
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CsPbBr3–CdS heterostructure: stabilizing perovskite nanocrystals for photocatalysis†
Anthony
Kipkorir
ab,
Jeffrey
DuBose
 ab,
Junsang
Cho
a and
Prashant V.
Kamat
*abc
ab,
Junsang
Cho
a and
Prashant V.
Kamat
*abc
aRadiation Laboratory, University of Notre Dame, Notre Dame, Indiana 46556, USA
bDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA
cDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: pkamat@nd.edu
First published on 22nd October 2021
Abstract
The instability of cesium lead bromide (CsPbBr3) nanocrystals (NCs) in polar solvents has hampered their use in photocatalysis. We have now succeeded in synthesizing CsPbBr3–CdS heterostructures with improved stability and photocatalytic performance. While the CdS deposition provides solvent stability, the parent CsPbBr3 in the heterostructure harvests photons to generate charge carriers. This heterostructure exhibits longer emission lifetime (τave = 47 ns) than pristine CsPbBr3 (τave = 7 ns), indicating passivation of surface defects. We employed ethyl viologen (EV2+) as a probe molecule to elucidate excited state interactions and interfacial electron transfer of CsPbBr3–CdS NCs in toluene/ethanol mixed solvent. The electron transfer rate constant as obtained from transient absorption spectroscopy was 9.5 × 1010 s−1 and the quantum efficiency of ethyl viologen reduction (ΦEV+˙) was found to be 8.4% under visible light excitation. The Fermi level equilibration between CsPbBr3–CdS and EV2+/EV+˙ redox couple has allowed us to estimate the apparent conduction band energy of the heterostructure as −0.365 V vs. NHE. The insights into effective utilization of perovskite nanocrystals built around a quasi-type II heterostructures pave the way towards effective utilization in photocatalytic reduction and oxidation processes.
Introduction
Semiconductor quantum dots are excellent building blocks for designing light-harvesting assemblies.1,2 The ability to chemically modify the surface with a functionalized ligand or to couple with another semiconductor particle offers a variety of ways to harvest visible photons.3–5 Since the 1990s, metal chalcogenide quantum dots (QDs), CdSe in particular, have served as the prototypical compound to elucidate excited state and charge transfer properties.6–8 In recent years, another quantum dot system, viz., perovskite nanocrystals (CsPbX3, X = Cl, Br, I), has emerged as a model semiconductor QD system to probe light induced optoelectronic and photocatalytic properties.9–13 We have recently elucidated the photocatalytic aspects of CsPbBr3 QDs by probing the interfacial electron transfer to methyl viologen14,15 and ferrocenium cation.16 To date, the use of these perovskite quantum dots in photocatalysis has been limited only to a few nonpolar solvents.17–21 The weakly-binding organic ligand shell around perovskite nanocrystals does not provide sufficient stability in polar solvents. In order to expand the scope of the perovskite nanocrystals to a wide range of photocatalytic applications (e.g., solar hydrogen production or CO2 reduction), it is important to provide protection against chemical transformation in the presence of a redox couple or in a polar medium.One simple approach to achieve stability in polar solvents is to cap the semiconductor nanocrystals with a thin inorganic shell. Design of such heterostructures has been successfully employed for binary and ternary semiconductors like CdSe/ZnS,22,23 InP/ZnS,24 and AgInS2/ZnS.25 The heterostructure with type I or type II band alignment offers strategies to enhance emission of the core QD or improve charge separation within the heterostructure. Although a few reports exist to-date of capping CsPbBr3 QDs with SiO2,26 CdS,27 or ZnS28,29 shells, none of these heterostructures have shown a major leap in achieving improved performance with a long-term stability in polar solvents. Ambiguity still exists whether the added material forms a continuous shell around the perovskite core or forms smaller discontinuous islands on the surface.26 Given the difficulty in imaging the thin inorganic shell around perovskite nanocrystals, because of the image contrast, one employs stability tests in a polar medium or its resistance to halide exchange to confirm surface modification.30–33
Designing perovskite heterostructures with metal chalcogenide shells can have several distinct advantages: (i) providing stability towards increased polarity of the solvent, (ii) remediating surface defects by directly interacting with the vacancies, and (iii) allowing for type I or quasi-type II band alignment to promote increased charge recombination in the core (increased emission yield) or improved charge separation.34–36 In this context, a CsPbBr3–CdS heterostructure offers an attractive means to tune the band energies, as their conduction bands are nearly isoenergetic (ECB of CsPbBr3 and quantized CdS ≈ −0.8 V versus NHE)37–39 and facilitate charge separation. In addition, cubic CsPbBr3 nanocrystals have a lattice constant (a) of 5.85 Å (ref. 40) while that of CdS (zinc blende structure) is 5.83 Å.41 The similarity of the two values signifies the possibility of having a less strained interface with reduced defect states.42 We have now successfully prepared CsPbBr3–CdS heterostructures in a two-step method, and the optical properties of these structures are discussed.
Results and discussion
CsPbBr3–CdS heterostructure
CsPbBr3 quantum dots (QDs) dispersed in octadecene (ODE) were prepared using a previously reported procedure.43 These QDs were then treated with cadmium diethyldithiocarbamate (Cd(DDTC)2) at 110 °C to obtain CdS capped CsPbBr3 QDs. Experimental details on the synthesis of CsPbBr3 and CsPbBr3–CdS QDs are presented in the ESI (Fig. S1†). The transmission electron microscopy (TEM) images of the two nanocrystals are shown in Fig. 1A and B. These cubic particles are similar in size showing particles of lengths 8–9 nm (see Fig. S2† for size distribution analysis). This shows that CdS capping in the heterostructure is relatively thin compared to the CsPbBr3 core and it is difficult to identify with the image contrast in TEM images. This observation is consistent with earlier work which reports difficulty in characterizing the shell in a CsPbBr3 heterostructure using TEM analysis.44,45 These studies have attributed the imaging difficulty to low electron density contrast of the shell. However, other techniques such as elemental analysis can be useful to overcome these limitations. | ||
| Fig. 1 TEM image for (A) pristine CsPbBr3 and (B) CsPbBr3–CdS. (C–G) Elemental maps showing presence of Cs, Pb, Br, Cd, and S, respectively. (H) Atomic percentage of different elements as measured by EDAX. | ||
We succeeded in establishing the presence of CdS in the CsPbBr3–CdS heterostructures through elemental analysis with TEM energy dispersive X-ray (EDX) spectroscopy. Fig. 1C–G which present elemental mapping, confirm the presence of Cd and S along with Cs, Pb, and Br. The elemental ratio (Fig. 1H) also suggests a relatively low concentration of CdS as compared to Cs, Pb and Br in the heterostructure. We can conclude that any CdS in the heterostructure is of the order of a monolayer. However, there is also the possibility of forming small clusters in and around the CsPbBr3 QDs.
Evidence of surface modification with CdS was also seen through the change in the surface charge. Zeta potential measurements indicated that CsPbBr3–CdS QDs suspended in toluene carry more negative surface charge (−37.4 mV) than pristine CsPbBr3 nanocrystals in toluene (−15.8 mV). This increased surface charge of CsPbBr3–CdS QDs enabled us to carry out electrophoretic deposition of a film under the influence of a DC field (see ESI† for details; Fig. S3†). The increased surface negativity and ability to be deposited as a film under applied bias indicates a modified surface around CsPbBr3. Similar electrophoretic deposition was also possible when CsPbBr3 nanocrystals were coated with a PbSO4–oleate shell.46 It should be noted that pristine CsPbBr3 QDs suspended in toluene cannot be deposited as film using electrophoresis as it does not carry sufficient surface charge.
In addition to CsPbBr3 and CsPbBr3–CdS heterostructures, we also synthesized CdS QDs with the same ligands following a similar experimental procedure (i.e. without CsPbBr3). Fig. 2A shows the absorption spectra of CsPbBr3, CdS and CsPbBr3–CdS QDs in toluene. The CdS and CsPbBr3 QDs exhibit characteristic excitonic peaks at 434 and 518 nm, respectively. Although the absorption spectrum of the CsPbBr3–CdS heterostructure shows two peaks that overlap with the absorption of individual QDs, the peak around 434 nm may also arise from the deposition of small size CdS particles on the surface of CsPbBr3 nanocrystals. Such a decoration of CdS particles, if any, would give rise to a CdS excitonic peak in the heterostructures. The excitonic peak at 518 nm (CsPbBr3) remains unaffected after heterostructure formation, thus ruling out any interference of exchange of metal ions. Similarly, the tail absorption at longer wavelengths arises from the scattering effects, similar to what is seen in PbSO4–oleate capped CsPbBr3.46
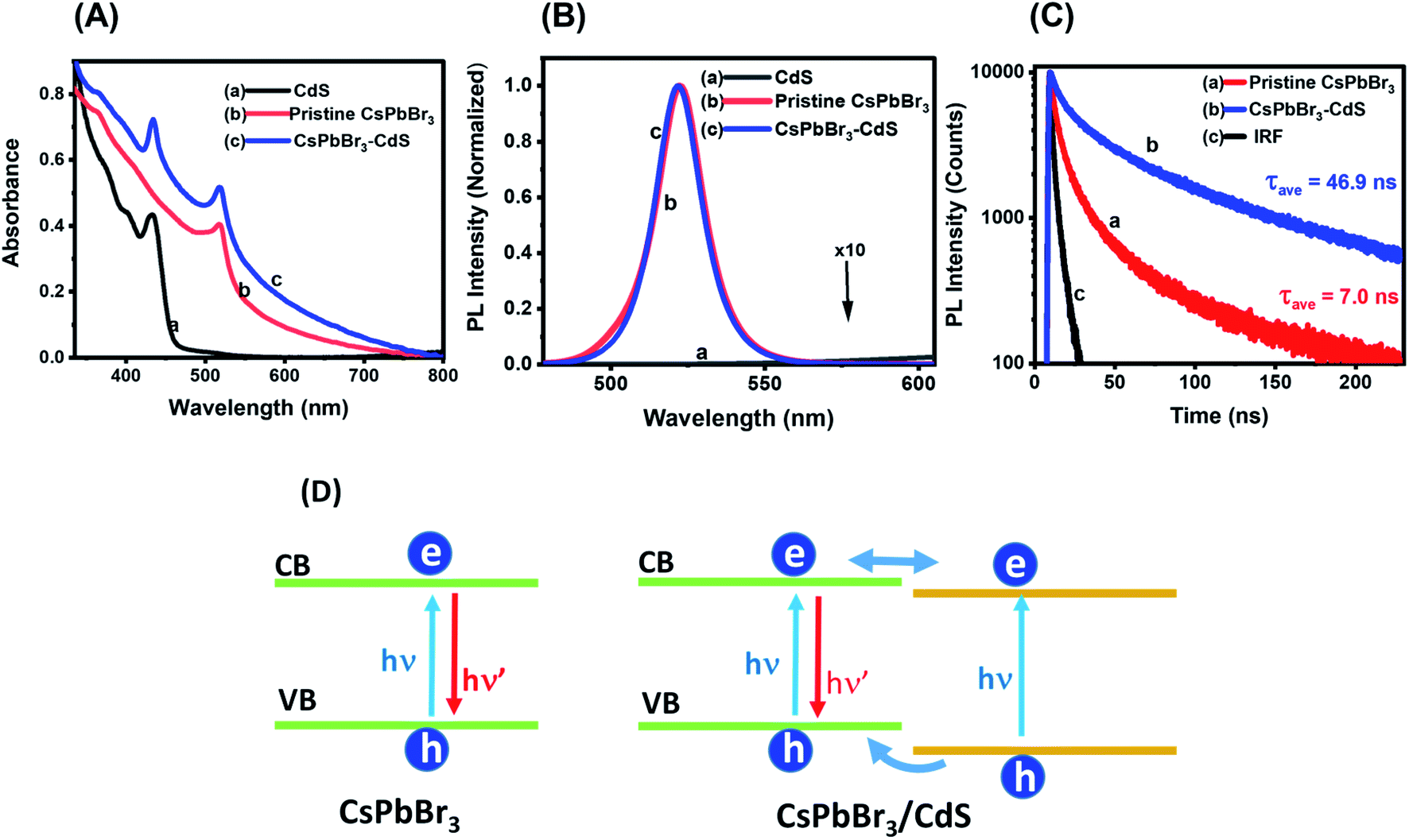 | ||
| Fig. 2 (A) Absorption spectra and (B) corresponding photoluminescence (PL) spectra of (a) CdS, (b) pristine CsPbBr3 and (c) CsPbBr3–CdS QD suspension in toluene. (C) PL decay traces monitored at 520 nm using 370 nm excitation: (a) pristine CsPbBr3 and (b) CsPbBr3–CdS nanocrystal suspensions in toluene, and (c) instrument response (IRF). The kinetic analysis is presented in the ESI.† (D) Schematic diagram illustrating charge separation and charge recombination in CsPbBr3 and CsPbBr3–CdS nanocrystals. | ||
The emission spectra of these three nanostructures are shown in Fig. 2B. Whereas CdS QDs remain the least emissive, both CsPbBr3 and CsPbBr3–CdS QDs are highly emissive. Additionally, the emission features of CsPbBr3 QDs remain unchanged following the deposition of CdS: the emission maximum (521 nm) and the full width at half maximum (18 nm) of CsPbBr3 are unaffected after CdS modification. These results confirm that there is no substitution of cations during the heterostructures synthesis, and thus the emission characteristics of the parent CsPbBr3 QDs are retained in the heterostructure. If there was any substitution of Pb2+ with Cd2+ we would expect a blue shift in the absorption and emission maxima of the perovskite QD.47 The excitation spectra recorded at different emission wavelengths confirm the origin of the emission to arise from the CsPbBr3 (Fig. S4 in the ESI†).
Another interesting aspect of the capping with CdS is the enhancement in emission yield. The emission quantum yields as determined using an integrating sphere were 39% and 60% for CsPbBr3 and CsPbBr3–CdS QDs, respectively (see ESI† for details). Passivation of surface defects by CdS is expected to suppress nonradiative processes and thus lead to increased emission yield. For example, capping of CdSe with CdS has resulted in the significant enhancement of emission yield.48–51 The flow of charge carriers from the CdS shell to CdSe core in these studies was established through emission and excitation spectral measurements. Control experiments were carried out to check whether introduction of Cd2+ ions alone can induce similar changes in the emission properties. Fig. S5† shows a decrease in emission yield and lifetime when CsPbBr3 was treated with cadmium acetylacetonate (Cd(acac)2) instead of Cd(DDTC)2. Additionally, treatment with Cd2+ did not change the absorption of the QDs. This further confirms that the observed optical properties are due to the presence of CdS in the heterostructure.
We employed time-resolved emission measurements to monitor the excited state behavior of CsPbBr3 before and after CdS deposition. The emission decay at 520 nm was monitored for CsPbBr3 and CsPbBr3–CdS samples (Fig. 2C). Each trace was analyzed using a biexponential decay fit and the fitting parameters are presented in Table S1.† Of interest is the increase in emission lifetime of CsPbBr3 upon capping with CdS. A nearly seven-fold increase in average lifetime of CsPbBr3–CdS QDs (τave = 46.9 ns) was observed over that of pristine CsPbBr3 QDs (τave = 7.0 ns). This shows that CdS deposition facilitates long-lived charge separation in CsPbBr3–CdS. In addition to surface passivation, we can also expect the formation of a quasi-type II heterojunction as shown in the scheme (Fig. 2D). Whereas direct charge carrier recombination is dominant in pristine CsPbBr3, the nearly isoenergetic conduction bands of CsPbBr3 and CdS can facilitate delocalization of electrons across the two semiconductors, thus improving charge separation. Since the CdS layer is relatively thin, its contribution to the emission is expected to be small. The excitation spectra (Fig. S4†) rule out the contribution from CdS to overall emission. The observed increase in lifetime parallels the emission yield enhancement seen in the CsPbBr3–CdS heterostructure.
Stability in polar environment
Attaining long term stability of CsPbBr3 nanocrystals in polar solvents remains a challenge. CsPbBr3 nanocrystals undergo rapid degradation in polar solvents, which has hampered their applications in photocatalysis. Even the addition of a small amount of polar solvents such as ethanol or water can induce chemical transformation/precipitation of CsPbBr3 QDs and thus a loss of photoactivity.9,52,53 Recently, it was reported that ZnS-capped CsPbBr3 QDs were stable in a toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water biphasic mixture. The contact with water was made by periodic shaking since the two solvents are immiscible.28 We also conducted a similar stability test of CsPbBr3 and CsPbBr3–CdS nanocrystals using a biphasic mixture of toluene and water with periodic shaking. The emission spectra of CsPbBr3 and CsPbBr3–CdS nanocrystals and the PL intensity variation recorded during 30 hour period are shown in Fig. S6.† CsPbBr3 nanocrystals became non emissive after 20 hours of exposure in biphasic mixture. On the other hand, CsPbBr3–CdS nanocrystals, after an initial drop in photoluminescence, maintained more than 40% emission even after 30 hours.
:water biphasic mixture. The contact with water was made by periodic shaking since the two solvents are immiscible.28 We also conducted a similar stability test of CsPbBr3 and CsPbBr3–CdS nanocrystals using a biphasic mixture of toluene and water with periodic shaking. The emission spectra of CsPbBr3 and CsPbBr3–CdS nanocrystals and the PL intensity variation recorded during 30 hour period are shown in Fig. S6.† CsPbBr3 nanocrystals became non emissive after 20 hours of exposure in biphasic mixture. On the other hand, CsPbBr3–CdS nanocrystals, after an initial drop in photoluminescence, maintained more than 40% emission even after 30 hours.
The biphasic solvent mixture approach does not represent an increase in the overall polarity of the medium. Ideally, an inorganic shell should prevent direct contact of CsPbBr3 with a polar environment and maintain its photostability. We checked the stability of CsPbBr3 and CsPbBr3–CdS by introducing a miscible polar solvent (ethanol) to a toluene suspension of the QDs and monitoring the absorption and emission spectra over time. Fig. 3A and B show the absorption and emission spectra recorded following addition of ethanol (15% v/v) to toluene solution over a period of 60 minutes. The absorption of pristine CsPbBr3 QDs shows enhanced absorbance with time due to scattering effects caused by turbidity as the ligands from QD surface become detached in the polar medium.54 A ∼85% decrease in the CsPbBr3 emission yield is seen immediately after the addition of ethanol to the toluene solution. In addition, upon exposure to ethanol we also see a change in the absorption of CsPbBr3 due to particle aggregation. These results confirm the susceptibility of CsPbBr3 QDs to polar environment (Fig. 3C). On the other hand, CsPbBr3–CdS QDs exhibit only a small decrease (∼15%) in emission with a relatively small change in the absorption during 60 min of exposure in toluene/ethanol mixed solvent. The CdS deposition provides the necessary protection for CsPbBr3, and thus decreases its susceptibility to ethanol-induced degradation.
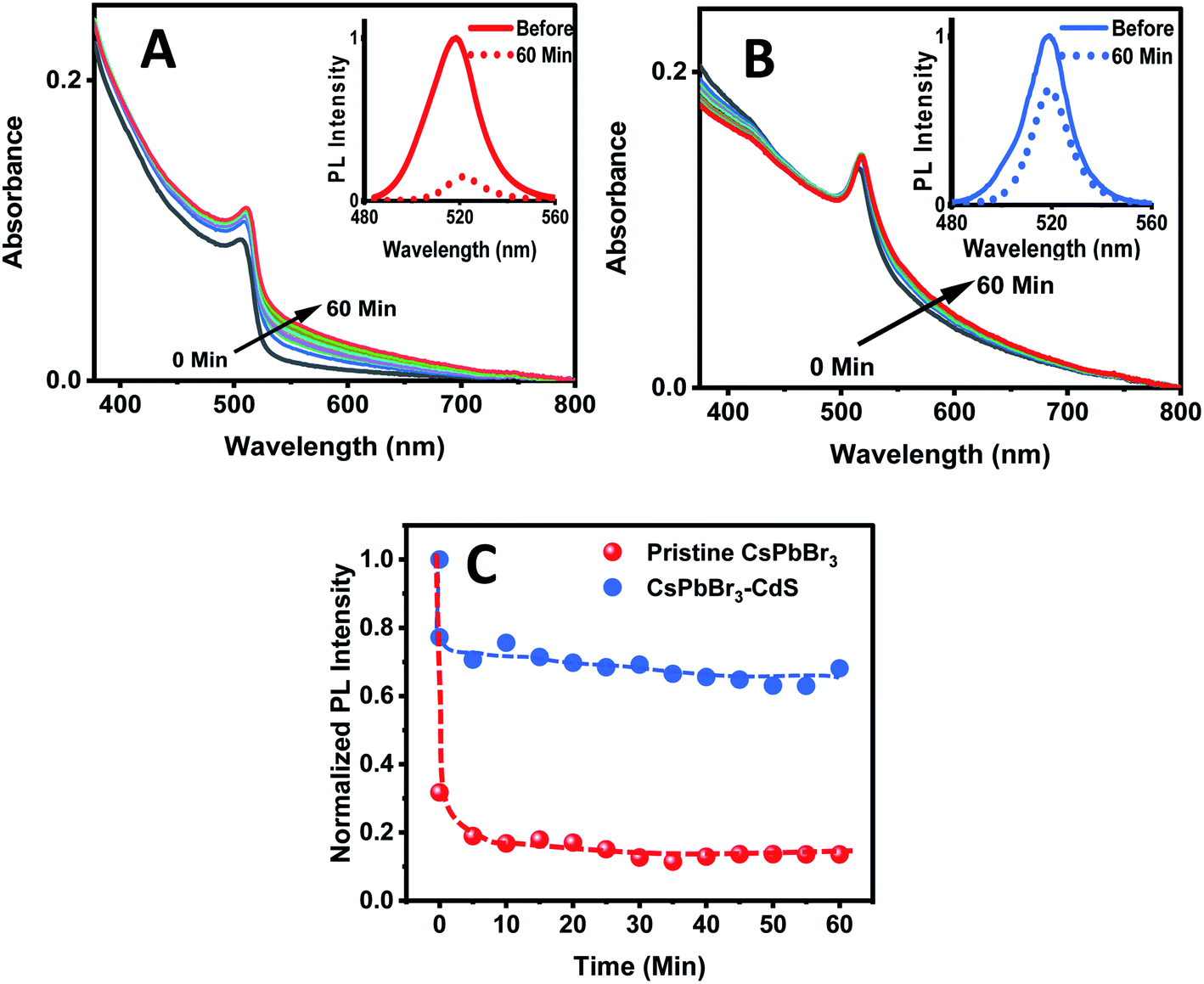 | ||
| Fig. 3 Absorption spectra of (A) CsPbBr3 and (B) CsPbBr3–CdS QDs in toluene recorded at different time intervals after addition of ethanol (15% v/v). Spectra were recorded at a time interval of 5 min. Insets: PL spectra of QDs before and 60 min after ethanol addition (excitation: 370 nm). (C) Changes in the normalized PL intensity of the CsPbBr3 and CsPbBr3–CdS QDs monitored at 520 nm as a function of time after addition of ethanol (15% v/v) to the toluene suspension. | ||
Excited state interactions with an electron acceptor
Since CsPbBr3–CdS QDs were stable in toluene/ethanol mixed solvent, we were able to probe the excited state interactions and interfacial electron transfer with a cationic electron acceptor, ethyl viologen, EV2+. Emission spectra recorded at different concentrations of EV2+ are shown in Fig. 4A. The quenching of photoluminescence confirmed the excited state interaction between CsPbBr3–CdS QDs and EV2+. As a control, we tested the solubility of EV2+ in the toluene/ethanol mixed solvent separately by recording absorption spectra and confirming the probe molecules are fully soluble at the concentrations employed in this study (Fig. S7†). Earlier studies have shown direct complexation between CsPbBr3 and methyl viologen and elucidated the role of surface bound ligands in dictating the complexation constant.14,15 Here, we were able to quench the emission at micromolar concentrations of EV2+, thus indicating a complex formation in the ground state between CsPbBr3–CdS and EV2+.51 The equilibrium of the bound and unbound EV2+ molecules (reaction (1)) can be expressed in terms of the apparent association constant, Kapp and emission yields (expression (2)).55 The observed quantum yield (ϕf(obs)) takes into account the emission arising from EV2+-bound (ϕf′) and pristine (ϕf0) CsPbBr3 QDs. With increasing concentration of EV2+, more CsPbBr3–CdS QDs bind to viologen and thus exhibit a decrease in the emission yield. | (1) |
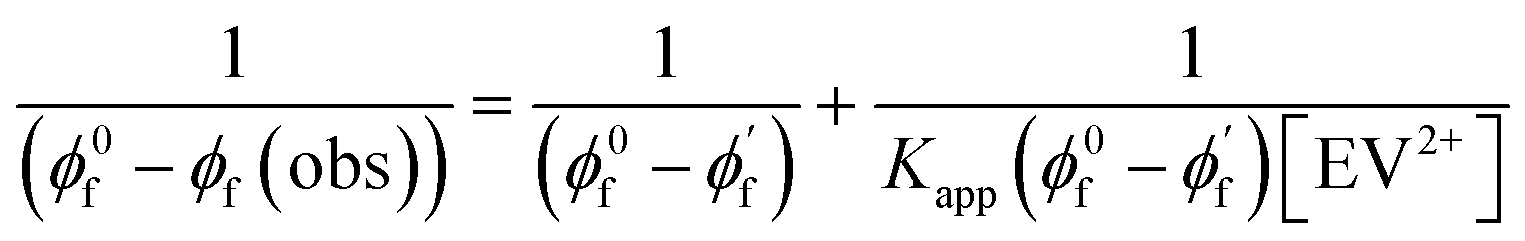 | (2) |
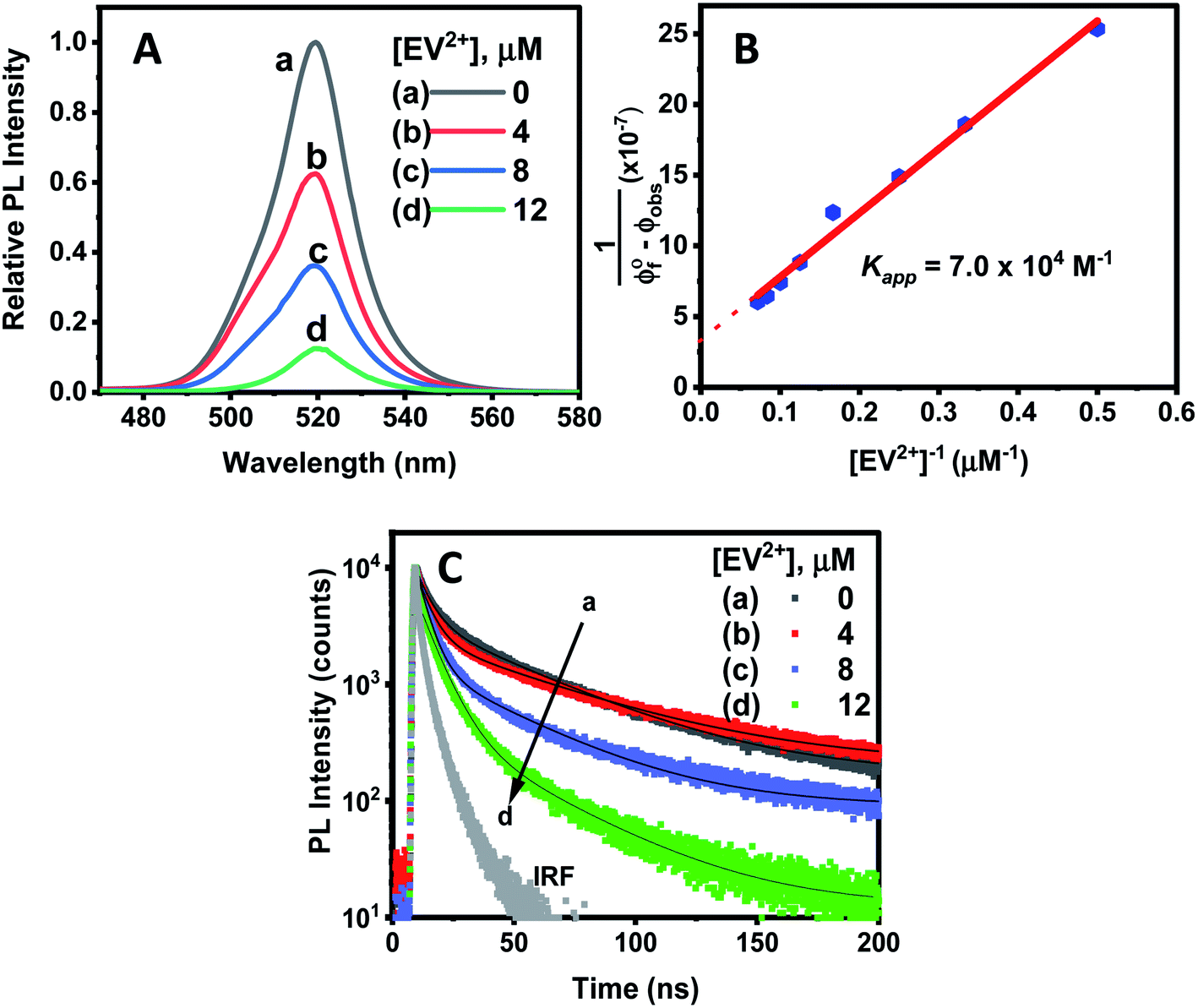 | ||
| Fig. 4 (A) PL quenching of CsPbBr3–CdS QDs (≈16 nM) in toluene:ethanol (90:10% v/v) upon additions of increasing concentration of ethyl viologen (EV2+) dissolved in ethanol. (B) Double reciprocal plot analysis of emission quenching of CsPbBr3–CdS in toluene:ethanol. The slope (3.3 × 10−7) and intercept (4.3 × 10−12 M−1) was used to determine the apparent association constant, Kapp, 7.0 (±0.8) × 104 M−1. (C) Photoluminescence decay at different EV2+ concentrations, recorded with 370 nm excitation and monitored at λmax = 520 nm. | ||
The photoluminescence quenching data was analyzed using expression (2). The emission intensity of the QDs at the emission maximum (which is proportional to quantum yield, (ϕf(obs))) was monitored at different concentration of EV2+.55,56 The linear dependence of the double reciprocal plot (1/(ϕ0f-ϕf(obs)) versus 1/[EV2+]) in Fig. 4B confirms the validity of the association between CsPbBr3–CdS and EV2+. The apparent association constant Kapp determined from the slope and intercept of the plot in Fig. 4B was 7.0 × 104 M−1. This complexation constant is 1–2 orders of magnitude smaller than the one observed for uncapped CsPbBr3 and viologen (0.8–7.0 × 106 M−1).15 The decrease in Kapp further indicates that the presence of CdS reduces the surface interactions with the viologen. The Kapp value we obtain in this study is in line with literature values of CdS interacting with viologens.57
To further establish the excited state interactions, we monitored the photoluminescence lifetime of the CsPbBr3–CdS QDs at different EV2+ concentrations. Time-resolved luminescence decay traces were recorded in toluene:ethanol (85:15% v/v) using an excitation source at 370 nm. The lifetimes were fitted to a biexponential kinetic expression (expression 3), and the fitting parameters are given in Table S2.†
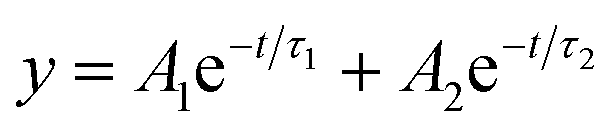 | (3) |
The average lifetime (τave) decreased with increasing concentration of EV2+ in accordance with the photoluminescence quenching seen in Fig. 4A. The decrease in average lifetime from 42.4 ns to 18.1 ns upon addition of 12 μM EV2+ is indicative of a competing excited state deactivation pathway involving electron transfer from the CsPbBr3–CdS QDs to EV2+. Since this electron transfer is likely to occur within the time resolution (∼1 ns) of our photoluminescence lifetime set up, we employed femtosecond transient absorption spectroscopy to resolve the electron transfer process.
Transient absorption spectra were recorded following laser pulse excitation at 400 nm (16 μJ cm−2). The transient absorption spectra of a representative CsPbBr3–CdS QD sample containing 0 and 4 μM EV2+ are shown in Fig. 5A and B respectively. The negative absorption (exciton bleach) feature centered at ∼527 nm corresponds to the charge separated state within the NCs.58–60 The charge separation which occurs within the laser pulse is seen in spectrum ‘a’ recorded with a probe delay of 1 ps. As electrons and holes recombine, a recovery in the bleached absorption is seen. The bleach recovery at 527 nm for the samples containing different amounts of EV2+ are presented in Fig. 5C (see Fig. S8† for longer time scale kinetics). With increasing EV2+ concentration we observe a quick recovery of the bleach feature, thus confirming the presence of an additional deactivation pathway for the photogenerated electrons, viz., electron transfer to EV2+ (reaction (4)).
| [CsPbBr3–CdS⋯EV2+] + hν → [(CsPbBr3–CdS)*⋯EV2+] − ket → [CsPbBr3–CdS (h)⋯EV+˙] | (4) |
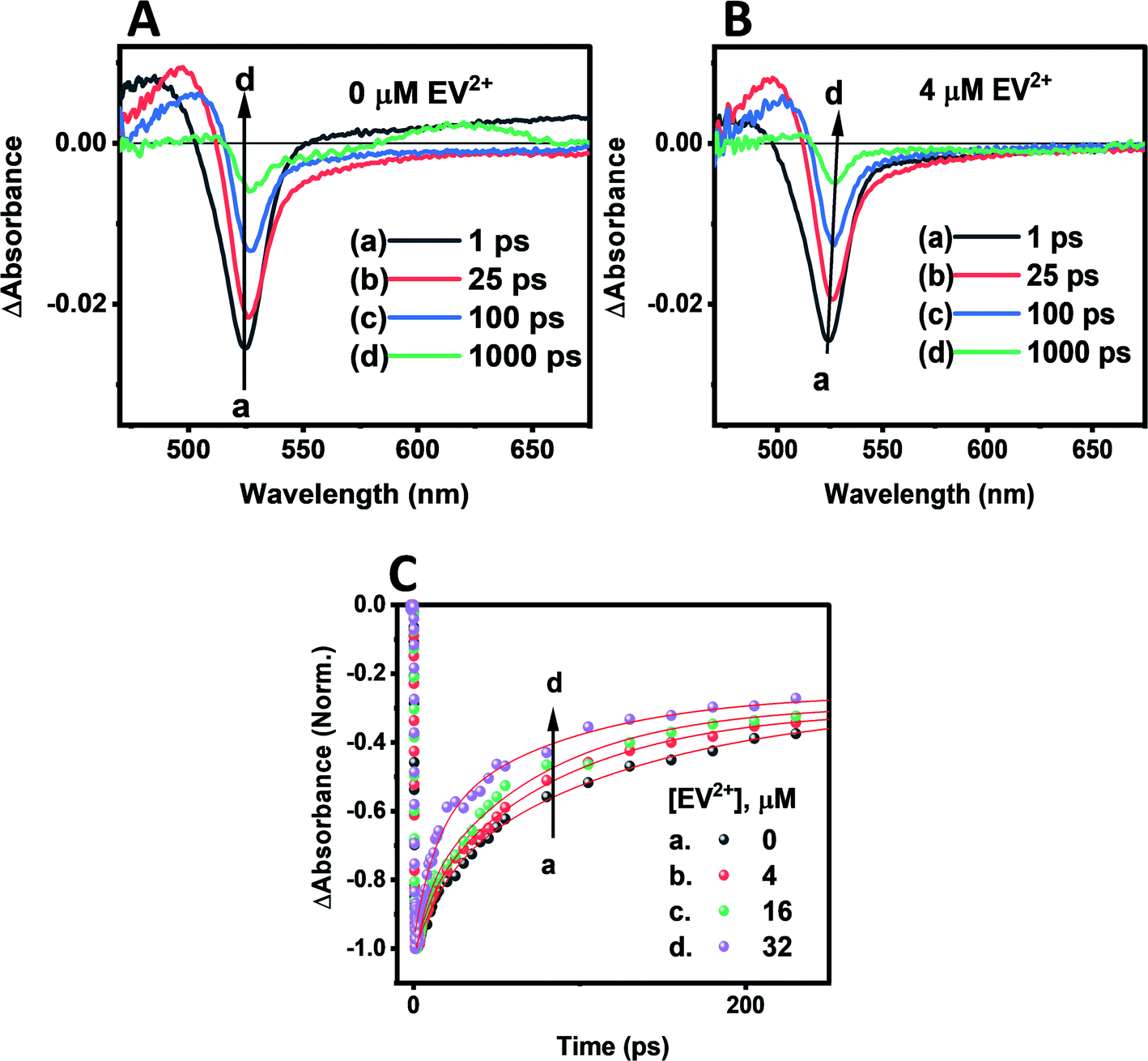 | ||
| Fig. 5 Transient absorption spectra of CsPbBr3–CdS suspension in toluene (≈16 nM): (A) without EV2+ and (B) with 4 μM EV2+. The difference absorption spectra were recorded following a 400 nm laser pulse (16 μJ cm−2) excitation. (C) Kinetic traces of the CsPbBr3–CdS bleach recovery at 527 nm in the absence and increasing concentrations of EV2+. | ||
The bleach recovery was analyzed using a biexponential kinetic fit35 and the fitting parameters (a1, τ1) and (a2, τ2) corresponding to fast and slow components are presented in Table S3.† While the fast component varied in the range 18.6–6.6 ps the long component varied in the range of 129.6–71.7 ps.
If we assign the decrease in the fast component to the electron transfer pathway from CsPbBr3–CdS to EV2+, we can obtain the rate constant for electron transfer through the expression (5).
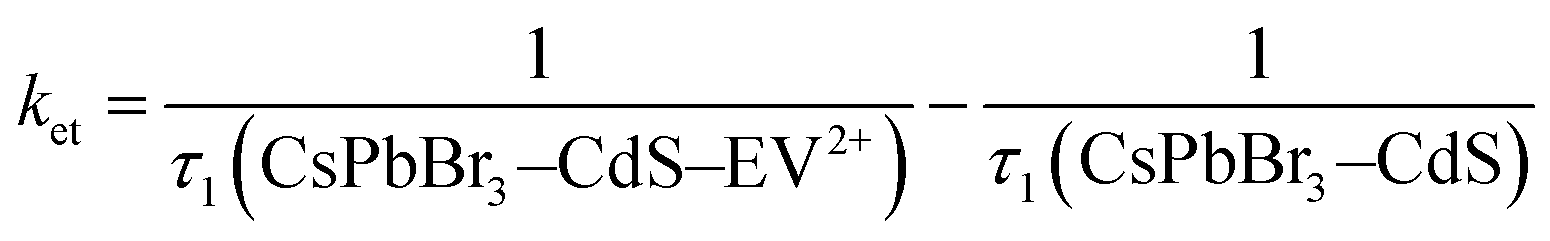 | (5) |
If we substitute the fast time components (τ1) of CsPbBr3–CdS and that of CsPbBr3–CdS with 32 μM EV2+ in expression 5 (Table S3†), we obtain rate constant (ket) of the electron transfer in the range of 3.8–9.8 × 1010 s−1 for three different EV2+ concentrations (4–32 μM) or an average rate constant of 6.5 × 1010 s−1. This rate constant for electron transfer is lower than the one obtained for electron transfer between oleic acid/oleylamine capped CsPbBr3 QDs and viologen (ket = 3.6 × 1011 s−1).15 As discussed in the emission quenching experiments, the surface interactions play a role in dictating the kinetics of interfacial electron transfer.
It is evident that modification of CsPbBr3 with CdS slows down the electron transfer rate. Schemes 1A and B illustrate the two scenarios for achieving electron transfer, viz., without and with CdS modification. As shown earlier, when methyl viologen is directly bound to CsPbBr3, the charge separation is extended, with electrons residing in the viologen moiety and holes residing within the CsPbBr3 QDs.14 This extended charge separation as a bound pair was manifested as a long-lived transient bleach component. In contrast, the electron transfer with CsPbBr3–CdS is mediated through the CdS layer as expected by the quasi-type II band alignment (Scheme 1C). The reduced ethyl viologen (EV+˙) is no longer directly bound to the CsPbBr3, but instead is now linked to the CdS layer. Similar CdS-mediated electron transfer has been observed in CdSe–CdS heterostructures.51 The bleaching recovery accelerates with increasing viologen concentrations as the electrons are depleted from the CsPbBr3 core, mediated through CdS. The distinct difference between the two bleach recovery kinetics observed with pristine CsPbBr3 and CsPbBr3–CdS QDs further highlights the electron mediation of CdS in promoting interfacial electron transfer in the heterostructure (Scheme 1C).
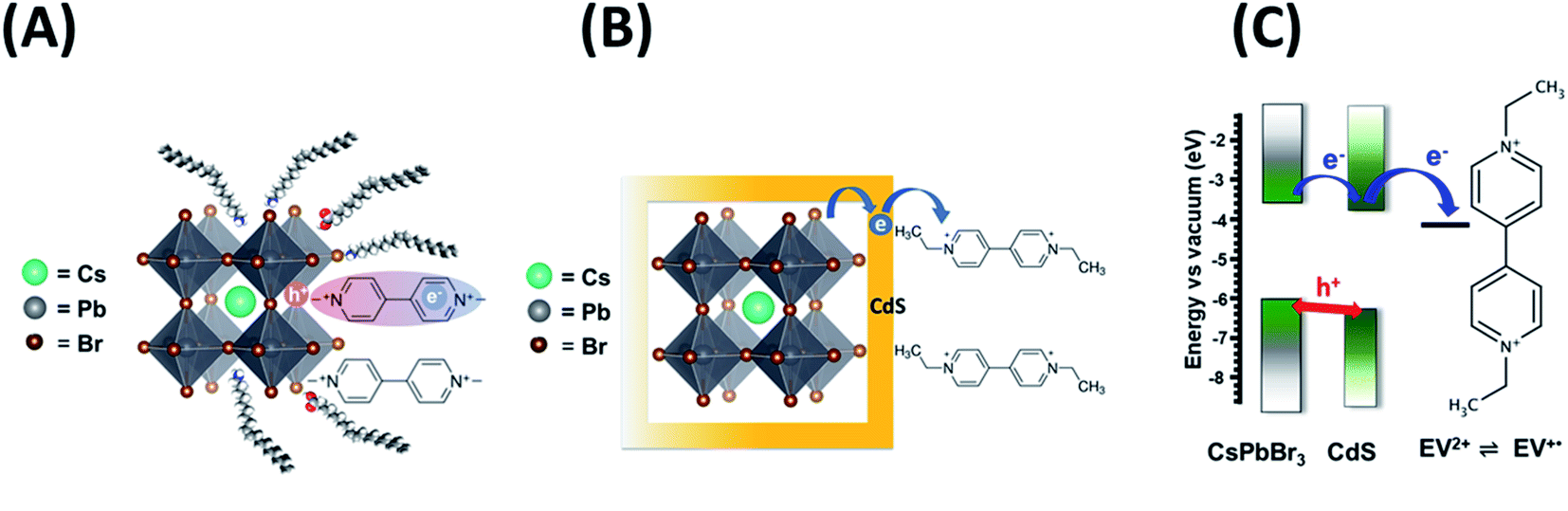 | ||
| Scheme 1 Photoinduced electron transfer between CsPbBr3 and viologen (A) without and (B) with mediation through a CdS layer. (C) Energetic diagram showing the flow of charge carriers and CdS mediated reduction of viologen. | ||
Steady state photolysis and Fermi level equilibration
Although there have been several studies that demonstrate the photocatalytic properties through product identification,17,18,36,61 direct spectroscopic identification of intermediates or electron transfer products is rather limited. If indeed the CsPbBr3–CdS heterostructure is responsible for photocatalytic reduction, we should be able to observe the buildup of stable viologen radical under continuous photoirradiation.62,63Deaerated CsPbBr3–CdS nanocrystal suspensions in toluene:ethanol (85:15 v/v%) mixed solvent containing 100 μM of EV2+ were subjected to steady-state visible light illumination (>400 nm; 200 mW cm−2). Absorbance spectra were recorded periodically during the steady state photolysis experiment. Fig. 6A shows a representative difference absorbance spectrum, which shows distinct peaks at 405 nm and 608 nm, corresponding to the absorbance of EV+˙.64Fig. 6B shows the growth of the 608 nm absorption over time, which attains a plateau after about 45 minutes. The steady state concentration of EV+˙ increased with increasing concentration of EV2+. From the extinction coefficient of EV+˙ at 608 nm (ε = 1.4 × 104 M−1 cm−1)63,64 we can determine the steady state concentration of the electron transfer product. The quantum yield (QY) of EV+˙ formation (ΦEV+˙) was determined using potassium ferrioxalate actinometry.51,65 Details of the actinometry experiments are presented in the ESI.† In the present case we obtain a maximum quantum efficiency (ΦEV+˙) of 8.4% for the electron transfer. Although the initial electron transfer yield as monitored from the transient absorption could be as high as 73%,15 the back-electron transfer during the steady state irradiation makes the net electron transfer yield lower than the value obtained immediately after laser pulse excitation.
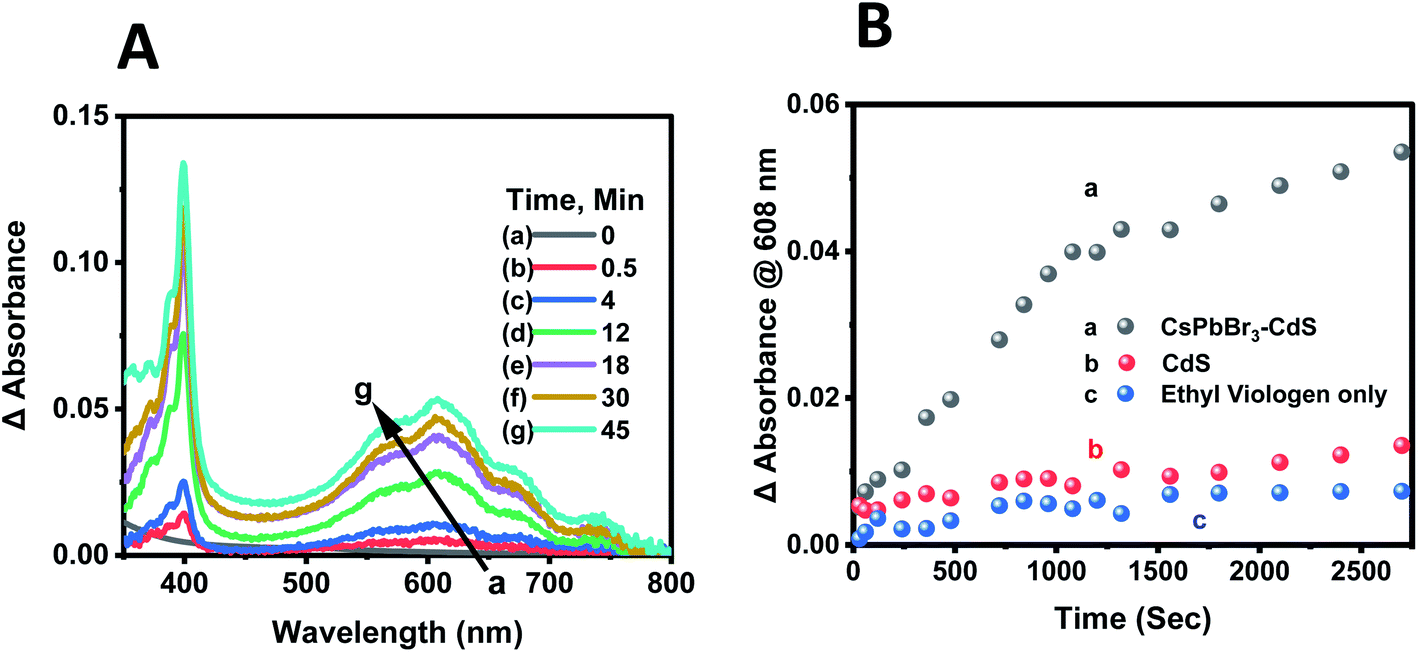 | ||
| Fig. 6 (A) Difference absorbance spectra recorded during steady state photolysis corresponding to EV+˙ formation. The [CsPbBr3–CdS + EV2+] sample before excitation was used as a reference. Absorption spectra were recorded at different light exposure times. (B) Evolution of EV+˙ formation monitored via the growth of absorbance at 608 nm with CsPbBr3–CdS. Control experiments with CdS photocatalyst and in the absence of any photocatalyst are also shown. The initial concentration of EV2+ was 100 μM. Experiments were carried out in deaerated toluene/ethanol 85:15 v/v% with visible light excitation (Xe lamp, 400 nm long pass filter, 200 mW cm−2). | ||
The steady state concentration of the reduction product EV+˙ is dictated by the forward and back electron transfer processes. Because of the use of ethanol as a hole scavenger, the back electron transfer rate constant (kbet) is significantly lower in the present experiments. The redox couple in contact with a semiconductor surface undergoes Fermi level equilibration that is dependent on the position of the conduction band and the potential of the redox couple. The equilibrium concentration of the reduced and oxidized species can be used to determine the flat band potential of the semiconductor. The steady state concentration of [EV+˙]ss is indicative of charge equilibration between the CsPbBr3–CdS nanocrystals and EV2+/EV+˙ couple. We employed the Nernst equation (expression (6)) to obtain the flat band potential of the CsPbBr3–CdS heterostructure.
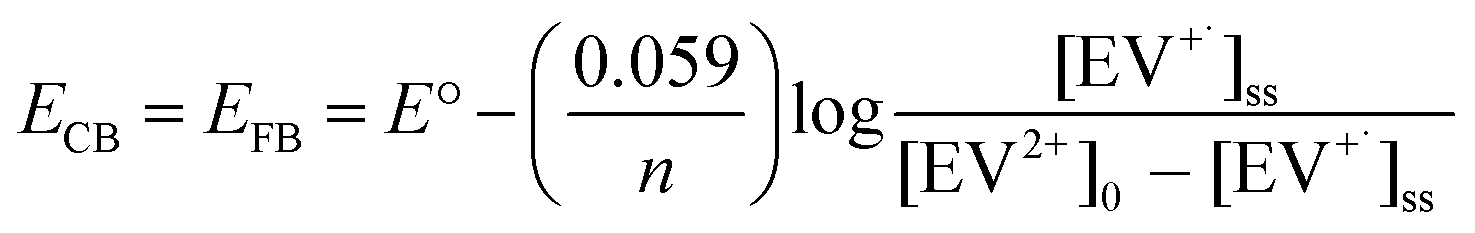 | (6) |
By substituting the redox potential of the EV2+/EV+˙ couple, E° = −0.449 V vs. NHE,64 and the steady state concentration values of the EV+˙ (3.65 μM) and EV2+ ([EV2+] = [EV2+]0 − [EV+˙]ss = 96.35 μM), we obtain EFB = −0.365 V vs. NHE. It is evident that the close lying conduction band potential of the CsPbBr3–CdS heterostructure limits the one electron transfer to EV2+ (Scheme 1C). It should be noted that the flat band potential of the CsPbBr3–CdS heterostructure obtained in this study is based on the charge equilibration between the semiconductor QD and redox couple and may differ from the values obtained from theoretical estimates or bulk material. The estimate of flat band potential provides an estimate of the energetics of the CsPbBr3–CdS heterostructure suspended in the solvent medium to execute photocatalytic processes.
Conclusions
The design of CsPbBr3–CdS heterostructure offers stabilization of perovskite nanocrystals for photocatalytic applications in polar medium. The salient feature of the CsPbBr3–CdS heterostructure is realized through its stability in mixed solvents, remediation of surface defects, and increased emission yield. The stability of the CsPbBr3 structure in mixed solvents with increased polarity has allowed us to accumulate electron transfer product (reduced viologen) under steady state irradiation conditions with a quantum efficiency of 8.4%. The relatively high electron transfer efficiency observed in the present study shows how a heterostructure design of perovskite nanocrystals plays a crucial role in dictating the photocatalytic properties of stable perovskite nanocrystals.Data availability
The data is available within the main text and ESI.†Author contributions
AK: conceptualization, synthesis, measurements, data analysis, validation, visualization, writing first draft and revision. JTD: data analysis, discussions, revision. JC: conceptualization of material synthesis, preliminary investigation; revision. PVK: conceptualization, method development, funding acquisition, resources, supervision, discussions, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research described herein is supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy, through award DE-FC02-04ER15533. This is contribution number NDRL No. 5331 from the Notre Dame Radiation Laboratory. J. T. D. acknowledges the support of the Forgash Fellowship for Solar Energy Research. We also acknowledge the University of Notre Dame Equipment Restoration and Renewal (ERR) program for the purchase of the Spectra Physics laser used for the transient absorption measurements.References
- P. V. Kamat, Semiconductor Surface Chemistry as Holy Grail in Photocatalysis and Photovoltaics, Acc. Chem. Res., 2017, 50, 527–531 CrossRef CAS PubMed
.
- Z. Xu, Z. Huang, T. Jin, T. Lian and M. L. Tang, Mechanistic Understanding and Rational Design of Quantum Dot/Mediator Interfaces for Efficient Photon Upconversion, Acc. Chem. Res., 2021, 54, 70–80 CrossRef CAS PubMed
- D. A. Hines and P. V. Kamat, Recent Advances in Quantum Dot Surface Chemistry, ACS Appl. Mater. Interfaces, 2014, 6, 3041–3057 CrossRef CAS PubMed
- Q. Shang, B. D. Piercy, M. D. Losego and T. Lian, Effect of Surface Ligand on Charge Separation and Recombination at CsPbI3 Perovskite Quantum Dot/TiO2 Interfaces, J. Phys. Chem. C, 2019, 123, 21415–21421 CrossRef CAS
- S. R. Smock, Y. Chen, A. J. Rossini and R. L. Brutchey, The Surface Chemistry and Structure of Colloidal Lead Halide Perovskite Nanocrystals, Acc. Chem. Res., 2021, 54, 707–718 CrossRef CAS PubMed
- C. Murray, D. Norris and M. Bawendi, Synthesis and Characterization of Nearly Monodisperse CdE (E = S, Se, Te) Semiconductor, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS
- P. Alivisatos, Perspectives on the Physical Chemistry of Semiconductor Nanocrystals, J. Phys. Chem., 1996, 100, 13226–13239 CrossRef
- M. Nirmal and L. Brus, Luminescence Photophysics in Semiconductor Nanocrystals, Acc. Chem. Res., 1999, 32, 407–414 CrossRef CAS
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Metal Halide Perovskite Nanocrystals: Synthesis, Post-Synthesis Modifications, and Their Optical Properties, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed
- Y. Wu, X. Li and H. Zeng, Highly Luminescent and Stable Halide Perovskite Nanocrystals, ACS Energy Lett., 2019, 4, 673–681 CrossRef CAS
- A. Dey,
et al. State of the Art and Prospects for Halide Perovskite Nanocrystals, ACS Nano, 2021, 15(7), 10775–10981 CrossRef CAS PubMed
- S. T. Ha, R. Su, J. Xing, Q. Zhang and Q. H. Xiong, Metal Halide Perovskite Nanomaterials: Synthesis and Applications, Chem. Sci., 2017, 8, 2522–2536 RSC
- Y. L. Li, R. C. Lai, X. Luo, X. Liu, T. Ding, X. Lu and K. F. Wu, On the Absenceof a Phonon Bottleneck in Strongly Confined CsPbBr3 Perovskite Nanocrystals, Chem. Sci., 2019, 10, 5983–5989 RSC
- S. M. Kobosko, J. T. DuBose and P. V. Kamat, Perovskite Photocatalysis. Methyl Viologen Induces Unusually Long-Lived Charge Carrier Separation in CsPbBr3 Nanocrystals, ACS Energy Lett., 2020, 5, 221–223 CrossRef CAS
- J. T. DuBose and P. V. Kamat, Surface Chemistry Matters. How Ligands Influence Excited State Interactions between CsPbBr3 and Methyl Viologen, J. Phys. Chem. C, 2020, 124, 12990–12998 CrossRef CAS
- J. T. DuBose and P. V. Kamat, Probing Perovskite Photocatalysis. Interfacial Electron Transfer between CsPbBr3 and Ferrocene Redox Couple, J. Phys. Chem. Lett., 2019, 10, 6074–6080 CrossRef CAS PubMed
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, Solar-Driven Metal Halide Perovskite Photocatalysis: Design, Stability, and Performance, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS
- J. S. Martin,
et al. A Nanocrystal Catalyst Incorporating a Surface Bound Transition Metal to Induce Photocatalytic Sequential Electron Transfer Events, J. Am. Chem. Soc., 2021, 143, 11361–11369 CrossRef CAS PubMed
- H. Huang,
et al. Efficient and Selective Photocatalytic Oxidation of Benzylic Alcohols with Hybrid Organic–Inorganic Perovskite Materials, ACS Energy Lett., 2018, 3, 755–759 CrossRef CAS
- Q. Li and T. Lian, Ultrafast Charge Separation in Two-Dimensional CsPbBr3 Perovskite Nanoplatelets, J. Phys. Chem. Lett., 2019, 10, 566–573 CrossRef PubMed
- S. Shyamal and N. Pradhan, Halide Perovskite Nanocrystal Photocatalysts for CO2 Reduction: Successes and Challenges, J. Phys. Chem. Lett., 2020, 11, 6921–6934 CrossRef CAS PubMed
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, Synthesis and Properties of Biocompatible Water-Soluble Silica-Coated CdSe/ZnS Semiconductor Quantum Dots, J. Phys. Chem. B, 2001, 105, 8861–8871 CrossRef CAS
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Highly Luminescent Monodisperse CdSe and CdSe/ZnS Nanocrystals Synthesized in a Hexadecylamine-Trioctylphosphine Oxide–Trioctylphospine Mixture, Nano Lett., 2001, 1, 207–211 CrossRef CAS
- E. Ryu, S. Kim, E. Jang, S. Jun, H. Jang, B. Kim and S.-W. Kim, Step-Wise Synthesis of InP/ZnS Core–Shell Quantum Dots and the Role of Zinc Acetate, Chem. Mater., 2009, 21, 573–575 CrossRef CAS
- T. Torimoto, T. Adachi, K.-i. Okazaki, M. Sakuraoka, T. Shibayama, B. Ohtani, A. Kudo and S. Kuwabata, Facile Synthesis of ZnS–AgInS2 Solid Solution Nanoparticles for a Color-Adjustable Luminophore, J. Am. Chem. Soc., 2007, 129, 12388–12389 CrossRef CAS PubMed
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, One-Pot Synthesis of Highly Stable CsPbBr3@SiO2 Core–Shell Nanoparticles, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed
- J. Shi, W. Ge, J. Zhu, M. Saruyama and T. Teranishi, Core–Shell CsPbBr3@CdS Quantum Dots with Enhanced Stability and Photoluminescence Quantum Yields for Optoelectronic Devices, ACS Appl. Nano Mater., 2020, 3, 7563–7571 CrossRef CAS
- V. K. Ravi, S. Saikia, S. Yadav, V. V. Nawale and A. Nag, CsPbBr3/ZnS Core/Shell Type Nanocrystals for Enhancing Luminescence Lifetime and Water Stability, ACS Energy Lett., 2020, 5, 1794–1796 CrossRef CAS
- W. Chen, J. Hao, W. Hu, Z. Zang, X. Tang, L. Fang, T. Niu and M. Zhou, Enhanced Stability and Tunable Photoluminescence in Perovskite CsPbX3/ZnS Quantum Dot Heterostructure, Small, 2017, 13, 1604085 CrossRef PubMed
- H. Huang, B. Chen, Z. Wang, T. F. Hung, A. S. Susha, H. Zhong and A. L. Rogach, Water Resistant CsPbX3 Nanocrystals Coated with Polyhedral Oligomeric Silsesquioxane and Their Use as Solid State Luminophores in All-Perovskite White Light-Emitting Devices, Chem. Sci., 2016, 7, 5699–5703 RSC
- J. Pan,
et al. Bidentate Ligand-Passivated CsPbI3 Perovskite Nanocrystals for Stable Near-Unity Photoluminescence Quantum Yield and Efficient Red Light-Emitting Diodes, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed
- H. Huang, B. K. Chen, Z. G. Wang, T. F. Hung, A. S. Susha, H. Z. Zhong and A. L. Rogach, Water Resistant CsPbX3 Nanocrystals Coated with Polyhedral Oligomeric Silsesquioxane and Their Use as Solid State Luminophores in All-Perovskite White Light-Emitting Devices, Chem. Sci., 2016, 7, 5699–5703 RSC
- M. Imran, B. T. Mai, L. Goldoni, M. Cirignano, H. B. Jalali, F. Di Stasio, T. Pellegrino and L. Manna, Switchable Anion Exchange in Polymer-Encapsulated APbX3 Nanocrystals Delivers Stable All-Perovskite White Emitters, ACS Energy Lett., 2021, 6, 2844–2853 CrossRef CAS PubMed
- P. V. Kamat, N. Pradhan, K. Schanze, P. S. Weiss, J. Buriak, P. Stang, T. W. Odom and G. Hartland, Challenges and Opportunities in Designing Perovskite Nanocrystal Heterostructures, ACS Energy Lett., 2020, 2253–2255 CrossRef CAS
- S. Bera and N. Pradhan, Perovskite Nanocrystal Heterostructures: Synthesis, Optical Properties, and Applications, ACS Energy Lett., 2020, 5, 2858–2872 CrossRef CAS
- S. K. Dutta, S. K. Mehetor and N. Pradhan, Metal Semiconductor
Heterostructures for Photocatalytic Conversion of Light Energy, J. Phys. Chem. Lett., 2015, 6, 936–944 CrossRef CAS PubMed
- J. H. Bang and P. V. Kamat, Quantum Dot Sensitized Solar Cells. A Tale of Two Semiconductor Nanocrystals: CdSe and CdTe, ACS Nano, 2009, 3, 1467–1476 CrossRef CAS PubMed
- V. K. Ravi, G. B. Markad and A. Nag, Band Edge Energies and Excitonic Transition Probabilities of Colloidal CsPbX3 (X = Cl, Br, I) Perovskite Nanocrystals, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS
- Q. A. Akkerman,
et al. Strongly Emissive Perovskite Nanocrystal Inks for High-Voltage Solar Cells, Nat. Energy, 2016, 2, 16194 CrossRef
- P. Cottingham and R. L. Brutchey, On the Crystal Structure of Colloidally Prepared CsPbBr3 Quantum Dots, Chem. Commun., 2016, 52, 5246–5249 RSC
- J. Zhang, S. Wageh, A. Al-Ghamdi and J. Yu, New Understanding on the Different Photocatalytic Activity of Wurtzite and Zinc-Blende CdS, Appl. Catal., B, 2016, 192, 101–107 CrossRef CAS
- A. Demortière,
et al. Strain-Driven Stacking Faults in CdSe/CdS Core/Shell Nanorods, J. Phys. Chem. Lett., 2018, 9, 1900–1906 CrossRef PubMed
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
- S. Bhaumik, S. A. Veldhuis, Y. F. Ng, M. Li, S. K. Muduli, T. C. Sum, B. Damodaran, S. Mhaisalkar and N. Mathews, Highly Stable, Luminescent Core–Shell Type Methylammonium–Octylammonium Lead Bromide Layered Perovskite Nanoparticles, Chem. Commun., 2016, 52, 7118–7121 RSC
- K. Xu, C. C. Lin, X. Xie and A. Meijerink, Efficient and Stable Luminescence from Mn2+ in Core and Core–Isocrystalline Shell CsPbCl3 Perovskite Nanocrystals, Chem. Mater., 2017, 29, 4265–4272 CrossRef CAS PubMed
- V. K. Ravi, R. A. Scheidt, J. DuBose and P. V. Kamat, Hierarchical Arrays of Cesium Lead Halide Perovskite Nanocrystals through Electrophoretic Deposition, J. Am. Chem. Soc., 2018, 140, 8887–8894 CrossRef CAS PubMed
- Y. Zhao, C. Shen, L. Ding, J. Liu, W. Xiang and X. Liang, Novel B-site Cd2+ doped CsPbBr3 Quantum Dot Glass Toward Strong Fluorescence and High Stability For wLED, Opt. Mater., 2020, 107, 110046 CrossRef CAS
- S. Christodoulou,
et al. Synthesis of Highly Luminescent Wurtzite CdSe/CdS Giant-Shell Nanocrystals Using a Fast Continuous Injection Route, J. Mater. Chem. C, 2014, 2, 3439–3447 RSC
- L. Carbone,
et al. Synthesis and Micrometer-Scale Assembly of Colloidal CdSe/CdS Nanorods Prepared by a Seeded Growth Approach, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Seeded Growth of Highly Luminescent CdSe/CdS Nanoheterostructures with Rod and Tetrapod Morphologies, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS PubMed
- V. L. Bridewell, R. Alam, C. J. Karwacki and P. V. Kamat, CdSe/CdS Nanorod Photocatalysts: Tuning the Interfacial Charge Transfer Process through Shell Length, Chem. Mater., 2015, 27, 5064–5071 CrossRef CAS
- Z. Zhu, Q. Sun, Z. Zhang, J. Dai, G. Xing, S. Li, X. Huang and W. Huang, Metal Halide Perovskites: Stability and Sensing-Ability, J. Mater. Chem. C, 2018, 6, 10121–10137 RSC
- X. Li,
et al. All Inorganic Halide Perovskites Nanosystem: Synthesis, Structural Features, Optical Properties and Optoelectronic Applications, Small, 2017, 13, 1603996 CrossRef PubMed
- L. J. Ruan, B. Tang and Y. Ma, Improving the Stability of CsPbBr3 Nanocrystals in Ethanol by Capping with PbBr2-Adlayers, J. Phys. Chem. C, 2019, 123, 11959–11967 CrossRef CAS
- P. V. Kamat, J. P. Chauvet and R. W. Fessenden, Photoelectrochemistry in Particulate Systems. 4. Photosensitization of a TiO2 Semiconductor with a Chlorophyll Analogue, J. Phys. Chem., 1986, 90, 1389–1394 CrossRef CAS
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy. Springer, New York, 2006 Search PubMed
- M. D. Peterson, S. C. Jensen, D. J. Weinberg and E. A. Weiss, Mechanisms for Adsorption of Methyl Viologen on CdS Quantum Dots, ACS Nano, 2014, 8, 2826–2837 CrossRef CAS PubMed
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, Ultrafast Interfacial Electron and Hole Transfer from CsPbBr3 Perovskite Quantum Dots, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed
- N. Mondal and A. Samanta, Complete Ultrafast Charge Carrier Dynamics in Photo-Excited All-Inorganic Perovskite Nanocrystals (CsPbX3), Nanoscale, 2017, 9, 1878–1885 RSC
- J. Aneesh, A. Swarnkar, V. Kumar Ravi, R. Sharma, A. Nag and K. V. Adarsh, Ultrafast Exciton Dynamics in Colloidal CsPbBr3 Perovskite Nanocrystals: Biexciton Effect and Auger Recombination, J. Phys. Chem. C, 2017, 121, 4734–4739 CrossRef CAS
- D. Cardenas-Morcoso, A. F. Gualdrón-Reyes, A. B. Ferreira Vitoreti, M. García-Tecedor, S. J. Yoon, M. Solis de la Fuente, I. Mora-Seró and S. Gimenez, Photocatalytic and Photoelectrochemical Degradation of Organic Compounds with All-Inorganic Metal Halide Perovskite Quantum Dots, J. Phys. Chem. Lett., 2019, 10, 630–636 CrossRef CAS PubMed
- F. Zhao, Q. Li, K. Han and T. Lian, Mechanism of Efficient Viologen Radical Generation by Ultrafast Electron Transfer from CdS Quantum Dots, J. Phys. Chem. C, 2018, 122, 17136–17142 CrossRef CAS
- C. T. Harris and P. V. Kamat, Photocatalysis with CdSe Nanoparticles in Confined Media: Mapping Charge Transfer Events in the Subpicosecond to Second Timescales, ACS Nano, 2009, 3, 682–690 CrossRef CAS PubMed
- C. L. Bird and A. T. Kuhn, Electrochemistry of the Viologens, Chem. Soc. Rev., 1981, 10, 49–82 RSC
- C. G. Hatchard and C. A. Parker, A New Sensitive Chemical Actinometer – II. Potassium Ferrioxalate as a Standard Chemical Actinometer, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CAS
Footnote |
| † Electronic supplementary information (ESI) available: The ESI includes experimental methods and procedures, actinometry, data of control experiments and kinetic analysis. See DOI: 10.1039/d1sc04305f |
| This journal is © The Royal Society of Chemistry 2021 |