans influence of NO49 observed in other compounds, such as nitrosyl-cobalamin and Fe2(dobdc), where the metal–ligand bond trans to NO increases to more than 2.3 Å in length.18,28
In contrast, dosing a sample of desolvated Co2(OH)2(bbta) with 2 mbar of NO gas at 298 K results in a much more drastic decrease in the framework unit cell volume, from 4427.7(3) to 4101.9(3) Å3, equivalent to a 7.3% contraction. Rietveld refinement against the diffraction data (Fig. 2c and S23†) led to a structural model with the formula Co2(OH)2(bbta)(NO)1.82 featuring a cobalt-bound NO and bent Co–N–O angle. As a result of a crystallographic two-fold axis along the Co–NNO bond, the NO-based oxygen atom is disordered over two positions, each with 45.7(5)% occupancy. The structural parameters are indicative of the presence of a cobalt(III)–nitrosyl species (Fig. 2c).7 Specifically, the Co–Ntriazolate distances decrease from 2.102(4) and 2.066(17) Å in the bare framework to 1.980(6) and 2.029(10) Å in the NO-dosed material, supporting either a spin-state change or charge transfer to generate cobalt(III). The slight elongation of the N–O bond distance to 1.134(16) Å, the Co–NNO distance of 1.864(16) Å, and the Co–N–O angle of 128.5(11)° are also consistent with those observed for other six-coordinate {CoNO}8 systems.18,50,51 For example, characterization of nitrosyl-cobalamin derivatives via single-crystal X-ray diffraction revealed Co–NNO bond lengths ranging from 1.907(2) to 1.940(8) Å and Co–N–O angles ranging from 118.9(8)° to 120.2(8)°.18 A lengthening of the metal–ligand bond trans to the NO is not observed for NO-dosed Co2(OH)2(bbta). The absence of such a perturbation in this case might be due to an inability of the highly contracted lattice of Co2(OH)2(bbta)(NO)1.82 to support substantial and asymmetric deviations in bond length. Finally, the NO-based oxygen atom is resolved at a distance of 3.079(19) Å from the oxygen atom of the nearest bridging hydroxo group, less than that of the combined crystallographic van der Waals radii of two oxygen atoms (3.1 Å).52 This proximity indicates that hydrogen bonding may contribute to the adsorption of NO at low pressures,40,41,44 as it is known to promote the reduction of NO to NO−.53
Powder X-ray diffraction data were also collected for Co2(OH)2(bbta) under 200 mbar of NO to investigate the structural origins of the additional gas uptake at higher pressures (Fig. 2d and S24†). Rietveld refinement against this data revealed a structural model wherein the occupancy of the NO oxygen increased from 50% to 60% of that of the NO nitrogen. This change, in tandem with the elongation of the N–O bond distance to 1.17(3) Å, suggests that the metal centers may be partially occupied by nitrite (NO2−) as well as nitrosyl, owing to NO disproportionation upon gas uptake at higher pressures. In this structure, the NO oxygen is also much closer to the framework hydroxo group than in the 2 mbar structure, with an O(H)⋯ONO distance of 2.85(4) Å and a smaller Co–N–O angle of 116.0(19)° (Fig. 2d). These structural differences further indicate that at higher NO pressures, hydrogen bonding can significantly stabilize the adsorbed species in Co2(OH)2(bbta).
In situ infrared spectra were collected for NO-dosed samples of both frameworks to further elucidate changes upon NO adsorption. In the case of Co2Cl2(bbta), these data suggest that there is little electronic perturbation of the cobalt center upon NO binding, consistent with the diffraction data. For example, dosing with 1 mbar of NO gas results in the appearance of a peak at 1857 cm−1 that shifts to 1841 cm−1 under increasing pressures of NO up to 1 bar (Fig. S17†), energies that are only slightly different from the value for free NO (1876 cm−1). Additionally, the bands associated with framework vibrations do not shift significantly upon NO binding (Fig. S16†), signifying that adsorption does not substantially perturb the framework structure. While it was not possible to heat the sample in the IR spectrometer to fully desorb NO after dosing with 1 bar, evacuation at 298 K over the course of eight hours resulted in a spectrum resembling that of the sample when dosed with 1 mbar of NO (Fig. S19†), indicative of some degree of reversible adsorption.
Like the gas adsorption and powder X-ray diffraction data, the infrared spectrum of Co2(OH)2(bbta) changes more dramatically upon dosing with NO, consistent with a change in the oxidation state of the cobalt and similar to what occurs upon dosing the material with O2.40 In order to deconvolute the bands arising due to NO adducts, infrared spectra were collected upon dosing with 14NO (up to 750 mbar) and 15NO (up to 176 mbar) at 298 K (Fig. 3a, b and S9†). Under 11 mbar of 14NO, two new broad features appear at 1814 and 1652 cm−1 and these shift to 1780 and 1624 cm−1, respectively, when the framework is dosed with the same pressure of 15NO (Fig. S9†). These features were assigned as the υ(NO) band of multiple cobalt(III)–NO− adducts. At a pressure of 750 mbar 14NO, these bands shift to higher energies of 1844 and 1730 cm−1, respectively, indicating less perturbation of the NO adduct. The appearance of multiple NO-derived species and their subsequent perturbation at higher loadings is consistent with earlier studies performed on this framework,40 which indicated that the bridging nature of the ligands, as well as the close proximity of neighboring cobalt centers, lowered the reduction potential of cobalt(II) centers situated next to cobalt(III) sites that had been oxidized upon gas binding. As a result, a variety of bound superoxide species, depending upon O2 loading, were observed. We propose an analogous phenomenon here, in which the cobalt(III)–NO− species generated upon binding of NO (υNO = 1652 cm−1) results in a lower reduction potential of adjacent cobalt(II) centers. These cobalt(II) sites still bind NO (υNO = 1814 cm−1), but are not able to reduce the gas to the same extent. As the electron density of the framework decreases at higher pressures due to increased oxidant loading and the formation of NO2− species (see below), these bands further undergo a blue shift as both types of bound NO become less reduced. These less reduced species may not be as liable to disproportionate, contributing to their continued presence in the IR spectra. Additionally, we postulate that at 750 mbar, the band at 1844 cm−1 may also represent some portion of NO bound to cobalt(III) sites that evolved N2O by disproportionation and became coordinatively unsaturated, allowing them to interact with NO gas further by physisorption. The complexity of the infrared spectra, and the presence of multiple NO-derived species, is thus reasonable.
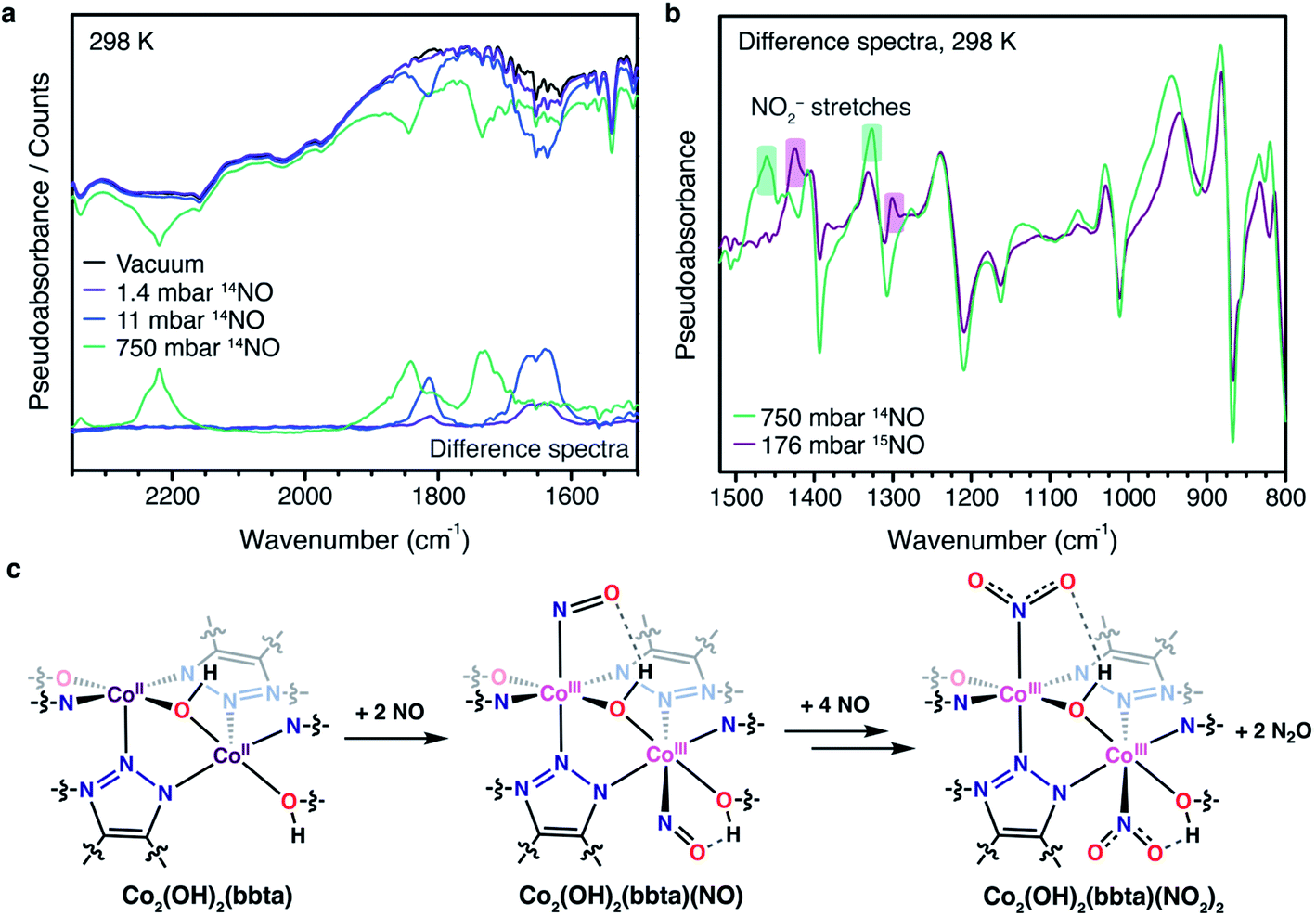 | ||
| Fig. 3 (a) Raw DRIFTS spectra collected at 298 K for desolvated Co2(OH)2(bbta) under vacuum (black trace) and dosed with 1.4, 11, and 750 mbar 14NO gas (blue, teal, and green traces, respectively) between 2350 and 1500 cm−1. This region features stretches that can be assigned to cobalt(III)–NO (1650–1840 cm−1) and N2O (2220 cm−1). The difference spectra relative to the desolvated framework for each gas pressure are shown at the bottom of the figure. (b) Difference spectra for Co2(OH)2(bbta) dosed with 750 mbar 14NO gas (green trace) and 176 mbar 15NO gas (purple trace) relative to the desolvated framework under vacuum, between 800 and 1500 cm−1. New bands at approximately 1460 and 1330 cm−1 in the spectrum for the 14NO-dosed sample and approximately 1424 and 1300 cm−1 in the spectrum for the 15NO-dosed sample are highlighted in green and purple, respectively, and can be distinguished from the non-highlighted framework shifts, which are consistent in the presence of both gases. We note that the data for 14NO and 15NO were collected using different pressures, which may account for the deviations in peak positions at lower wavenumbers. (c) Proposed scheme for NO uptake in Co2(OH)2(bbta), which proceeds first through initial binding of 1 equiv. of NO per cobalt center to form a cobalt(III)–nitrosyl species (NO pressures up to at least 11 mbar) followed by subsequent disproportionation in the presence of additional NO at higher pressures (176 or 200 mbar as indicated by the infrared and diffraction data, respectively) to form a cobalt(III)–bound NO2− and gaseous N2O. | ||
Additionally, at this higher pressure new bands appear at 1460 and 1326 cm−1 (1424 and 1300 cm−1, respectively, for the material dosed with 176 mbar 15NO), consistent with the asymmetric and symmetric NO stretching mode of an NO2− (nitro) moiety bound to cobalt(III) (Fig. 3b and S14†).54,55 Another new peak at 2220 cm−1 corresponds to free N2O (Fig. 3a and S11, S12†).56 Due to poor scattering at lower wavenumbers, it was not possible to observe the NO2 bend or Co–N stretch, which are predicted to appear at approximately 830 and 500 cm−1.54,55,57 We further note that there is no distinct isotopically-sensitive NO band between 1100 and 1050 cm−1, which would indicate the presence of a nitrito rather than a nitro moiety (i.e., O rather than N-linkage).57 This observation is in contrast to that recently noted in two copper(I)-based frameworks.34,35 Notable changes also occur to the υ(OH) stretch of the bridging hydroxo unit upon NO dosing: the band of the desolvated framework at 3647 cm−1 diminishes substantially upon dosing with NO pressures of 750 mbar (14NO) or 176 mbar (15NO), while a new set of bands appears at 3598 cm−1 under 11 mbar of 14/15NO (Fig. S8 and S10†). At the highest pressures examined for each gas, a second, even more redshifted band is present at 3517 cm−1 (Fig. S8†), which was assigned to formation of a hydrogen bond between the bridging hydroxo group and the adsorbed NO (or NO2−) species.
Dc magnetic susceptibility data were collected for Co2Cl2(bbta) and Co2(OH)2(bbta), both as desolvated and NO-dosed samples, to further probe changes occurring upon NO binding in each material. In the case of Co2Cl2(bbta), at 300 K the molar magnetic susceptibility temperature product (χMT) is 3.39 emu K mol−1 per cobalt atom, in accord with an S = 3/2 (g = 2.69) complex (Fig. 4a). Upon dosing with one equiv. of NO, the value of χMT at 300 K decreases to 1.23 emu K mol−1 per cobalt, in accord with an S = 1 (g = 2.22) complex. While the high degree of covalency in metal nitrosyl complexes complicates the assignment of formal oxidation states, and thus the interpretation of magnetic data,50 the S = 1 state is likely best described as arising from strong antiferromagnetic coupling between a high-spin S = 3/2 cobalt(II) center and an S = 1/2 NO molecule. Upon cooling from approximately 160 to 120 K, a decrease in the magnetic susceptibility is observed. This may be attributed to cobalt(II)-based spin-crossover from S = 3/2 to S = 1/2, in agreement with the variable-temperature powder X-ray diffraction data (Fig. S28†). A magnetic state arising from an uncoupled S = 1/2 (g = 2.00) cobalt(II) center and an S = 1/2 (g = 2.00) NO molecule gives an expected χMT value of 0.75 emu K mol−1 per cobalt, which is near the χMT value of 0.86 emu K mol−1 per cobalt at 120 K. Indeed, taken in concert with the gas adsorption, powder X-ray diffraction, and infrared spectroscopy data described above, NO appears to undergo only a minor perturbation upon binding to the metal sites of Co2Cl2(bbta), consistent with a lack of electron transfer from cobalt to NO.
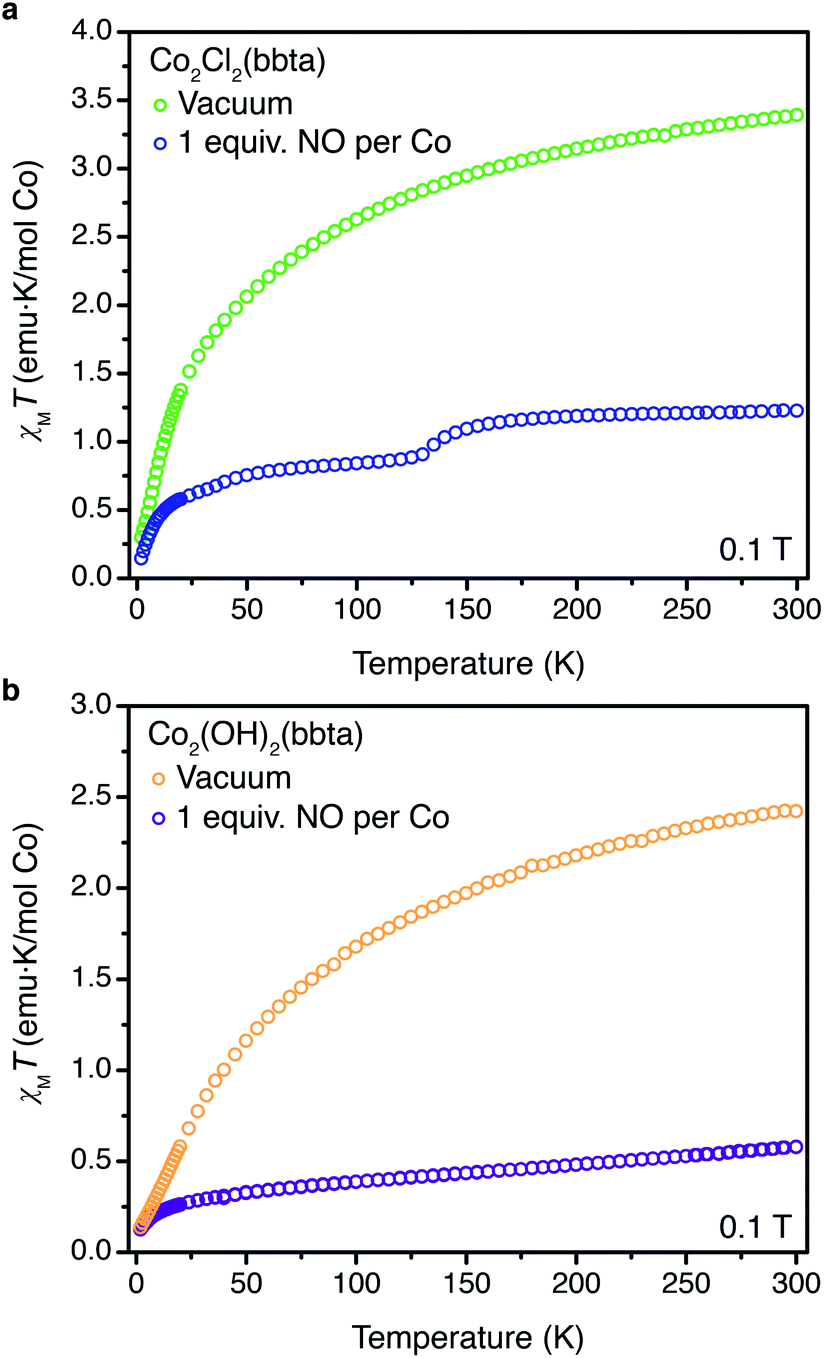 | ||
| Fig. 4 (a) Plot of χMT versus T for desolvated Co2Cl2(bbta) (green symbols) and desolvated Co2Cl2(bbta) dosed with 1 equiv. of NO per cobalt (blue symbols). (b) Plot of χMT versus T for desolvated Co2(OH)2(bbta) (yellow symbols) and desolvated Co2(OH)2(bbta) dosed with 1 equiv. of NO per cobalt (purple symbols). All data were collected under a dc field of 0.1 T. Data for desolvated Co2Cl2(bbta) and Co2(OH)2(bbta) are reproduced from ref. 40. | ||
Dc magnetic susceptibility data collected for NO-dosed Co2(OH)2(bbta) are also distinct from data for the desolvated framework (Fig. 4b). At 300 K, the desolvated framework has a χMT value of 2.42 emu K mol−1 per cobalt atom, corresponding to an S = 3/2 (g = 2.27) complex. The increased ligand field strength of the hydroxide ligand set is likely responsible for a decreased orbital contribution to the magnetic moment and a lower g-value relative to the desolvated Co2Cl2(bbta) framework.48 Dosing Co2(OH)2(bbta) with one equiv. of NO resulted in a significant decrease in χMT at 300 K, from 2.42 to 0.575 emu K mol−1 per cobalt. At 300 K, the moment of the NO-dosed framework is lower than the value predicted for both a coupled or uncoupled high-spin S = 3/2 cobalt(II) and an S = 1/2 NO unit (1 and 2.25 emu K mol−1, respectively) as well as that predicted for an S = 0 cobalt(III) center and an S = 1 NO− (1 emu K mol−1). Measurements at multiple applied fields reveal no evidence for temperature-independent paramagnetism or ferromagnetic impurities (Fig. S32†). Taken together with the clear shift of the υ(NO) and framework-based bands in the infrared spectroscopy data and the changes in structural parameters of both the framework and NO adduct in the powder X-ray diffraction data, the magnetic data generally support the reduction of NO by Co2(OH)2(bbta) upon adsorption. Indeed, the lower than expected moment may be due to further disproportionation of cobalt(III)-bound NO− to form cobalt(III)-bound NO2− and N2O gas (see below).
Overall, the foregoing data reveal that adsorption of NO in Co2(OH)2(bbta) is accompanied by more drastic structural and electronic changes than in the case of Co2Cl2(bbta). These changes are indicative of electron transfer from cobalt(II) to NO in Co2(OH)2(bbta) and are consistent with the more basic ligand set in this framework relative to Co2Cl2(bbta), which can stabilize cobalt(III) over cobalt(II).58 Significantly, as evidenced by infrared spectroscopy data at pressures above 176 mbar and diffraction data obtained at 200 mbar NO, charge transfer to adsorbed NO appears to further facilitate some degree of disproportionation to form cobalt(III)-bound NO2− and N2O gas. In a gas adsorption isotherm measurement, the formed N2O is likely to desorb into the headspace of the sample holder such that, for a given pressure, the equivalency of NO as measured by the gas adsorption analyzer will appear less than the actual NO that is required to generate the corresponding proportion of cobalt(III)–NO2− adducts. Taking this into account, and assuming that at 1 bar of NO the adsorbed species is exclusively NO2−, a capacity of 10.1 mmol g−1 (1.57 equiv. NO per Co, Fig. 1e and f) would suggest as many as 78.5% of the cobalt sites have reacted with coordinated NO to form bound NO2− and gaseous N2O, a similar level of oxidative reactivity to that observed previously with O2.33
The occurrence of a disproportionation reaction at higher NO pressures also explains why Co2(OH)2(bbta) cannot be fully regenerated after NO uptake at pressures beyond a few hundred mbar. Indeed, attempted regeneration of a sample of Co2(OH)2(bbta) following collection of adsorption data yielded a crystalline solid with a powder X-ray diffraction pattern distinct from that of desolvated Co2(OH)2(bbta) (Fig. S27†). In particular, the resulting unit cell volume is contracted relative to that of the parent desolvated material, which indicates that the material remains partially oxidized, as would be expected upon formation of cobalt(III)-bound NO2− and N2O gas. While a satisfactory Rietveld refinement could not be obtained, Fourier difference maps suggest residual electron density over the metal centers and near the hydroxo group. These data are suggestive of bound NO2− species that would likely display strong interactions with the bridging hydroxo groups. Additionally, these differences are consistent with those observed in the infrared spectra of the framework upon NO dosing at high pressures, such as the generation of a cobalt(III)–nitro species and dramatic shifts in the hydroxo group bands. Thus, hydrogen bonding likely plays an important role in facilitating NO disproportionation, much like with other examples of nearby Lewis acids and hydrogen bond donors aiding in transition metal complex reactivity with NO.59–61 In particular, the hydrogen bond may help to further polarize the NO adduct, promoting its reaction with additional equivalents of nitric oxide to form a putative Co(N2O2) species. We postulate that the cobalt centers are then close enough across the pore to enable Co(N2O2) and Co(NO) species to interact and disproportionate, generating N2O gas and cobalt(III)-bound NO2− (Fig. S30†).
Conclusions
The foregoing results demonstrate that both Co2Cl2(bbta) and Co2(OH)2(bbta) are capable of strongly binding NO, and notably these materials constitute only the second and third examples of cobalt-based metal–organic frameworks for NO capture. In particular, Co2Cl2(bbta) displays reversible nitric oxide binding and a high adsorption capacity at room temperature, making it a promising material for nitric oxide delivery. Additionally, data collected on NO-dosed Co2Cl2(bbta) at low temperatures suggest spin-crossover behavior to afford low-spin NO-bound cobalt(II). We note that while data on the toxicity of Co2Cl2(bbta) and Co2(OH)2(bbta) are unavailable, one study that examined the effects of a wide range of metal–organic frameworks in human cell lines and zebrafish embryo found that the cobalt-based framework Co2(dobdc) showed minimal toxicity in vitro and in vivo, and that health effects of frameworks in vitro and in vivo appeared to be related to metal ion identity.62 These results suggest that Co2Cl2(bbta) might be similarly benign.The introduction of a more basic bridging hydroxo ligand in the material Co2(OH)2(bbta) promotes charge transfer from cobalt(II) to NO, and at higher pressures (>0.1 bar) of NO, disproportionation is clearly favored, with formation of N2O and generation of cobalt(III)-nitro species, as observed by in situ infrared spectroscopy. Additionally, powder X-ray diffraction and spectroscopic data indicate that the hydroxo moiety of the framework engages in hydrogen bonding to the adsorbed NO, which seemingly promotes the observed disproportionation reactivity at higher pressures. As in the case of Fe-MOF-5 and Cu-MFU-4l, it is anticipated that further study of NO adsorption in this material at lower temperatures, as well as computational validation, could elucidate the identities of intermediates of NO disproportion (i.e., Co(N2O2)), to corroborate the mechanism of NO disproportionation in this material. This knowledge would be useful for tuning and controlling denitrification reactions within metal–organic frameworks,37,63 and future studies could contribute critical insights in this regard.21,34,35,64–67
Data availability
Full experimental details, supplementary figures and tables, gas sorption data, refinement information from powder X-ray diffraction analyses, infrared spectra, and magnetometry data are provided in the ESI. CCDC: 2098007–2098011.†Author contributions
J. O. and J. R. L. formulated the project. J. O. synthesized the materials and measured and analyzed the gas adsorption data. J. O. and H. Z. H. J. collected the powder X-ray diffraction data, and J. O. analyzed the data. H. Z. H. J. collected and analyzed the infrared spectra, with contributions from J. O. A. B. T. collected and analyzed the magnetic susceptibility data. J. O. and J. R. L. wrote the paper, and all authors contributed to revising the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy Office of Basic Energy Sciences (DE-SC0019992). Powder X-ray diffraction data were collected on Beamline 17-BM-B at the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated by Argonne National Laboratory, supported by the U.S. Department of Energy Office of Basic Energy Sciences (DE-AC02-06CH11357). We additionally thank the National Science Foundation for graduate fellowship support of J. O. and A. B. T. (DGE 1752814), Dr Andrey Yakovenko, Dr Wenqian Xu, Dr Benjamin Trump, and Maria Paley for assistance with powder X-ray diffraction measurements, Dr Miguel Gonzalez for assistance with manifold construction, Dr Andy Nguyen for helpful discussions, and Dr David Harris and Dr Katie Meihaus for editorial assistance.References
- E. Culotta and D. E. Koshland, Science, 1992, 258, 1861–1865 CrossRef PubMed.
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265–9269 CrossRef CAS PubMed.
- M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305–321 CrossRef CAS PubMed.
- J. Martel, Y.-F. Ko, J. D. Young and D. M. Ojcius, Microbes Infect., 2020, 22, 168–171 CrossRef CAS PubMed.
- L. Chen, P. Liu, H. Gao, B. Sun, D. Chao, F. Wang, Y. Zhu, G. Hedenstierna and C. G. Wang, Clin. Infect. Dis., 2004, 39, 1531–1535 CrossRef CAS PubMed.
- S. Åkerström, V. Gunalan, C. T. Keng, Y. J. Tan and A. Mirazimi, Virology, 2009, 395, 1–9 CrossRef PubMed.
- J. A. McCleverty, Chem. Rev., 2004, 104, 403–418 CrossRef CAS PubMed.
- J. A. McCleverty, Chem. Rev., 1979, 79, 53–76 CrossRef CAS.
- A. R. Butler and I. L. Megson, Chem. Rev., 2002, 102, 1155–1165 CrossRef CAS PubMed.
- R. Radi, Chem. Res. Toxicol., 1996, 9, 828–835 Search PubMed.
- S. H. Francis, J. L. Busch and J. D. Corbin, Pharmacol. Rev., 2010, 62, 525–563 CrossRef CAS PubMed.
- G. C. Brown, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 46–57 CrossRef CAS.
- R. Gardiner, T. G. Traylor, V. S. Sharma and H. Mizukami, Biochemistry, 1987, 26, 3837–3843 CrossRef PubMed.
- J. F. Deatherage and K. Moffat, J. Mol. Biol., 1979, 134, 401–417 CrossRef CAS PubMed.
- M. Brouwer, W. Chamulitrat, G. Ferruzzi, D. L. Sauls and J. B. Weinberg, Blood, 1996, 88, 1857–1864 CrossRef CAS PubMed.
- L. Hannibal, C. A. Smith, D. W. Jacobsen and N. E. Brasch, Angew. Chem., Int. Ed., 2007, 46, 5140–5143 CrossRef PubMed.
- M. Wolak, A. Zahl, T. Schneppensieper, G. Stochel and R. Van Eldik, J. Am. Chem. Soc., 2001, 123, 9780–9791 CrossRef CAS PubMed.
- H. A. Hassanin, M. F. El-Shahat, S. DeBeer, C. A. Smith and N. E. Brasch, Dalton Trans., 2010, 39, 10626 RSC.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed.
- S. Zhang, M. M. Melzer, S. N. Sen, N. Çelebi-Ölçüm and T. H. Warren, Nat. Chem., 2016, 8, 663–669 CrossRef CAS PubMed.
- P. Tavares, A. S. Pereira, J. J. G. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087–2100 CrossRef CAS PubMed.
- R. V. Pinto and M. L. Pinto, in Therapeutic Application of Nitric Oxide in Cancer and Inflammatory Disorders, Elsevier, 2019, pp. 277–305 Search PubMed.
- N. J. Hinks, A. C. McKinlay, B. Xiao, P. S. Wheatley and R. E. Morris, Microporous Mesoporous Mater., 2010, 129, 330–334 CrossRef CAS.
- T. Yang, A. N. Zelikin and R. Chandrawati, Adv. Sci., 2018, 5, 1701043 CrossRef PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, S. Chavan, P. S. Wheatley, J. M. Zadrozny, R. Morris, C. M. Brown, C. Lamberti, S. Bordiga and J. R. Long, J. Am. Chem. Soc., 2015, 137, 3466–3469 CrossRef CAS PubMed.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson, R. E. Morris, A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS PubMed.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059–14067 RSC.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, R. Castledine, N. Parkinson, I. Haley and R. E. Morris, Dalton Trans., 2016, 45, 618–629 RSC.
- R. R. Haikal, C. Hua, J. J. Perry, D. O'Nolan, I. Syed, A. Kumar, A. H. Chester, M. J. Zaworotko, M. H. Yacoub and M. H. Alkordi, ACS Appl. Mater. Interfaces, 2017, 9, 43520–43528 CrossRef CAS PubMed.
- A. M. Wright, C. Sun and M. Dinca, J. Am. Chem. Soc., 2021, 143, 681–686 CrossRef CAS PubMed.
- C. Sun, L. Yang, M. A. Ortuño, A. M. Wright, T. Chen, A. R. Head, N. López and M. Dincă, Angew. Chem., Int. Ed., 2021, 2–8 Search PubMed.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592–1599 CrossRef CAS.
- Z. Cai, W. Tao, C. E. Moore, S. Zhang and C. R. Wade, Angew. Chem., Int. Ed., 2021, 2–7 Search PubMed.
- C. K. Brozek, J. T. Miller, S. A. Stoian and M. Dincă, J. Am. Chem. Soc., 2015, 137, 7495–7501 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincǎ, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- J. Oktawiec, H. Z. H. Jiang, J. G. Vitillo, D. A. Reed, L. E. Darago, B. A. Trump, V. Bernales, H. Li, K. A. Colwell, H. Furukawa, C. M. Brown, L. Gagliardi and J. R. Long, Nat. Commun., 2020, 11, 3087 CrossRef CAS PubMed.
- A. S. Rosen, M. R. Mian, T. Islamoglu, H. Chen, O. K. Farha, J. M. Notestein and R. Q. Snurr, J. Am. Chem. Soc., 2020, 142, 4317–4328 CrossRef CAS PubMed.
- P. Q. Liao, H. Chen, D. D. Zhou, S. Y. Liu, C. T. He, Z. Rui, H. Ji, J. P. Zhang and X. M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- X. F. Lu, P. Q. Liao, J. W. Wang, J. X. Wu, X. W. Chen, C. T. He, J. P. Zhang, G. R. Li and X. M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- Y. Wang, N. Y. Huang, J. Q. Shen, P. Q. Liao, X. M. Chen and J. P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- R. D. Jones, D. A. Summerville and F. Basolo, Chem. Rev., 1979, 79, 139–179 CrossRef CAS.
- H. A. Goodwin, in Topics in Current Chemistry, 2004, pp. 23–47 Search PubMed.
- P. Thuery, J. Zarembowitch, A. Michalowicz and O. Kahn, Inorg. Chem., 1987, 26, 851–855 CrossRef CAS.
- Z. Dori and H. B. Gray, Inorg. Chem., 1968, 7, 889–892 CrossRef CAS.
- T. G. Traylor and V. S. Sharma, Biochemistry, 1992, 31, 2847–2849 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- Here is referenced notation developed by Enemark and Feltham (50) for NO-bound transition metal complexes, given that NO is a non-innocent ligand capable of multiple possible oxidation states. Specifically, {M(NO)x}n, where M is the element, n is the sum of the initial metal d and the NO π* orbitals, and x is the number of NO ligands.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. D. Bartberger, W. Liu, E. Ford, K. M. Miranda, C. Switzer, J. M. Fukuto, P. J. Farmer, D. A. Wink and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10958–10963 CrossRef CAS PubMed.
- K. Nakamoto, J. Fujita and H. Murata, J. Am. Chem. Soc., 1958, 80, 4817–4823 CrossRef CAS.
- R. B. Penland, T. J. Lane and J. V. Quagliano, J. Am. Chem. Soc., 1956, 78, 887–889 CrossRef CAS.
- R. L. Hudson, M. J. Loeffler and P. A. Gerakines, J. Chem. Phys., 2017, 146, 1–9 CrossRef PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 2009, 1–273 Search PubMed.
- E. C. Niederhoffer, J. H. Timmons and A. E. Martell, Chem. Rev., 1984, 84, 137–203 CrossRef CAS.
- E. G. Abucayon, R. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, Angew. Chem., Int. Ed., 2019, 2–8 Search PubMed.
- C. H. Chuang, W. F. Liaw and C. H. Hung, Angew. Chem., Int. Ed., 2016, 55, 5190–5194 CrossRef CAS PubMed.
- G. B. Wijeratne, M. Bhadra, M. A. Siegler and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 17962–17967 CrossRef CAS PubMed.
- A. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Lorenzo, I. Imaz and D. Maspoch, Chem.–Eur. J., 2015, 21, 2508–2518 CrossRef CAS PubMed.
- X. Han, S. Yang and M. Schröder, Nat. Rev. Chem., 2019, 3, 108–118 CrossRef CAS.
- J. Jover, C. K. Brozek, M. Dincǎ and N. López, Chem. Mater., 2019, 31, 8875–8885 CrossRef CAS.
- A. Shiotari, S. Hatta, H. Okuyama and T. Aruga, Chem. Sci., 2014, 5, 922–926 RSC.
- N. Yeung, Y. W. Lin, Y. G. Gao, X. Zhao, B. S. Russell, L. Lei, K. D. Miner, H. Robinson and Y. Lu, Nature, 2009, 462, 1079–1082 CrossRef CAS PubMed.
- S. Matsumoto, K. Yokota, H. Doi, M. Kimura, K. Sekizawa and S. Kasahara, Catal. Today, 1994, 22, 127–146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2098007–2098011. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03994f |
| This journal is © The Royal Society of Chemistry 2021 |