
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConverting molecular luminescence to ultralong room-temperature phosphorescence via the excited state modulation of sulfone-containing heteroaromatics†
Zetong
Ma
a,
Zhiqiang
Yang
c,
Lan
Mu
 a,
Lisong
Deng
a,
Liangjian
Chen
a,
Bohan
Wang
a,
Xianfeng
Qiao
a,
Dehua
Hu
*ab,
Bing
Yang
c,
Dongge
Ma
a,
Junbiao
Peng
a and
Yuguang
Ma
*a
a,
Lisong
Deng
a,
Liangjian
Chen
a,
Bohan
Wang
a,
Xianfeng
Qiao
a,
Dehua
Hu
*ab,
Bing
Yang
c,
Dongge
Ma
a,
Junbiao
Peng
a and
Yuguang
Ma
*a
aInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: msdhhu@scut.edu.cn; ygma@scut.edu.cn
bSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China
cState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun 130012, China
First published on 19th October 2021
Abstract
Manipulating the molecular orbital properties of excited states and the subsequent relaxation processes can greatly alter the emission behaviors of luminophores. Herein we report a vivid example of this, with luminescence conversion from thermally activated delayed fluorescence (TADF) to ultralong room-temperature phosphorescence (URTP) via a facile substituent effect on a rigid benzothiazino phenothiazine tetraoxide (BTPO) core. Pristine BTPO with multiple heteroatoms shows obvious intramolecular charge transfer (ICT) excited states with small exchange energy, featuring TADF. Via delicately functionalizing the BTPO core with peripheral moieties, the excited states of the BTPO derivatives become a hybridized local and charge transfer (HLCT) state in the S1 state and a local excitation (LE) dominated HLCT state in the T1 state, with enlarged energy bandgaps. Upon dispersion in a polymer matrix, the BTPO derivatives exhibit a persistent bright green afterglow with long lifetimes of up to 822 ms and decent quantum yields of up to 11.6%.
Introduction
A molecule excited by photons usually loses its energy through various relaxation processes, including internal conversion (IC), intersystem crossing (ISC), energy transfer, and radiative emission. Manipulating these competitive relaxation processes to obtain a certain photophysical property can make molecules suitable for targeted applications.1–3 For example, reversed intersystem crossing (RISC) from a triplet to a singlet state (both from the lowest triplet and high-lying triplet states) has been purposively enhanced via molecule design to realize 100% exciton utilization in organic light-emitting diodes (OLEDs).4–7 In addition, organic room-temperature phosphorescence (RTP), radiative emission from a triplet state promoted via enhancing the singlet–triplet ISC process, is also of considerable interest due to its innovative application prospects in the fields of bioimaging, information encryption, displays, etc.8–12 In particular, phosphors exhibiting ultralong room-temperature phosphorescence (URTP) with a luminescence lifetime of over 0.1 s, also referred to as afterglow or persistent luminescence, have gained increased attention because of their revolutionary potential in terms of naked-eye detection13. Nevertheless, such organic phosphors often present moderate phosphorescence quantum efficiency of less than 10%, which indicates the urgent need to develop high performance URTP materials.11In accordance with the Jablonski diagram and emission mechanism of phosphorescence, two key factors should be taken into consideration in order to obtain highly efficient RTP emitters:14 (i) strengthening the ISC from the excited singlet state (Sm) to the excited triplet state (Tn) via efficient spin–orbit coupling (SOC) and (ii) suppressing the nonradiative dissipation of long-lived triplets caused by vibration, molecular oxygen quenching, or other factors. In this regard, molecular design strategies based on halogen bonding,15–17 n–π transitions involving heteroatoms (S, P, etc.),18–20 and H-aggregation,21,22 and RTP reinforcement approaches based on crystal engineering,23–25 host–guest systems,26–28 copolymerization,29,30 and supramolecular interactions31,32 have been developed and have allowed the more-effective realization of pure organic RTP.
Though great achievements relating to RTP have been achieved through crystal engineering, one of the most commonly adopted methods, the practical application of resultant phosphors is largely hindered by the repeatability of crystal synthesis and the crystal quality. Very recently, Bin Liu et al. found that carbazole isomers existing as impurities in commercial carbazole sources are responsible for the ultralong organic phosphorescence of some carbazole-derivative crystalline solids.33 These impurities still exist even after recrystallization. In contrast, embedding monomeric phosphors into amorphous rigid polymer matrices, for instance polymethyl methacrylate (PMMA),34–40 polyvinyl alcohol (PVA),41–43 and Zeonex,44–46 provides a more facile approach for device fabrication and processing.47 The rigid polymer matrix environment minimizes phosphor energy dissipation via reducing molecular vibration and rotation and intermolecular collisions, resulting in effective RTP. In addition, through the appropriate selection of polymers and the design of device structures, the quenching effects of oxygen and moisture from the ambient environment can be largely diminished, resulting in triplet excitons with prolonged lifetimes.37,48 Also, the photophysical properties of monomers are better maintained at a low doping ratio in the polymer matrix. So far, this strategy has been well utilized in some monomeric phosphorescence systems, such as aromatic phosphonate,49 thianthrene,50etc.
Herein, we demonstrate the URTP properties of a new category of organic derivatives based on a hetero[4]helicene of benzo[5,6][1,4]thiazino[2,3,4-kl]phenothiazine 5,5,9,9-tetraoxide (BTPO) in amorphous polymer matrices. Through the delicate functionalization of the BTPO core, three compounds with different electron-donating/-withdrawing substituent groups (benzonitrile (BCN), phenylcarbazole (BCZ), and triphenylamine (TPA)) were designed and synthesized, as shown in Scheme 1. The photophysical results confirm that the BTPO core presents TADF with a small energy bandgap (ΔEST) of 0.16 eV between the lowest triplet state (T1) and the lowest singlet state (S1), while the BTPO derivatives exhibit excellent ultralong RTP with broadened ΔEST values of 0.44–0.70 eV. An in-depth study reveals that the delicate modification results in these emitters undergoing obvious changes in their excited states, from an ICT state to a HLCT state or LE-dominated HLCT state, resulting in different arrangements of electronic orbitals. All the BTPO derivatives embedded in a PMMA matrix show prolonged luminescence lifetimes of up to 822 ms with decent phosphorescence quantum yields of 7.4% to 11.6%. These findings not only provide us with brand-new building blocks for phosphor design, but this is also a fascinating molecular design strategy that combines a TADF molecular core with different peripheral moieties to modulate the nature of the excited states and thus fine-tune the singlet and triplet exciton populations.
 | ||
| Scheme 1 The molecular structures of BTPO and BTPO derivatives. | ||
Results and discussion
The synthetic routes to BTPO and BTPO derivatives are outlined in Schemes S1 and S2.† As shown in Scheme S1,† 4-bromo-N,N-bis(4-(tert-butyl)phenyl)aniline reacts with phthalimidesulfenyl chloride to give the bis-sulfenylated derivative. Following intramolecular cyclization using the Lewis acid AlCl3, the bis-sulfenylated derivative is converted to 3-bromo-7,11-ditertbutylbenzo[5,6][1,4]thiazino[2,3,4-kl]phenothiazine (BTP-Br) with a yield of 77%.51 Sequentially, oxidizing BTP-Br with H2O2 affords the key intermediate 3-bromo-7,11-di-tert-butylbenzo[5,6][1,4]thiazino[2,3,4-kl]phenothiazine 5,5,9,9-tetraoxide (BTPO-Br). Through Suzuki coupling reactions between BTPO-Br and the respective boronic acid ester, the three target compounds (BTPO-BCN, BTPO-BCZ, and BTPO-TPA) are obtained in quite good yields. All compounds were unambiguously characterized via1H and 13C NMR spectroscopy. Detailed synthetic procedures are described in the ESI.† Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) show that these BTPO derivatives are thermally stable, with thermal decomposition temperature values (Td, corresponding to 5% weight loss) above 366 °C and glass transition temperature (Tg) values above 195 °C (see Fig. S2†). All compounds were further purified via vacuum sublimation before use. High performance liquid chromatography was carried out to confirm the high purity of the target compounds (see Fig. S3†).The photophysical properties of the BTPO and BTPO derivatives were systematically studied in both dilute solution and as a doped thin film. In dilute toluene solution (1 × 10−5 M) at room temperature, all BTPO derivatives possess a maximum absorption band at around 360 nm with a bathochromic shift of 14 nm in comparison with BTPO (Fig. S4a†). Due to the increased degree of conjugation as a result of the attached substituent groups, the absorption onsets of the BTPO derivatives exhibit slight red shifts, resulting in narrower optical bandgaps. As the fluorescence spectra in Fig. S4a† show, strong fluorescence is observed at 377 nm with a narrower full-width at half-maximum (FWHM) of 28 nm for BTPO-BCN, while BTPO-BCZ and BTPO-TPA exhibit wider emission bands centered at 388 nm and 429 nm, respectively, with FWHM values of 38 and 52 nm, respectively. With respect to BTPO, enhanced photoluminescence quantum yields (PLQYs) of 15.3%, 29.3%, and 38.8% and increased fluorescence lifetimes of 1.14 ns, 1.49 ns, and 2.14 ns are observed for BTOP-BCN, BTPO-BCZ, and BTPO-TPA, respectively. The BTPO derivatives in toluene solution did not display any phosphorescence at room temperature due to the fast vibrational and rotational quenching of triplet excitons. Therefore, steady-state and time-resolved PL spectra were recorded in glassy matrices of the same solution at 77 K. As shown in Fig. S5,† all the compounds exhibit dual emission with different intensities. For BTPO-BCN and BTPO-BCZ, phosphorescence peaks with vibrational characteristics were clearly found at around 450–650 nm, with intense proportions that were more than twice those of the fluorescence peaks. However, BTPO-TPA displayed moderate phosphorescence signals at 501 and 531 nm, with one third the proportion of the fluorescence peaks. The time-resolved PL spectra plotted using red lines (delayed for 0.5–3 ms) further confirm the phosphorescence nature of the luminescence.
A rigid PMMA matrix is adopted to confine the emitters and prevent the nonradiative deactivation of triplets. Similar spectral features are also present in the doped thin film (5 wt% doped in PMMA) with broader FWHM values in the emission spectra, probably owing to polarity variations in the environment (Fig. S4b†). The similarities between the absorption and emission spectra from dilute solutions and doped thin films indicate that the photophysical properties of these compounds in the monomer state are well preserved at a low doping ratio in PMMA films. Intriguingly, as shown in Fig. 1a, all of the BTPO derivatives exhibit obvious RTP features, with well-resolved vibronic bands in the doped PMMA film under vacuum conditions as expected. Relative to the behaviors in toluene at 77 K, biluminescent behaviors are also observed for the BTPO derivatives at room temperature in the doped films, while the phosphorescence signals are relatively weakened. The overall phosphorescence proportion for BTPO-BCN drops to being almost equal to the fluorescence one, while the overall phosphorescence levels of BTPO-BCZ and BTPO-TPA are reduced by a greater extent to one half or less. Yet high phosphorescence quantum yields of 11.6% for BTPO-BCN, 10.6% for BTPO-BCZ, and 7.4% for BTPO-TPA are obtained, where the PLQYs of fluorescence are 14.2% for BTPO-BCN, 21.3% for BTPO-BCZ, and 29.4% for BTPO-TPA under ambient conditions (Table 1). In addition, the phosphorescence lifetimes of the BTPO derivatives in doped PMMA film under vacuum conditions were measured and are depicted in Fig. 1b. Luminescence lifetimes in the range of hundreds of milliseconds are derived via biexponentially fitting the time-resolved PL decay curves. It is noteworthy that BTPO-BCN in 5 wt% PMMA film shows ultralong averaged lifetimes of 822 ms at 485 nm and 760 ms at 518 nm. The phosphorescence lifetimes of BTPO-BCZ are slightly lower: 730 m at 486 nm and 747 ms at 514 nm. For BTPO-TPA, obvious reductions in the phosphorescence lifetimes are observed, with lifetimes of 340 ms at 510 nm and 284 ms at 545 nm. To the best of our knowledge, the phosphorescence quantum yields of these BTPO derivatives are some of the highest values for organic phosphors with phosphorescence lifetimes exceeding 0.1 s, especially in a polymer matrix11,19 (see Scheme S3, Fig. S1, and Table S1†). The above results also imply that the diverse peripheral groups of the BTPO derivatives could significantly manipulate the phosphorescence lifetime and quantum yield. Unlike other host–guest systems that involve H-bonds or other intermolecular interactions,42,52,53 the RTP of BTPO derivatives can be easily achieved in rigid environments in addition to PMMA, such as when using polystyrene (PS) (see Fig. S8†).
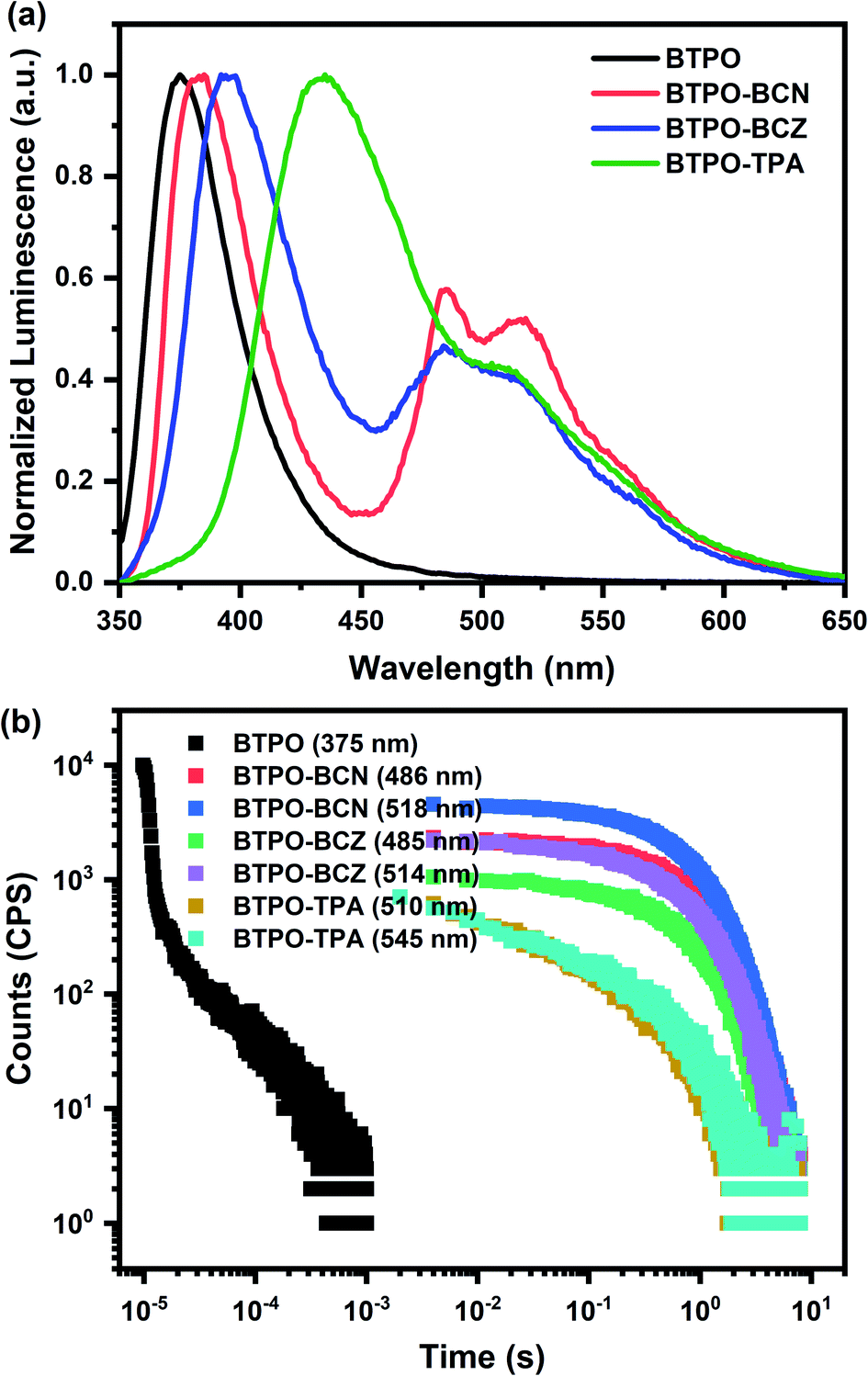 | ||
| Fig. 1 The photophysical properties of 5 wt% BTPO and BTPO derivatives doped into PMMA films. (a) Steady-state PL spectra of BTPO (black), BTPO-BCN (red), BTPO-BCZ (blue), and BTPO-TPA (green) under vacuum. (b) Time-resolved PL decay curves of BTPO and BTPO derivatives at different wavelengths. | ||
| Compound | λ abs [nm] | λ fluo [nm] | λ phos [nm] | E S1 [eV] | E T1 [eV] | ΔESTa [eV] | τ phos [ms] | Φ fluo [%] | Φ phos [%] | Φ total [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| a E S1 and ET1 were estimated from the highest energy peaks of fluorescence and phosphorescence (1240/λ), respectively. The energy gap is calculated according to ΔEST = ES1 − ET1. b Averaged phosphorescence lifetime. | ||||||||||
| BTPO-BCN | 356 | 382 | 486 | 3.25 | 2.55 | 0.70 | 822 | 14.2 | 11.6 | 25.8 |
| BTPO-BCZ | 356 | 394 | 485 | 3.15 | 2.56 | 0.59 | 730 | 21.3 | 10.6 | 31.9 |
| BTPO-TPA | 358 | 432 | 510 | 2.87 | 2.43 | 0.44 | 340 | 29.4 | 7.4 | 36.8 |
To elucidate the relationship between the BTPO core and the BTPO derivatives, the photophysical characteristics of BTPO in toluene solution and in doped thin film form are investigated as a reference. As shown in Fig. S10a,† BTPO in toluene in the cryogenic state exhibits intense vibrational phosphorescence peaks at 387 and 410 nm with a relative weak fluorescence peak at 363 nm. In contrast to the BTPO derivatives, BTPO in 5 wt% doped PMMA film shows no phosphorescence at room temperature under vacuum. Hence, temperature-dependent steady PL measurements of BTPO in doped film under vacuum were conducted (Fig. S11†). The phosphorescence intensity variations with temperature further confirm the disappearance of RTP. As shown in Fig. S10b,† derived from the highest energy peak of the PL spectrum of BTPO doped thin film at 100 K, the energy levels of the S1 and T1 states are calculated to be 3.31 eV and 3.15 eV, respectively, leading to a small ΔEST value of 0.16 eV. In striking contrast, BTPO derivatives in doped film show broadened ΔEST values of 0.70 eV for BTPO-BCN, 0.59 eV for BTPO-BCZ, and 0.44 eV for BTPO-TPA. These results suggest a possible reversed intersystem crossing (RISC) process between the singlet and triplet states of the BTPO core. The PL decay curve of 5 wt% BTPO in PMMA film at 375 nm at room temperature was measured (see Fig. S12† and 1b). The fitting results show a combination of a short prompt lifetime of 1.04 ns and a long delayed lifetime of 41 μs, verifying the TADF features of the BTPO core. Moreover, as shown in Fig. S13,† the temperature-dependent fluorescence decay spectra of BTPO measured in doped PMMA film for a time range of 10 ms under vacuum demonstrate a declined trend of delayed fluorescence lifetime with the temperature but an increased proportion of delayed component to prompt one, further confirming the TADF nature of the BTPO emission behavior.7 In other words, modifying the BTPO core through the introduction of such aromatic substituent groups could effectively modulate the energy levels of excited states and alter the nature of luminescence from TADF to RTP.
To verify the above molecular design strategy, a phenyl group was also introduced to the BTPO core to produce BTPO-Ph as a reference material, and the photophysical properties are summarized in Fig. S14 and Table S7.† BTPO-Ph in 5 wt% PMMA under vacuum at room temperature presents dual emission, with a prompt lifetime of 1.27 ns at 391 nm and averaged phosphorescence lifetimes of 658 ms at 465 nm and 657 ms at 493 nm. This further confirms that the introduction of a peripheral aromatic substituent group can easily modulate the energy difference between singlet and triplet states, greatly reducing the triplet energy and thus realizing phosphorescent luminescence.
Theoretical calculations were performed using the TD-M062X/6-31G(d,p) method to uncover the underlying RTP mechanism of the BTPO derivatives; the natural transition orbitals (NTOs) of the BTPO and BTPO derivatives in the S1 and T1 states are depicted in Fig. 2a and S18.† With regard to BTPO, the NTOs of both the S1 and T1 states show a similar mix of (n, π*) and (π, π*) transition configurations. In addition to the separative distributions of holes and particles on the phenyl rings, the holes are predominately localized on the nitrogen atom while the particles are mainly distributed on the sulfonyl groups. This distribution feature endows the BTPO core with the obvious characteristic of short-range ICT in the excited state and a small oscillator strength of 0.0811 for the S1 → S0 transition, which leads, to a certain extent, to a small ΔEST value and a low fluorescence quantum yield. These theoretical calculations are consistent with the experimental results given above. A small energy gap of 0.16 eV between the S1 and T1 states, weak S1 emission with a low PLQY of 6.7%, and a delayed lifetime of 41 μs were observed for the BTPO core, which are indicative of TADF emission via a RISC process. On the contrary, all the BTPO derivatives show a HLCT state or a LE-dominated HLCT state in the S1 and T1 states, tending to enhance the quantum yield and enlarge ΔEST. As shown in Fig. 2a and S18,† the holes are mainly located on the external substituents and the attached benzene rings of the core while the particles predominately occupy the interphase sites of the attached phenyl groups. With the weakening of the electron-donating properties of the peripheral groups from TPA to BCZ, and further to BCN, the ratio of the CT component in the mixed HLCT state gradually decreases, especially for the T1 state of BTPO-BCN, in which the T1 state almost becomes a LE-like state. The solvatochromic effect of BTPO derivatives in different solvents further confirms the existence of a HLCT state for emission transitions (see Fig. S19–S22†).54 These apparent changes of the excited states from an ICT state to a HLCT state or LE-dominated HLCT state play a significant role in the modulation of energy levels, which can result in an enlargement of ΔEST. As shown in Fig. 2b, taking BTPO-BCN as an example, a large energy difference of 0.70 eV between the S1 and T1 states is determined based on experimental results. Combining this with a SOC constant of 0.303 between S1 and T1, as obtained from theoretical calculations, efficient ISC from S1 to T1 with rare RISC from T1 to S1 will result in an abundant T1 exciton population. Based on the NTO analysis, all BTPO derivatives possess an almost pure (π, π*) configuration in the T1 state, which is considered to have an extremely slow decay rate, facilitating URTP. With additional strong SOC effects between the S0 and T1 states, these T1 excitons with (π, π*) character could radiatively decay to the ground state in a rigid environment and generate URTP.
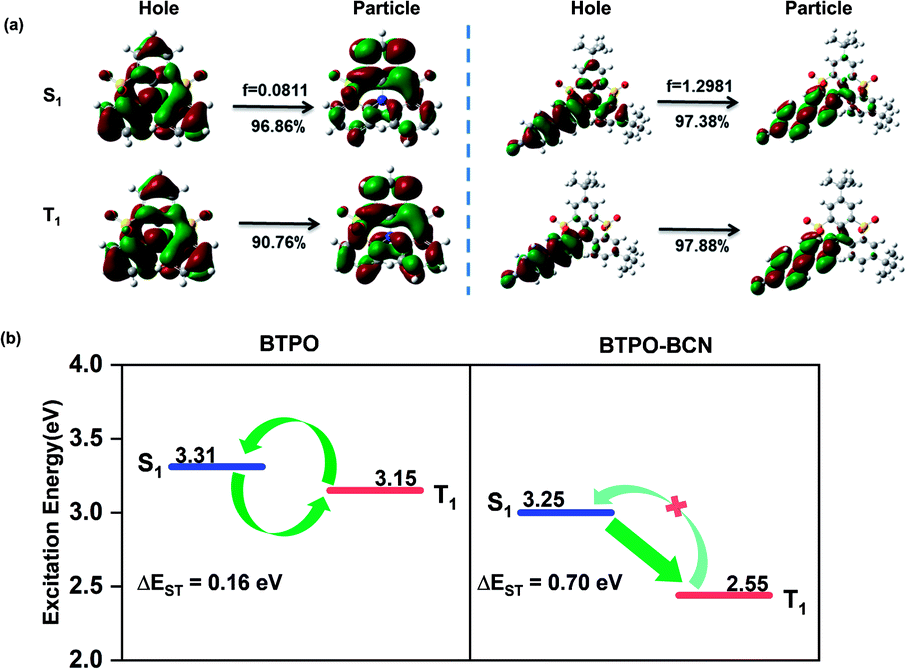 | ||
| Fig. 2 (a) The natural transition orbitals of BTPO and BTPO-BCN in singlet and triplet excited states. (b) The energy levels of BTPO and BTPO-BCN determined based on experimental results. | ||
Fig. S23 and Videos 1–4† show photographs and the evolution process of BTPO derivatives embedded in polymer matrices upon excitation and in its absence. Upon irradiation using a 365 nm UV lamp in inert gas, the BTPO-BCN-doped PMMA film exhibits bluish-green emission with the Commission Internationale de l'Éclairage (CIE) coordinates of (0.20, 0.38), while the BTPO-BCZ- and BTPO-TPA-doped films present blue luminescence with CIE coordinates of (0.19, 0.26) and (0.18, 0.18), respectively. After the removal of the excitation source, these thin films show persistent green emission with a long duration of several seconds. Due to the excellent insulating properties of oxygen, PVA was deposited on top of the doped PMMA film as an O2 barrier to ensure phosphorescence generation under ambient conditions. Taking BTPO-BCN as an example, BTPO-BCN-doped PMMA film coated with a PVA layer demonstrated dual emission features under ambient conditions with slightly weakened phosphorescence compared with under vacuum (Fig. S15†). In comparison to the monolayer film, this bilayer film possesses an approximately close total PLQY of 23.1% with a phosphorescence quantum yield of 8.6% in air. As expected, similar emission changes from bluish-green to green are observed for the BTPO-BCN bilayer film in air (Fig. S23b†).
In view of the extraordinary RTP features of the BTPO derivatives in the monomer state, their preliminary application to confidential encryption is demonstrated in Fig. 3 and Videos 5 and 6.† Using 5 wt% BTPO-BCN-doped PMMA mixed organic solution as an ink, complicated patterns could be easily fabricated through an ink-jet printing technique, such as the logo of South China University of Technology (SCUT), the simplified Chinese and English names of SCUT, and the abbreviation of State Key Laboratory of Luminescent Materials and Devices (SKLLMD). The patterns can barely be seen under sunlight, but they can be clearly observed using a 365 nm UV lamp. After ceasing the UV irradiation, green patterns could be seen for a few seconds under vacuum. Also, a pattern spelling SCUT covered with a PVA layer was fabricated, as shown in Fig. 3b. Under ambient conditions, the bluish-green SCUT writing shifts to green after switching off the UV light, and green emission is maintained for at least 3 s.
 | ||
| Fig. 3 A demonstration of confidential encryption: photographs of BTPO-BCN in PMMA film (a) in an inert gas and (b) covered by PVA in air under a UV lamp (left) and after the removal of the UV source (right). | ||
Conclusions
To summarize, a novel family of ultralong-lifetime organic phosphors based on the BTPO skeleton was delicately designed, and ultralong phosphorescence in the monomer state could be well preserved upon scaffolding the BTPO derivatives into a PMMA matrix. Among these compounds, BTPO-BCN exhibits a long-lived phosphorescence lifetime of 822 ms with a high quantum yield of 11.6%, representing some of the best phosphorescent properties seen in terms of URTP materials in polymer matrices to date. Moreover, a strategy based on the modulation of the excited state from a CT state to a HLCT state or LE-dominated HLCT state was proposed to achieve efficient URTP. Systematic theoretical calculations revealed that this modulation of the excited states of the BTPO derivatives enlarges ΔEST and thus reduces the likelihood of a RISC process. In the meantime, a pure (π, π*) configuration of the T1 state was obtained for the BTPO derivatives, enabling ultralong phosphorescence. The preliminary application of these highly efficient URTP materials via ink-jet printing shows the great application prospects in anti-counterfeiting and other fields. This work extends the scope of organic RTP materials and also provides a new molecular design approach for obtaining efficient RTP materials.Data availability
All the experimental, crystallographic and computational data are available in the ESI.†Author contributions
The manuscript was written through contributions from all authors. Y. Ma and D. Hu designed the experiments, reviewed and edited the manuscript, and acquired financial support; Z. Ma synthesized and characterized the target compounds, analyzed the experimental data, and wrote the original draft; Z. Yang and B. Yang conducted the theoretical calculations; L. Mu and J. Peng performed the confidential encryption study; L. Chen, X. Qiao, and D. Ma contributed to characterization and analyzing the photophysical properties of the compounds; L. Deng synthesized and characterized BTPO; and B. Wang carried out the cyclic voltammetry measurements and provided the electrochemical results. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was received from National Key R&D Program of China (2020YFA0714604), Natural Science Foundation of China (91833304, 21973081 and 51521002), the Basic and Applied Basic Research Major Program of Guangdong Province (No. 2019B030302007), Research and Development Funds for Science and Technology Program of Guangzhou (No. 202007020004), Natural Science Foundation of Guangdong Province (2019B121205002), Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates (2019B030301003) and Fundamental Research Funds of State Key Laboratory of Luminescent Materials and Devices (Skllmd-2021-07).Notes and references
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- N. A. Kukhta and M. R. Bryce, Mater. Horiz., 2021, 8, 33–55 RSC.
- P. Data and Y. Takeda, Chem.–Asian J., 2019, 14, 1613–1636 CrossRef CAS PubMed.
- X. Y. Cai and S. J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
- J. Zhi, Q. Zhou, H. Shi, Z. An and W. Huang, Chem.–Asian J., 2020, 15, 947–957 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS PubMed.
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai and S. Minakata, Chem. Commun., 2018, 54, 6847–6850 RSC.
- S. Tian, H. Ma, X. Wang, A. Lv, H. Shi, Y. Geng, J. Li, F. Liang, Z. M. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, C. Wang, X. Zheng, Y. Zheng, L. Gao, C. Yang, Y. Li, L. Qu and Y. Zhao, Adv. Mater., 2020, 32, 1907355 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed.
- W. Jia, Q. Wang, H. Shi, Z. An and W. Huang, Chem.–Eur. J., 2020, 26, 4437–4448 CrossRef CAS PubMed.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- Q. Liao, Q. Gao, J. Wang, Y. Gong, Q. Peng, Y. Tian, Y. Fan, H. Guo, D. Ding, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2020, 59, 9946–9951 CrossRef CAS PubMed.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 CrossRef CAS PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- T. Zhang, X. Ma and H. Tian, Chem. Sci., 2020, 11, 482–487 RSC.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- H. Bhatia, I. Bhattacharjee and D. Ray, J. Phys. Chem. Lett., 2018, 9, 3808–3813 CrossRef CAS PubMed.
- H. Bhatia and D. Ray, J. Phys. Chem. C, 2019, 123, 22104–22113 CrossRef CAS.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- S. Reineke, N. Seidler, S. R. Yost, F. Prins, W. A. Tisdale and M. A. Baldo, Appl. Phys. Lett., 2013, 103, 093302 CrossRef.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef PubMed.
- S. Garain, S. Kuila, B. C. Garain, M. Kataria, A. Borah, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 12323–12327 CrossRef CAS PubMed.
- L. Xiao, Y. Wu, J. Chen, Z. Yu, Y. Liu, J. Yao and H. Fu, J. Phys. Chem. A, 2017, 121, 8652–8658 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 2101719 CrossRef CAS.
- Y. Su, Y. Zhang, Z. Wang, W. Gao, P. Jia, D. Zhang, C. Yang, Y. Li and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 9967–9971 CrossRef CAS PubMed.
- S. Kuila, S. Garain, S. Bandi and S. J. George, Adv. Funct. Mater., 2020, 30, 2003693 CrossRef CAS.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS.
- H. F. Higginbotham, M. Okazaki, P. de Silva, S. Minakata, Y. Takeda and P. Data, ACS Appl. Mater. Interfaces, 2021, 13, 2899–2907 CrossRef CAS PubMed.
- R. Huang, J. Avó, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- P. Pander, A. Swist, J. Soloducho and F. B. Dias, Dyes Pigm., 2017, 142, 315–322 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- M. Gmelch, H. Thomas, F. Fries and S. Reineke, Sci. Adv., 2019, 5, eaau7310 CrossRef PubMed.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, X. Feng and S. Reineke, Adv. Mater., 2020, 32, 2000880 CrossRef CAS PubMed.
- Y. Wen, H. Liu, S.-T. Zhang, G. Pan, Z. Yang, T. Lu, B. Li, J. Cao and B. Yang, CCS Chem., 2020, 2, 1940–1948 Search PubMed.
- S. Menichetti, C. Faggi, M. Onori, S. Piantini, M. Ferreira, S. Rocchi, M. Lupi, I. Marin, M. Maggini and C. Viglianisi, Eur. J. Org. Chem., 2019, 2019, 168–175 CrossRef CAS.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. adv., 2018, 4, eaas9732 CrossRef PubMed.
- Y. Zhang, L. Gao, X. Zheng, Z. Wang, C. Yang, H. Tang, L. Qu, Y. Li and Y. Zhao, Nat. Commun., 2021, 12, 2297 CrossRef CAS PubMed.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2079614. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04118e |
| This journal is © The Royal Society of Chemistry 2021 |